

Abstract
In airway smooth muscle (ASM) cells, excitation-contraction coupling is accomplished via a cascade of events that connect an elevation of cytosolic Ca2+ concentration ([Ca2+]cyt) with cross-bridge attachment and ATP-consuming mechanical work. Excitation-energy coupling is mediated by linkage of the elevation of [Ca2+]cyt to an increase in mitochondrial Ca2+ concentration, which in turn stimulates ATP production. Proximity of mitochondria to the sarcoplasmic reticulum (SR) and plasma membrane is thought to be an important mechanism to facilitate mitochondrial Ca2+ uptake. In this regard, mitochondrial movement in ASM cells may be key in establishing proximity. Mitochondria also move where ATP or Ca2+ buffering is needed. Mitochondrial movement is mediated through interactions with the Miro-Milton molecular complex, which couples mitochondria to kinesin motors at microtubules. We examined mitochondrial movement in human ASM cells and hypothesized that, at basal [Ca2+]cyt levels, mitochondrial movement is necessary to establish proximity of mitochondria to the SR and that, during the transient increase in [Ca2+]cyt induced by agonist stimulation, mitochondrial movement is reduced, thereby promoting transient mitochondrial Ca2+ uptake. We further hypothesized that airway inflammation disrupts basal mitochondrial movement via a reduction in Miro and Milton expression, thereby disrupting the ability of mitochondria to establish proximity to the SR and, thus, reducing transient mitochondrial Ca2+ uptake during agonist activation. The reduced proximity of mitochondria to the SR may affect establishment of transient “hot spots” of higher [Ca2+]cyt at the sites of SR Ca2+ release that are necessary for mitochondrial Ca2+ uptake via the mitochondrial Ca2+ uniporter.
Keywords: TNFα, airway, asthma, mitochondria, Milton, Miro
mitochondrial motility is a feature in a variety of cell types, and research in this area has expanded over the past decade from curiosity to questions concerning the role of mitochondrial movement in mitochondrial function and in many cellular functions (6, 8, 9, 11–16, 32, 52, 78, 93). It is usually thought that mitochondria move to sites where ATP production and Ca2+ buffering are required (3, 6, 8–10, 14, 16, 41, 45, 53, 60, 63, 82, 84).
Two types of mitochondrial motility have been described: movement in a directed trajectory and a kinetic, more random motion (58, 78, 90). Most available information concerns directed trajectory movement of mitochondria, e.g., in neuronal axons, where ~70% of mitochondria are stationary while the remaining mitochondria move in a directed anterograde and/or retrograde trajectory (78). Such directed trajectory movement of mitochondria is primarily mediated through interactions with microtubule filaments (58, 78). Of particular interest is the interaction of Milton, a trafficking protein kinesin (TRAK), with Miro, a mitochondrial Rho GTPase protein (also called RhoT1/2), which couples mitochondria to microtubules via kinesin heavy chains and dynein/dynactin (34, 51, 55, 80, 87). Cytosolic Ca2+ binding to the EF-hand domains of Miro disrupts the Miro-Milton interaction and, thus, the coupling of mitochondria to microtubule filaments (51, 78). Thus, transient elevation of cytosolic Ca2+ ([Ca2+]cyt) in neurons and other cell types reduces mitochondrial movement (14, 66, 82, 92). Actin filaments might also play a role in tethering mitochondria, thereby halting mitochondrial motility and regulating the pool of mitochondria in a stationary state (78, 81). Far less is known about regulation of the kinetic, more random movement of mitochondria; however, it is likely that microtubules are involved and that it is regulated by the same Ca2+-sensitive Miro-Milton pathway.
It is widely believed that it is important for mitochondria to stop in areas of transient Ca2+ flux, e.g., agonist-induced release from the sarcoplasmic reticulum (SR) or influx through the plasma membrane (PM). A transient increase in [Ca2+]cyt, or “hot spot,” allows activation of the mitochondrial Ca2+ uniporter (MCU) and mitochondrial Ca2+ uptake, thereby transiently increasing mitochondrial Ca2+ concentration ([Ca2+]mito) to facilitate ATP production through oxidative phosphorylation (9, 20, 21, 55, 73, 78, 89). The MCU has relatively low affinity for Ca2+ (e.g., in the 5–10 µM range) (7, 19, 32, 36, 37, 39, 68, 70); therefore, proximity of mitochondria to the SR or PM hot spots may be a determining factor in mitochondrial Ca2+ uptake (7, 19, 32, 36, 37, 39, 68, 70). Proximity of mitochondria to the SR is indicated in many cell types, including ASM cells, in that a transient increase in [Ca2+]cyt leads to a transient increase in [Ca2+]mito (17, 22, 26, 31, 42, 59, 71, 83). In muscle, coupling between transient [Ca2+]cyt and [Ca2+]mito responses may reflect an “excitation-energy coupling” that mirrors the increased energy demand resulting from excitation-contraction coupling (9, 11, 23, 24, 27, 38, 40, 62, 77, 85). During rest at basal [Ca2+]cyt, when Miro/Milton is not disrupted, mitochondrial movement may be important in establishing proximity of mitochondria to the SR or PM. Previously, we found that the normal coupling of transient [Ca2+]cyt and [Ca2+]mito responses to agonist stimulation in human airway smooth muscle (ASM) cells was blunted by exposure to inflammatory cytokines (e.g., TNFα and IL-13), even though the [Ca2+]cyt response was enhanced (22).
In the present study we hypothesized that, at basal [Ca2+]cyt levels, mitochondrial movement is necessary to establish proximity of mitochondria to the SR and that, during the transient increase in [Ca2+]cyt induced by agonist stimulation, mitochondrial movement is reduced, thereby promoting transient mitochondrial Ca2+ uptake. We further hypothesized that airway inflammation disrupts basal mitochondrial movement via a reduction in Miro and Milton expression, thereby disrupting the ability of mitochondria to establish proximity to the SR and, thus, reducing transient mitochondrial Ca2+ uptake during agonist activation.
MATERIALS AND METHODS
Isolation of human ASM cells.
The method used to isolate human ASM cells is described elsewhere (64, 65). Lung samples (from 3rd- to 6th-generation bronchi) were obtained incidental to patient surgery at the Mayo Clinic under a protocol reviewed and approved by the Institutional Review Board of the Mayo Clinic. The human bronchial samples were placed in ice-cold Hanks’ balanced salt solution (HBSS; Invitrogen, Carlsbad, CA) supplemented with 10 mM HEPES and 2 mM Ca2+. ASM tissue was dissected, and cells were dissociated using papain and collagenase with ovomucoid/albumin separation according to the manufacturer's instructions (Worthington Biochemical, Lakewood, NJ). Human ASM cells were maintained at 37°C (5% CO2-95% air) using phenol red-free DMEM/F-12 medium (Invitrogen) supplemented with 10% FBS. Before experiments, culture medium was changed to serum-free DMEM/F-12 medium for 48 h. Experiments were performed with freshly isolated cells or from passages 1–3 of subculture. Cell viability (tested by exclusion of Trypan blue) and ASM phenotype (smooth muscle actin and myosin and agonist receptors, as well as lack of fibroblast markers) were periodically evaluated as previously described (5, 46).
Real-time fluorescence imaging.
For visualization of mitochondria, human ASM cells, with or without exposure to TNFα for 24 h, were plated on eight-well glass-bottomed Lab-Tek chamber slides (Nalge Nunc, Rochester, NY) and loaded with 300 nM MitoTracker Red CMXRos (Invitrogen; 568 nm excitation/590 nm emission) for 5 min and then washed extensively in dye-free HBSS. For assessment of mitochondrial motility, human ASM cells were visualized using an epifluorescence imaging system (MetaFluor, Universal Imaging, Downingtown, PA) on a Nikon Diaphot inverted microscope (Fryer Instruments, Edina, MN) with a ×60/1.3 NA oil-immersion lens and a 12-bit Photometrics Cascade digital camera system (Roper Scientific, Tucson, AZ), as previously described (22, 64, 75). For simultaneous visualization of SR and mitochondria, human ASM cells were loaded with 1 µM BODIPY FL thapsigargin (endoplasmic reticulum Ca2+ pumps; Invitrogen; 503 excitation/512 nm emission) for 30 min and then with 300 nM MitoTracker Red CMXRos for 5 min. Cells were visualized using a laser scanning confocal system (Eclipse A1, Nikon Instruments, Melville, NY) equipped with a ×60/1.4 NA oil-immersion lens. Optical sectioning was performed with a 0.5-µm step size. Human ASM cells were imaged at 37°C using a stable z-stage heater (Bioptechs, Butler, PA) and perfusion with warm 2 mM Ca2+ HBSS for an average of 5 min (time lapse of 5 s). In other experiments, ASM cells were initially imaged at 20°C, and the temperature was raised to 30°C and then to 37°C. In selected experiments, ASM cells were imaged at 20°C or 30°C. The time lapse and duration of imaging were experimentally determined so as not to induce motility changes as a result of photobleaching or phototoxicity.
Image preprocessing.
Fluorescence images were subjected to background correction, thresholding, and skeletonization using ImageJ software (imagej.nih.gov/ij/) and MATLAB (MathWorks, Natick, MA), as previously described (48–50).
Mitochondrial motility.
Overall general mitochondrial motility (all types of mitochondrial movement) was quantified by a difference image protocol using time-lapse sequence images (time lapse of 5 s for an average duration of 5 min). After image preprocessing, the fluorescence of each pixel was calculated by subtraction of sequential images. The velocity of mitochondria was measured by “kymographic analysis,” i.e., tracking of mitochondria over time using the ImageJ Manual Tracking plugin. Because mitochondria accelerated and decelerated, an average velocity (μm/s) was computed. The number of mitochondria for which a velocity was estimated is indicated as events per minute.
Proximity of mitochondria to the SR.
The z-stacks images were analyzed using the ImageJ Coloc 2 plugin, as previously described (28). Manders' colocalization coefficients M1 (mitochondria colocalization with the SR) and M2 (SR colocalization with mitochondria) were used to estimate proximity of mitochondria to the SR (28, 56).
Western blot analysis.
Proteins were separated by SDS-PAGE (Criterion Gel System, Bio-Rad, Hercules, CA; 10% or 4–15% gradient gels) and transferred to polyvinylidene fluoride membranes (Bio-Rad) for 60 min (paired samples from control and TNFα-exposed ASM cells were run on the same gel). Membranes were blocked for 1 h with 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween and then incubated overnight at 4°C with anti-Miro-2 or anti-Milton-2 antibody (Abcam, Cambridge, MA). Primary antibody was detected using horseradish peroxidase-conjugated secondary antibody, and signals were developed using SuperSignal West Dura chemiluminescent substrate (Pierce Chemical, Rockford, IL). The membranes were also probed against glyceraldehyde 3-phosphate dehydrogenase (Abcam) as a loading control and porin (voltage-dependent anion-selective anion channel 1; Abcam) as a mitochondrial loading control.
[Ca2+]cyt and [Ca2+]mito measurements.
ASM cells were grown in 96-well plates and incubated with 5 µM fluo 4-AM and/or Rhod 2-AM (Invitrogen) for 40 min. Basal [Ca2+]cyt and [Ca2+]mito and transient [Ca2+] responses to agonists [acetylcholine (ACh) and histamine] were assessed using a fluorescence imaging plate reader with robotic pipetting capabilities (FlexStation 3, Molecular Devices, Sunnyvale, CA), as previously described (43).
Statistical analysis.
Eight human bronchial samples were used to obtain ASM cells. Movement of the mitochondria of approximately one to two cells per visual field was analyzed, and all experiments were repeated at least three times for each ASM sample. The number of ASM samples used for each specific experiment is represented by N (N ≥ 5 for each experimental group), and n is the number of ASM cells analyzed. In experiments where mitochondrial movement was compared in the presence or absence of cytokines, paired t-test was used, whereas population studies were compared using unpaired t-test or one-way ANOVA with repeated measures as appropriate. Bonferroni’s correction was applied for multiple comparisons. Statistical significance was established at P < 0.05. Values are means ± SE.
RESULTS
Characterization of mitochondrial movement.
Healthy human ASM cells generally exhibit a fusiform shape with a centrally located nucleus. As shown in Fig. 1A, mitochondria are not uniformly distributed in ASM cells, and they can be divided into two groups with varying density and morphology: perinuclear and distal mitochondria (Fig. 1A). The perinuclear mitochondria are more densely arranged in a somewhat concentric pattern around the nucleus (Fig. 1A). The distal mitochondria are more linearly aligned along the cytoskeleton and exist in longer, less branched networks than the shorter perinuclear mitochondria (Fig. 1A).
Fig. 1.

Characterization of overall mitochondrial motility. A: human airway smooth muscle (ASM) cells were loaded with 300 nM MitoTracker Red CMXRos for 5 min to visualize mitochondria. ASM cells were imaged at 20°C, 30°C, and 37°C. Mitochondrial motility was assessed using time-lapse vital imaging (sequence of images obtained at 5-s intervals across a 5-min period). B: images were converted to binary to indicate mitochondrial location and differences in location from image to image used to index overall mitochondrial motility. Overall mitochondrial motility was significantly reduced at 20°C compared with 30°C and 37°C. Values are means ± SE. *Significant effect (P < 0.05, N = 8) compared with 20°C. †Significant effect compared with 30°C.
While the extent of mitochondrial movement varied, it could be separated into two general types of movement: mitochondria moving with a random/wiggling motion and, less frequently, mitochondria moving with directed motion trajectories toward or away from the nucleus. These two general types of mitochondrial motility were assessed using a difference image protocol or overlap metric (Fig. 1A). The infrequent occurrence of mitochondrial movements with directed motion trajectories (see below) indicates that changes in overall mitochondrial motility were mainly driven by changes in random/wiggling mitochondrial motion.
To fully characterize mitochondrial movements in human ASM cells, we examined their temperature sensitivity. If these are physiological processes, they should be temperature-sensitive. At 20°C, mitochondrial movement was severely impaired (5.1 ± 0.76% motility, N = 8 patients, n = 51 ASM cells; Fig. 1B). When temperature was raised to 30°C, overall mitochondrial motility was tripled (15.8 ± 0.82%). Nearly six times more mitochondria were motile in experiments performed at 37°C than in experiments performed at 20°C (29.2 ± 1.02%, N = 8 patients, n = 51 ASM cells, P < 0.05 vs. 20°C or 30°C; Fig. 1B).
For mitochondria exhibiting directed motion trajectories, the average velocity of motion was computed using kymographic analysis (Fig. 2, A and B). Mitochondria accelerated and decelerated within a relatively short time frame, and the distance covered by mitochondria was variable, from several micrometers to >250 µm. At 20°C, an average of nine mitochondria per ASM cell exhibited a directed motion trajectory (Fig. 2C). The average velocity of mitochondria at 20°C was 1.38 ± 0.31 µm/s (Fig. 2D). The number of mitochondria (∼11 per ASM cell) and their average velocity (1.36 ± 0.22 µm/s) were not significantly increased at 30°C (Fig. 2, C and D). However, when the temperature was raised to 37°C, we noted a significant increase in the number of mitochondria with directed motion trajectories (21 per ASM cell) and average velocity (1.8 ± 0.1 µm/s, N = 8 patients, n = 51 ASM cells, P < 0.05 vs. 20°C or 30°C; Fig. 2, C and D).
Fig. 2.

Characterization of mitochondrial movements with directed motion trajectories. A: fluorescence image of human ASM cells loaded with 300 nM MitoTracker Red CMXRos for 5 min to visualize mitochondria. Velocity of mitochondrial movement was measured by “kymographic analysis.” Arrow indicates direction of mitochondrial movement. B: fluorescence images showing mitochondria moving over time. C: effect of temperature on the number of mitochondria with directed motion trajectories. Number of mitochondria with directed motion trajectories was not significantly different between 20°C and 30°C but significantly higher at 37°C. Values are means ± SE. *Significant effect (P < 0.05). D: effect of temperature on average velocity of mitochondrial movement. Average velocity of mitochondrial movement was not significantly different between 20°C and 30°C but significantly higher at 37°C. Values are means ± SE. *Significant effect (P < 0.05, N = 8).
Mitochondrial movement following histamine stimulation.
In human ASM cells, agonists such ACh and histamine induce a transient elevation of [Ca2+]cyt and [Ca2+]mito (Fig. 3A) (21, 22). The overall mitochondrial motility (random/wiggling motion and directed motion) in human ASM cells was examined at 37°C following stimulation with 10 µM histamine (Fig. 3A). Under resting conditions, 29.2 ± 5.4% of mitochondria were motile (Fig. 3), and overall mitochondrial motility decreased significantly after histamine stimulation (to 26.6 ± 3.4%, N = 8 patients, n = 45 ASM cells, P < 0.05; Fig. 3). By superposing overall mitochondrial motility and [Ca2+]mito response to 10 µM histamine, we noted that an increase in [Ca2+]mito correlated with a decrease in mitochondrial motility (Fig. 3A).
Fig. 3.

Overall mitochondrial motility following histamine stimulation. A: representative trace showing overall mitochondrial motility over a 5-min period. Stimulation of human ASM cells with histamine (10 µM) reduced overall mitochondrial motility. Mitochondrial Ca2+ response (dashed gray trace) obtained from ASM cells loaded with Rhod 2 is superposed on analysis of mitochondrial motility and shows that an increase in mitochondrial Ca2+ concentration ([Ca2+]mito) is associated with a decrease in mitochondrial motility. B: summary data showing that overall mitochondrial motility is decreased following histamine stimulation. Values are means ± SE. *Significant effect (P < 0.05, N = 8).
Fewer mitochondria were found to have directed motion trajectories after histamine stimulation than in the resting state (11 vs. 20 mitochondria per ASM cell, N = 8 patients, n = 45 ASM cells, P < 0.05; Fig. 4A). However, the average velocity of mitochondrial movement increased after histamine stimulation compared with the resting state (2.5 ± 0.3 vs. 1.8 ± 0.2 µm/s, N = 8 patients, n = 45 ASM cells, P < 0.05; Fig. 4A).
Fig. 4.
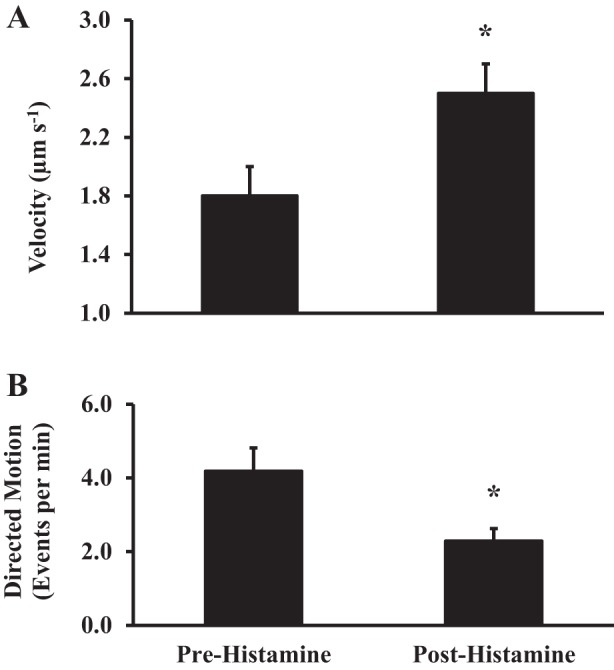
Effect of histamine stimulation on mitochondrial movement with directed motion trajectories. A: summary data showing an increase in velocity of mitochondrial movement following histamine stimulation. B: after histamine stimulation, there was a decrease in the number of mitochondria with directed motion trajectories. Values are means ± SE. *Significant effect (P < 0.05, N = 8).
Role of microtubules in mitochondrial movement.
To examine whether mitochondrial movement in human ASM cells is mediated through interactions with microtubule filaments, ASM cells were incubated for 30 min with 10 µM nocodazole to inhibit microtubule polymerization. Experiments were performed at 37°C. In human ASM cells exposed to nocodazole, overall mitochondrial motility was severely impaired compared with control ASM cells (4.80 ± 0.92% vs. 28.01 ± 1.76%, N = 5 patients, n = 31 ASM cells, P < 0.05; Fig. 5A). Similarly, the incidence of mitochondria with directed motion trajectories was also greatly reduced in human ASM cells exposed to nocodazole compared with control ASM cells (1 vs. 20 mitochondria per ASM cell, N = 5 patients, n = 31 ASM cells, P < 0.05; Fig. 5B). Because of the low number of mitochondria with directed motion trajectories in ASM cells exposed to nocodazole, no average velocity was estimated in this experimental condition.
Fig. 5.
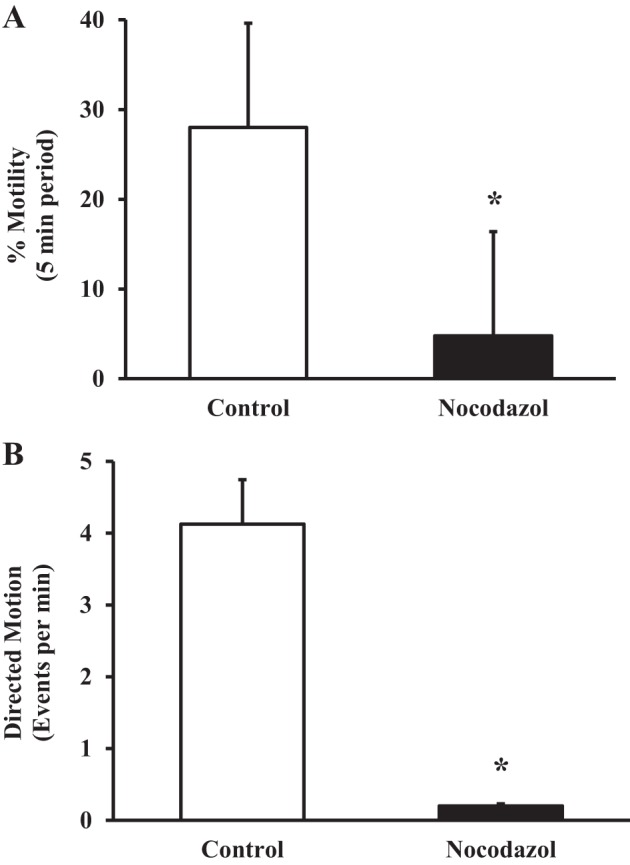
Effect of nocodazole on mitochondrial movement. A: summary data showing a decrease in mitochondrial motility in ASM cells exposed to 10 µM nocodazole for 30 min. B: nocodazole exposure markedly reduced the number of mitochondria with directed motion trajectories. Values are means ± SE. *Significant effect (P < 0.05, N = 5).
TNFα reduces mitochondrial motility.
To examine whether TNFα exposure affects mitochondrial motility, human ASM cells were exposed to 20 ng/ml TNFα for 24 h, and mitochondrial motility was examined at 37°C before and after stimulation with 10 µM histamine. In human ASM cells exposed to TNFα, overall mitochondrial motility under resting conditions was significantly reduced compared with control ASM cells (20.4 ± 2.3% vs. 29.2 ± 3.1%, N = 8 patients, n = 51 ASM cells, P < 0.05; Fig. 6A). Similar to control ASM cells, histamine stimulation further reduced overall mitochondrial motility in TNFα-exposed ASM cells (17.1 ± 2.1%, N = 8 patients, n = 51 ASM cells, P < 0.05; Fig. 6A).
Fig. 6.
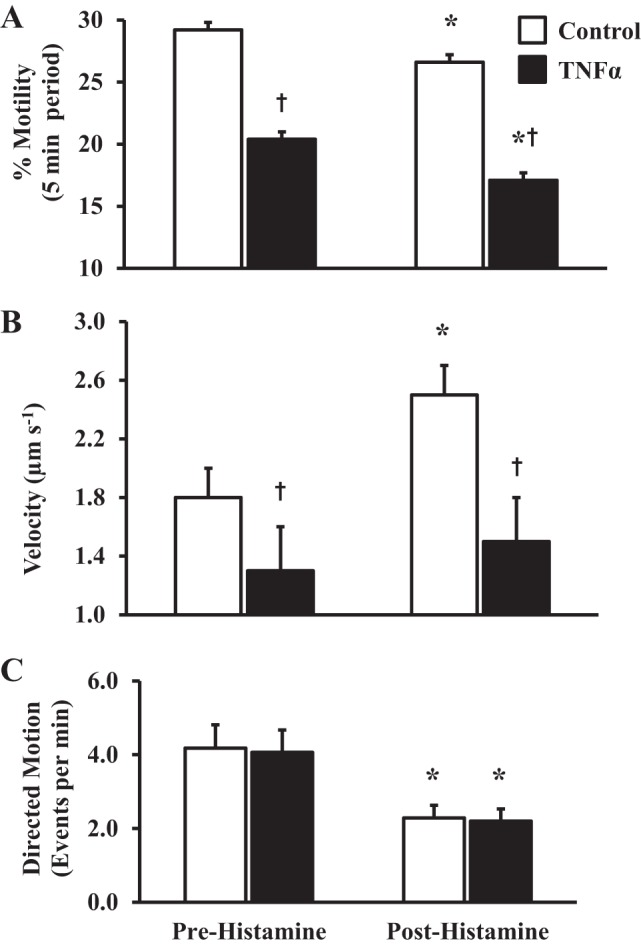
Effect of TNFα on overall mitochondrial motility. A: summary data showing significantly reduced overall mitochondrial motility in human ASM cells exposed to 20 ng/ml TNFα for 24 h compared with control ASM cells. Values are means ± SE. †Significant effect (P < 0.05) compared with control. *Significant effect of 10 µM histamine stimulation (P < 0.05) compared with resting conditions. B: summary data showing decrease in average velocity of directed trajectory mitochondrial movement in ASM cells exposed to TNFα for 24 h compared with control ASM cells. Values are means ± SE. †Significant effect (P < 0.05) compared with control. *Significant effect of 10 µM histamine stimulation (P < 0.05) compared with resting conditions. C: summary data showing no significant reduction in the number of mitochondria with directed motion trajectories after 24 h of TNFα exposure but an increase in the number of mitochondria with directed motion trajectories in cells stimulated with 10 µM histamine. Values are means ± SE. *Significant effect of histamine stimulation (P < 0.05, N = 8) compared with resting conditions.
Under resting conditions, TNFα exposure did not affect the incidence of mitochondria with directed motion trajectories compared with control ASM cells (20 and 21 mitochondria per ASM cell, respectively, N = 8 patients, n = 51 ASM cells; Fig. 6C). Similar to controls, in TNFα-exposed ASM cells, histamine reduced the number of mitochondria displaying directed motion trajectories (11 and 11 mitochondria in TNFα-exposed and control ASM cells, N = 8 patients, n = 51 ASM cells; Fig. 6C). TNFα exposure also slowed the average velocity of mitochondrial-directed motion trajectories during resting conditions compared with controls (1.3 ± 0.3 vs. 1.8 ± 0.2 µm/s, N = 8 patients, n = 51 ASM cells, P < 0.05; Fig. 6B). Unlike the histamine-induced increase in average velocity of directed motion trajectories in control ASM cells, histamine stimulation did not affect the average velocity of directed motion trajectories in TNFα-exposed ASM cells (1.5 ± 0.3 µm/s, N = 8 patients, n = 51 ASM cells, P < 0.05; Fig. 6B).
TNFα reduces Miro and Milton protein expression.
The Miro-Milton complex has been identified as a major regulator of mitochondrial motility along the microtubules. We found that both Miro and Milton proteins are expressed in human ASM cells (Fig. 7A) and that 24 h of TNFα exposure reduced expression of both Miro (89 ± 11% decrease) and Milton (86 ± 14% decrease) compared with control ASM cells (N = 5 patients, P < 0.05; Fig. 7).
Fig. 7.
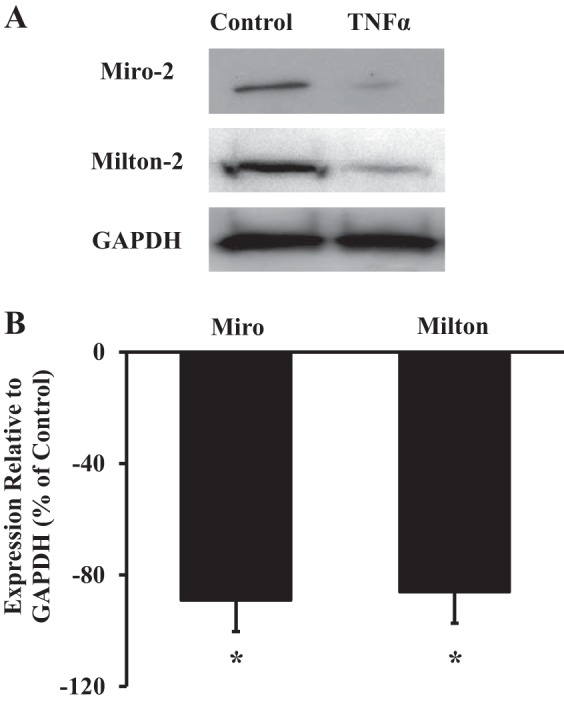
Effect of TNFα on Miro and Milton protein expression. A: Western blot analysis of Miro-2 and Milton-2 expression in human ASM cells with and without 24 h of exposure to 20 ng/ml TNFα. GAPDH expression was used as a loading control. B: summary data showing that TNFα decreases both Miro-2 and Milton-2 expression in human ASM cells. Values are means ± SE. *Significant effect (P < 0.05, N = 5).
TNFα decreases proximity of mitochondria to the SR.
Proximity of mitochondria to the SR was examined in human ASM cells using the Manders' colocalization coefficients M1 (mitochondria colocalization with the SR) and M2 (SR colocalization with mitochondria) (Fig. 8). In TNFα-exposed human ASM cells, mitochondria colocalization with the SR was reduced by ~62% compared with control ASM cells (M1 = 0.27 ± 0.14 vs. 0.72 ± 0.26, N = 5 patients, n = 49 ASM cells, P < 0.05). In contrast, TNFα did not affect SR colocalization with mitochondria (M2 = 0.45 ± 0.29 and 0.47 ± 0.15 for TNFα-exposed and control ASM cells, respectively, N = 5 patients, n = 49 ASM cells, P > 0.05; Fig. 8).
Fig. 8.

Effect of TNFα on proximity of mitochondria to the sarcoplasmic reticulum (SR). Top: fluorescence images of human ASM cells loaded simultaneously with BODIPY FL thapsigargin (endoplasmic reticulum Ca2+ pumps, green) to visualize SR and MitoTracker Red CMXRos to visualize mitochondria (red). Bottom: Manders' colocalization coefficients M1 (mitochondria colocalization with the SR) and M2 (SR colocalization with mitochondria) were used to estimate proximity of mitochondria to the SR. In ASM cells exposed to TNFα, proximity of mitochondria to the SR (M1) was significantly decreased. Values are means ± SE. *Significant effect (P < 0.05, N = 5).
TNFα enhances [Ca2+]cyt but reduces [Ca2+]mito responses to histamine.
Under resting conditions, TNFα exposure did not affect basal [Ca2+]cyt or [Ca2+]mito compared with control human ASM cells (N = 5 patients, P > 0.05; Fig. 9). In TNFα-exposed cells, transient [Ca2+]cyt responses induced by histamine significantly increased compared with control ASM cells (N = 5 patients, P < 0.05; Fig. 9). However, the transient [Ca2+]mito response to histamine was not increased in TNFα-exposed ASM cells (Fig. 9).
Fig. 9.
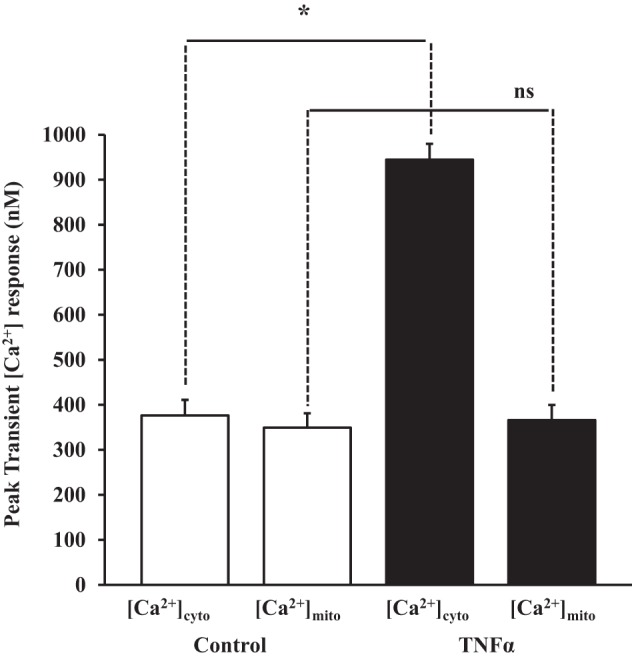
Effect of TNFα on cytosolic and mitochondrial Ca2+ ([Ca2+]cyt and [Ca2+]mito) response to histamine. Summary data show significant increases in the peak of the transient [Ca2+]cyt response to histamine in TNFα-exposed compared with control human ASM cells. Peak of the transient [Ca2+]mito response to histamine was comparable in ASM cells with or without exposure to TNFα. Values are means ± SE. *Significant effect (P < 0.05, interaction). ns, Not significant.
DISCUSSION
The results of the present study indicate that, in human ASM cells at basal [Ca2+]cyt levels, there are two forms of mitochondrial movement, random and directed, that are affected by temperature and, thus, reflect energy-requiring physiological processes. The basal movement of mitochondria appears to be necessary to establish proximity of mitochondria to the SR. During the transient increase in [Ca2+]cyt induced by histamine, mitochondrial movement is transiently reduced; this is associated with the transient increase in [Ca2+]mito. Exposure of human ASM cells to TNFα for 24 h reduced expression of Miro and Milton, which was associated with a decrease in basal mitochondrial movement and reduced proximity of mitochondria to the SR. TNFα exposure increased the [Ca2+]cyt response to both histamine and ACh but had reduced the transient [Ca2+]mito response.
Mitochondrial movement.
Most available information about mitochondrial movement comes from other cell types, e.g., neuronal axons, where ~30% of mitochondria are motile and the remaining mitochondria are stationary (53, 78). A similar proportion of motile mitochondria was found in human ASM cells in the present study, and two types of mitochondrial movement were characterized: movement in a directed trajectory and a kinetic, more random motion. In human ASM cells, mitochondrial motility was found to be primarily a kinetic/random type of movement. The smaller fusiform shape of the ASM cell may explain the relatively low incidence of mitochondria exhibiting movement in a directed trajectory. In comparison, neurons have elongated axons and dendrites extending for millimeters. It is possible that, in ASM cells, mitochondria are already relatively localized where they are needed, whereas mitochondrial transport may be required in neurons.
Mitochondrial movement is primarily mediated through interactions with microtubule filaments (58, 78). Accordingly, mitochondrial movement in human ASM cells was severely disrupted in the presence of nocodazole, which interferes with the polymerization of microtubules. Of particular interest is the Milton-Miro interaction, which couples mitochondria to microtubules (34, 51, 55, 80, 87). Both Milton and Miro proteins are expressed in human ASM cells and may underlie mitochondrial movement at basal [Ca2+]cyt levels and the slowing of mitochondrial movement when [Ca2+]cyt levels were transiently increased during agonist stimulation. In addition to microtubules, actin filaments might regulate the pool of mitochondria in a stationary state by tethering mitochondria, thereby halting mitochondrial motility (78, 81). In this regard, agonist stimulation of ASM cells increases actin polymerization, which is also important in force generation (47, 57).
The exact mechanism by which mitochondrial movement is regulated under basal [Ca2+]cyt conditions is still under investigation, but our results implicate mitochondrial interaction with Miro and Milton. In other cell types, several studies have shown that, with a transient elevation of [Ca2+]cyt, there is slowing of mitochondrial movement (14, 66, 82, 92). The exact mechanism is not known, but it has been proposed that the elevated cytosolic Ca2+ binds to the EF-hand domains of Miro and, thus, disrupts the Miro-Milton interaction (51, 78). Accordingly, several studies have shown that mitochondrial movement is halted when [Ca2+]cyt transiently increases in cells (14, 66, 82, 92). This modulation of mitochondrial movement is believed to be involved in a transient increase in [Ca2+]mito and, with it, ATP production (excitation-energy coupling) (9, 11, 13, 15, 20, 32, 52, 77, 78, 93). In human ASM cells, agonist stimulation induces a coupled transient elevation in both [Ca2+]cyt and [Ca2+]mito (21, 22). The results of the present study suggest that a slowing of mitochondrial movement may be important to enhance the coupling of [Ca2+]cyt and [Ca2+]mito during agonist stimulation.
Temperature sensitivity of mitochondrial movement.
The temperature sensitivity of mitochondrial movement in human ASM cells in the present study is consistent with that reported in other cell types (29, 54). It has been suggested that the reduced mitochondrial movement at lower temperatures is a consequence of increased [Ca2+]cyt, which would disrupt the Miro-Milton interaction. Changes in [Ca2+]cyt are also likely to impact a large number of Ca2+-dependent enzymatic reactions. In addition to [Ca2+]cyt, temperature is likely to affect a large number of enzymatic reactions, including the motor-adaptor complex, which mediates transport of mitochondria along microtubules independent of changes in [Ca2+]cyt. We found that only a small fraction (5%) of mitochondria were motile at 20°C in human ASM cells. By comparison, ∼30% of mitochondria were motile at 37°C, which is comparable to the proportion of motile mitochondria reported in neurons (53).
Mitochondrial movement and the “hot-spot theory” for mitochondrial Ca2+ uptake.
As mentioned previously, a transient elevation in [Ca2+]cyt in response to agonist stimulation reduces mitochondrial movement, which is believed to facilitate mitochondrial Ca2+ uptake and ATP production. Mitochondrial Ca2+ uptake is mediated by MCU, which has relatively low affinity for Ca2+ (e.g., in the 5–10 µM range) (7, 19, 32, 36, 37, 39, 68, 70). Such high [Ca2+]cyt levels (in the µM range) are only achieved within localized microdomains (i.e., hot spots) of the cell in the vicinity of Ca2+ flux sites at the SR and PM. Proximity of mitochondria to the SR and PM, where Ca2+ release occurs, is therefore a determining factor in mitochondrial Ca2+ uptake (32, 36, 37, 39, 67, 68, 70). As in other cell types (61, 69, 79), mitochondria are often located near the SR or PM in human ASM cells (18, 22). Changes in basal mitochondrial movement are likely to alter mitochondrial distribution within the cell and the proximity of the mitochondria to the SR and PM with downstream effects on the transient regulation of mitochondrial Ca2+ uptake and ATP production. The influence of other factors in mitochondrial distribution and proximity of mitochondria to the SR, including heterogeneity of the [Ca2+]cyt response or cytoskeleton remodeling associated with ASM contraction, poses interesting questions for further study.
Link between excitation-contraction coupling and energy-contraction coupling
Mitochondria provide the energy source to meet most of the energetic demands of the cell by replenishing ATP levels through oxidative phosphorylation: consumption of both intramitochondrial NADH and O2 to synthesize ATP from ADP and Pi. There is substantial evidence to indicate that increasing [Ca2+]mito plays an important role in cellular energetics (24, 25, 33, 35, 85). In many cell types (31, 42, 71), including smooth muscle cells (17, 22, 26, 59, 83), an increase in [Ca2+]cyt is temporally coupled to an increase in [Ca2+]mito (i.e., excitation-energy coupling). A transient increase in [Ca2+]cyt is the initial event in a multistep cascade, called excitation-contraction coupling, which causes muscle to generate force or contract in response to agonist stimulation and, thereby, increase ATP consumption. Given that [Ca2+]mito can influence mitochondrial ATP production, it is important to couple the transient elevation of [Ca2+]cyt to a transient increase in [Ca2+]mito (20, 21, 77).
Inflammation and mitochondrial movement.
Inflammatory cytokines such as TNFα increase both [Ca2+]cyt and force responses to agonist stimulation in human ASM cells (1, 2, 22, 44, 72, 74–76, 86, 91). However, TNFα paradoxically blunts the transient [Ca2+]mito response to agonist stimulation (20–22). In the present study we showed that TNFα reduces basal mitochondrial movement in human ASM cells, which would be consistent with a reduction in Miro and Milton expression. Furthermore, we found that TNFα exposure reduced proximity of mitochondria to the SR, which is also consistent with the uncoupling of transient [Ca2+]cyt and [Ca2+]mito responses to agonist stimulation due to an inability to establish hot spots for mitochondrial Ca2+ influx via the MCU.
In human ASM cells, TNFα increases force generation in response to agonist stimulation and, therefore, should be accompanied by an increase in mitochondrial production of ATP, which is stimulated by mitochondrial Ca2+ uptake. The effect of TNFα exposure on mitochondrial movement and proximity of mitochondria to the SR will impact excitation-energy coupling in human ASM cells, such that the increased metabolic demands of airway hyperreactivity are not met by an increase in mitochondrial production of ATP, leading to metabolic/oxidative stress.
Physiological role of mitochondrial movement.
Mitochondrial movement is required to supply ATP in distant sites of the cell (6, 8, 9, 11, 13, 15, 32, 52, 78, 93). The results of the present study suggest that mitochondrial movement under basal [Ca2+]cyt conditions is necessary to establish initial proximity of mitochondria to areas where there will be transient Ca2+ flux during agonist stimulation, e.g., Ca2+ release from the SR or influx through the PM. Then the higher transient levels of [Ca2+]cyt disrupt movement and lock mitochondria to these Ca2+ flux sites for excitation-energy coupling. Furthermore, mitochondrial proximity may also be associated with Ca2+ buffering and shaping the transient Ca2+ signals (32, 36, 37, 39, 67, 68, 70). Importantly, the close proximity of mitochondria to the hot spots near the SR and PM facilitates Ca2+ flux into the mitochondria and enhances oxidative metabolism (8, 9, 11–16, 24, 25, 32, 33, 35, 52, 78, 85, 93). The mitochondria-SR relationship is also particularly important in formation of the mitochondria-associated endoplasmic reticulum membrane (MAM) (30, 88). Recent studies have shown the importance of this interorganelle communication for many cellular functions such as Ca2+ homeostasis and regulation of lipid metabolism (30, 88). In this regard, how mitochondrial movement plays a role in the regulation of MAM is under scrutiny. Mitochondrial movement is also involved in the quality control of mitochondria and is closely associated with mitochondrial fission and fusion (8, 16). We recently reported that mitochondrial fusion was altered in ASM cells from asthmatic patients, resulting in a more fragmented mitochondrial network (4). However, mitochondrial movement was not examined in this study.
In conclusion, two types of mitochondrial motion are observed in human ASM cells: a kinetic/random type of overall motility and mitochondrial movement in a directed trajectory. Both types of mitochondrial motion were affected by temperature, nocodazole-induced disruption of microtubule polymerization, and transient elevation of [Ca2+]cyt induced by histamine. TNFα exposure for 24 h reduced overall mitochondrial motility and directed trajectory mitochondrial movements, most likely due to reduced expression of Miro and Milton proteins. A decrease in mitochondrial movement and proximity of mitochondria to the SR in ASM cells induced by proinflammatory cytokines may be involved in reduced mitochondrial Ca2+ uptake. This effect of TNFα exposure on mitochondrial movement and proximity of mitochondria to the SR will impact excitation-energy coupling in human ASM cells, such that the increased metabolic demands of airway hyperreactivity are not met by an increase in ATP synthesis, which may result in metabolic/oxidative stress.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-126451 (to G. C. Sieck).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.F.D., V.A.Z., and M.A.T. performed experiments; P.F.D., V.A.Z., and M.A.T. analyzed data; P.F.D., Y.S.P., and G.C.S. interpreted results of experiments; P.F.D., M.A.T., Y.S.P., and G.C.S. prepared figures; P.F.D., Y.S.P., and G.C.S. drafted manuscript; P.F.D., Y.S.P., and G.C.S. edited and revised manuscript; P.F.D., V.A.Z., M.A.T., Y.S.P., and G.C.S. approved final version of manuscript; Y.S.P. and G.C.S. conceived and designed research.
REFERENCES
- 1.Amrani Y, Chen H, Panettieri RA Jr. Activation of tumor necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma? Respir Res 1: 49–53, 2000. doi: 10.1186/rr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amrani Y, Panettieri RA Jr. Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol 2: 39–45, 2002. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta 1757: 692–699, 2006. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YS. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306: L840–L854, 2014. doi: 10.1152/ajplung.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aravamudan B, Thompson M, Pabelick C, Prakash YS. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J Cell Mol Med 16: 812–823, 2012. doi: 10.1111/j.1582-4934.2011.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnhart EL. Mechanics of mitochondrial motility in neurons. Curr Opin Cell Biol 38: 90–99, 2016. doi: 10.1016/j.ceb.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 7.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol 17: 502–510, 2007. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 10.Brough D, Schell MJ, Irvine RF. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem J 392: 291–297, 2005. doi: 10.1042/BJ20050738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carafoli E. The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta 1797: 595–606, 2010. doi: 10.1016/j.bbabio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252, 2006. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22: 79–99, 2006. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 14.Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26: 7035–7045, 2006. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 14: R283–R289, 2005. 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 18: R169–R176, 2009. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T, Zhu L, Wang T, Ye H, Huang K, Hu Q. Mitochondria depletion abolishes agonist-induced Ca2+ plateau in airway smooth muscle cells: potential role of H2O2. Am J Physiol Lung Cell Mol Physiol 298: L178–L188, 2010. doi: 10.1152/ajplung.00134.2009. [DOI] [PubMed] [Google Scholar]
- 18.Dai J, Kuo KH, Leo JM, van Breemen C, Lee CH. Rearrangement of the close contact between the mitochondria and the sarcoplasmic reticulum in airway smooth muscle. Cell Calcium 37: 333–340, 2005. doi: 10.1016/j.ceca.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 19.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delmotte P, Jia L, Sieck G. The role of mitochondria in calcium regulation in airway smooth muscle. In: Calcium Signaling in Airway Smooth Muscle Cells, edited by Wang Y-X. New York: Springer International, 2014, p. 211–234. doi: 10.1007/978-3-319-01312-1_11 [DOI] [Google Scholar]
- 21.Delmotte P, Sieck GC. Interaction between endoplasmic/sarcoplasmic reticulum stress (ER/SR stress), mitochondrial signaling and Ca2+ regulation in airway smooth muscle (ASM). Can J Physiol Pharmacol 93: 97–110, 2015. doi: 10.1139/cjpp-2014-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delmotte P, Yang B, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am J Physiol Cell Physiol 303: C244–C256, 2012. doi: 10.1152/ajpcell.00414.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demaurex N, Poburko D. Cell biology. A revolving door for calcium. Science 326: 57–58, 2009. doi: 10.1126/science.1180482. [DOI] [PubMed] [Google Scholar]
- 24.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787: 1309–1316, 2009. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol 52: 451–466, 1990. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- 26.Drummond RM, Tuft RA. Release of Ca2+ from the sarcoplasmic reticulum increases mitochondrial [Ca2+] in rat pulmonary artery smooth muscle cells. J Physiol 516: 139–147, 1999. doi: 10.1111/j.1469-7793.1999.139aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28: 339–348, 2000. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 28.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–C742, 2011. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson IB, Reid MS, Romani RJ. Effects of low temperature and respiratory inhibitors on calcium flux in plant mitochondria. Plant Physiol 77: 877–880, 1985. doi: 10.1104/pp.77.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 62: 1–15, 2017. doi: 10.1016/j.ceca.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Filippin L, Magalhães PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem 278: 39224–39234, 2003. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- 32.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38: 280–290, 2010. doi: 10.1016/j.molcel.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51: 2959–2973, 2012. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires Milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173: 545–557, 2006. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787: 1324–1333, 2009. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 36.Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28: 285–296, 2000. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- 37.Gunter TE, Gunter KK. Uptake of calcium by mitochondria: transport and possible function. IUBMB Life 52: 197–204, 2001. doi: 10.1080/15216540152846000. [DOI] [PubMed] [Google Scholar]
- 38.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol 258: C755–C786, 1990. [DOI] [PubMed] [Google Scholar]
- 39.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta 1787: 1291–1308, 2009. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett 567: 96–102, 2004. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 41.Hajnóczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4,5-trisphosphate receptors by Ca2+. J Biol Chem 274: 14157–14162, 1999. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- 42.Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415–424, 1995. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 43.Hartman WR, Smelter DF, Sathish V, Karass M, Kim S, Aravamudan B, Thompson MA, Amrani Y, Pandya HC, Martin RJ, Prakash YS, Pabelick CM. Oxygen dose responsiveness of human fetal airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 303: L711–L719, 2012. doi: 10.1152/ajplung.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter I, Cobban HJ, Vandenabeele P, MacEwan DJ, Nixon GF. Tumor necrosis factor-α-induced activation of RhoA in airway smooth muscle cells: role in the Ca2+ sensitization of myosin light chain20 phosphorylation. Mol Pharmacol 63: 714–721, 2003. doi: 10.1124/mol.63.3.714. [DOI] [PubMed] [Google Scholar]
- 45.Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep 7: 390–396, 2006. doi: 10.1038/sj.embor.7400620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia L, Delmotte P, Aravamudan B, Pabelick CM, Prakash YS, Sieck GC. Effects of the inflammatory cytokines TNF-α and IL-13 on stromal interaction molecule-1 aggregation in human airway smooth muscle intracellular Ca2+ regulation. Am J Respir Cell Mol Biol 49: 601–608, 2013. doi: 10.1165/rcmb.2013-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones KA, Perkins WJ, Lorenz RR, Prakash YS, Sieck GC, Warner DO. F-actin stabilization increases tension cost during contraction of permeabilized airway smooth muscle in dogs. J Physiol 519: 527–538, 1999. doi: 10.1111/j.1469-7793.1999.0527m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koopman WJ, Distelmaier F, Esseling JJ, Smeitink JA, Willems PH. Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods 46: 304–311, 2008. doi: 10.1016/j.ymeth.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 49.Koopman WJ, Visch HJ, Smeitink JA, Willems PH. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A 69: 1–12, 2006. doi: 10.1002/cyto.a.20198. [DOI] [PubMed] [Google Scholar]
- 50.Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289: C881–C890, 2005. doi: 10.1152/ajpcell.00104.2005. [DOI] [PubMed] [Google Scholar]
- 51.Lee KS, Lu B. The myriad roles of Miro in the nervous system: axonal transport of mitochondria and beyond. Front Cell Neurosci 8: 330, 2014. doi: 10.3389/fncel.2014.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 53.Lovas JR, Wang X. The meaning of mitochondrial movement to a neuron’s life. Biochim Biophys Acta 1833: 184–194, 2013. doi: 10.1016/j.bbamcr.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luvisetto S, Schmehl I, Intravaia E, Conti E, Azzone GF. Mechanism of loss of thermodynamic control in mitochondria due to hyperthyroidism and temperature. J Biol Chem 267: 15348–15355, 1992. [PubMed] [Google Scholar]
- 55.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61: 541–555, 2009. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103: 857–862, 1992. [DOI] [PubMed] [Google Scholar]
- 57.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519: 829–840, 1999. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 104: 917–927, 1993. [DOI] [PubMed] [Google Scholar]
- 59.Nassar A, Simpson AW. Elevation of mitochondrial calcium by ryanodine-sensitive calcium-induced calcium release. J Biol Chem 275: 23661–23665, 2000. doi: 10.1074/jbc.M000457200. [DOI] [PubMed] [Google Scholar]
- 60.Nicholls DG. Mitochondria and calcium signaling. Cell Calcium 38: 311–317, 2005. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 61.Pacher P, Csordás P, Schneider T, Hajnóczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol 529: 553–564, 2000. doi: 10.1111/j.1469-7793.2000.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrústegui J, Palmieri F. Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J 20: 5060–5069, 2001. doi: 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poburko D, Liao CH, van Breemen C, Demaurex N. Mitochondrial regulation of sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ Res 104: 104–112, 2009. doi: 10.1161/CIRCRESAHA.108.180612. [DOI] [PubMed] [Google Scholar]
- 64.Prakash YS, Iyanoye A, Ay B, Sieck GC, Pabelick CM. Store-operated Ca2+ influx in airway smooth muscle: interactions between volatile anesthetic and cyclic nucleotide effects. Anesthesiology 105: 976–983, 2006. doi: 10.1097/00000542-200611000-00019. [DOI] [PubMed] [Google Scholar]
- 65.Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am J Physiol Cell Physiol 272: C966–C975, 1997. [DOI] [PubMed] [Google Scholar]
- 66.Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23: 7881–7888, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262: 744–747, 1993. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 68.Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta 1787: 1342–1351, 2009. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766, 1998. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 70.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408, 2006. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 71.Rudolf R, Mongillo M, Magalhães PJ, Pozzan T. In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol 166: 527–536, 2004. doi: 10.1083/jcb.200403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakai Y, Kwan CY. Calcium regulation and contractile dysfunction of smooth muscle. Biol Signals 2: 305–312, 1993. doi: 10.1159/000109511. [DOI] [PubMed] [Google Scholar]
- 73.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105: 20728–20733, 2008. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sathish V, Delmotte PF, Thompson MA, Pabelick CM, Sieck GC, Prakash YS. Sodium-calcium exchange in intracellular calcium handling of human airway smooth muscle. PLoS One 6: e23662, 2011. doi: 10.1371/journal.pone.0023662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sathish V, Leblebici F, Kip SN, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Regulation of sarcoplasmic reticulum Ca2+ reuptake in porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 294: L787–L796, 2008. doi: 10.1152/ajplung.00461.2007. [DOI] [PubMed] [Google Scholar]
- 76.Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 297: L26–L34, 2009. doi: 10.1152/ajplung.00026.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schaible N, Delmotte P, Sieck G. Mitochondrial excitation-energy coupling in airway smooth muscle. In: Mitochondrial Function in Lung Health and Disease, edited by Natarajan V, Parinandi NL. Totowa, NJ: Humana, 2014, p. 96–116. doi: 10.1007/978-1-4939-0829-5_5 [DOI] [Google Scholar]
- 78.Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5: 5, 2013. doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32: 97–104, 2000. doi: 10.1023/A:1005520714221. [DOI] [PubMed] [Google Scholar]
- 80.Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 36: 1063–1077, 2002. doi: 10.1016/S0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- 81.Stürmer K, Baumann O, Walz B. Actin-dependent light-induced translocation of mitochondria and ER cisternae in the photoreceptor cells of the locust Schistocerca gregaria. J Cell Sci 108: 2273–2283, 1995. [DOI] [PubMed] [Google Scholar]
- 82.Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta 1763: 442–449, 2006. doi: 10.1016/j.bbamcr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 83.Szado T, Kuo KH, Bernard-Helary K, Poburko D, Lee CH, Seow C, Ruegg UT, van Breemen C. Agonist-induced mitochondrial Ca2+ transients in smooth muscle. FASEB J 17: 28–37, 2003. doi: 10.1096/fj.02-0334com. [DOI] [PubMed] [Google Scholar]
- 84.Szalai G, Csordás G, Hantash BM, Thomas AP, Hajnóczky G. Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem 275: 15305–15313, 2000. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- 85.Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 52: 28–35, 2012. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tirumurugaan KG, Jude JA, Kang BN, Panettieri RA, Walseth TF, Kannan MS. TNF-α induced CD38 expression in human airway smooth muscle cells: role of MAP kinases and transcription factors NF-κB and AP-1. Am J Physiol Lung Cell Mol Physiol 292: L1385–L1395, 2007. doi: 10.1152/ajplung.00472.2006. [DOI] [PubMed] [Google Scholar]
- 87.van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77: 485–502, 2013. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 88.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843: 2253–2262, 2014. doi: 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 89.Wang CC, Lin WN, Lee CW, Lin CC, Luo SF, Wang JS, Yang CM. Involvement of p42/p44 MAPK, p38 MAPK, JNK, and NF-κB in IL-1β-induced VCAM-1 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288: L227–L237, 2005. doi: 10.1152/ajplung.00224.2004. [DOI] [PubMed] [Google Scholar]
- 90.Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136: 163–174, 2009. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.White TA, Xue AL, Sieck GC, Wylam ME. CD38 involvement in TNF-α induced changes in airway smooth muscle Ca2+ homeostasis (Abstract). FASEB J 20: A1243–A1244, 2006. [Google Scholar]
- 92.Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167: 661–672, 2004. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]