

Abstract
In dividing cells, DNA replication occurs in a precise order, but many questions remain regarding the mechanisms of replication timing establishment and regulation. We now have generated genome-wide, high-resolution replication timing maps throughout zebrafish development. Unexpectedly, in the rapid cell cycles preceding the midblastula transition, a defined timing program was present that predicted the initial wave of zygotic transcription. Replication timing was thereafter progressively and continuously remodeled across the majority of the genome, and epigenetic changes involved in enhancer activation frequently paralleled developmental changes in replication timing. The long arm of Chromosome 4 underwent a dramatic developmentally regulated switch to late replication during gastrulation, reminiscent of mammalian X Chromosome inactivation. This study reveals that replication timing is dynamic and tightly linked to epigenetic and transcriptional changes throughout early zebrafish development. These data provide insight into the regulation and functions of replication timing and will enable further mechanistic studies.
In dividing cells, each region of the genome replicates at a particular time during S-phase, resulting in a defined DNA replication timing program (Rhind and Gilbert 2013). Early replication is associated with active gene expression, yet how DNA replication is coordinated with the dynamic changes in transcription and epigenetic marks during early development is not fully understood (Schübeler et al. 2002; MacAlpine et al. 2004; Hiratani et al. 2009). Furthermore, the replication timing program correlates with chromatin conformation, as early and late replicating regions correspond to open and closed chromatin, respectively (Ryba et al. 2010; Pope et al. 2014). Therefore, replication timing can also be used to assess changes in chromatin organization during development. The timing program is modulated when embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are differentiated in culture, but it is unknown what replication timing changes occur in vivo during vertebrate development (Hiratani et al. 2010; Rivera-Mulia et al. 2015).
The zebrafish is an ideal model of vertebrate development, because its embryos are transparent, develop externally, can be produced in large numbers, and proceed rapidly through well-defined stages with precise kinetics (Kimmel et al. 1995; Lessman 2011). The zebrafish is also a powerful model to study how the embryonic cell cycle is remodeled, when cells undergo numerous profound transcriptional, epigenetic, and structural changes necessary for normal development (Newport and Kirschner 1982; Farrell and O'Farrell 2014; Siefert et al. 2015). Prior to the midblastula transition (MBT), the embryonic cells undergo 10 rapid cell cycles consisting only of S/M phases, and the entire genome (1.4 Gb) is replicated in <15 min (Kane and Kimmel 1993; Vastenhouw et al. 2010; Siefert et al. 2015). Pre-MBT embryos lack a G1-phase, which is thought to be necessary for the establishment of the replication timing program (Dimitrova and Gilbert 1999; Lu et al. 2010). Prior to the MBT, zebrafish embryos rely on maternally deposited mRNA, and there is little or no transcription throughout most of the genome (Lee et al. 2014). After the MBT, widespread transcription begins, and certain histone modifications associated with transcriptional activation and repression are added (Vastenhouw et al. 2010). Cell cycle remodeling also commences at the MBT, with initial S-phase lengthening and the appearance of a G2-phase (Siefert et al. 2015). Furthermore, zebrafish display a conserved fate map after the blastula stage, and like other vertebrates, undergo cell fate restrictions to form germ layers during gastrulation (Kimmel et al. 1990). By 28 h post fertilization (hpf), zebrafish embryos have formed a basic vertebrate body plan, and their proliferating cells have a prototypical cell cycle profile (Kimmel et al. 1995). Therefore, zebrafish provide an excellent in vivo model to study how replication timing is altered coincident with cell fate specification and with widespread transcriptional, epigenetic, and structural changes transpiring during normal vertebrate development.
Here, we report the first genome-wide, high-resolution replication timing profiles generated in vivo across multiple stages of zebrafish development.
Results
High-resolution replication timing analysis in zebrafish embryos and adults
Because DNA replication timing profiles had not been generated in zebrafish embryos, we first sought to establish the presence of a replication timing program in zebrafish. To determine replication timing, we used an established method that is based on the premise that early replicating DNA will be present at a higher copy number than late replicating DNA in proliferating cells (Koren et al. 2012, 2014). For this approach, cells in G1-phase are used for a 2N DNA copy number reference, and replication timing is inferred from copy number differences between G1 and S-phase cells (Fig. 1A; Supplemental Fig. S1A). We initially sorted cells from 28 hpf zebrafish embryos, as they display a typical cell cycle profile (Fig. 1A). We then performed whole-genome next-generation sequencing on genomic DNA purified from G1 and S-phase cells, which enabled the generation of high-resolution replication timing profiles for the entire zebrafish genome. Normalized timing profiles were generated by transforming S/G1 copy number ratios across the genome into Z-scores (Fig. 1A,B; Supplemental Fig. S2). The long arm of Chromosome 4 was late replicating in 28 hpf embryos, which is consistent with previously published cytological data (Daga et al. 1996; Sola and Gornung 2001; Anderson et al. 2012). To further validate our results, an independent experimental replicate of 28 hpf embryos was generated. The timing data were highly reproducible, as the replicate profiles were qualitatively indistinct and the timing values genome-wide were highly correlated (r = 0.95) (Fig. 1C; Supplemental Fig. S1B). We assessed the degree to which the timing program was structured using autocorrelation, a measure of pattern continuity. The autocorrelation of timing values along the lengths of the chromosomes (excluding Chromosome 4) in each of the 28 hpf samples indicated the timing program was highly structured (Fig. 1D). Collectively, these results demonstrate that a defined and structured replication timing program can be reproducibly detected in 28 hpf zebrafish.
Figure 1.

High-resolution replication timing analysis in zebrafish. (A) Experimental outline: cells were FACS sorted based on DNA content into G1 and S-phase fractions, DNA was purified and sequenced, and replication timing was calculated based on variations in DNA copy number between G1 and S-phase cells (S/G1 ratio). To normalize, the genome mean replication timing value was set to 0, and the standard deviation was set to 1 (for full details, see Supplemental Figure S1A; Methods and Supplemental Methods). (B) Whole-genome replication timing profile for 28 hpf zebrafish embryos: (blue/yellow) odd/even chromosomes. (C) Replication timing profiles for two experimental replicates of 28 hpf embryos. (D) Autocorrelation of replication timing values along the length of the chromosomes (except Chromosome 4) for two experimental replicates of 28 hpf embryos.
Characteristics of the zebrafish replication timing program
To define and validate the characteristics of the 28 hpf replication timing program, we compared the 28 hpf embryos to an isolated adult zebrafish cell type. We generated a primary adult zebrafish tailfin fibroblast cell line (ZTF cells) and performed replication timing analysis as described above. We first assessed the distribution of timing values throughout the genome, as replication timing in mammals is bimodal, with most DNA replication occurring either in early or late S-phase (Goldman et al. 1984; Chambers et al. 2013). Unexpectedly, the distribution of replication timing throughout the zebrafish genome in 28 hpf embryos and ZTF cells was unimodal, indicating DNA replication is not biased toward early or late S-phase (Fig. 2A,B). Like the 28 hpf embryos (Fig. 1C), replication timing profiles in ZTF cells also consisted primarily of sharp peaks and valleys, with continuous fluctuations between early and late replication (Supplemental Fig. S3A). Yeast and mammalian replication timing profiles are also similarly composed of a peak and valley structure. In yeast, where replication origin positions have been mapped, peaks in the replication timing profiles represent the precise locations of origins (Raghuraman et al. 2001). In mammals, peaks represent either isolated origins or clusters of coordinately regulated origins (Koren et al. 2014). Therefore, the sharp peaks of the zebrafish replication timing profiles likely represent the positions of highly efficient origins or origin clusters. Despite differences between the replication timing profiles of 28 hpf embryos and ZTF cells (r = 0.56) (Supplemental Fig. S3B), peak locations were significantly conserved, suggesting that efficient origins are consistently utilized in different cell types (Supplemental Fig. S3C,D).
Figure 2.
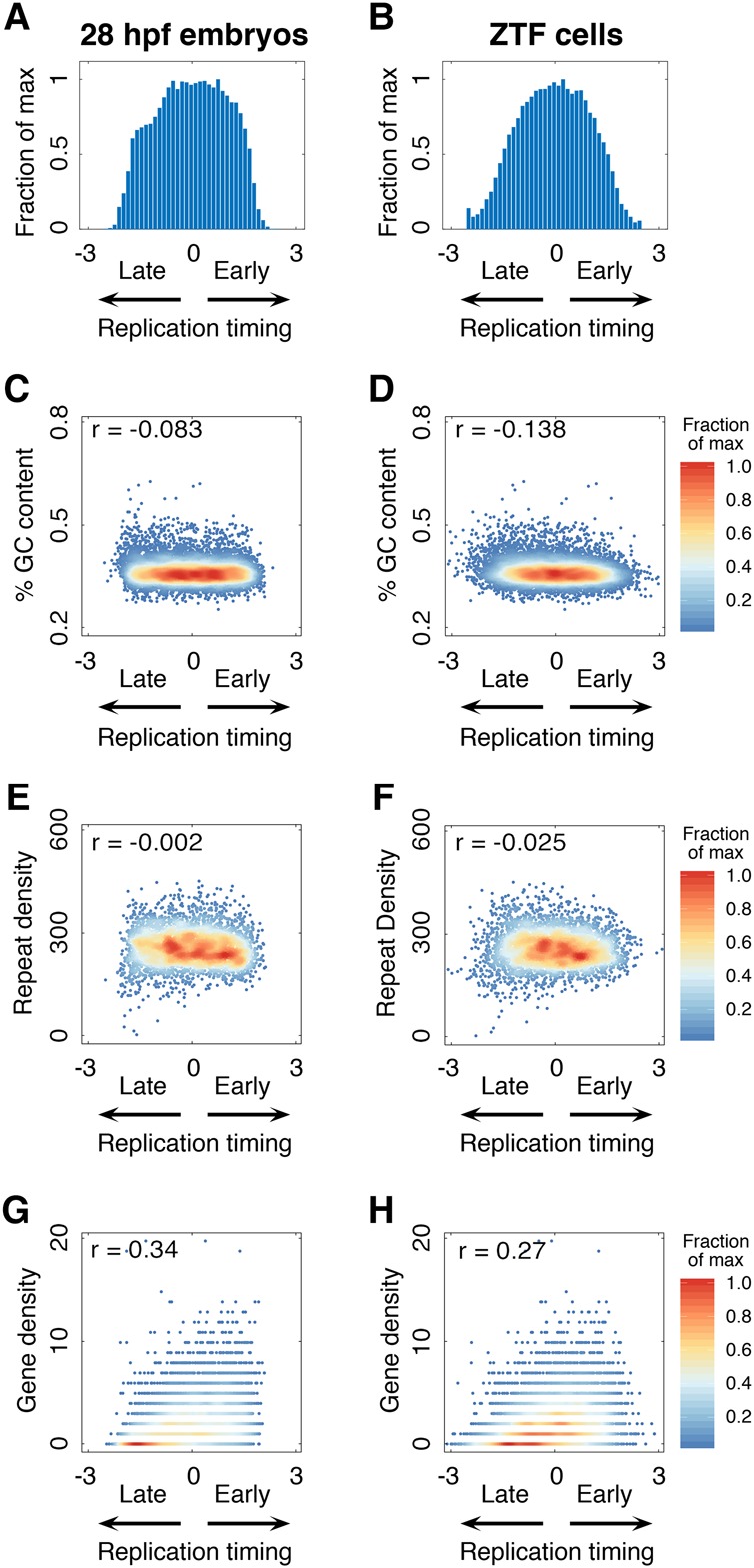
Characteristics of the zebrafish replication timing program. (A,B) Timing distribution histograms for 28 hpf zebrafish and ZTF cells. (C–H) Replication timing does not correlate with the percentage of GC content in 28 hpf embryos (C) or ZTF cells (D), or with repeat density in 28 hpf embryos (E) or ZTF cells (F), but it does correlate with gene density in 28 hpf embryos (G) and ZTF cells (H). Color bars represent the fraction (percentage) of maximum density plotted.
Replication timing in mammals correlates with DNA sequence features including GC content, repeat density, and gene density (Rhind and Gilbert 2013). Furthermore, replication timing was proposed to affect human genome sequence composition through GC-biased substitutions and gene conversions (Galtier et al. 2001; Stamatoyannopoulos et al. 2009; Chen et al. 2010; Kenigsberg et al. 2016). Therefore, we determined whether zebrafish replication timing was related to DNA sequence features. Despite the relationships between GC content and replication timing in mammals, there was no correlation observed in zebrafish (Fig. 2C,D), suggesting that GC content variation is neither a conserved cause nor a consequence of the replication timing program across vertebrate species. The density of repetitive elements in zebrafish also did not correlate with replication timing (Fig. 2E,F). In contrast, a positive correlation between replication timing and gene density was observed (Fig. 2G,H). The broad evolutionary conservation of the correlation between replication timing and gene density, but not GC or repeat content, supports a link between DNA replication and functional aspects of the genome rather than sequence composition.
Early replication is associated with actively transcribed genes and active enhancers
Replication timing positively correlates with transcriptional activity in cultured cells from multicellular organisms (Hiratani et al. 2009); thus, we examined whether a link between replication timing and transcription could be observed in vivo using 28 hpf embryos. Indeed, we found that transcription start sites (TSSs) were significantly early replicating (Fig. 3A). Next, we analyzed published RNA sequencing (RNA-seq) data of 28 hpf embryos and classified genes based on whether their expression level was not detectable, low, or moderate-to-high (Pauli et al. 2012). There was a link between expression level and replication timing, as nonexpressed genes were significantly late replicating, low-level expressed genes were significantly early replicating, and moderate-to-high-level expressed genes were even earlier replicating (Fig. 3B). Further separating the moderate-to-highly expressed genes into additional expression level categories did not reveal a stronger relationship between earlier replication timing and higher expression (Supplemental Fig. S4A).
Figure 3.

Early replication is associated with actively transcribed genes and active enhancers. (A) Replication timing of transcription start sites (TSSs) versus all genomic regions (Genome). (B) Replication timing of genes in three expression level bins: not detectable (0 FPKM), low (1–10 FPKM), and moderate-to-high (>10 FPKM): (FPKM) fragments per kilobase of transcript per million mapped reads (Pauli et al. 2012). (C) Replication timing of H3K4me3+ and H3K4me3− TSSs (Aday et al. 2011). (D) Actively transcribed genes (H3K36me3 marked) replicate significantly earlier with increasing H3K36me3 (Vastenhouw et al. 2010). (E) All genomic regions marked by H3K4me3, H3K4me1, and H3K27ac peaks replicate early, and enhancers defined by either PDREs or DMRs replicate even earlier (Bogdanović et al. 2012; Lee et al. 2015). t-test with Bonferroni corrected P-values: (*) P < 10−8; (#) P < 10−8 compared to H3K4me3, H3K4me1, and H3K27ac peaks. Box plots show the median (line), 95% confidence interval (notch), 25th–75th percentile (box), and 10th–90th percentile (whiskers).
Steady-state transcript abundance may not necessarily indicate active transcription in early embryos, which have a significant maternal mRNA contribution. Therefore, we used publicly available ChIP-seq data sets from 24 hpf embryos to identify genes exhibiting epigenetic marks associated with active transcription and assessed their replication timing (Ulitsky et al. 2011; Bogdanović et al. 2012). TSSs marked with H3K4me3, a modification associated with transcriptionally active promoters (Aday et al. 2011), were significantly earlier replicating than TSSs lacking H3K4me3 (Fig. 3C). In addition, genes with higher levels of H3K36me3, a mark for actively transcribed gene bodies (Hon et al. 2009; Vastenhouw et al. 2010), displayed increasingly earlier replication timing (Fig. 3D). These results further support that transcribed genes are significantly earlier replicating than nontranscribed genes.
To determine whether the correlation between early replication timing and gene expression extended beyond active genes and to genomic regulatory elements, we broadened our analysis to include H3K4me3 sites genome-wide, as well as marks of enhancers (H3K4me1 and H3K27ac). We found that sites marked by H3K4me3, H3K4me1, or H3K27ac were significantly early replicating (Fig. 3E). H3K4me1 and H3K27ac sites were especially notable, as they are distal from genes and replicated significantly earlier than H3K4me3 sites, which are predominantly located at TSSs (Fig. 3E). Regions marked by both H3K4me1 and H3K27ac, termed putative distal regulatory elements (PDREs) in zebrafish, reliably mark active transcriptional enhancers (Bogdanović et al. 2012; Shlyueva et al. 2014). These enhancers replicated significantly earlier than all regions marked with either H3K4me1 or H3K27ac alone, indicating that replication timing specifically correlates with active transcriptional enhancers rather than these histone modifications generally (Fig. 3E). It was shown that developmentally regulated enhancers undergo DNA demethylation during zebrafish development (Lee et al. 2015), so we investigated whether differentially methylated regions (DMRs) that were demethylated by 24 hpf were early replicating. Indeed, similar to PDREs, these DMRs were significantly early replicating (Fig. 3E). In fact, both DMRs and PDREs replicated significantly earlier than highly expressed genes (Supplemental Fig. S4B), suggesting the relationship between replication timing and transcription may be through enhancer regulation. Collectively, these results suggest that early replication may influence enhancer activity, or that active enhancers may promote early replication.
A pre-MBT replication timing program anticipates initial zygotic transcription
Although work with Drosophila melanogaster embryos and Xenopus laevis egg extracts has suggested that the replication timing program is not established until the MBT (Hyrien et al. 1995; Sasaki et al. 1999), a genome-wide quantitative analysis of replication timing in pre-MBT embryos has not been done. We collected cells from pre-MBT zebrafish embryos (2.75 hpf), which are almost entirely in S-phase (Siefert et al. 2015), and compared them to the 28 hpf G1 reference as described above to assess replication timing. Considering that the genome (1.4 Gb) is replicated in <15 min during the pre-MBT cell cycles and pre-MBT embryos lack a G1-phase, we anticipated that the timing program would be random and unstructured (Siefert et al. 2015). To demonstrate the difference in temporal resolution, we scaled the replication timing data to the length of S-phase and plotted the temporal order based on actual time in S-phase. When the pre-MBT embryos (15 min S-phase) were compared to ZTF cells (8 h S-phase), the pre-MBT timing profile appeared compressed and resembled random data (Fig. 4A; Kane and Kimmel 1993; Kuriya et al. 2015). However, consistent timing profiles between biological replicates were evident when the data were scaled to normalized units (Fig. 4A). Furthermore, a high correlation among timing values genome-wide for biological replicates (r = 0.85) indicate a defined temporal order is present in pre-MBT embryos (Fig. 4B). Additionally, assessing the pattern of the timing program by autocorrelation revealed that the pre-MBT timing program has structure and is not random (Fig. 4C). Collectively, these results demonstrate the presence of a defined and nonrandom replication timing program in pre-MBT embryos, which is compressed due to the short length of S-phase.
Figure 4.

A pre-MBT replication timing program anticipates initial zygotic transcription. (A) Replication timing values scaled to S-phase length (left) for pre-MBT (blue line) and ZTF cells (gray line) and scaled to normalized units (right) for pre-MBT biological replicates. (B) Pearson's correlation for genome-wide replication timing values from biological replicates of pre-MBT embryos. (C) Autocorrelation for biological replicates of pre-MBT replication timing data (blue lines) and randomly permutated data (black lines). (D) Replication timing of the genome overall (white), all genes (gray), the genes transcribed pre-MBT (blue), or the first genes transcribed after zygotic genome activation (first wave, red). ANOVA with Bonferroni post-hoc correction: (**) P < 0.0001; (*) P = 0.0067. Box plots show the median (line), 95% confidence interval (notch), 25th–75th percentile (box), and 10th–90th percentile (whiskers).
Comparison of pre-MBT and 28 hpf embryo timing profiles revealed an overall similar structure despite many differences in replication timing (Supplemental Fig. S5A). We hypothesized that the replication timing program in pre-MBT embryos would be shaped primarily by sequence features, since they display little transcription, and many chromatin marks are not added until after the MBT (Vastenhouw et al. 2010). Interestingly, pre-MBT replication timing showed a negative correlation with repeat density (r = −0.44); however, this correlation was almost entirely due to late replication of the highly repetitive chromosome ends (Supplemental Fig. S5C–E). Pre-MBT replication timing also had a negative correlation with GC content (r = −0.228), but this correlation was also largely due to elevated GC content at chromosome ends (Supplemental Fig. S5F–H). Thus, our data indicate that pre-MBT replication timing is related to repeat density or GC-content only near the chromosome ends.
The replication timing of genes pre-MBT was of interest because there is little transcription throughout most of the genome until after the MBT (Lee et al. 2014). Despite the lack of bulk transcription in pre-MBT embryos, genes were significantly enriched in early replicating areas of the genome (Fig. 4D). Recently, it was demonstrated that a small number of genes are transcribed prior to the zebrafish MBT, so we assessed the replication timing of those genes (Heyn et al. 2014). The pre-MBT-expressed gene set was significantly earlier replicating than genes overall, consistent with a relationship between their transcription and early replication (Fig. 4D). Finally, we examined replication timing of the first genes to be transcribed after the MBT (Lee et al. 2013). Unexpectedly, these unexpressed “first-wave” genes were already early replicating in pre-MBT embryos and were even significantly earlier replicating in these embryos than the pre-MBT transcribed genes (Fig. 4D). After removing chromosome ends, genes were still significantly early replicating, and first-wave genes were significantly earlier replicating than genes (Supplemental Fig. S5B). These results demonstrate that early replication of zygotic genes precedes their transcription and suggest either that DNA replication is controlled by the same regulators that poise “first-wave” genes for transcription, or that the pre-MBT replication timing program itself plays a role in priming the zygotic genome for transcriptional activation.
Replication timing is dynamically regulated throughout development
Having demonstrated the presence of a replication timing program in pre-MBT embryos, we next identified changes in replication timing that occur throughout early development and their relationship with genetic and epigenetic features of the genome. We profiled replication timing at three additional stages of development—Dome (4.3 hpf), Shield (6 hpf), and Bud (10 hpf)—to uncover changes across several important developmental milestones, including the MBT and gastrulation (Fig. 5A). In the early stages of development, cells are highly proliferative and primarily in S-phase (Supplemental Fig. S6; Siefert et al. 2015); therefore, the early embryos were processed as S-phase samples and compared to the G1 reference as described above. Genome-wide changes in replication timing throughout development were assessed by comparing replication timing values at each developmental stage in a Pearson's correlation matrix (Fig. 5B). The correlation coefficients between replicates were very high for each developmental stage, ranging from r = 0.85 at pre-MBT to r = 0.96 at 28 hpf. The correlation between biological replicates increased at each stage of development. This suggests that the timing program progressively gained definition throughout development, but could also be due in part to a greater degree of difference between biological replicates in the rapid stages of early development. Initially, we anticipated major timing program restructuring at the MBT. Instead, this analysis revealed that replication timing changes occurred continuously throughout early development, with gradual changes occurring at each developmental stage and widespread changes occurring across the developmental time course. However, the strongest differences observed were between Shield and Bud stage, when gastrulation occurs and germ layer fates are determined. Despite embryos at 28 hpf being composed of a much more diverse array of differentiated cell types than embryos at the Bud stage, the replication timing values genome-wide were highly correlated between these two stages (r = 0.8) (Fig. 5B). It will be of great future interest to isolate individual cell types from zebrafish embryos and determine to what extent replication timing differs between cell types in vivo during development.
Figure 5.

Replication timing is dynamically regulated throughout development. (A) Developmental time points used in this study. (B) Pearson's correlation matrix comparing genome-wide replication timing values from biological replicates of each developmental time point to all other samples. (C) Using a two-state hidden Markov model (HMM) the whole genome was segmented into 1620 contiguous regions that either change timing from early-to-late or late-to-early throughout development. Representative genomic regions that switch from early-to-late and late-to-early are shown. (D) Using k-means clustering, the HMM-defined changing regions were grouped based on the patterns of their timing changes. (E) Replication timing plotted as a function of distance from the telomere for each developmental time point. (F) Distances between the location of peaks in the 28 hpf sample and peaks in all other developmental samples (P-values from Kolmogorov-Smirnov test). (G) Autocorrelation of replication timing for each developmental sample (colored lines) and randomly permutated data (black lines).
To describe in more detail how replication timing changes during zebrafish development, we analyzed changes at defined genomic regions. Because replication timing fluctuates across regions that are hundreds of kilobases long, we used a two-state hidden Markov model (HMM) to consolidate the genome into regions with similar levels of replication timing change throughout development. The HMM segmented the entire genome into 1620 regions with a median size of 520 kb (Fig. 5C). This is within the range reported for the size of mammalian timing domains that switch timing during development (Pope et al. 2014). For each of these regions, we calculated a mean replication timing value for each developmental time point and determined whether the mean values changed significantly during development. Almost half the regions (761/1620) underwent significant developmental changes in replication timing (P < 0.05), consistent with the percentage of mammalian genomes that changes during development (Hiratani et al. 2010). Nearly equal numbers of the regions underwent early-to-late (382/761) or late-to-early (379/761) replication timing shifts. When k-means clustering was used to group the early-to-late or late-to-early regions based on replication timing across all developmental time points, patterns with unique trends of replication timing changes became apparent: some with gradual shifts across all stages of early development and others with sharp changes between specific developmental stages (Fig. 5D; Supplemental Fig. S7). This suggests that the replication timing of different regions of the genome may be influenced by distinct developmental cues.
We also investigated timing changes specifically at chromosome ends, which appeared to be late replicating at the pre-MBT stage (Fig. 4C; Supplemental Fig. S5) but were not obviously late replicating in 28 hpf embryos (Fig. 1B). The chromosome ends gradually shifted toward earlier replication at each developmental stage, until ultimately there was no bias for early or late replication (Fig. 5E). Because the correlations between late replication timing and GC content or repeat density in pre-MBT embryos was due to the chromosome ends (Supplemental Fig. S5), this result is consistent with the lack of correlation between replication timing and GC content or repeat density in 28 hpf embryos (Fig. 2).
Despite widespread changes in replication timing, we observed that the positions of many peaks in the profiles appeared to be constant. This was consistent with our previous observation that the locations of peaks were conserved between 28 hpf embryos and ZTF cells (Supplemental Fig. S3C,D). To quantitatively evaluate peak positions during development, we measured the distances between peaks in the 28 hpf sample and peaks in all other developmental samples. Surprisingly, despite widespread changes in replication timing throughout development, peak locations were significantly conserved at all stages (Fig. 5F). This provides further evidence that many highly efficient origins or origin clusters are consistently utilized throughout development in multiple cell types.
Finally, to assess the structure of the timing program throughout development, we determined the autocorrelation of timing values along the lengths of the chromosomes at each time point. This revealed that the autocorrelation gradually increased at each stage, indicating that the timing program progressively gained structure throughout development (Fig. 5G). Collectively, these results support a model in which the replication timing program is shaped during development both by progressive maturation and by sharp changes at specific genomic regions.
Enhancer activation parallels late-to-early replication timing changes during development
Based on the finding that enhancers were especially early replicating in 28 hpf embryos (Fig. 3E), we investigated whether enhancer activation paralleled any of the sharp changes in replication timing that we observed at specific genomic regions. Enhancers that are acetylated at different times during early zebrafish development, termed differentially acetylated regions (DARs), have been identified (Bogdanović et al. 2012). Plotting the replication timing profiles with H3K27ac ChIP-seq data revealed that increases in DAR acetylation, indicative of enhancer activation, frequently coincided with developmental shifts in replication timing at these regions (Fig. 6A). To determine if enhancer activation paralleled replication timing changes across the entire genome, we calculated replication timing at all stages for every DAR. Indeed, the stages at which DARs are acetylated mirrored changes in their replication timing; DARs acetylated between 4.3–8 hpf became significantly earlier replicating between the Dome (4.3 hpf) and Bud (10 hpf) stages, whereas DARs acetylated between 8–24 hpf became significantly earlier replicating between Bud (10 hpf) and 28 hpf stages (Fig. 6B). This effect was not simply due to a general shifting of gene-containing regions to earlier replication, as the median replication timing of all TSSs did not undergo the same developmental changes (Fig. 6B). To assess whether this phenomenon occurred genome-wide or was restricted to specific genomic regions, we looked at the distribution of DARs across all of the nonchanging (No Change) or changing timing regions (Late-to-Early or Early-to-Late) that were defined by the HMM segmentation (Fig. 5C). DARs were significantly enriched in late-to-early replication timing regions and depleted from early-to-late regions (Fig. 6C). In contrast, TSSs were slightly depleted from late-to-early regions, so the enrichment of DARs in those regions was not related to their proximity to genes in general (Fig. 6C).
Figure 6.

Enhancer activation parallels late-to-early replication timing changes during development. (A) Replication timing plots at Dome (4.3 hpf; orange), Bud (10 hpf; purple), and 28 hpf (green), with dashed lines indicating genomic region subject to developmental change. Circles mark the genomic positions of differentially acetylated regions (DARs) and have diameters proportionate to the acetylation levels at similar developmental stages (4.3 hpf, 8 hpf, and 24 hpf, respectively) (Bogdanović et al. 2012). (B) Replication timing at each developmental stage for the transcription start sites (TSSs), DARs that become acetylated between 4.3 and 8 hpf or 8 and 24 hpf, and differentially methylated regions (DMRs) that become demethylated between 6 and 24 hpf: (*) paired t-test with Bonferroni corrected P < 0.0001 (Bogdanović et al. 2012; Lee et al. 2015). Box plots show the median (line), 95% confidence interval (notch), 25th–75th percentile (box), and 10th–90th percentile (whiskers). (C) Observed/expected counts of TSSs, DARs, or DMRs in all genomic regions classified by hidden Markov model (HMM) segmentation as either undergoing developmental replication timing changes from late-to-early, early-to-late, or no change: (*) binomial test with Bonferroni corrected P-value <5 × 10−12.
As transcriptional enhancers are often DNA demethylated during development, we also analyzed the replication timing and enrichment of differentially methylated regions (DMRs) that were demethylated between 6 and 24 hpf (Lee et al. 2015). Like the DARs, replication timing of DMRs shifted from late-to-early during the same time frame in which they were demethylated (Fig. 6B). Furthermore, DMRs were significantly enriched in late-to-early regions and depleted from early-to-late regions (Fig. 6C). The majority (311/442, 70%) of late-to-early regions contained at least one demethylated DMR or acetylated DAR. Collectively these data show that epigenetic activation of enhancers frequently parallels replication timing changes during development.
The long arm of Chromosome 4 undergoes a developmentally regulated switch to late replication during gastrulation
The most remarkable replication timing change that occurred during early zebrafish development was on Chromosome 4. Nearly the entire long arm of Chromosome 4 (>42 Mb) changed replication timing coordinately between the Shield and Bud stages, a period when gastrulation occurs and germ layers are determined (Fig. 7A). From pre-MBT to Shield stage, the long arm of Chr 4 (Chr 4q) was composed of continuous fluctuations between early and late replication (Supplemental Fig. S8A). However, at Bud stage, almost the entire long arm underwent an abrupt switch to late replication, and it remained late replicating in 28 hpf embryos and the differentiated ZTF cells. The developmental timing and scale of the Chr 4q change are similar to mammalian X Chromosome inactivation, which is marked by a nearly chromosome-wide switch to late replication timing that similarly occurs when cell lineages are specified (Aanes et al. 2011).
Figure 7.

The long arm of Chromosome 4 undergoes a developmentally regulated switch to late replication. (A) Replication timing of Chromosome 4 plotted at Shield (gold) and Bud (purple) stages. (B) Autocorrelation of replication timing for a 25-Mb segment of the late replicating long arm of Chromosome 4 (Chr 4q, red), a 25-Mb segment of the short arm of Chromosome 4 (Chr 4p, gray), and randomly permutated data (black lines). (C) Correlation between biological replicates of 28 hpf embryos. Blue and yellow denote alternating chromosomes, red highlights Chromosome 4. (D) Replication timing profiles for 28 hpf embryos show a low consistency in replication timing between experimental replicates along the long arm of Chromosome 4. The gray box indicates a region of low correlation between experimental replicates (average r = 0.16). (E) mRNA levels (log2 ratio of fold change from transcript counts at the two-cell stage) for RefSeq genes on all chromosomes except Chromosome 4 (All), genes on the short arm of Chromosome 4 (Chr 4p), or genes on the late replicating long arm of Chromosome 4 (Chr 4q): (*) comparison to All genes at the matching developmental stage, two-way ANOVA with Bonferroni corrected P-value <0.005. Box plots show the median (line), 95% confidence interval (notch), 25th–75th percentile (box), and 10th–90th percentile (whiskers). (F) Average H3K27ac signal ±1 kb from all Dome peak apexes on the short (4p) and long (4q) arms of Chromosome 4 at Dome (orange), Bud (purple), and 28 hpf (green) (Pauli et al. 2012).
In human cells, the late-replicating inactive X Chromosome (Xi) lacks a defined spatial pattern of replication (Koren and McCarroll 2014). To determine whether this was also true for Chr 4q, the autocorrelation of replication timing along Chr 4q was compared to the autocorrelation of the short arm of Chromosome 4 (Chr 4p) in 28 hpf embryos. Although the short arm was highly structured, the autocorrelation of the long arm was similar to randomized data, indicating that it lacks a structured timing program (Fig. 7B). An unstructured replication timing profile should also display low consistency between biological replicates; indeed, Chr 4q was unique in having a weak correlation between replication timing profiles from 28 hpf biological replicates (mean r = 0.16) (Fig. 7C,D). Interestingly, the distal tip of Chr 4q is the only region of the long arm that has a high correlation between biological replicates and is not entirely late replicating, drawing similarity to the pseudoautosomal region (PAR) at the distal tip of the Xi (Fig. 7D; Koren and McCarroll 2014).
Mammalian X inactivation is associated with chromosome-wide epigenetic changes and transcriptional silencing, so we examined whether similar changes occur on Chr 4q. First we determined the expression levels of genes on Chr 4q before the timing change occurred. Between the pre-MBT and Shield stages, the median levels of mRNAs expressed from Chr 4q increased 26-fold, whereas there was no change in the expression of the genome as a whole, indicating that Chr 4q was unusually transcriptionally active upon zygotic genome activation (Fig. 7E). The switch to late replication during gastrulation coincided with a reduction in transcript levels from Chr 4q (Fig. 7E). The median level of mRNAs expressed from Chr 4q decreased 21-fold between Shield stage and 5 d post-fertilization (dpf), whereas there was a slight increase (1.4-fold) in the levels of all other expressed genes over the same period. Given our observation that changes in replication timing often occurred with changes in H3K27 acetylation at enhancers (Fig. 6), we analyzed H3K27ac levels before and after the switch to late replication. Consistent with the overall transcriptional silencing of Chr 4q, H3K27ac levels across Chr 4q decreased coincident with the switch to late replication (Fig. 7F). Overall, the long arm of Chromosome 4 is a remarkable example of developmentally regulated changes in replication timing that occur in vivo during vertebrate development and exemplifies the relationship between replication timing, chromatin structure, and transcription.
Discussion
This study provides the first genome-wide, high-resolution analysis of replication timing throughout zebrafish development. A compressed but defined replication timing program was evident before the MBT that appears to poise the genome for the initial wave of zygotic transcription. The chromosome ends are late replicating in the pre-MBT embryos and gradually shift earlier throughout development. After the MBT, replication timing is dynamic across most of the genome, as it changed progressively and gained structure throughout development. The gradual increase in structure coincided with a stretching of the timing program, as regions became later replicating throughout development with progressive S-phase lengthening. Peaks in the replication timing profiles were significantly conserved at different stages, suggesting that the positions of many highly efficient origins or origin clusters are maintained throughout development, including before the MBT. In addition to the progressive changes in the replication timing profiles, specific genomic regions displayed sharp timing changes at distinct stages that often coincided with regional histone acetylation and DNA demethylation involved in enhancer activation. The long arm of Chromosome 4 underwent an abrupt switch to late replication during gastrulation, when germ layer fates are determined, and it remained late replicating throughout subsequent stages of development.
Previous studies examining DNA replication prior to the MBT have produced conflicting results regarding its structure. Work from Xenopus egg extracts suggested that DNA replication in early embryos initiates from random sequences and does not follow spatial patterns characteristic of the timing program of somatic cells, whereas other work demonstrated that large chromosomal domains are replicated in a reproducible manner but origins and origin clusters fire stochastically (Mills et al. 1989; Hyrien and Méchali 1993; Dimitrova and Gilbert 1999; Labit et al. 2008). This study established that at low temporal resolution the program appears random; however, at high temporal and spatial resolution allowed by the sequencing-based, genome-wide approach, a fine yet significantly structured program emerges and is visually evident with normalized replication timing profiles. The temporal order to DNA replication timing in pre-MBT embryos may be required to poise the embryos for transcription after the MBT, suggesting evolutionary selection for a temporal order of DNA replication that favors transcriptional competence of genes immediately expressed upon zygotic genome activation.
A significant finding of this study is the high correlation between early replication and enhancer activation. Acetylation of multiple sites on H3 and H4 histones was previously shown to influence origin activity, and hypoacetylation of these histones is thought to be necessary to maintain late replication timing (Unnikrishnan et al. 2010; Casas-Delucchi et al. 2012). Furthermore, there is evidence that replication timing itself can influence histone acetylation levels, and thereby transcription and nucleosome structure (Zhang et al. 2002; Lande-Diner et al. 2009). The results presented in this study are in agreement with the relationship between replication timing and histone acetylation, and further suggest a role for replication timing in influencing H3K27ac levels at transcriptional enhancers during development. Another possibility is that acetylation may regulate DNA replication by influencing the recruitment or activity of DNA replication initiation factors on chromatin.
Finally, the dramatic replication timing switch that occurs along Chr 4q draws many interesting parallels to mammalian X Chromosome inactivation. These include the switch to late replication coincident with cell fate specification, replication without a clear spatial pattern, and deacetylation and transcriptional silencing occurring contemporaneously with the switch. Sex chromosomes have not been identified in zebrafish, and the genetic basis for their gender determination is unclear. The existence of a sex-associated region (sar4) on Chr 4q has led to the suggestion that it may be an evolving sex chromosome (Anderson et al. 2012; Howe et al. 2013). Overall, Chr 4q provides a stunning example of developmentally regulated changes in replication timing that occur during vertebrate development. Further studies on the replication timing switch of Chr 4q are likely to yield new insight into the mechanisms of large-scale replication timing and chromatin conformation changes that occur during embryogenesis. The data presented here establish the zebrafish as a promising in vivo model for studying the mechanisms of replication timing change and the effects of deregulating replication timing on development and disease.
Methods
Sample preparation and replication timing analysis
Hundreds of zebrafish embryos were collected at indicated time points, and disaggregated cells were fixed in 70% ethanol. ZTF cells were derived from a caudal fin clip of adult zebrafish, cultured, expanded and fixed as before. Fixed 28 hpf embryos and ZTF cells were treat with RNase A and stained with propidium iodide, then sorted based on DNA content to isolate G1 and S-phase fractions. DNA was purified from these populations and from the ethanol-fixed cells of embryos from earlier time points. All DNA samples were treated with RNase A and then deep sequenced on the Illumina platform. Replication timing data were generated based on copy-number variations between filtered G1 and S-phase read counts (Koren et al. 2014). Briefly, 200 read count windows were defined in the G1 sample, and S-phase read depth was determined in the same windows. Raw data were smoothed with a cubic smoothing spline using a parameter of 10–16, and data were transformed to Z-score by normalizing to a genome mean of 0 and standard deviation of 1. To scale data to the length of S-phase, timing data were normalized to a maximum value of 0 and a minimum value to 1, then multiplied by S-phase length.
All animals were handled in strict accordance with protocols approved by the OMRF Institutional Animal Care and Use Committee.
Analysis of CHIP-seq and RNA-seq data
For the analyses of H3K27me3, H3K27me1, and H3K27ac marks, raw ChIP-seq sequencing files were obtained from recent studies: GSE32483 (Bogdanović et al. 2012), GSE20600 (Aday et al. 2011), and GSE20023 (Vastenhouw et al. 2010). The ChIP-seq reads were mapped to the GRCz10/danRer10 zebrafish genome assembly using BWA-MEM, and duplicate reads as well as reads mapping to multiple positions in the genome were removed. For the H3K27me3, H3K27me1, and H3K27ac ChIP-seq data, peaks were called using MACS2 with a Q-value cutoff of 0.01 (Zhang et al. 2008; Li and Durbin 2010). H3K4me3-positive RefSeq genes were defined as having at least one H3K4me3 peak within the 5 kb surrounding the gene start. To identify PDREs in 24 hpf embryos, the published criteria were used (Bogdanović et al. 2012). Briefly, PDREs were defined as H3K27ac peaks that overlapped with H3K4me1 peaks farther than 1 kb away from either RefSeq or Ensembl gene transcription start sites or H3K4me3 peaks. Overlapping features were identified using BEDOPS (Neph et al. 2012). For calculating the mean H3K36me3 read counts across RefSeq gene exons, reads previously aligned to the Zv9 zebrafish genome were used (Ulitsky et al. 2011).
For the analysis of developmentally regulated enhancers, published lists of differentially acetylated PDREs and differentially methylated DMRs were used (Bogdanović et al. 2012; Lee et al. 2015). The PDREs and DMRs were originally mapped to the Zv9 zebrafish genome, so the UCSC liftOver tool was used to remap the coordinates onto GRCz10/danRer10.
For the analysis of mRNA expression, we used RNA-seq data from a recent study (Pauli et al. 2012). Raw RNA-seq reads for two biological replicates for each time point were mapped to GRCz10/danRer10 using TopHat, then raw counts of mapped RNA-seq reads and mean FPKM values were calculated for every RefSeq gene using Cufflinks (Trapnell et al. 2012). For the analysis of gene expression changes, two biological replicates for each time point were used in the analysis. The number of RNA-seq reads mapped to each RefSeq transcript were counted using HTSeq, and differential expression between time points was called using DESeq2 (Love et al. 2014; Anders et al. 2015).
Statistical processing
All correlations between samples were performed using Pearson's correlation in Matlab (Figs. 2C–H, 5B; Supplemental Figs. S1B, S3B, S5B,C,E,F). Autocorrelation was performed in 300-bp increments along the length of indicated chromosomes using the autocorr function in Matlab and plotted at 3-kb resolution (Figs. 1D, 4B, 5G, 7B). Randomized autocorrelation was performed on 10 iterations of randomly permutated data using the autocorr function in Matlab (Figs. 1D, 4B, 5G, 7B). Correlation between biological replicates along the length of the chromosome was performed with Pearson's correlation in a 3-Mb sliding window using Matlab (Fig. 7C). Average Pearson's correlation along Chr 4q was computed based on the average of 3-Mb sliding window correlations in Matlab (Fig. 7D). Significance P-values for difference in distribution for the distances between sample peaks and the 28 hpf peaks were computed with a Kolmogorov-Smirnov test in Matlab (Fig. 5F; Supplemental Fig. S3D). All box plots show the median (line), 95% confidence interval (notch), 25th–75th percentile (box), and 10th–90th percentile (whiskers) (Figs. 3A–E, 4D, 6B, 7E; Supplemental Figs. S4A,B, S7). All additional statistical analyses were performed using R (R Core Team 2015): Student's t-test with Bonferroni corrected P-values (Figs. 3A–E; Supplemental Fig. S4A,B); one-way ANOVA with Bonferroni post hoc correction (Fig. 4D; Supplemental Figs. S4A, S7); paired t-test with Bonferroni corrected P-values (Fig. 6B); binomial test with Bonferroni corrected P-value (Fig. 6C); and two-way ANOVA with Bonferroni corrected P-value (Fig. 7E).
Data access
The raw and processed sequencing data produced in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE85713.
Supplementary Material
Acknowledgments
We thank Linda Thompson and Dean Dawson for their thorough critiques of the manuscript; James Robertson, Graham Wiley, and Pat Gaffney for assistance with library preparation and sequencing; Stuart Glen and Gene Chao for assistance with cluster computing; and Angela Andersen of Life Science Editors for manuscript editing. This research was supported by the National Institutes of Health (NIH) 4P20GM103636-04, NIH 1R01GM121703-01, and Oklahoma Center for Adult Stem Cell Research Grants (OCASCR).
Author contributions: C.L.S. and J.C.S. conceptualized the study. C.L.S. and A.K. supervised the work. J.C.S. performed the experiments. A.K. developed the replication timing analysis procedure. A.K. and J.C.S. processed the replication timing data. J.C.S., C.G., J.D.W., A.K., and C.L.S. analyzed data and performed statistics. J.C.S., A.K., and C.L.S. wrote and prepared the manuscript and figures. J.C.S., C.G., J.D.W., A.K., and C.L.S. reviewed and edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.218602.116.
References
- Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, Lee SG, Lim AY, Hajan HS, Collas P, Bourque G, et al. 2011. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21: 1328–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aday AW, Zhu LJ, Lakshmanan A, Wang J, Lawson ND. 2011. Identification of cis regulatory features in the embryonic zebrafish genome through large-scale profiling of H3K4me1 and H3K4me3 binding sites. Dev Biol 357: 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Rodríguez Marí A, Braasch I, Amores A, Hohenlohe P, Batzel P, Postlethwait JH. 2012. Multiple sex-associated regions and a putative sex chromosome in zebrafish revealed by RAD mapping and population genomics. PLoS One 7: e40701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanović O, Fernandez-Miñán A, Tena JJ, de la Calle-Mustienes E, Hidalgo C, Van Kruysbergen I, van Heeringen SJ, Veenstra GJ, Gómez-Skarmeta JL. 2012. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res 22: 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Delucchi CS, Van Bemmel JG, Haase S, Herce HD, Nowak D, Meilinger D, Stear JH, Leonhardt H, Cardoso MC. 2012. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res 40: 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers EV, Bickmore WA, Semple CA. 2013. Divergence of mammalian higher order chromatin structure is associated with developmental loci. PLoS Comput Biol 9: e1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d'Aubenton-Carafa Y, Arneodo A, Hyrien O, et al. 2010. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 20: 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daga RR, Thode G, Amores A. 1996. Chromosome complement, C-banding, Ag-NOR and replication banding in the zebrafish Danio rerio. Chromosome Res 4: 29–32. [DOI] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM. 1999. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell 4: 983–993. [DOI] [PubMed] [Google Scholar]
- Farrell JA, O'Farrell PH. 2014. From egg to gastrula: how the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu Rev Genet 48: 269–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159: 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MA, Holmquist GP, Gray MC, Caston LA, Nag A. 1984. Replication timing of genes and middle repetitive sequences. Science 224: 686–692. [DOI] [PubMed] [Google Scholar]
- Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, Neugebauer KM. 2014. The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep 6: 285–292. [DOI] [PubMed] [Google Scholar]
- Hiratani I, Takebayashi S, Lu J, Gilbert DM. 2009. Replication timing and transcriptional control: beyond cause and effect—part II. Curr Opin Genet Dev 19: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. 2010. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20: 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Ren B. 2009. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet 18: R195–R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, Mclaren K, Matthews L, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496: 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Méchali M. 1993. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J 12: 4511–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Maric C, Méchali M. 1995. Transition in specification of embryonic metazoan DNA replication origins. Science 270: 994–997. [DOI] [PubMed] [Google Scholar]
- Kane DA, Kimmel CB. 1993. The zebrafish midblastula transition. Development 119: 447–456. [DOI] [PubMed] [Google Scholar]
- Kenigsberg E, Yehuda Y, Marjavaara L, Keszthelyi A, Chabes A, Tanay A, Simon I. 2016. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res 44: 4222–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Warga RM, Schilling TF. 1990. Origin and organization of the zebrafish fate map. Development 108: 581–594. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. 1995. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- Koren A, McCarroll SA. 2014. Random replication of the inactive X chromosome. Genome Res 24: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. 2012. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlic R, Ghosh S, Polak P, Eggan K, McCarroll SA. 2014. Genetic variation in human DNA replication timing. Cell 159: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriya K, Higashiyama E, Avsar-Ban E, Tamaru Y, Ogata S, Takebayashi S, Ogata M, Okumura K. 2015. Direct visualization of DNA replication dynamics in zebrafish cells. Zebrafish 12: 432–439. [DOI] [PubMed] [Google Scholar]
- Labit H, Perewoska I, Germe T, Hyrien O, Marheineke K. 2008. DNA replication timing is deterministic at the level of chromosomal domains but stochastic at the level of replicons in Xenopus egg extracts. Nucleic Acids Res 36: 5623–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande-Diner L, Zhang J, Cedar H. 2009. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol Cell 34: 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Takacs CM, Bazzini AA, Divito KR, Fleming ES, Giraldez AJ. 2013. Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature 503: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Giraldez AJ. 2014. Zygotic genome activation during the maternal-to-zygotic transition. Annu Rev Cell Dev Biol 30: 581–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Lowdon RF, Maricque B, Zhang B, Stevens M, Li D, Johnson SL, Wang T. 2015. Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat Commun 6: 6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessman CA. 2011. The developing zebrafish (Danio rerio): a vertebrate model for high-throughput screening of chemical libraries. Birth Defects Res C Embryo Today 93: 268–280. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Li F, Murphy CS, Davidson MW, Gilbert DM. 2010. G2 phase chromatin lacks determinants of replication timing. J Cell Biol 189: 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Rodríguez HK, Bell SP. 2004. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev 18: 3094–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills AD, Blow JJ, White JG, Amos WB, Wilcock D, Laskey RA. 1989. Replication occurs at discrete foci spaced throughout nuclei replicating in vitro. J Cell Sci 94(Pt 3): 471–477. [DOI] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. 2012. BEDOPS: high-performance genomic feature operations. Bioinformatics 28: 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J, Kirschner M. 1982. A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell 30: 675–686. [DOI] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, et al. 2012. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22: 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. 2014. Topologically associating domains are stable units of replication-timing regulation. Nature 515: 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. 2001. Replication dynamics of the yeast genome. Science 294: 115–121. [DOI] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Med 3: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Buckley Q, Sasaki T, Zimmerman J, Didier RA, Nazor K, Loring JF, Lian Z, Weissman S, Robins AJ, et al. 2015. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 25: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Sawado T, Yamaguchi M, Shinomiya T. 1999. Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolα-dE2F locus of Drosophila melanogaster. Mol Cell Biol 19: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M. 2002. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat Genet 32: 438–442. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15: 272–86. [DOI] [PubMed] [Google Scholar]
- Siefert JC, Clowdus EA, Sansam CL. 2015. Cell cycle control in the early embryonic development of aquatic animal species. Comp Biochem Physiol C Toxicol Pharmacol 178: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola L, Gornung E. 2001. Classical and molecular cytogenetics of the zebrafish, Danio rerio (Cyprinidae, Cypriniformes): an overview. Genetica 111: 397–412. [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. 2009. Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. 2011. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147: 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnikrishnan A, Gafken PR, Tsukiyama T. 2010. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 17: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, Schier AF. 2010. Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464: 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xu F, Hashimshony T, Keshet I, Cedar H. 2002. Establishment of transcriptional competence in early and late S phase. Nature 420: 198–202. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.