

ABSTRACT
Regulatory T cells (Tregs) are immunosuppressive cells of the immune system that control autoimmune reactivity. Tregs also respond during immune reactions to infectious agents in order to limit immunopathological damage from potent effectors such as CD8+ cytolytic T lymphocytes. We have used the Friend virus (FV) model of retroviral infection in mice to investigate how viral infections induce Tregs. During acute FV infection, there is significant activation and expansion of thymus-derived (natural) Tregs that suppress virus-specific CD8+ T cell responses. Unlike conventional T cells, the responding Tregs are not virus specific, so the mechanisms that induce their expansion are of great interest. We now show that B cells provide essential signals for Treg expansion during FV infection. Treg responses are greatly diminished in B cell-deficient mice but can be restored by adoptive transfers of B cells at the time of infection. The feeble Treg responses in B cell-deficient mice are associated with enhanced virus-specific CD8+ T cell responses and accelerated virus control during the first 2 weeks of infection. In vitro experiments demonstrated that B cells promote Treg activation and proliferation through a glucocorticoid-induced receptor superfamily member 18 (GITR) ligand-dependent mechanism. Thus, B cells play paradoxically opposing roles during FV infection. They provide proliferative signals to immunsosuppressive Tregs, which slows early virus control, and they also produce virus-specific antibodies, which are essential for long-term virus control.
KEYWORDS: B cells, regulatory T cells, retroviruses, tumor necrosis factor receptors
IMPORTANCE
When infectious agents invade a host, numerous immunological mechanisms are deployed to limit their replication, neutralize their spread, and destroy the host cells harboring the infection. Since immune responses also have a strong capacity to damage host cells and tissues, their magnitude, potency, and duration are under regulatory control. Regulatory T cells are an important component of this control, and the mechanisms that induce them to respond and exert immunosuppressive regulation are of great interest. In the current report, we show that B cells, the cells responsible for making pathogen-specific antibodies, are also involved in promoting the expansion of regulatory T cells during a retroviral infection. In vitro studies demonstrated that they do so via stimulation of the Tregs through interactions between cell surface molecules: GITR interactions with its ligand (GITRL) on B cells and GITR on regulatory T cells. These findings point the way toward therapeutics to better treat infections and autoimmune diseases.
INTRODUCTION
CD4+ FOXP3+ regulatory T cells (Tregs) are immunomodulatory cells necessary for the prevention of both autoimmune disorders (1, 2) and deleterious inflammatory reactions during immune responses to infections (3–8). On the other hand, Treg activity during infections can have detrimental effects when their potent immunosuppressive effects prevent the clearance of pathogens and contribute to the establishment and maintenance of chronic infections (9–11). During acute Friend virus (FV) infection of mice, antigen-presenting cells (12) activate CD8+ T cells, which are essential for early virus control (12–16). FV infection also induces Tregs, which expand and inhibit the antiviral CD8+ T cell responses and thereby contribute to virus persistence (10, 17–20). Tregs can arise from conversion of conventional T cells into “peripherally derived” Tregs, which can be foreign antigen specific. Diverse mechanisms drive the activation of peripherally derived Tregs, some of which have been elucidated for certain pathogens (7). For example, Treg conversion can occur directly, such as through secretion of transforming growth factor beta (TGF-β) mimics (21) or polysaccharide cross-linking of T cell receptors (TCRs) (22). Treg conversion can also occur indirectly such as through pathogen-induced polarization of dendritic cells (DCs) through Toll-like receptors (TLRs) to induce production of interleukin-10 (IL-10) (23–25). However, the Tregs that respond to FV infection are “natural” Tregs or “thymus-derived” Tregs (tTregs), and conversion of conventional CD4+ T cells into Tregs during FV infection does not occur (26). A strong Treg effect on viral immunity can be observed when Tregs are transiently depleted during either acute or chronic FV infection. Such depletion leads to elevated CD8+ T cell responses and reduced infection levels (11, 18). Thus, Tregs play an important role during FV infection, but the mechanisms by which viral infections induce Tregs are still not fully understood. FV-induced Tregs are not FV specific (27), so their mechanisms of induction are different from those that induce conventional Th1 and Th2 type responses. Studies have shown that FV-activated CD8+ T cells upregulate membrane-bound tumor necrosis factor alpha (TNF-α), which binds to tumor necrosis factor (TNF) receptor II (TNFRII) on a subset of endogenous retroviral antigen-specific Tregs and stimulates their proliferation (26, 28). In this way, the virus-specific CD8+ T cells provide the context of infection and the second signal for Treg activation and expansion. However, that mechanism accounts for only about 10% of the total Treg response. Understanding the remaining induction mechanisms will provide the basis for the rational design of therapeutics that can fine tune the Treg response, either downward to enhance T cell immunity or upward to suppress immunopathological responses.
In the current study, we investigated whether B cells play a role in the induction of Treg responses during Friend virus (FV) infection. Several studies have demonstrated that B cells can affect Treg responses to autoimmune diseases (29–32). In mouse studies using both genetically B cell-deficient and anti-CD20-treated animals, B cells have been reported to have both positive and negative effects on the size and function of the Treg subset (reviewed in references 33 and 34). Some of the differences in experimental outcomes can be attributed to the time during autoimmune disease when the depleting antibodies (Abs) were given, to mouse strain differences (NOD [33] and BALB/c [35] mice give results different from those given by C57BL/6 mice), and/or to whether the Tregs induced were peripherally derived or thymus derived. In the C57BL/6 genetic background, similarly to the results seen with the mice used in the current study (C57BL/10), genetic deficiency in B cells (μMT) or depletion of B cells with anti-CD20 antibody causes significant reductions in Treg numbers in spleen and Peyer’s patches which could be restored by adoptive transfers of B cells (36). This homeostatic maintenance of B cell-dependent peripheral Treg numbers was shown to rely on glucocorticoid-induced receptor superfamily member 18 (GITR) on Tregs and GITR ligand (GITRL) on B cells (36). These interactions with Tregs likely explain the protective effect of B cells in experimental autoimmune encephalomyelitis (EAE) (36–38) as well. B cells also help maintain Treg homeostasis in gut-associated lymphoid tissues to prevent dextran sulfate sodium-induced colitis (39). In stark contrast, B cells have been shown to suppress Treg activity in a mouse model of rheumatoid arthritis (35). In that study, the authors found that B cells inhibited the expansion and function of Tregs. In the current study, we investigated whether B cells also play a role in the induction of thymus-derived Tregs during infectious disease, specifically, during Friend retrovirus infection of mice. The experiments demonstrated a significant role for B cells in promoting the in vivo Treg response to retroviral infection.
RESULTS
Fewer Tregs in B cell-deficient mice.
Results from a previous study suggested that CD8+ T cell responses to Friend virus (FV) infection were more robust in B10.μMT (B cell-deficient) mice than in wild-type (wt) mice (40). Since it is known that FV-specific CD8+ T cell responses are strongly suppressed by regulatory T cells (Tregs) (17, 19, 20) and that naive B cell-deficient mice have reduced levels of Tregs (36, 41), it was of interest to determine if B cell-deficient mice had normal FV-induced Treg responses. Analysis of Treg levels in naive B cell-deficient B10.μMT mice compared with wt C57BL/10 mice confirmed that B10.μMT mice had significantly decreased proportions of CD4+ T cells expressing the FOXP3+ Treg phenotype (Fig. 1A), averaging 3.8% in B10.μMT mice compared to 6.8% in wt mice (Fig. 1B). The mean levels of Treg proliferation (Ki-67+) in the two mouse strains were not significantly different (Fig. 1C). We also examined the proportions of naive CD8+ T cells in the two strains. Since a lack of B cells in B10.μMT mice results in spleens only about half the size of normal spleens, it is not appropriate to directly compare either the proportions or absolute numbers of lymphocytes expressing CD8. To make a relevant comparison, we analyzed the proportion of non-B spleen cells in each strain that expressed CD8 and found very similar levels (Fig. 1D). The mean levels of CD8+ T cell proliferation (Ki-67+) in the two mouse strains were also equivalent (Fig. 1E). Thus, the reduced proportions of Tregs in the B cell-deficient mice prior to FV infection did not affect the proportion of T cells expressing CD8 or their homeostatic level of proliferation.
FIG 1 .

Fewer Tregs in B cell-deficient mice. Spleens were harvested from naive C57BL/10 mice (A to E) or B cell-deficient μMT mice (B to E), and CD4, CD8, CD19, Ki-67, and FOXP3 expression was analyzed by flow cytometry. (A) Representative gating strategy for CD4+ T cells (top) or CD8+ T cells (bottom). (B) Quantification of the percentage of FOXP3+ cells within the CD4+ T cell gate. (C) Quantification of the percentage of Ki-67+ cells within the Treg (CD4+ FOXP3+) gate. (D) Quantification of the percentage of CD8+ cells within the non-B cell lymphocyte gate (CD19− lymphocytes). (E) Quantification of the percentage of Ki-67+ cells within the CD8+ T cell gate. Each dot in panels B to E represents an individual mouse pooled from experiments performed with 4 to 5 mice per group. Lines indicate the means of data for the group. In panels B to E, statistical significance was determined with a Student’s t test; ns, = P > 0.05.
B cell-deficient mice have impaired Treg induction during FV infection.
Next, the Treg responses to FV infection were investigated in a kinetic manner. Cohorts of mice were infected with FV, and levels of splenic Tregs (Fig. 2A) were analyzed at weekly intervals for the first 3 weeks of infection. Peak induction of Tregs typically occurs at 2 weeks postinfection (wpi) (26, 28). Absolute numbers of Tregs in the spleens were determined, not for direct comparison between B10 and B10.μMT mice but to analyze the kinetics of Treg expansion within each strain during FV infection. By 2 weeks postinfection, Tregs in wt mice expanded to approximately 20% of the CD4+ T cell level (Fig. 2B). In contrast, Tregs in B10.μMT mice expanded to only about 12% of the CD4+ T cell level in the spleens (Fig. 2B). A two-way analysis of variance (ANOVA) comparing the kinetics of the expansion of absolute numbers of Tregs in each mouse strain indicated that Tregs in wt mice expanded at a significantly higher rate than Tregs in B10.μMT mice (P value of <0.0001, Fig. 2C), and the difference is obvious by visual assessment of the graph. Treg numbers in wt mice continued to expand between 1 and 2 weeks postinfection, whereas Treg numbers in μMT mice decreased during that time period (Fig. 2C). Although fewer Tregs were present during FV infection of μMT mice, a higher proportion of their Tregs appeared to have been activated at both 1 and 2 weeks postinfection, as determined by CD43 expression (Fig. 2D and E). Analysis of activation by CD11a expression gave an equivalent result (data not shown). Thus, the Tregs in μMT mice appeared to have activated and proliferated for the first week of infection but were unable to sustain proliferation between 1 and 2 weeks postinfection. It appeared that the Tregs were missing a signal for continued proliferation, possibly a signal from the missing B cells.
FIG 2 .

Impaired Treg induction during FV infection of B cell-deficient mice. (A to E) C57BL/10 (B10) and μMT mice were infected with 20,000 SFFU of FV i.v. or left naïve, spleens were harvested at 1 week (B to E), 2 weeks (A to E), or 3 weeks (B and C) postinfection, and CD4, FOXP3, and CD43 expression was analyzed by flow cytometry. (A) Representative gating strategy for Tregs (CD4+ FOXP3+) in B10 mice (left) or μMT mice (right) at 2 wpi. (B to E) Quantification of the percentage of Tregs among CD4+ T cells (B), the total number of Tregs (C), the percentage of CD43+ Tregs (D), and the total number of CD43+ Tregs per spleen (E). In panels B to E, each dot represents an average of 5 to 12 mice from pooled experiments and error bars represent ± standard errors of the means (SEM). ***, P < 0.001; ****, P < 0.0001; ns, P > 0.05 (comparing B10 to μMT mice at the indicated time point, as determined by one-way ANOVA with Tukey's posttest for multiple comparisons).
B cells stimulate Treg proliferation and stabilize FOXP3 expression in vitro.
To determine whether B cells were providing a stimulus for Treg proliferation, untouched Tregs were obtained from FOXP3-GFP (FOXP3-green fluorescent protein) mice by fluorescence-activated cell sorter (FACS) analysis of GFP+ cells, labeled with CellTrace proliferation dye, and cocultured in vitro with or without B cells from naive or FV-infected mice. After 2.5 days of stimulation with anti-CD3 and IL-2, Treg proliferation was analyzed by flow cytometry. Figure 3A shows representative flow cytometry plots of two measures of proliferation, Ki-67 expression and dilution of CellTrace fluorescence. There was almost no proliferation in unstimulated Tregs (without anti-CD3 or IL-2) and only minimal proliferation in stimulated Tregs in the absence of B cells (Fig. 3B). In contrast, when B cells from either naive or FV-infected mice were cocultured with the Tregs, the rate of proliferation was over 60% (Fig. 3B). In addition, there was no significant expression of the CD43 activation marker on the Tregs in the absence of B cells (Fig. 3C). We also found that, in the absence of B cells, the presence of anti-CD3 and IL-2 was insufficient to induce Treg activation, as measured by an increase in CD43 mean fluorescence intensity (MFI). However, a significant increase in Treg activation was observed with the addition of B cells (Fig. 3C). Finally, stimulated Tregs cultured in the absence of B cells had reduced expression of FOXP3 (Fig. 3D). Thus, B cells promoted the stabilization of FOXP3 expression in Tregs as well as their activation and proliferation. B cells from naive mice worked as well as B cells from infected mice.
FIG 3 .

B cells induce Treg proliferation and activation in vitro. (A to E) Untreated Tregs were harvested by FACS analysis of GFP+ cells from FOXP3-GFP mice and labeled with CellTrace Violet. B cells were harvested from naive or FV-infected mice at 1 wpi with CD19+ beads and stained with CellTrace Far Red. Tregs were cultured in the absence of B cells or with B cells at a B cell/Treg ratio of either 10:1 (A to D) or 1:1 (E) in the presence of 10 µg/ml anti-CD3 and 10 ng/ml IL-2. As a control, unstimulated (Unstim) Tregs were cultured in the absence of anti-CD3 and IL-2. For panel E, naive B cells were either left untreated or treated with 10 µg/ml anti-GITRL for 1 h prior to coculture. After 2.5 days of coculture, cells were harvested and CD4+ CellTrace Far Red− Tregs were analyzed for FOXP3, Ki-67, CD43, and CellTrace violet expression by flow cytometry. (A) Representative dot plots of Ki-67 and CellTrace Violet expression on Tregs (CD4+ CellTrace Far Red−). The gate shows proliferating cells (Ki-67+ CellTrace Violet−). (B to E) Quantification of proliferation (% Ki-67+ CellTrace Violet−) (B and E), CD43 expression (C), and FOXP3 (D) expression among Tregs (CD4+ CellTrace Far Red−). In panels B to E, bars represent means ± SEM of data from 5 to 12 mice in pooled experiments. **, P > 0.01; ****, P > 0.0001 (as determined by one-way ANOVA with Tukey's posttest for multiple comparisons).
B cell “help” is partially dependent on GITR-GITRL interactions.
It has previously been shown that stimulation of Tregs via glucocorticoid-induced TNF receptor-related protein (GITR) interactions with its ligand (GITRL) promotes Treg proliferation (42). Furthermore, GITRL expression specifically on B cells has been shown to promote Treg proliferation in the context of experimental autoimmune encephalomyelitis (EAE) (36). To determine if blocking GITRL would reduce the capacity for B cells to induce the proliferation of Tregs, the proliferation experiments were repeated in the presence of either control antibody or anti-GITRL blocking antibody. Cocultures containing anti-GITRL still showed Treg proliferation, but there was significantly less seen than with control antibody (Fig. 3D). These results indicated that GITRL was a significant factor in B cell enhancement of Treg proliferation.
Enhanced CD8+ T cell responses in B cell-deficient mice.
Since Tregs have been shown to inhibit critical CD8+ T cell responses during FV infection (11, 20), it was of interest to determine if the decreased Treg responses in B10.μMT mice were associated with improved CD8+ T cell responses. CD8+ T cells from FV-infected B10 B10.μMT mice were analyzed using dextramers to detect T cells with receptors specific for the FV immunodominant Db-GagL epitope (43) (Fig. 4A). At 2 wpi, when Db-GagL responses become detectable, B10.μMT mice had a significantly higher proportion of dextramer-positive cells than wt B10 mice (Fig. 4A). Furthermore, they also had higher percentages (Fig. 4B) and absolute numbers of dextramer-positive effector cells making granzyme B cytotoxic granules (Fig. 4D). The proportion of all splenic CD8+ T cells that were activated and producing granzyme B at 2 wpi was significantly higher in B10.μMT mice (Fig. 4E), and despite their having significantly smaller spleens than wt B10 mice, the absolute numbers of activated, granzyme B-producing CD8+ T cells per spleen were equivalent in the two strains (Fig. 4F). These results indicated that impaired Treg responses in B cell-deficient mice were indeed associated with heightened activation and expansion of virus-specific CD8 T cells.
FIG 4 .

Enhanced anti-FV CD8+ T cell responses in B cell-deficient mice. (A to F) C57BL/10 (B10) mice and μMT mice were infected with 20,000 SFFU of FV i.v. or left naïve, spleens were harvested at 2 weeks (A to F) or 3 weeks (B to F) postinfection, and CD8, GrB, CD11a, and Db-GagL dextramer binding was analyzed by flow cytometry. (A) Representative gating strategy for CD8+ T cells at 2 wpi. (B to F) Quantification of the percentage of dextramer+ (B), percentage of dextramer+ GrB+ (C), absolute number of dextramer+ GrB+ (D), percentage of CD11a+ GrB+ (E), and absolute number of CD11a+ GrB+ (F) cells among splenic CD8+ T cells. In panels B to F, each dot represents an average of 5 to 12 mice from pooled experiments and error bars show ± SEM. *, P < 0.05; **, P < 0.01 (comparing B10 mice to μMT mice at the indicated time point, as determined by one-way ANOVA with Tukey's posttest for multiple comparisons).
Improved early virus clearance in B10.μMT mice.
Since CD8+ T cells have been shown to be important for early clearance of FV-infected cells (14), it was possible that the B cell-deficient mice might control FV infection better than wt mice. A predominant site of FV replication is the spleen, which contains an abundance of the Ter119+ erythroid cells that are the primary targets for infection (44). At 2 wpi, the total number of FV infectious centers (ICs) per spleen was significantly lower in B10.μMT mice than in wt B10 mice (Fig. 5A), but, as noted previously, their spleens were only about half of normal size. Therefore, the level of infection was also calculated as the number of infectious centers per gram of spleen. By 2 wpi, infection levels in spleens were more than 1 log10 lower in B10.μMT mice than in wt B10 mice (Fig. 5B). FV infection stimulates the proliferation of Ter119+ erythroid cells (45), and a reduction in erythroid cell proliferation was observed between 1 and 2 weeks postinfection in B10.μMT mice but not until between 2 and 3 weeks postinfection in wt B10 mice (Fig. 5C). Infection levels in monocytes, as determined by cell surface expression of FV glycosylated Gag (glycogag) antigen (44), was also lower in B10.μMT mice than in wt mice at 2 wpi (Fig. 5D). It is known that virus-specific antibodies are required for long-term control over FV (46, 47), which likely explains why B10.μMT mice failed to maintain control over FV infection beyond 2 wpi (Fig. 5B).
FIG 5 .

More-rapid control of FV in B cell-deficient mice. (A to D) C57BL/10 (B10) mice and μMT mice were infected with 20,000 SFFU of FV i.v. or left naïve, and spleens were harvested at 1, 2, or 3 weeks postinfection for infectious center assays. (A and B) Flow cytometry analysis of Ter119, CD11b, and FV glycogag (mAb34+) expression (see panels C and D). (A) Total FV infectious centers per spleen. (B) FV infectious centers per gram of spleen. (C) Percent Ter119+ (erythroid progenitor) cells within the total splenocyte gate. (D) Percent glycogag+ (% Infected, mAb34+) CD11b+ monocytes. Each dot represents an average of 5 to 12 mice from pooled experiments, and error bars show ± SEM. **, P < 0.01; ****, P < 0.0001 (comparing B10 mice to μMT mice at the indicated time point, as determined by one-way ANOVA with Tukey's posttest for multiple comparisons).
Adoptive transfer of B cells into B10.μMT mice restores Tregs.
We next sought to determine whether adoptive transfer of B cells into B cell-deficient μMT mice could restore FV-induced Treg responses. CD19+ B cells were enriched from naive wt B10 spleens by the use of magnetic bead columns, and 5 × 107 donor B cells were infused into B10.μMT recipients the day before infection with FV. Treg responses in the recipients were then analyzed at 2 wpi. Not only did the adoptive transfer of B cells into B10.μMT mice restore their Treg numbers to levels equivalent to those seen with wt B10 mice, but the B transfers also significantly improved the expansion of the Tregs in response to FV infection (Fig. 6). These results indicated that the FV-induced expansion of Tregs was dependent on the presence of B cells at the time of infection.
FIG 6 .
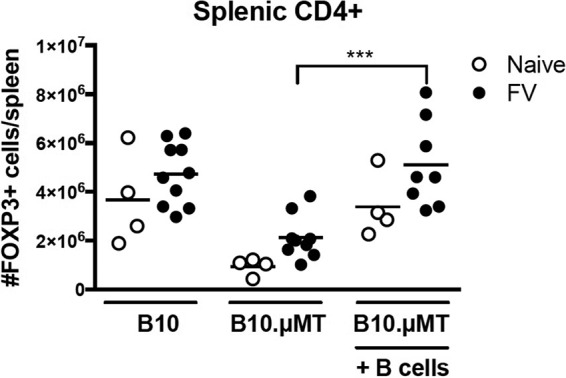
Induction of Tregs following adoptive transfer of B cells. Donor B cells were purified from naive B10 mice, and 5 × 107 B cells/mouse were injected i.v. into μMT mice 1 day before infection with 20,000 SFFU of FV i.v. or left naive. B10 mice or uMT mice without B cell adoptive transfer were used as controls. At 2 weeks postinfection, spleens were harvested and the absolute number of CD4+ FOXP3+ cells per spleen was analyzed by flow cytometry. Each dot represents an individual mouse pooled from experiments performed with 2 to 4 mice per group. Bars indicate means. ***, P < 0.001 (as determined by one-way ANOVA with Tukey's posttest for multiple comparisons).
DISCUSSION
It has been known for some time that B cells and antiviral antibodies are essential for control of FV infections (particularly for long-term control) (46, 48). The current results reveal an additional role for B cells in promoting the response of thymus-derived Treg responses to infection. These immunosuppressive Treg responses suppress both CD8+ T cells (17, 18) and NK cells (49), diminishing early virus control. Our in vitro experiments showing that Treg responsiveness to TCR stimulation in the presence of IL-2 is largely dependent on B cells and GITR ligand suggest that this B cell regulatory activity during infection is related to the previously described function of B cells in promoting the homeostatic maintenance of Tregs, also shown previously to be dependent on GITR ligand (36, 50). Our discovery that B cells promote Treg responses to infection adds to the increasing number of roles for B cells as modulators of the immune system. The current results indicate that GITR/GITRL interactions account for approximately half of the B cell effects on Treg expansion in vitro (Fig. 3). Further experiments will be required to determine the mechanisms involved in the remaining effects.
Regulatory functions for B cells have been attributed to certain subsets of B cells (termed Bregs) that expand and typically function via IL-10 production to suppress immunopathological cells and promote regulation (51–53). For example, high levels of IL-10-producing Bregs correlate with high HIV loads and there is evidence that Bregs directly suppress HIV-specific CD8+ T cell function (54, 55). Also, infection with Schistosoma mansoni worms induced expansion of the IL-10-producing B cells that recruit Tregs to the lungs and suppressed allergic airway inflammation in an asthma model (56). Helicobacter pylori infections activate B cells via TLR-2 to convert conventional T cells to the IL-10-producing Tr-1 cells that suppress gastric immunopathology (57). Inflammation-induced Bregs such as these are different from what we have observed in FV infections. IL-10 does not play an immunosuppressive role in FV infections (58), and there is no evidence of virus-specific (27), converted, or IL-10-producing Tr-1 Tregs in FV infection. Rather, FV-induced Tregs are thymus derived (26) and are most commonly specific for self-antigens.
If FV-induced Tregs are specific for self-antigens rather than FV antigens, how does FV infection drive Treg activation and proliferation? We previously showed that for the specific subset of Tregs possessing a Vβ5 TCR, which accounts for about 8% to 10% of the Treg response to FV infection, reactivity was specific for an endogenous retroviral superantigen that provided signal 1 through the TCR. Signal 2, or costimulation, was provided by CD8+ T cells in the following way. Upon FV infection, virus-specific CD8+ T cells become activated and transiently upregulate expression of the membrane-bound form of TNF-α. This membrane-bound TNF-α then reacts with TNFRII on Vβ5+ Tregs to provide signal 2 (28). Because superantigens provide exceptionally strong TCR signals (59–61), TNFRII signaling provides a second signal sufficient to induce proliferation of the Vβ5+ Tregs in the absence of IL-2. Thus, the Vβ5+ Treg response is dependent on CD8+ T cells but, interestingly, independent of IL-2 (26). The remaining 90% of the FV-induced Treg response was found to be IL-2 dependent (26). We now show that it is also B cell dependent. Full FV-induced Treg expansion could be restored by adoptive transfer of B cells into B10.μMT mice (Fig. 6), indicating that the defect in Treg expansion was indeed attributable to the lack of B cells at the time of infection and not to a problem of Treg development in a B cell-deficient mouse. In vitro experiments indicated that the Tregs needed not only T cell receptor stimulation and IL-2 for proliferation but also stimuli from B cells, which promoted both Treg proliferation and the maintenance of FoxP3 expression (Fig. 3). Interestingly, B cells from either naive or FV-infected mice were able to promote anti-CD3/IL2-induced Treg proliferation in vitro (Fig. 2). Thus, infection-induced changes in a B cell phenotype such as activation or differentiation were not required for their ability to promote the virus-induced expansion of Tregs. Rather, this ability appeared to be a constitutive property of the B cells. B cells constitutively express GITRL, the ligand for GITR, which is expressed on the cell surface of Tregs (62, 63) and which we found to provide a strong signal for Treg proliferation in vitro. Thus, we propose a model for FV-induced Treg proliferation in which Tregs receive signal 1 via TCR stimulation from normal self-antigens. Under homeostatic conditions, they also receive a second signal from GITR signaling through resting B cells (36). These two signals maintain the low level of Treg activity necessary for self-tolerance. Like other T cells, the responsiveness of Tregs appears to be tightly controlled and expansion of the Treg subset requires additional signals. In FV infections, the additional signal comes from IL-2, which is secreted by a number of responding cells, including dendritic cells, NK cells, and antigen-specific CD4+ and CD8+ T cells. Thus, Tregs, which constitutively express the high-affinity IL-2 receptor CD25, receive a potent cytokine signal for proliferation during FV infection.
It is interesting to speculate whether B cells promote Treg responses during infections with viruses in humans. Since treatment of infected patients with B cell-depleting therapeutics such as rituximab is not typical, the effect of such therapy on Tregs has been sparsely investigated. However, in a study of 21 hepatitis C virus-infected mixed cryoglobulinemia vasculitis patients treated with rituximab, complete responders to therapy exhibited expansions of the regulatory T cell subsets (64). Increases in levels of Tregs following rituximab therapy in noninfectious diseases such as systemic lupus erythematosus have also been shown (33). Thus, B cells strongly affect Tregs in humans, although the clinical results thus far appear to be opposite those that we observed in the mice. In contrast, in vitro studies have shown that B cells can stimulate potent expansion of human Tregs (65, 66), similarly to what we observed in the mice. At this point, it will take significantly more investigation to determine what factors produce these differential outcomes. In HIV infections, Tregs play complex roles, postulated to be both beneficial in downregulating immune hyperactivation and detrimental in suppressing antiviral immune-mediated virus clearance (67, 68). A recent study using FOXP3 and Helios to label Tregs indicated that HIV-1-infected children had expanded FOXP3+ Helios+ subsets and that increased Treg levels were associated with advanced disease (69). Of course, it is still completely unknown whether B cells play any role in Treg expansion during HIV infections or even whether it would be beneficial to patients to modulate Treg responses either up or down. What is becoming increasingly clear, however, is that individual subsets of lymphocytes play multifunctional roles, including regulatory as well as effector functions. Thus, the modulation of one subset through immunotherapy can have unexpected consequences for other subsets unless the interconnections and mechanisms of intercellular communications between all subsets are fully understood. The current study revealed that B cells not only provide important regulatory control over homeostatic Treg levels and autoimmune disease-related expansions of Tregs but also regulate the virus-induced expansion of this important subset. Understanding this opens the possibility of designing therapeutics that can alter the regulation of immune responses during infections in a manner that is beneficial to the host.
MATERIALS AND METHODS
Mice.
Experiments were conducted using female C57BL/10 (B10) mice, C57BL/10SgSnAi-[KO]μMT N13 (B10.μMT) mice (obtained from NIAID/Taconic and bred at the Rocky Mountain Laboratories, Hamilton, MT), and B6.FOXP3-DTR-GFP (FDG) mice (obtained from Jackson Laboratory and bred at the Rocky Mountain Laboratories, Hamilton, MT) (70). All of the mice were 10 to 20 weeks old at the beginning of the experiments and were treated in accordance with Animal Care and Use Committee-approved protocols 2015-024 and 2015-039 under the regulations and guidelines of the National Institutes of Health.
Virus, injections, and infectious center assay.
The FV stock used in these experiments was an FV complex containing replication-competent B-tropic Friend murine leukemia helper retrovirus and replication-defective polycythemia-inducing spleen focus-forming retrovirus (free of lactate dehydrogenase-elevating virus) (44). Mice were infected by intravenous (i.v.) injection of 20,000 spleen focus-forming units (SFFU) in 0.2 ml via the retroorbital sinus. For analysis of in vivo responses, spleens were harvested at 7, 14, or 21 days postinfection, homogenized, processed, and stained for flow cytometric analysis following red blood cell lysis with ammonium chloride potassium (ACK) buffer for 5 min. The IC assays were performed as described previously by seeding dilutions of splenocyte suspensions onto susceptible Mus dunni cells (44, 71).
Surface and intracellular antibody staining and flow cytometry.
The following antibodies were used for cell surface staining: monoclonal antibody 34 (mAb34) against FV glycogag, anti-CD19 (1D3; BioLegend), anti-CD8 (53-6.7; BD), anti-CD43 (1B11; BioLegend), anti-CD11a (LFA1; BioLegend), anti-CD44 (1M7; BioLegend), anti-Ter119 (TER-119; eBio), anti-CD11b (M1/70; eBio), and Db GagL dextramer (Immudex). Following surface staining, intracellular staining was performed with a FOXP3 permeabilization and fixation kit from eBiosciences according to the recommendations of the manufacturer using the following antibodies: Ki-67 (SolA15; BD) and GrB (GB11; Invitrogen). Lymphocyte populations were initially gated on the basis of forward scatter versus side scatter. Flow cytometric data were collected with an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star). FACS analysis was performed on a FACSAria instrument (BD Biosciences).
Adoptive transfers.
Donor B cells were purified from naive B10 mice using CD19+ magnetically activated cell sorting (MACS) columns per the manufacturer’s recommendations. B cells were pooled and resuspended in heparinized phosphate-buffered balanced salt solution (PBBS). Recipient mice received 5 × 107 B cells i.v. 1 day before infection.
In vitro coculture assays.
Splenocytes were harvested from naive FDG mice, and CD19+ cells were depleted with MACS LD columns according to the manufacturer’s instructions. Using the remaining CD19− fraction, untouched Tregs were enriched by FACS analysis of GFP+ (FOXP3+) cells and stained with CellTrace Violet (Pacific Blue [PB]) dye according to the manufacturer’s recommendations. B cells were harvested from naive or FV-infected B10 mice at 1 wpi by positive selection with CD19 MACS columns. B cells were incubated with or without 10 μg/ml anti-GITRL for 1 h, stained with CellTrace Far Red (allophycocyanin [APC]), and cocultured with Tregs at a 10:1 B cell/Treg ratio for 2.5 days in the presence or absence of 10 ng/ml IL-2 and 10 μg/ml anti-CD3. Proliferation was determined by Ki-67 expression and loss of CellTrace PB dye by flow cytometry. CellTrace-APC+ cells were excluded from the analysis.
ACKNOWLEDGMENT
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA.
Footnotes
Citation Moore TC, Gonzaga LM, Mather JM, Messer RJ, Hasenkrug KJ. 2017. B cell requirement for robust regulatory T cell responses to Friend retrovirus infection. mBio 8:e01122-17. https://doi.org/10.1128/mBio.01122-17.
REFERENCES
- 1.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164. [PubMed] [Google Scholar]
- 2.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. 2008. Regulatory T cells and immune tolerance. Cell 133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. 2004. CD4+ CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol 172:4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 4.Guilliams M, Oldenhove G, Noel W, Hérin M, Brys L, Loi P, Flamand V, Moser M, De Baetselier P, Beschin A. 2007. African trypanosomiasis: naturally occurring regulatory T cells favor trypanotolerance by limiting pathology associated with sustained type 1 inflammation. J Immunol 179:2748–2757. doi: 10.4049/jimmunol.179.5.2748. [DOI] [PubMed] [Google Scholar]
- 5.Hesse M, Piccirillo CA, Belkaid Y, Prufer J, Mentink-Kane M, Leusink M, Cheever AW, Shevach EM, Wynn TA. 2004. The pathogenesis of schistosomiasis is controlled by cooperating IL-10-producing innate effector and regulatory T cells. J Immunol 172:3157–3166. doi: 10.4049/jimmunol.172.5.3157. [DOI] [PubMed] [Google Scholar]
- 6.McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. 2006. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol 177:6215–6226. doi: 10.4049/jimmunol.177.9.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maizels RM, Smith KA. 2011. Regulatory T cells in infection. Adv Immunol 112:73–136. doi: 10.1016/B978-0-12-387827-4.00003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boer MC, Joosten SA, Ottenhoff TH. 2015. Regulatory T-cells at the interface between human host and pathogens in infectious diseases and vaccination. Front Immunol 6:217. doi: 10.3389/fimmu.2015.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, Sacks DL. 2001. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med 194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwashiro M, Messer RJ, Peterson KE, Stromnes IM, Sugie T, Hasenkrug KJ. 2001. Immunosuppression by CD4+ regulatory T cells induced by chronic retroviral infection. Proc Natl Acad Sci U S A 98:9226–9230. doi: 10.1073/pnas.151174198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietze KK, Zelinskyy G, Gibbert K, Schimmer S, Francois S, Myers L, Sparwasser T, Hasenkrug KJ, Dittmer U. 2011. Transient depletion of regulatory T cells in transgenic mice reactivates virus-specific CD8+ T cells and reduces chronic retroviral set points. Proc Natl Acad Sci U S A 108:2420–2425. doi: 10.1073/pnas.1015148108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bánki Z, Posch W, Ejaz A, Oberhauser V, Willey S, Gassner C, Stoiber H, Dittmer U, Dierich MP, Hasenkrug KJ, Wilflingseder D. 2010. Complement as an endogenous adjuvant for dendritic cell-mediated induction of retrovirus-specific CTLs. PLoS Pathog 6:e1000891. doi: 10.1371/journal.ppat.1000891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dittmer U, Race B, Peterson KE, Stromnes IM, Messer RJ, Hasenkrug KJ. 2002. Essential roles for CD8+ T cells and gamma interferon in protection of mice against retrovirus-induced immunosuppression. J Virol 76:450–454. doi: 10.1128/JVI.76.1.450-454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joedicke JJ, Zelinskyy G, Dittmer U, Hasenkrug KJ. 2014. CD8+ T cells are essential for controlling acute Friend retrovirus infection in C57BL/6 mice. J Virol 88:5200–5201. doi: 10.1128/JVI.00312-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasenkrug KJ. 1999. Lymphocyte deficiencies increase susceptibility to Friend virus-induced erythroleukemia in Fv-2 genetically resistant mice. J Virol 73:6468–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bila C, Oberhauser V, Ammann CG, Ejaz A, Huber G, Schimmer S, Messer R, Pekna M, von Laer D, Dittmer U, Hasenkrug KJ, Stoiber H, Bánki Z. 2011. Complement opsonization enhances Friend virus infection of B cells and thereby amplifies the virus-specific CD8+ T cell response. J Virol 85:1151–1155. doi: 10.1128/JVI.01821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zelinskyy G, Dietze K, Sparwasser T, Dittmer U. 2009. Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog 5:e1000406. doi: 10.1371/journal.ppat.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zelinskyy G, Dietze KK, Hüsecken YP, Schimmer S, Nair S, Werner T, Gibbert K, Kershaw O, Gruber AD, Sparwasser T, Dittmer U. 2009. The regulatory T-cell response during acute retroviral infection is locally defined and controls the magnitude and duration of the virus-specific cytotoxic T-cell response. Blood 114:3199–3207. doi: 10.1182/blood-2009-03-208736. [DOI] [PubMed] [Google Scholar]
- 19.Robertson SJ, Messer RJ, Carmody AB, Hasenkrug KJ. 2006. In vitro suppression of CD8+ T cell function by Friend virus-induced regulatory T cells. J Immunol 176:3342–3349. doi: 10.4049/jimmunol.176.6.3342. [DOI] [PubMed] [Google Scholar]
- 20.Dittmer U, He H, Messer RJ, Schimmer S, Olbrich ARM, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, Evans LH, Peterson KE, Yang GJ, Hasenkrug KJ. 2004. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity 20:293–303. doi: 10.1016/S1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- 21.Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, Finney CA, Greenwood EJ, Knox DP, Wilson MS, Belkaid Y, Rudensky AY, Maizels RM. 2010. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med 207:2331–2341. doi: 10.1084/jem.20101074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mertens J, Fabri M, Zingarelli A, Kubacki T, Meemboor S, Groneck L, Seeger J, Bessler M, Hafke H, Odenthal M, Bieler JG, Kalka C, Schneck JP, Kashkar H, Kalka-Moll WM. 2009. Streptococcus pneumoniae serotype 1 capsular polysaccharide induces CD8CD28 regulatory T lymphocytes by TCR crosslinking. PLoS Pathog 5:e1000596. doi: 10.1371/journal.ppat.1000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Kleij D, Latz E, Brouwers JF, Kruize YC, Schmitz M, Kurt-Jones EA, Espevik T, de Jong EC, Kapsenberg ML, Golenbock DT, Tielens AG, Yazdanbakhsh M. 2002. A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates Toll-like receptor 2 and affects immune polarization. J Biol Chem 277:48122–48129. doi: 10.1074/jbc.M206941200. [DOI] [PubMed] [Google Scholar]
- 24.McGuirk P, Mills KH. 2002. Pathogen-specific regulatory T cells provoke a shift in the Th1/Th2 paradigm in immunity to infectious diseases. Trends Immunol 23:450–455. doi: 10.1016/S1471-4906(02)02288-3. [DOI] [PubMed] [Google Scholar]
- 25.Higgins SC, Lavelle EC, McCann C, Keogh B, McNeela E, Byrne P, O’Gorman B, Jarnicki A, McGuirk P, Mills KH. 2003. Toll-like receptor 4-mediated innate IL-10 activates antigen-specific regulatory T cells and confers resistance to Bordetella pertussis by inhibiting inflammatory pathology. J Immunol 171:3119–3127. doi: 10.4049/jimmunol.171.6.3119. [DOI] [PubMed] [Google Scholar]
- 26.Myers L, Joedicke JJ, Carmody AB, Messer RJ, Kassiotis G, Dudley JP, Dittmer U, Hasenkrug KJ. 2013. IL-2-independent and TNF-alpha-dependent expansion of Vbeta5+ natural regulatory T cells during retrovirus infection. J Immunol 190:5485–5495. doi: 10.4049/jimmunol.1202951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antunes I, Tolaini M, Kissenpfennig A, Iwashiro M, Kuribayashi K, Malissen B, Hasenkrug K, Kassiotis G. 2008. Retrovirus-specificity of regulatory T cells is neither present nor required in preventing retrovirus-induced bone marrow immune pathology. Immunity 29:782–794. doi: 10.1016/j.immuni.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joedicke JJ, Myers L, Carmody AB, Messer RJ, Wajant H, Lang KS, Lang PA, Mak TW, Hasenkrug KJ, Dittmer U. 2014. Activated CD8+ T cells induce expansion of Vbeta5+ regulatory T cells via TNFR2 signaling. J Immunol 193:2952–2960. doi: 10.4049/jimmunol.1400649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dass S, Vital EM, Emery P. 2007. Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum 56:2715–2718. doi: 10.1002/art.22811. [DOI] [PubMed] [Google Scholar]
- 30.El Fassi D, Nielsen CH, Kjeldsen J, Clemmensen O, Hegedüs L. 2008. Ulcerative colitis following B lymphocyte depletion with rituximab in a patient with Graves’ disease. Gut 57:714–715. doi: 10.1136/gut.2007.138305. [DOI] [PubMed] [Google Scholar]
- 31.Goetz M, Atreya R, Ghalibafian M, Galle PR, Neurath MF. 2007. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflamm Bowel Dis 13:1365–1368. doi: 10.1002/ibd.20215. [DOI] [PubMed] [Google Scholar]
- 32.Ray A, Mann MK, Basu S, Dittel BN. 2011. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol 230:1–9. doi: 10.1016/j.jneuroim.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lund FE, Randall TD. 2010. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol 10:236–247. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellis JS, Braley-Mullen H. 2017. Mechanisms by which B cells and regulatory T cells influence development of murine organ-specific autoimmune diseases. J Clin Med 6. doi: 10.3390/jcm6020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamel KM, Cao Y, Ashaye S, Wang Y, Dunn R, Kehry MR, Glant TT, Finnegan A. 2011. B cell depletion enhances T regulatory cell activity essential in the suppression of arthritis. J Immunol 187:4900–4906. doi: 10.4049/jimmunol.1101844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. 2012. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol 188:3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf SD, Dittel BN, Hardardottir F, Janeway CA Jr. 1996. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med 184:2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. 2007. B cell regulation of CD4+ CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol 178:3447–3456. doi: 10.4049/jimmunol.178.6.3447. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Ray A, Jiang X, Wang JY, Basu S, Liu X, Qian T, He R, Dittel BN, Chu Y. 2015. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol 8:1297–1312. doi: 10.1038/mi.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Messer RJ, Dittmer U, Peterson KE, Hasenkrug KJ. 2004. Essential role for virus-neutralizing antibodies in sterilizing immunity against Friend retrovirus infection. Proc Natl Acad Sci U S A 101:12260–12265. doi: 10.1073/pnas.0404769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiang Y, Peng J, Tai N, Hu C, Zhou Z, Wong FS, Wen L. 2012. The dual effects of B cell depletion on antigen-specific T cells in BDC2. 5NOD mice. J Immunol 188:4747–4758. doi: 10.4049/jimmunol.1103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, Hamann J, van Lier RA, Nolte MA. 2009. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol 182:7490–7500. doi: 10.4049/jimmunol.0802751. [DOI] [PubMed] [Google Scholar]
- 43.Chen W, Qin H, Chesebro B, Cheever MA. 1996. Identification of a gag-encoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J Virol 70:7773–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson SJ, Ammann CG, Messer RJ, Carmody AB, Myers L, Dittmer U, Nair S, Gerlach N, Evans LH, Cafruny WA, Hasenkrug KJ. 2008. Suppression of acute anti-Friend virus CD8+ T-cell responses by coinfection with lactate dehydrogenase-elevating virus. J Virol 82:408–418. doi: 10.1128/JVI.01413-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li JP, D’Andrea AD, Lodish HF, Baltimore D. 1990. Activation of cell growth by binding of Friend spleen focus-forming virus gp55 glycoprotein to the erythropoietin receptor. Nature 343:762–764. doi: 10.1038/343762a0. [DOI] [PubMed] [Google Scholar]
- 46.Chesebro B, Wehrly K. 1979. Identification of a non-H-2 gene (Rfv-3) influencing recovery from viremia and leukemia induced by Friend virus complex. Proc Natl Acad Sci U S A 76:425–429. doi: 10.1073/pnas.76.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC. 2008. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 321:1343–1346. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doig D, Chesebro B. 1979. Anti-Friend virus antibody is associated with recovery from viremia and loss of viral leukemia cell-surface antigens in leukemic mice. Identification of Rfv-3 as a gene locus influencing antibody production. J Exp Med 150:10–19. doi: 10.1084/jem.150.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Littwitz-Salomon E, Akhmetzyanova I, Vallet C, Francois S, Dittmer U, Gibbert K. 2015. Activated regulatory T cells suppress effector NK cell responses by an IL-2-mediated mechanism during an acute retroviral infection. Retrovirology 12:66. doi: 10.1186/s12977-015-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim YH, Shin SM, Choi BK, Oh HS, Kim CH, Lee SJ, Kim KH, Lee DG, Park SH, Kwon BS. 2015. Authentic GITR signaling fails to induce tumor regression unless Foxp3+ regulatory T cells are depleted. J Immunol 195:4721–4729. doi: 10.4049/jimmunol.1403076. [DOI] [PubMed] [Google Scholar]
- 51.Rosser EC, Mauri C. 2015. Regulatory B cells: origin, phenotype, and function. Immunity 42:607–612. doi: 10.1016/j.immuni.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Mauri C, Menon M. 2015. The expanding family of regulatory B cells. Int Immunol 27:479–486. doi: 10.1093/intimm/dxv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, Ehrenstein MR, Flavell RA, Mauri C. 2011. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol 186:5569–5579. doi: 10.4049/jimmunol.1100284. [DOI] [PubMed] [Google Scholar]
- 54.Siewe B, Stapleton JT, Martinson J, Keshavarzian A, Kazmi N, Demarais PM, French AL, Landay A. 2013. Regulatory B cell frequency correlates with markers of HIV disease progression and attenuates anti-HIV CD8 T cell function in vitro. J Leukoc Biol 93:811–818. doi: 10.1189/jlb.0912436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siewe B, Wallace J, Rygielski S, Stapleton JT, Martin J, Deeks SG, Landay A. 2014. Regulatory B cells inhibit cytotoxic T lymphocyte (CTL) activity and elimination of infected CD4 T cells after in vitro reactivation of HIV latent reservoirs. PLoS One 9:e92934. doi: 10.1371/journal.pone.0092934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG. 2010. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol 125:1114–1124.e8. doi: 10.1016/j.jaci.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 57.Sayi A, Kohler E, Toller IM, Flavell RA, Müller W, Roers A, Müller A. 2011. TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J Immunol 186:878–890. doi: 10.4049/jimmunol.1002269. [DOI] [PubMed] [Google Scholar]
- 58.Strestik BD, Olbrich AR, Hasenkrug KJ, Dittmer U. 2001. The role of IL-5, IL-6 and IL-10 in primary and vaccine-primed immune responses to infection with Friend retrovirus (murine leukaemia virus). J Gen Virol 82:1349–1354. doi: 10.1099/0022-1317-82-6-1349. [DOI] [PubMed] [Google Scholar]
- 59.Arad G, Levy R, Nasie I, Hillman D, Rotfogel Z, Barash U, Supper E, Shpilka T, Minis A, Kaempfer R. 2011. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol 9:e1001149. doi: 10.1371/journal.pbio.1001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bueno C, Lemke CD, Criado G, Baroja ML, Ferguson SS, Rahman AK, Tsoukas CD, McCormick JK, Madrenas J. 2006. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity 25:67–78. doi: 10.1016/j.immuni.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 61.Zamoyska R. 2006. Superantigens: supersignalers? Sci STKE 2006:pe45. doi: 10.1126/stke.3582006pe45. [DOI] [PubMed] [Google Scholar]
- 62.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 63.Nocentini G, Riccardi C. 2009. GITR: a modulator of immune response and inflammation. Adv Exp Med Biol 647:156–173. doi: 10.1007/978-0-387-89520-8_11. [DOI] [PubMed] [Google Scholar]
- 64.Saadoun D, Rosenzwajg M, Landau D, Piette JC, Klatzmann D, Cacoub P. 2008. Restoration of peripheral immune homeostasis after rituximab in mixed cryoglobulinemia vasculitis. Blood 111:5334–5341. doi: 10.1182/blood-2007-11-122713. [DOI] [PubMed] [Google Scholar]
- 65.Chen LC, Delgado JC, Jensen PE, Chen X. 2009. Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol 183:4094–4102. doi: 10.4049/jimmunol.0901081. [DOI] [PubMed] [Google Scholar]
- 66.Sun JB, Flach CF, Czerkinsky C, Holmgren J. 2008. B lymphocytes promote expansion of regulatory T cells in oral tolerance: powerful induction by antigen coupled to cholera toxin B subunit. J Immunol 181:8278–8287. doi: 10.4049/jimmunol.181.12.8278. [DOI] [PubMed] [Google Scholar]
- 67.Chevalier MF, Weiss L. 2013. The split personality of regulatory T cells in HIV infection. Blood 121:29–37. doi: 10.1182/blood-2012-07-409755. [DOI] [PubMed] [Google Scholar]
- 68.Moreno-Fernandez ME, Presicce P, Chougnet CA. 2012. Homeostasis and function of regulatory T cells in HIV/SIV infection. J Virol 86:10262–10269. doi: 10.1128/JVI.00993-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khaitan A, Kravietz A, Mwamzuka M, Marshed F, Ilmet T, Said S, Ahmed A, Borkowsky W, Unutmaz D. 2016. FOXP3+Helios+ regulatory T cells, immune activation, and advancing disease in HIV-Infected children. J Acquir Immune Defic Syndr 72:474–484. doi: 10.1097/QAI.0000000000001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim JM, Rasmussen JP, Rudensky AY. 2007. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 71.Morrison RP, Earl PL, Nishio J, Lodmell DL, Moss B, Chesebro B. 1987. Different H-2 subregions influence immunization against retrovirus and immunosuppression. Nature 329:729–732. doi: 10.1038/329729a0. [DOI] [PubMed] [Google Scholar]