

Abstract
Background
Maternal and inherited (i.e., case) genetic factors likely contribute to the etiology of congenital heart defects, but it is unclear whether individual common variants confer a large risk.
Methods and Results
To evaluate the relationship between individual common maternal/inherited genotypes and risk for heart defects, we conducted genome-wide association studies (GWAS) in five cohorts. Three cohorts were recruited at the Children’s Hospital of Philadelphia: 670 conotruncal heart defect (CTD) case-parent trios; 317 left ventricular obstructive tract defect (LVOTD) case-parent trios; and 406 CTD cases and 2,976 pediatric controls. Two cohorts were recruited through the Pediatric Cardiac Genomics Consortium: 355 CTD trios and 192 LVOTD trios. We also conducted meta-analyses using the GWAS results from the CTD cohorts, the LVOTD cohorts and from the combined CTD and LVOTD cohorts. In the individual GWAS, several genome-wide significant associations (p≤5×10−8) were observed. In our meta-analyses, one genome-wide significant association was detected: the case genotype for rs72820264, an intra-genetic SNP associated with LVOTDs (p=2.1×10−8).
Conclusions
We identified one novel candidate region associated with LVOTDs and report on several additional regions with suggestive evidence for association with CTD and/or LVOTD. These studies were constrained by the relatively small samples sizes and thus have limited power to detect small to moderate associations. Approaches that minimize the multiple testing burden (e.g. gene- or pathway-based) may, therefore, be required to uncover common variants contributing to the risk of these relatively rare conditions.
Journal Subject Terms: Epidemiology, Genetic, Association Studies, Congenital Heart Disease, Functional Genomics
Keywords: Genome Wide Association Study, congenital cardiac defect, left ventricular outflow tract obstruction, epidemiology
Introduction
Congenital heart defects (CHDs) are the most common birth defect, with a prevalence of nearly 1 in 100 live births,1 and account for nearly 40% of infant deaths in North America.2 Although few specific risk factors have been identified for CHDs, a genetic contribution to risk is expected based on narrow sense heritability estimates (h2 ~0.8)3–5 and the occurrence of CHDs as part of specific genetic syndromes (e.g., 22q11 deletion syndrome).6, 7 Moreover, familial recurrence patterns indicate that the genetic contribution to non-syndromic CHDs is likely to be complex and involve multiple, inherited variants of low to moderate effect.8, 9 Both maternal and inherited (i.e., case) genetic factors (as well as non-genetic factors) are thought to contribute to the risk of CHDs.10–12 In addition, different groups of heart defects (e.g., conotruncal defects, left ventricular outflow tract defects) may share some genetic risk factors while also having group and even lesion-specific genetic etiologies.10, 13, 14
While much remains to be learned about the genetic contribution to CHDs, genomic approaches, including genome-wide association studies (GWAS), have identified novel candidate genes and loci with relatively strong effects. For example, genome-wide significant and/or suggestive associations have been reported for conotruncal defects (CTDs),15 tetralogy of Fallot,16 left ventricular obstructive tract defects (LVOTDs)17 and septal defects.18–20 Most of the reported associations have yet to be independently replicated. Hence, while GWAS have provided some evidence that the risk of CHDs is influenced by common genetic variants, further studies are warranted. Though the majority of GWAS of adult-onset traits have identified weak to moderate effects, stronger effects have been reported for birth defects and other early-onset traits.21 Thus, we hypothesized that common individual variants contribute to the genetic architecture of CHD risk.
To agnostically identify a subset of common candidate SNPs for future study, we conducted GWAS in multiple CHD cohorts, as well as meta-analyses of the combined data. Our analyses evaluated associations of CHD with both the maternal and inherited (case) genotypes.
Methods
Study Subjects
Children’s Hospital of Philadelphia – CTD Trios
CTD case-parent trios were recruited from The Cardiac Center at The Children’s Hospital of Philadelphia (CHOP) from 1992–2010.15 Individuals of all races/ethnicities were eligible to participate. All subjects provided informed consent under a protocol approved by the CHOP Institutional Review Board for the Protection of Human Subjects. The CTD diagnoses included: tetralogy of Fallot, D-transposition of the great arteries, ventricular septal defects (conoventricular, posterior malalignment and conoseptal hypoplasia), double outlet right ventricle, isolated aortic arch anomalies, truncus arteriosus, and interrupted aortic arch. Cardiac medical records were reviewed (e.g., echocardiography, cardiac MRI, cardiac catheterization, operative notes) to ensure accuracy of the cardiac phenotype, and cases with suspected genetic syndromes were excluded. Further, cases were screened for 22q11.2 deletion syndrome using fluorescence in situ hybridization and/or multiplex ligation-dependent probe amplification.22, 23
Children’s Hospital of Philadelphia – LVOTD Trios
LVOTD case-parent trios were recruited from the CHOP Cardiac Center, 1997–2007, using the same approach as described for the CTD trios.17 Individuals of all races/ethnicities were eligible to participate. The LVOTD diagnoses included: hypoplastic left heart syndrome, coarctation of the aorta with or without bicuspid aortic valve, and aortic valve stenosis. We excluded those with variants of hypoplastic left heart syndrome, such as mal-aligned atrioventricular canal defects and double outlet right ventricle with mitral valve atresia. Cardiac medical records were reviewed to ensure accuracy of the cardiac phenotype. Cases with suspected genetic syndromes were excluded.
Children’s Hospital of Philadelphia – CTD cases and pediatric controls
Additional cases with CTDs were recruited through the CHOP Cardiac Center, 1999–2010, using the same approach as described for CTD trios. Controls were also recruited from CHOP, during well child visits, as previously described.24 Cases and controls were Caucasian by self-report.
Pediatric Cardiac Genomics Consortium
CTD and LVOTD case-parent trios were recruited as part of the Pediatric Cardiac Genomics Consortium’s (PCGC) Congenital Heart Disease GEnetic NEtwork Study. The recruitment of these subjects has been described.25 Briefly, subjects provided informed consent and were recruited into the PCGC cohort at ten clinical study sites from 2010–2012. Individuals of all races/ethnicities were eligible to participate. Cardiac diagnoses were classified based on medical record abstraction by trained study coordinators. Although the PCGC cohort included a broad range of CHDs, our analyses were restricted to those with CTDs or LVOTDs.
Genetic Methods
Study subjects in all cohorts provided either blood or saliva samples, and DNA was extracted directly from these samples (or from cell lines in a minority of subjects) using standard methods, as previously described. 20, 22, 25 All samples were array genotyped using Illumina arrays (Figure 1) in the CHOP Center for Applied Genomics.
Figure 1.
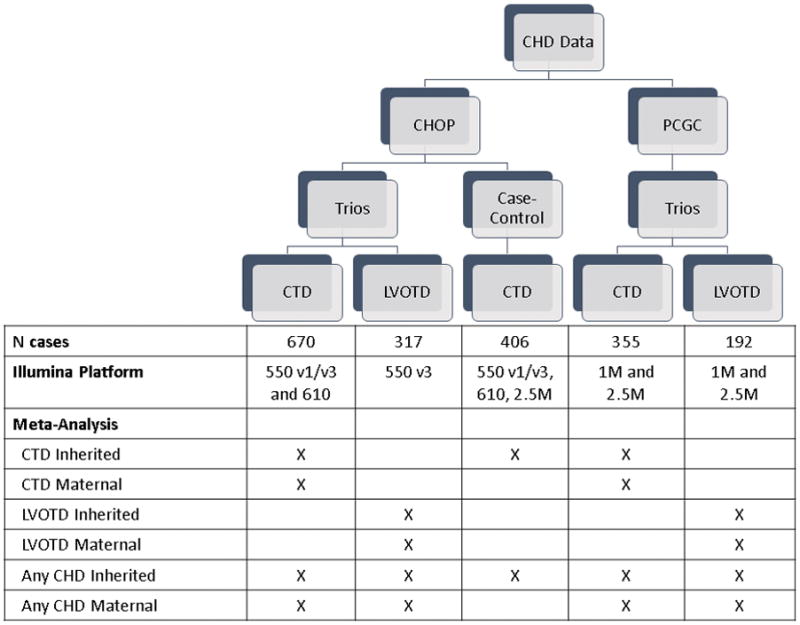
Schematic of the five independent cohorts and summary of which cohorts were analyzed in each meta-analysis.
The array genotype data from the CHOP CTD and LVOTD trios have previously been used to impute additional genotypes against Hapmap reference data26 and these genotyped and imputed data were used in our previous GWAS.15, 17 For the analyses presented here, data for all cohorts were imputed (or re-imputed) using data from the 1,000 Genomes Project reference data.27 Given differences in the timing of the availability of data from CHOP and PCGC, data from these two sources were imputed separately. However, data from all of the CHOP cohorts were imputed together. Likewise, data from PCGC CTD and LVOTD trios were imputed together. Both sets of imputations (i.e., CHOP and PCGC) were based on the genotype data for SNPs that overlapped across all genotyping platforms (i.e., SNPs in common to the 550K, 610K and 2.5M arrays for the CHOP imputation and the 1M and 2.5M arrays for the PCGC imputation). The overlapping set of SNPs was used to minimize differences in imputation accuracy across the cohorts that could arise if imputations were based on different SNPs for each cohort. The imputation and all subsequent analyses were restricted to autosomal variants.
Before imputation, we used PLINK v1.0628 to perform standard quality control procedures on each study cohort (e.g., CHOP CTD trios). We excluded: trios with Mendelian error rate >1%; SNPs with minor allele frequency <1%, genotyping rate <90%, or deviation from Hardy Weinberg equilibrium in controls/parents (p≤1×10−5); and suspected duplicate samples based on pairwise identity by descent pi-hat >0.6. Following these exclusions, data from the three CHOP cohorts were combined, haplotypes were pre-phased using SHAPEIT2 v2.72729 and genotypes for additional variants were imputed using Impute 2 v2.3.0,30 and pre-phased data from the 1,000 Genomes Project (Phase-I integrated v3 variants set).27, 31 Post-imputation quality control procedures included removing variants that were: indels, poorly imputed (r2< 0.8), did not have rs numbers, or rare (minor allele frequency <5%). Variants and individuals with a genotyping rate <90% were also removed. Data from the PCGC CTD and LVOTD trios were combined and processed using the same procedures.
Statistical Analysis
Analytic groups
We conducted separate GWAS for the five independent cohorts: CHOP CTD trios, CHOP LVOTD trios, CHOP CTD cases/controls, PCGC CTD trios, and PCGC LVOT trios (Figure 1). We assessed the inherited (case) genotype in all cohorts, and the maternal genotype in the trios.
Family-based analyses
The analyses of case-parent trios were conducted using a multinomial likelihood approach32 implemented in the EMIM software package.33 Because the assessment of inherited genotypes using this approach is immune to population stratification bias,34, 35 all trios were included in the analyses of inherited genotypes. However, the assessment of maternal genotypes using trio data can be biased by differences in the frequency of mating types as defined by maternal and paternal race/ethnicity (e.g. matings of non-Hispanic Caucasian males and Hispanic females are more frequent than the reciprocal mating type)36. Consequently, analyses of maternal genotypes were restricted to trios in which both parents were reported to be non-Hispanic Caucasian.
We used a likelihood ratio test to compare a full model, with terms for both the maternal and inherited genotypes, to a reduced model that excluded the term being tested. We used an additive (one degree of freedom) genetic risk model for the genotype being assessed (e.g., inherited genotype) and an unrestricted (two-degrees of freedom) model for the other genotype (e.g., maternal genotype). Manhattan plots were generated for each comparison using R version 3.03 (http://www.r-project.org/). SNPs with p-values ≤5×10−8 were considered significantly associated, and SNPs with p-values between 10−5 to 5×10−8 were considered to have suggestive evidence of association with CTDs and/or LVOTDs.15, 37
Case-control analysis
Using data from the CHOP CTD cases and controls, the association between CTDs and the inherited genotype was assessed using logistic regression, based on an additive genetic risk model. These analyses were adjusted for the first two principal components of race/ethnicity and were conducted using Golden Helix v8.1 (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com), which implements principal component adjustment. The results from this analysis were visualized and interpreted using the same approach described for the family-based analyses.
Replication
For each SNP with at least a suggestive association with CHDs in one or more cohorts, we examined the p-values and effect size estimate from the additional, comparable cohorts. For example, if there was suggestive evidence for an association with the inherited genotype for a SNP in the CHOP CTD case-control cohort, we examined the p-values and effect size estimates for that SNP in the analyses of the CHOP and PCGC CTD trios. We deemed associations to be “replicated” if a SNP had at least suggestive evidence of association (p≤10−5) in more than one dataset assessing the same genetic effect (maternal or inherited) and phenotype and consistent direction of association in two or more comparable cohorts.
Meta-analysis
Because there is evidence that sub-groups of CHDs (e.g. CTDs and LVOTDs) are likely to have overlapping but not identical genetic profiles, we conducted three separate meta-analyses of inherited effects, based on the following groupings (Figure 1): CTDs only (CHOP CTD trios, CHOP CTD case-control, and PCGC CTD analyses); LVOTD only (CHOP and PCGC LVOTD trios); and CTD+LVOTD (all five cohorts). Likewise, we conducted three separate meta-analyses of maternal effects: CTDs only (CHOP and PCGC CTD trios); LVOTD only (CHOP and PCGC LVOTD trios); and, CTD+LVOTD (all four trio cohorts). Each meta-analysis was performed using a fixed-effects model, unless there was evidence of heterogeneity (based on Cochran’s heterogeneity p≤0.1), in which case a random-effects model was used. We conducted meta-analyses using GWAMA v2.1 (http://www.well.ox.ac.uk/gwama/).38 Manhattan plots were generated using R version 3.03 (http://www.r-project.org/) and regional association plots were constructed for top hits using Locus Zoom v1.1 (http://locuszoom.sph.umich.edu/locuszoom/).39 Variants were annotated with Combined Annotation Dependent Depletion (CADD) scores.40 CADD scores predict the potential deleteriousness of individual variants, such that variants with CADD>10 are predicted to fall in the top 10% of the most deleterious variants in the genome. Variants were also annotated with GWAVA scores, which are scores for variants and corresponding regions that focus on predicting the functional impact of non-coding variants/regions https://www.sanger.ac.uk/sanger/StatGen_Gwava).41
Results
After quality control exclusions were applied, there were 670 CHOP CTD trios, 317 CHOP LVOTD trios, 406 CHOP CTD cases/2,976 pediatric controls, 355 PCGC CTD trios, and 192 PCGC LVOTD trios. The cohorts were characterized using counts and frequencies for race/ethnicity, sex, and heart defect lesion (Table 1). The most frequent CTD was tetralogy of Fallot (30–40%) and the most common LVOTD was hypoplastic left heart syndrome (44–50%). Consistent with known epidemiological characteristics of LVOTDs and certain CTDs,42 there was an excess of males. After quality control exclusions, data were available for 4,756,722 SNPs (N=4,483,243 imputed) in the CHOP cohorts and for 5,737,343 SNPs (n=5,112,962 imputed) in the PCGC cohorts.
Table 1.
Characteristics of cases by study sample.
| N (%)
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| CHOP CTD trios(N=670 trios) | CHOP LVOTD (N=317 trios) | CHOP CTD cases*(N=406 cases) | PCGC†(N=547 trios) | |||||
| Race/ethnicity | ||||||||
|
| ||||||||
| Non-Hispanic Caucasian | 483 | (72.1) | 247 | (77.9) | 406 | (100.0) | 389 | (71.1) |
| Other | 187 | (27.9) | 70 | (22.1) | 0 | (0.0) | 158 | (28.9) |
|
| ||||||||
| Sex | ||||||||
|
| ||||||||
| Male | 410 | (61.2) | 201 | (63.4) | 236 | (58.1) | 341 | (62.3) |
| Female | 260 | (38.8) | 116 | (36.6) | 170 | (41.9) | 206 | (37.7) |
|
| ||||||||
| Lesion | ||||||||
|
| ||||||||
| CTDs | 670 | (100.0) | - | - | 406 | (100.0) | 355 | - |
| Tetralogy of Fallot | 267 | (39.9) | - | - | 134 | (33.0) | 106 | (29.9) |
| D-transposition of the great arteries | 128 | (19.1) | - | - | 80 | (19.7) | 70 | (19.7) |
| Ventricular septal defects | 136 | (20.3) | - | - | 109 | (26.9) | 45 | (12.7) |
| Double outlet right ventricle | 76 | (11.3) | - | - | 25 | (6.2) | 57 | (16.1) |
| Isolated aortic arch anomalies | 30 | (4.5) | - | - | 22 | (5.4) | 7 | (2.0) |
| Truncus arteriosus | 20 | (3.0) | - | - | 19 | (4.7) | 13 | (3.7) |
| Interrupted aortic arch | 13 | (1.9) | - | - | 10 | (2.5) | 9 | (2.5) |
| Other | 0 | (0.0) | - | - | 7 | (1.7) | 48 | (13.5) |
| LVOTDs | - | - | 317 | (100.0) | - | - | 192 | - |
| Hypoplastic left heart syndrome | - | - | 157 | (49.5) | - | - | 85 | (44.3) |
| Coarctation of the aorta | - | - | 97 | (30.6) | - | - | 67 | (34.9) |
| Aortic stenosis | - | - | 63 | (19.9) | - | - | 28 | (14.6) |
| Other | - | - | 0 | (0.0) | - | - | 12 | (6.3) |
From case-control analysis
Percent of PCGC CTDs/LVOTs are listed for lesions
GWAS of Individual Cohorts
A GWAS was conducted separately for each cohort (Figure 1). The total numbers of genomewide significant and suggestive associations with SNPs observed in each cohort are summarized in Table 2. Genome-wide significant associations were only identified in the CHOP CTD case-control GWAS. In this analysis, 52 inherited genotypes were significantly associated with CTDs (Table 2, Supplemental Table 1), and 49 of these SNPs were in introns of MGAT4C. Multiple suggestive associations (p≤10−5) with the inherited and maternal genotypes were detected in the analysis of each cohort (Table 2). However, there was no overlap in the SNPs with at least suggestive evidence of association across comparable cohorts (data not shown). Thus, there was no evidence of replication for any of the suggestive or significant associations identified in the individual cohorts.
Table 2.
Summary of analytic groups and results for inherited or maternal variants associated with heart defects
| CTD
|
LVOTD
|
CTD+LVOTD
|
||||||
|---|---|---|---|---|---|---|---|---|
| CHOP trios | CHOP case/control | PCGC trios | Meta-Analysis | CHOP trios | PCGC trios | Meta-Analysis | Meta-Analysis | |
| Inherited | ||||||||
| Significant* | 0 | 52 | 0 | 0 | 0 | 0 | 1 | 0 |
| Suggestive† | 87 | 45 | 59 | 36 | 25 | 40 | 32 | 70 |
| Maternal | ||||||||
| Significant* | 0 | - | 0 | 0 | 0 | 0 | 0 | 0 |
| Suggestive† | 80 | - | 39 | 133 | 122 | 41 | 67 | 84 |
p≤5×10−8
5×10−8<p≤10−5
Meta-Analyses
There was suggestive evidence of associations with the maternal genotype for multiple SNPs in the meta-analysis of CTD only (133 SNPs in 11 regions), LVOTD only (67 SNPs in 10 regions, including one imputed SNP with borderline significant results, rs55788414, p=5.5×10−8, Figure 2) and CTD+LVOTD (84 SNPs in 20 regions) (Table 3: SNPs with p<10−6; Supplemental Tables 2–4: SNPs with p<10−5). However, no genome-wide significant associations with the maternal genotype were identified (Supplemental Figure 1). Further, the maternal genotypes for SNPs in linkage disequilibrium with rs55788414, the SNP with the smallest association p-value, were not associated with LVOTD (Figure 2).
Figure 2.

Regional association plots for the meta-analysis between LVOTDs and: A) maternal SNPs near rs55788414 B) inherited SNPs near rs72820264. Each pane shows the association statistic from the meta-analysis (−log10 p) on the left y-axis for the variant with the lowest combined p-value (purple diamond) and nearby markers (circles). The red shading indicates the amount of linkage disequilibrium (r2) between this variant and the nearby markers. The right y-axis indicates recombination rates from 1000 Genomes CEU data (blue lines). The x-axis indicates the chromosomal position (hg19) and the location of nearby genes.
Table 3.
Summary data for variants in the meta-analysis with p<10−6
| SNP | Chr* | Position† (bp) | Reference allele | Gene | Function | P-value‡ | Odds ratio§(95% CI) | CADD|| | GWAVA-TSS score# |
|---|---|---|---|---|---|---|---|---|---|
|
Inherited
| |||||||||
| CTDs+LVODTs | |||||||||
| rs11895588 | 2 | 141962157 | T | LRP1B | Intronic | 7.3 ×10−7 | 0.54 (0.43–0.69) | 0.85 | 0.06 |
| rs56409046 | 6 | 9320306 | T | - | Intergenic | 2.7 ×10−7 | 1.34 (1.20–1.50) | 1.00 | 0.23 |
| rs72820285 | 6 | 9318701 | A | - | Intergenic | 6.6 ×10−7 | 1.38 (1.21–1.56) | 2.74 | 0.22 |
| rs56100674 | 6 | 9310391 | T | - | Intergenic | 7.8 ×10−7 | 1.37 (1.21–1.56) | 3.48 | 0.41 |
| rs111792270 | 6 | 9327938 | A | - | Intergenic | 8.1 ×10−7 | 1.38 (1.22–1.58) | 2.00 | 0.11 |
| rs55720740 | 6 | 9309819 | T | - | Intergenic | 8.2 ×10−7 | 1.37 (1.21–1.55) | 1.18 | 0.27 |
| rs1176869 | 14 | 41276426 | G | - | Intergenic | 9.2 ×10−7 | 1.35 (1.20–1.52) | 5.41 | 0.19 |
| CTDs | |||||||||
| rs6886261 | 5 | 43022764 | C | LOC648987 | Non-coding | 1.7×10−7 | 1.78 (1.43–2.21) | 1.71 | 0.20 |
| rs585302 | 5 | 43034142 | T | LOC648987 | Non-coding | 1.7×10−7 | 1.78 (1.43–2.20) | 5.68 | 0.55 |
| LVOTDs | |||||||||
| rs11894932 | 2 | 99265186 | C | MGAT4A | Intronic | 1.5 ×10−7 | 0.44 (0.33–0.61) | 6.43 | 0.14 |
| rs72820264 | 6 | 9291883 | C | - | Intergenic | 2.1 ×10−8 | 2.05 (1.59–2.63) | 1.50 | 0.19 |
| rs72820266 | 6 | 9292316 | C | - | Intergenic | 4.9 ×10−7 | 1.86 (1.46–2.36) | 0.87 | 0.35 |
|
| |||||||||
| Maternal | |||||||||
|
| |||||||||
| CTDs+LVOTDs | |||||||||
| rs114952830 | 6 | 30850113 | A | - | Intergenic | 6.2 ×10−7 | 1.35 (1.20–1.52) | 5.08 | - |
| rs75661265 | 8 | 60187791 | T | - | Intergenic | 5.0 ×10−7 | 1.17 (1.10–1.25) | 2.99 | 0.47 |
| rs55884872 | 8 | 60192814 | A | - | Intergenic | 5.5 ×10−7 | 1.18 (1.11–1.26) | 1.17 | 0.15 |
| rs9643366 | 8 | 60190505 | A | - | Intergenic | 7.7 ×10−7 | 1.18 (1.10–1.25) | 11.35 | 0.56 |
| rs1941023 | 11 | 60275901 | G | - | Intergenic | 4.4 ×10−7 | 1.25 (1.15–1.36) | 1.10 | 0.38 |
| rs1245314 | 14 | 27492640 | A | - | Intergenic | 8.6×10−7 | 1.12 (1.07–1.17) | 0.80 | 0.11 |
| CTDs | |||||||||
| rs74461473 | 3 | 50250747 | T | SLC38A3 | Intronic | 4.2×10−7 | 0.80 (0.73–0.87) | 14.85 | 0.44 |
| rs116734199 | 6 | 30808762 | T | - | Non-coding | 2.8×10−7 | 1.17 (1.10–1.25) | 13 | - |
| rs114952830 | 6 | 30850113 | A | - | Intergenic | 3.0×10−7 | 1.17 (1.10–1.25) | 5.08 | - |
| rs114414726 | 6 | 30811164 | A | - | Intergenic | 5.2×10−7 | 1.11 (1.06–1.15) | 0.14 | - |
| rs2844660 | 6 | 30823760 | C | - | Intergenic | 7.8×10−7 | 1.41 (1.23–1.61) | 12.23 | 0.46 |
| rs115588720 | 6 | 30923527 | T | - | Intergenic | 8.5×10−7 | 1.04 (1.03–1.06) | 6.77 | - |
| rs115946461 | 6 | 30837538 | C | - | Intergenic | 9.0×10−7 | 1.06 (1.03–1.08) | 0.71 | - |
| rs116583468 | 6 | 30842674 | T | - | Intergenic | 9.7×10−7 | 1.07 (1.04–1.09) | 4.08 | - |
| rs55884872 | 8 | 60192814 | A | - | Intergenic | 1.7×10−7 | 1.04 (1.02–1.06) | 1.18 | 0.15 |
| rs75661265 | 8 | 60187791 | T | - | Intergenic | 2.2×10−7 | 1.19 (1.11–1.27) | 2.99 | 0.47 |
| rs9643366 | 8 | 60190505 | A | - | Intergenic | 3.4×10−7 | 1.39 (1.22–1.57) | 11.35 | 0.56 |
| LVOTDs | |||||||||
| rs1642645 | 1 | 42502281 | A | HIVEP3 | 5’ upstream | 7.8×10−7 | 0.53 (0.41–0.68) | 7.41 | 0.55 |
| rs141978609 | 4 | 168835335 | G | - | Intergenic | 8.6×10−7 | 0.50 (0.38–0.66) | 0.33 | - |
| rs55788414 | 16 | 81184939 | T | PKD1L2 | Intronic | 5.5×10−8 | 5.82 (3.08–10.97) | 2.08 | 0.13 |
Chromosome
Hg19/NCBI build 37
P-values only shown for top inherited or maternal effects (p<1 × 10−6)
Odds ratio estimate for carrying one copy of the high-risk allele compared to no copies, and corresponding 95% confidence interval
Scaled Combined Annotation Dependent Depletion (CADD) score (variants with CADD>10 are predicted to fall in the top 10% of the most deleterious variants in the genome)
Genome Wide Annotation of VAriants (GWAVA) score for predicting the functional impact of non-coding variants/regions (range 0–1)
Meta-analyses of the inherited genotype for CTDs only and CTDs+LVOTDs also did not identify any genome-wide significant associations (Supplemental Figure 1, Supplemental Figure 2). However, there was suggestive evidence of associations with the inherited genotype for multiple SNPs in the meta-analysis of CTDs only (36 SNPs in 11 regions) and CTD+LVOTD (70 SNPs in 9 regions) (Supplemental Tables 5–7).
One genome-wide significant association was identified in the meta-analysis of the inherited genotype for LVOTDs (rs72820264, p=2.1×10−8, minor allele frequency: 0.12 [1,000 Genomes release 17]). Seven additional SNPs in this region (6p24.3) also had suggestive evidence for association (p≤10−5) (Figure 2). The LVOTD meta-analysis also identified 24 additional SNPs, in 14 regions, with suggestive evidence of associations (Table 3, Supplemental Figure 1, Supplemental Table 7).
Discussion
Our GWAS of maternal and inherited genotypes and CTDs/LVOTDs in five independent cohorts identified several potential candidate regions. The majority of these signals were present in our case-control analysis, which was likely better powered than our family-based comparisons given the relatively large number of controls. It is noteworthy that nearly all of the genomewide significant SNPs in the CHOP case-control comparison were intronic variants in MGAT4C, which is involved in the biosynthesis of an N-glycan precursor. There are several congenital disorders of N-linked glycosylation, and there are reports of individuals with these conditions that have a CHD.43, 44 Two of the SNPs in MGAT4C had CADD scores >10 (rs863392 and rs839163) and are thus predicted to be highly deleterious. However, the GWAVA-TSS scores for intronic SNPs in MGAT4C were all less than the median value for GWAS SNPs that have been externally replicated. Moreover, as these variants were not significantly related to CTDs in the meta-analysis, it may be that associations identified in our case-control represent false-positive findings arising from uncontrolled confounding (e.g., due to inadequate control for population stratification).
No association met our pairwise replication threshold of p≤10−5 in more than one dataset assessing the same genetic effect (maternal or inherited) and phenotype. However, our meta-analyses identified several genes/regions of interest. We identified one inherited SNP (rs72820264, imputed) that was significantly associated with LVOTDs in our meta-analysis. This SNP is in an intergenic region on chromosome 6p24.3, 416kb from the nearest upstream gene (MRDS1/OFCC1) and 638kb from the nearest downstream gene (HULC). There were several additional SNPs in the region in strong linkage disequilibrium with rs72820264 that also had low p-values (Figure 2), including typed variants (e.g., rs7762096, p=6.2×10-5). There is a strong signal from an ENCODE clustered DNase I hypersensitivity site over 24 tissues, located at chr6:9293081-9293290, about 1.5kb away from this SNP (chr6:9291650). A DNAse I hypersensitivity site may indicate a promoter location within this primate-conserved region, because regulatory regions, especially promoters, tend to be DNase-sensitive.45, 46 However, GWAVA scores do not support a functional role for this SNP (0.2) or region (0.3). Also, the scaled CADD score was 1.5 for rs72820264, which suggests that this variant is not highly deleterious. The nearest downstream gene, OFCC1 (Orofacial Cleft 1), has been associated with orofacial clefts in several studies.47–49 Further, this region (6p24.3-21.2) has been linked to a rare autosomal dominant syndrome that includes heart anomalies (non-compaction of the ventricular myocardium, bradycardia, pulmonary valve stenosis, secundum atrial septal defect, left isomerism, heterotaxy).50 Thus, in silico data neither negates nor defines the potential contribution of this statistically associated SNP to disease risk.
The top suggestive associations from our meta-analyses included inherited variants in LRP1B (LDL receptor protein) and MGAT4A (regulates Golgi apparatus formation), as well as maternal variants in SLC38A3 (glutamine transporter), HIVEP3 (HIV enhancer-binding protein), and PKD1L2 (polycystin protein involved in transmembrane domains). Of these, the intronic variant in SLC38A3 was predicted to be within the most 10% of deleterious variants in the genome (scaled CADD score: 14.85) and had a GWAVA-TSS score that was within the third quartile of scores for GWAS SNPs that have been externally replicated. This gene is thought to play an important role in fetal development, as it likely is the main supplier of glutamine (necessary for fetal growth) to the fetus in early gestation.51 The maternal intronic variant in PKD1L2 had a borderline significant association with LVOTDs (p=5.5×10-8), but is not predicted to be deleterious based on CADD or GWAVA-TSS scores. However, no SNP in linkage disequilibrium with this PKD1L2 SNP was also associated.
The maternal SNP with the smallest association p-value, rs55788414, likely reflects a false positive finding, given that the maternal genotype for SNPs in linkage disequilibrium with the SNP with were not associated with LVOTD (Figure 2).
We had results for 16 SNPs that were previously identified in CHD GWAS, but none were significantly associated with the corresponding phenotype/effect in our meta-analysis (Supplementary Table 8). However, some of the phenotypes included in previous GWAS did not directly correspond to those in our cohorts (e.g., tetralogy of Fallot versus conotruncal defects; septal defects versus CTDs+LVOTDs). For other traits (e.g., adult-onset conditions), it has not been uncommon for GWAS findings to fail to replicate in subsequent GWAS.52 Potential reasons for lack of replication across GWAS studies include population differences (e.g., three of the previous CHD GWAS were conducted among Chinese or European samples), genotyping platform/imputation differences, modeling differences, gene-environment or gene-gene interactions, and differences in case phenotypes (e.g., CTDs versus tetralogy of Fallot).
Our results should be considered in light of several limitations. We conducted nine GWAS analyses and six meta-analyses, using the typical GWAS threshold for significance of p<5×10−8 in each analysis. Given that we conducted multiple analyses, this threshold may have been too lenient. In addition, some of our cohorts were small, which limited our ability to detect SNPs that are only modestly associated with CHDs. In addition, our study subjects were recruited from a clinical rather than population-based setting and, thus, may not be representative of the general population of individuals affected by CHDs. Despite these limitations, our study also had several strengths including a relatively large sample size, as compared to other GWAS of CHDs, and the evaluation of both inherited and maternal genetic effects.
In summary, we report on a number of potential candidate regions for some of the most common, severe heart defects. Our studies suggest that few, if any, single SNPs confer large risk of non-syndomic CTDs and/or LVOTDs. More work is needed to address the contribution of common SNPs that confer weak or moderate risk. For example, results from recent GWAS suggest that gene- or pathway-based studies may be required to identify genes that influence risk of CHDs via common variants.53 Future work might therefore focus on the candidate loci highlighted by these meta-analyses and/or on gene-sets defined by functional or developmental pathways. Otherwise, substantially larger sample sets will be required to identify individual common variants that confer a small to moderate risk for CHDs and to identify risks specific to cardiac subtypes.
Supplementary Material
Clinical Perspective.
Congenital heart defects (CHDs) are complex traits and familial recurrence patterns suggest that multiple, inherited genetic variants of low to moderate effect are likely to influence risk. The maternal genotype may also influence the risk of CHDs in offspring via an effect on the in utero environment. However, relatively little is known about the specific inherited or maternal genes that are associated with the risk of CHDs. In this study, we performed genome-wide association studies (GWAS) in five CHD cohorts, followed by meta-analyses of combined data. We evaluated inherited and maternal genotypes separately. In addition, cases with conotruncal defects and left ventricular obstructive defects were evaluated separately and together. While a number of suggestive associations were identified in the individual GWAS, none of these associations reached a GWAS level of significance. However, in our meta-analysis, one intra-genic, inherited SNP, rs72820264, was associated with left ventricular obstructive defects at a GWAS level of significance. The exact molecular mechanisms and pathways involved in this association are not yet clear. The results of this study will help guide future genomic efforts and will assist in narrowing down candidate regions of importance for the etiology of congenital heart defects.
Acknowledgments
We thank the Pediatric Cardiac Genomics Consortium (members listed in Supplemental Document 1), Sharon Edman, Jennifer Garbarini, Stacy Woyciechowski, and Brande Latney for technical assistance, as well as the families who consented to participate in this study. Results for additional data (e.g., from the analyses within each individual cohort) may be requested by contacting the authors.
Sources of Funding: This study was supported by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01HD070454), the National Heart, Lung, and Blood Institute including the Pediatric Cardiac Genomics Consortium (R21HL-098844, R01HL-076773, R01-HL74094, P50-HL74731, U01-HL098188, U01-HL098147, U01-HL098153, U01-HL098163, U01-HL098123, U01-HL098162), the Cardiovascular Development Consortium (U01-HL098166), the National Human Genome Research Institute (U54HG006504), the National Research Science Foundation (Award 1F30HL123238), and the National Center for Research Resources (M01-RR-000240, RR024134 [now the National Center for Advancing Translational Sciences (UL1TR000003)]). GWAS genotyping was funded by an Institutional Development Fund to The Center for Applied Genomics from The Children’s Hospital of Philadelphia. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosures: None
References
- 1.Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol. 2007;79:714–727. doi: 10.1002/bdra.20403. [DOI] [PubMed] [Google Scholar]
- 2.Rosano A, Botto LD, Botting B, Mastroiacovo P. Infant mortality and congenital anomalies from 1950 to 1994: An international perspective. J Epidemiol Community Health. 2000;54:660–666. doi: 10.1136/jech.54.9.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry LW, Neill CA, Ferencz C, Rubin JD, Loffredo CA. Infants with congenital heart disease: The cases. In: Ferencz C, Rubin JD, Loffredo CA, Magee CA, editors. Epidemiology of congenital heart disease: The baltimore-washington infant study 1981–1989. Mount Kisco: Futura Publishing Company, Inc; 1993. pp. 33–62. [Google Scholar]
- 4.Nora JJ, Nora AH. Genetic epidemiology of congenital heart diseases. Prog Med Genet. 1983;5:91–137. [PubMed] [Google Scholar]
- 5.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44:138–143. doi: 10.1016/j.jacc.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 6.Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 7.McElhinney DB, Driscoll DA, Levin ER, Jawad AF, Emanuel BS, Goldmuntz E. Chromosome 22q11 deletion in patients with ventricular septal defect: Frequency and associated cardiovascular anomalies. Pediatrics. 2003;112:e472. doi: 10.1542/peds.112.6.e472. [DOI] [PubMed] [Google Scholar]
- 8.Kwiatkowska J, Wierzba J, Aleszewicz-Baranowska J, Erecinski J. Genetic background of congenital conotruncal heart defects--a study of 45 families. Kardiol Pol. 2007;65:32–37. discussion 38–39. [PubMed] [Google Scholar]
- 9.Oyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009;120:295–301. doi: 10.1161/CIRCULATIONAHA.109.857987. [DOI] [PubMed] [Google Scholar]
- 10.Goldmuntz E, Woyciechowski S, Renstrom D, Lupo PJ, Mitchell LE. Variants of folate metabolism genes and the risk of conotruncal cardiac defects. Circ Cardiovasc Genet. 2008;1:126–132. doi: 10.1161/CIRCGENETICS.108.796342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu H, Yang W, Lu W, Etheredge AJ, Lammer EJ, Finnell RH, et al. Gene variants in the folate-mediated one-carbon metabolism (focm) pathway as risk factors for conotruncal heart defects. Am J Med Genet A. 2012;158A:1124–1134. doi: 10.1002/ajmg.a.35313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobbs CA, Cleves MA, Karim MA, Zhao W, MacLeod SL. Maternal folate-related gene environment interactions and congenital heart defects. Obstet Gynecol. 2010;116:316–322. doi: 10.1097/AOG.0b013e3181e80979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw GM, Lu W, Zhu H, Yang W, Briggs FB, Carmichael SL, et al. 118 snps of folate-related genes and risks of spina bifida and conotruncal heart defects. BMC Med Genet. 2009;10:49. doi: 10.1186/1471-2350-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell LE, Long J, Garbarini J, Paluru P, Goldmuntz E. Variants of folate metabolism genes and risk of left-sided cardiac defects. Birth Defects Res A Clin Mol Teratol. 2010;88:48–53. doi: 10.1002/bdra.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agopian AJ, Mitchell LE, Glessner J, Bhalla AD, Sewda A, Hakonarson H, et al. Genome-wide association study of maternal and inherited loci for conotruncal heart defects. PLoS ONE. 2014;9:e96057. doi: 10.1371/journal.pone.0096057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordell HJ, Topf A, Mamasoula C, Postma AV, Bentham J, Zelenika D, et al. Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of fallot. Hum Mol Genet. 2013;22:1473–1481. doi: 10.1093/hmg/dds552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell LE, Agopian AJ, Bhalla A, Glessner JT, Kim CE, Swartz MD, et al. Genome-wide association study of maternal and inherited effects on left-sided cardiac malformations. Hum Mol Genet. 2015;24:265–73. doi: 10.1093/hmg/ddu420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet. 2013;45:822–824. doi: 10.1038/ng.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Z, Shi Y, Mo X, Xu J, Zhao B, Lin Y, et al. A genome-wide association study identifies two risk loci for congenital heart malformations in han chinese populations. Nat Genet. 2013;45:818–821. doi: 10.1038/ng.2636. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Guo X, Zhao B, Liu J, Da M, Wen Y, et al. Association analysis identifies new risk loci for congenital heart disease in chinese populations. Nat Commun. 2015;6:8082. doi: 10.1038/ncomms9082. [DOI] [PubMed] [Google Scholar]
- 21.Agopian AJ, Eastcott LM, Mitchell LE. Age of onset and effect size in genome-wide association studies. Birth Defects Res A Clin Mol Teratol. 2012;94:908–11. doi: 10.1002/bdra.23066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 23.Peyvandi S, Lupo PJ, Garbarini J, Woyciechowski S, Edman S, Emanuel BS, et al. 22q11.2 deletions in patients with conotruncal defects: Data from 1,610 consecutive cases. Pediatr Cardiol. 2013;34:1687–1694. doi: 10.1007/s00246-013-0694-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White PS, Xie HM, Werner P, Glessner J, Latney B, Hakonarson H, et al. Analysis of chromosomal structural variation in patients with congenital left-sided cardiac lesions. Birth Defects Res A Clin Mol Teratol. 2014;100:951–964. doi: 10.1002/bdra.23279. [DOI] [PubMed] [Google Scholar]
- 25.Gelb B, Brueckner M, Chung W, Goldmuntz E, Kaltman J, Kaski JP, et al. The congenital heart disease genetic network study: Rationale, design, and early results. Circ Res. 2013;112:698–706. doi: 10.1161/CIRCRESAHA.111.300297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International HapMap Consortium. The international hapmap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 27.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 30.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1:457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ainsworth HF, Unwin J, Jamison DL, Cordell HJ. Investigation of maternal effects, maternal-fetal interactions and parent-of-origin effects (imprinting), using mothers and their offspring. Genet Epidemiol. 2011;35:19–45. doi: 10.1002/gepi.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howey R, Cordell HJ. Premim and emim: Tools for estimation of maternal, imprinting and interaction effects using multinomial modelling. BMC Bioinformatics. 2012;13:149. doi: 10.1186/1471-2105-13-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilcox AJ, Weinberg CR, Lie RT. Distinguishing the effects of maternal and offspring genes through studies of “case-parent triads”. Am J Epidemiol. 1998;148:893–901. doi: 10.1093/oxfordjournals.aje.a009715. [DOI] [PubMed] [Google Scholar]
- 35.Benyamin B, Visscher PM, McRae AF. Family-based genome-wide association studies. Pharmacogenomics. 2009;10:181–190. doi: 10.2217/14622416.10.2.181. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell LE, Weinberg CR. Evaluation of offspring and maternal genetic effects on disease risk using a family-based approach: The “pent” design. Am J Epidemiol. 2005;162:676–685. doi: 10.1093/aje/kwi249. [DOI] [PubMed] [Google Scholar]
- 37.Duggal P, Gillanders EM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics. 2008;9:516. doi: 10.1186/1471-2164-9-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magi R, Morris AP. Gwama: Software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. Locuszoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long J, Ramadhani T, Mitchell LE. Epidemiology of nonsyndromic conotruncal heart defects in texas, 1999–2004. Birth Defects Res A Clin Mol Teratol. 2010;88:971–979. doi: 10.1002/bdra.20724. [DOI] [PubMed] [Google Scholar]
- 43.Kranz C, Basinger AA, Gucsavas-Calikoglu M, Sun L, Powell CM, Henderson FW, et al. Expanding spectrum of congenital disorder of glycosylation ig (cdg-ig): Sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet A. 2007;143A:1371–1378. doi: 10.1002/ajmg.a.31791. [DOI] [PubMed] [Google Scholar]
- 44.Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, et al. Srd5a3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142:203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. Encode data in the ucsc genome browser: Year 5 update. Nucleic Acids Res. 2013;41:D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salahshourifar I, Halim AS, Sulaiman WA, Zilfalil BA. Contribution of 6p24 to non-syndromic cleft lip and palate in a malay population: Association of variants in ofc1. J Dent Res. 2011;90:387–391. doi: 10.1177/0022034510391798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen CP, Tzen CY, Chern SR, Tsai FJ, Hsu CY, Lee CC, et al. A 12 mb deletion of 6p24.1-->pter in an 18-gestational-week fetus with orofacial clefting, the dandy-walker malformation and bilateral multicystic kidneys. Eur J Med Genet. 2009;52:59–61. doi: 10.1016/j.ejmg.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 49.Scapoli L, Pezzetti F, Carinci F, Martinelli M, Carinci P, Tognon M. Evidence of linkage to 6p23 and genetic heterogeneity in nonsyndromic cleft lip with or without cleft palate. Genomics. 1997;43:216–220. doi: 10.1006/geno.1997.4798. [DOI] [PubMed] [Google Scholar]
- 50.Wessels MW, De Graaf BM, Cohen-Overbeek TE, Spitaels SE, de Groot-de Laat LE, Ten Cate FJ, et al. A new syndrome with noncompaction cardiomyopathy, bradycardia, pulmonary stenosis, atrial septal defect and heterotaxy with suggestive linkage to chromosome 6p. Hum Genet. 2008;122:595–603. doi: 10.1007/s00439-007-0436-x. [DOI] [PubMed] [Google Scholar]
- 51.Balkrishna S, Broer A, Welford SM, Hatzoglou M, Broer S. Expression of glutamine transporter slc38a3 (snat3) during acidosis is mediated by a different mechanism than tissue-specific expression. Cell Physiol Biochem. 2014;33:1591–1606. doi: 10.1159/000358722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greene CS, Penrod NM, Williams SM, Moore JH. Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS ONE. 2009;4:e5639. doi: 10.1371/journal.pone.0005639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagg S, Ganna A, Van Der Laan SW, Esko T, Pers TH, Locke AE, et al. Gene-based meta-analysis of genome-wide association studies implicates new loci involved in obesity. Hum Mol Genet. 2015;24:6849–6860. doi: 10.1093/hmg/ddv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.