

ABSTRACT
Glycoprotein VI is a platelet-specific collagen receptor critical for in vivo formation of arterial thrombosis. It is also considered as an attractive target for the development of anti-thrombotic drugs because blocking glycoprotein (GP)VI inhibits platelet aggregation without inducing detrimental effects on physiologic hemostasis.
Here, we present data on the identification, in vitro and ex vivo pharmacology of a humanized Fab fragment designated as ACT017. ACT017 was selected out of 15 humanized variants based upon structural and functional properties. It was produced under GMP-like conditions followed by detailed physico-chemical analysis and functional characterization indicating high antigen-binding specificity and affinity. In addition, we demonstrate, in a dose-escalation study, that ACT017 has a high capacity to specifically inhibit collagen-induced platelet aggregation ex vivo after injection to the macaque without inducing thrombocytopenia, GPVI depletion or bleeding side effects as is the case for conventional anti-platelets. Therefore, ACT017 is a promising therapeutic candidate for the development of a new generation of safe and efficient anti-thrombotic drugs.
KEYWORDS: Cardiovascular disease, glycoprotein VI, humanization, platelet aggregation, therapeutic antibody, thrombosis, stroke
Abbreviations
- AA
amino acid
- CDR
complementary-determining region
- Fd
heavy chain fragment
- FR
framework region
- GP
glycoprotein
- HRP
horseradish peroxidase
- Mr
relative molecular weight
- PBS
phosphate-buffered saline
- PpL
Peptostreptococcus protein L
- PPP
platelet-poor plasma
- PRP
platelet-rich plasma
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEC-HPLC
size-exclusion high performance liquid chromatography
- SPR
surface plasmon resonance
- VH
heavy variable domain
- VL
light variable domain
Introduction
Arterial thrombosis is a common disorder usually caused by the erosion or rupture of an atherosclerotic plaque and, through platelet-mediated thrombi, can cause ischemic injury. When thrombotic occlusion occurs in coronary or cerebral arteries, it results in myocardial infarction and stroke, respectively, which are leading causes of death worldwide.1
Arterial thrombus formation is a complex process initiated by transient bridging of the platelet glycoprotein (GP) Ib-V-IX complex to collagen via the von Willebrand factor, while the integrin α2β1 and GPVI stabilize further adhesion, activation and accumulation of platelets.2 Then, platelets secrete adenosine diphosphate (ADP) and synthesize thromboxane A2 (TXA2), which are both soluble agonists that favor recruitment of new circulating platelets. Activated platelets aggregate via activated integrin αIIbβ3 (glycoprotein GPIIbIIIa) and fibrinogen. They also provide the procoagulant surface for thrombin generation, which in turns stabilizes the thrombus via fibrin formation.3
The treatment of arterial thrombosis by antiplatelet agents has been a burning issue over the past decades and several classes of drugs are now available for solving the problem.4-6 The current standard for the treatment of acute coronary syndrome is mainly based on dual antiplatelet therapy with aspirin (inhibition of platelet TXA2 formation) and ADP-receptor P2Y12 blockers (thienopyridine or direct inhibitors) or antagonists of the integrin αIIbβ3 (abciximab, eptifibatide). However, these drugs target mechanisms common to hemostasis and thrombosis, and so their use leads to an inherent antihemostatic effects and an increased risk of uncontrolled bleeding complications that correlates with their antithrombotic efficacy. For these reasons, pharmacological treatment of acute ischemic stroke remains restricted to fibrinolysis and the administration of the recombinant tissue plasminogen activator (rtPA) that binds to fibrin in the thrombus and converts the entrapped plasminogen to plasmin. Plasmin in turn degrades the fibrin matrix of the thrombus, thereby exerting its thrombolytic action, but with a major risk of cerebral bleeding when rtPA is not injected within a short therapeutic window (4.5 hours post-initial clinical manifestation). Today, fibrinolysis, whose success outcome does not exceed 40%, is challenged by mechanical thrombectomy, which improves clinical outcome. However, a very recent study did not reveal any clinical benefit with combined intravenous thrombolysis and thrombectomy treatment.7
In this context, there is a need for new safe and efficient treatments without systemic side-effects such as hemorrhage. GPVI is increasingly recognized as a new potential target for the treatment of arterial thrombosis.8-10 Indeed, GPVI is known to be the main platelet-collagen receptor involved in arterial thrombus formation. GPVI has also been identified as a functional platelet receptor for fibrin, and thus contributes to thrombus growth and stabilization.11 However, GPVI is an accessory in physiologic hemostasis, as indicated by the observation of patients presenting a congenital or autoimmune GPVI deficiency who do not present a bleeding phenotype.12,13 In this way, blocking GPVI is now considered a potentially valuable alternative to conventional treatments of arterial thrombosis. Several traditional small molecule drugs and biologics are in the pipeline for further developments.14,15
One promising and suitable strategy is the design of GPVI-specific antibodies with functional inhibitory properties. Several antibody molecules directed against GPVI have been prepared.16-20 However, few are capable of blocking GPVI-collagen interaction, some induce GPVI shedding and most of them have a weak affinity for GPVI, making them unsuitable for clinical investigation.21 Mouse monoclonal IgG 9O12 satisfies all the criteria required in terms of specificity, selectivity, affinity and inhibitory properties.18 However, 2 main hindrances must be overcome: 1) a whole bivalent IgG would bridge platelet membrane GPVI with the low affinity FcγRIIA receptor, leading to platelet activation and possibly induce GPVI deficiency because of internalization or shedding; and 2) the potential immunogenicity of antibodies from murine origin is a major barrier to injection in humans. These drawbacks can be overcome by using re-engineered monovalent humanized antibody fragments. Initiated by Muzard et al.,22 this strategy failed because of improper humanization and the inability to produce sufficient quantities, purify and characterize the recombinant molecule, regardless of the engineered format (scFv or Fab). Here, we designed and selected a humanized Fab (ACT017), without loss of specificity and affinity for its target, that fulfills all the criteria required for further clinical developments.
Results
Design of humanized anti-GPVI Fabs
Fab 9O12 prepared after limited papainolysis of mouse IgG 9O12 was previously shown to have a great potential to inhibit in vivo collagen-induced platelet aggregation.23 However, it is unsuitable for therapeutic application in humans because of its rodent origin, and also because it does not bind either protein A or Peptostreptococcus protein L (PpL), making purification extremely difficult. For these reasons we designed a set of humanized Fabs with the aim of creating non-immunogenic monovalent antibody fragments suitable for injection in humans, carrying an internal affinity tag (PpL tag) and with the ability to preserve specific GPVI binding activity.
9O12 canonical class and sub-class complementary-determining regions (CDRs) were identified as L1–4/16A; L2–1/7A; L3–1/9A; H1–1/10A; H2–2/10A. As commonly accepted, CDR H3 does not adopt canonical conformations, and thus was analyzed independently.
Sequence database searches allowed us to select 4 templates for each VH and VL upon the following 4 independent criteria: 1) human germline sequences most similar to 9O12 IGKV and IGHV (IGKV2–29*02 and IGHV1–3*01, respectively); 2) high sequence identity and identical canonical sub-class CDRs (bevacizumab and IGHV1–3*01 for VH); 3) same antibody template for both V-domains even at the cost of a less optimal template for one of the chains (bevacizumab); and 4) high sequence identity and identical or closely related canonical class CDRs and secondary structure (canakinumab and REI for VL) (Table 1). We also selected the human myeloma antibodies NEW (for the VH) and REI (for the VL) because they are well characterized in terms of stability and expression and they are frequently used in a “fixed framework” strategy of humanization where no database search is used.24 In some cases, the selected templates fit several criteria at the same time, and this was considered beneficial.
Table 1.
Main features of the 9O12 V-domains and the humanized V-domains designed for generation of Fabs ACT004 to ACT018. Template and criteria: (a) human germline most similar to 9O12; (b) high sequence identity and identical canonical subclasses; (c) same antibody template for both V-domains; (d) high sequence identity and closely related canonical classes. T-cell epitope score represents the number of 9 mers with a score higher than arbitrary selected cut-off of 10 and 1. Canonical classes indicated here are those from the template. * indicates that L1–4/16A canonical subclass was restored after humanization. Packing angle is a prediction after pairing humanized variants of VL and VH. Z-score: a value in the [−1.0; +1.0] range indicates a high degree of humanness.
| Antibody V-domain |
Template & criteria |
T-cell epitope score |
Canonical class |
Packing angle |
Z-score |
Closest human germline gene and % identity |
| 9O12-VL | IGKV2-29*02 (a) | 1>10 ; 3>1 | L1-4/16A | −45.3 | −1.7 | IGKV2-29*02 |
| L2-1/7A | 69.0% - 100 overlap | |||||
| L3-1/9A | ||||||
| VL2b | bevacizumab (c) (d) | 1>10 ; 2>1 | L1-2/11A* | −45.3 (H1, 3, 4) | 0.5 | IGKV1-39*01 |
| L2-1/7A | −43.8 (H2) | 81.0% - 100 overlap | ||||
| L3-1/9A | ||||||
| VL3b | IGKV2-29*02 (a) (b) | 1>10 ; 3>1 | L1-4/16A | −45.3 (H1, 3, 4) | −0.3 | IGKV2-29*02 |
| L2-1/7A, | −43.8 (H2) | 80.0% - 100 overlap | ||||
| L3-1/9A | ||||||
| VL7b | canakinumab (d) | 1>10 ; 3>1 | L1-2/11A* | −45.3 | −0.2 | IGKV6-21*01 |
| L2-1/7A | (H1,3,4) | 72.0% - 100 overlap | ||||
| L3-1/9A | −46.9 (H2) | |||||
| VL8b | REI (d) | 1>10 ; 2>1 | L1-2/11A* | −45.3 | −0.1 | IGKV1-33*01 |
| L2-1/7A | (H1,3,4) | 80.0% - 100 overlap | ||||
| L3-1/9A | −43.8 (H2) | |||||
| 9O12-VH | IGHV1-3*01 (a) | 2>10 ; 3>1 | H1-1/10A | -45.3 | −1.1 | IGHV1-3*01 |
| H2-2/10A | 78.6% - 98 overlap | |||||
| VH1 | IGHV1-3*01 (a) (b) | 2>10 ; 4>1 | H1-1/10A | −45.3 | −0.2 | IGHV1-3*01 |
| H2-2/10A | −47 (VL7b) | 80.6% - 98 overlap | ||||
| VH2 | bevacizumab (b) (c) | 3>10 ; 0>1 | H1-1/10A | −43.8 | 1 | IGHV3-23*04 |
| H2-2/10A | −46.9 (VL7b) | 77.8% - 99 overlap | ||||
| VH3 | IGHV1-46*01 (b) | 2>10 ; 3>1 | H1-1/10A | −45.3 | 0.1 | IGHV1-46*01 |
| H2-2/10A | −47 (VL7b) | 92.9% - 98 overlap | ||||
| VH4 | NEW (b) | 1>10 ; 3>1 | H1-1/10A H2-2/10A |
−45.3 −47 (VL7b) |
−0.6 | IGHV4-38-2*02 77.8% - 99 overlap |
We first grafted 9O12 CDRs on the template and then went through each substitution in the framework regions (FR) considering its physico-chemical characteristics, the isoelectric point (pI) and the effect such change could have after inspecting the 3D-structure model (Fig. 1). Following this manual curated analysis, a limited number of residues belonging to the human FR were reverted to mouse to restore the expected antigen-binding activity of the original antibody.
Figure 1.

Design of humanized 9O12 V-domains. (A) Sequence alignment of the mouse 9O12 IGHV (9VH) (most similar human germline sequence being IGHV1–3*01) with 4 human or humanized IGHV templates (TH1: IGHV1–3*01; TH2: bevacizumab; TH3: IGHV1–46*01; TH4: NEW), the humanized 9O12 IGHV variants (VH1, VH2, VH3 and VH4) and the framework regions of their most similar human germline genes (CH1: IGHV1–3*01; CH2: IGHV3–23*04; CH3: IGHV1–46*01; CH4: IGHV4–38–2*02). The humanized variants that retained antigen-binding activity are highlighted in green. (B) Sequence alignment of the mouse 9O12 IGKV (9VL) (most similar human germline sequence being IGKV2–29*02) with 4 human or humanized IGKappaV templates (TL2b: bevacizumab; TL3b: IGKV2–29*02; TL7b: canakinumab; TL8b: REI), the humanized 9O12 IGKV (variants VL2b, VL3b, VL7b and VL8b) and the framework regions of their most similar human germline genes (CL2b: IGKV1–39*01; CL3b: IGKV2–29*02; CL7b: IGKV6–21*01; CL8b: IGKV1–33*01). The humanized variants that retained antigen-binding activity and acquired PpL recognition site are highlighted in green (or in blue if retaining antigen binding activity but not PpL recognition). CDRs are in italic, underlined, gray. Residues at key sites28,49 for canonical structures are highlighted in blue. Residues buried in VH/VL interfaces are underlined in yellow. Residues critical for PpL binding are highlighted in green. Based on the physico-chemical classes of the amino acids (AA), differences in the framework regions of mouse 9O12 and its humanized variants are classified into very similar AA (green), similar AA (blue), dissimilar AA (orange) and very dissimilar AA (red).
In the same way, the structurally defined CDRs L1-L3 and H1-H2, and key residues presumed to ensure the conformation assumed by the hypervariable loops in the donor Fv (9O12) were inspected.25-28 A limited number of residues belonging to Kabat's CDRs, but not to the hypervariable loops, were substituted because they were likely not to be involved into the antigen-binding site but exposed to the solvent. This was the case for residues H59 (F59>Y) and H64 (K 64 > Q) in CDR H2 (template VH3) (Fig. 2). We paid attention to maintaining cohesion between residues at 10 positions in VL and VH buried in the interface between the domains, as indicated in Fig. 1 and 2.29
Figure 2.
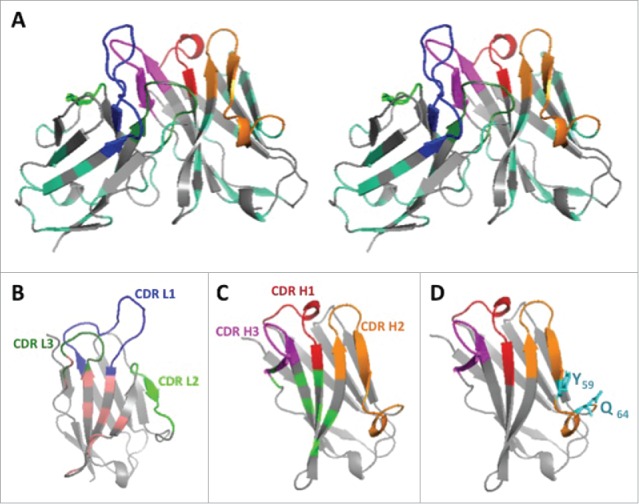
3D structure of the humanized V-domains of ACT017 (VL8b and VH3 variants). (A) Stereo view with colored CDRs. Mutated residues exposed at the surface are in cyan. (B) VL domain with residues buried at the interface with VH in light pink. (C) VH domain with residues buried at the interface with VL in light green. (D) VH domain with the 2 mutated residues from CDR H2 in cyan.
To create a PpL binding site that is highly dependent on several residues belonging to V-kappa FR 1 and residue L74 as well, we introduced the critical S 12 > P substitution in all 4 V-kappa domain variants while maintaining P8 and R18, which are essential, but not sufficient, for a strong interaction with PpL.30,31
During the entire process, the modifications were approved by a detailed analysis to confirm the ongoing improvement of the humanness Z score, the retention of a VH/VL packing angle identical or close to the one predicted for 9012 Fv, and the decrease of the residual immunogenicity (Table 1).
Thus, 4 humanized variants were designed for each V-domains (VL2b, 3b, 7b, 8b; VH1–4) requiring 16 to 32 substitutions for VL and 21 to 42 for VH domains. Their closest human germline counterpart was identified (Table 1). Referring to ACT017, Fig. 2 shows a stereo 3D-structural model and the 2 V-domains (VH3, VL8b), which reached the best Z-scoring (0.1 and −0.1, respectively) and very high percent of identity (92.9% and 80%, respectively) with human germline genes.
Primary screening of humanized anti-GPVI Fabs
The Fab variants were transiently expressed by CHO-S cells and culture supernatants were submitted to a multi-step analysis for ranking and selection of the lead (Table 2).
Table 2.
V-domains pairing for generation of humanized antibody fragments and functional characteristics.
| Variant |
VL domain |
VH domain |
Binding to GPVI-Fc |
| ACT 001 | 9O12 VL | 9O12 VH | Functional |
| ACT 002 | humanized | first generation | Functional |
| ACT 004 | VL2b | VH1 | Not functional |
| ACT 005 | Binds PpL | VH2 | Functional |
| ACT 006 | VH3 | Functional | |
| ACT 007 | VH4 | Not functional | |
| ACT 008 | VL3b | VH1 | Not functional |
| ACT 009 | Does not bind PpL | VH2 | Not functional |
| ACT 010 | VH3 | Functional | |
| ACT 011 | VH4 | Not functional | |
| ACT 012 | VL7b | VH2 | Not functional |
| ACT 013 | VH3 | Not functional | |
| ACT 014 | VH4 | Not functional | |
| ACT 015 | VL8b | VH1 | Not functional |
| ACT 016 | Binds PpL | VH2 | Functional |
| ACT 017 | VH3 | Functional | |
| ACT 018 | VH4 | Not functional |
Western blot analysis indicated that all the Fab variants were secreted in the supernatants as a 50 kDa protein even if an additional weak band of 25 kDa corresponding to free light chain was observed for some of them (Fig. 3A-B). Rapid ELISA screening allowed us to identify the variants that satisfied both criteria (GPVI binding and PpL recognition) (Fig. 3C-D). The best binders were the recombinant Fabs made of VH2 or VH3 paired with either VL2b or VL8b (ACT005, ACT006, ACT016 and ACT017). They satisfied both criteria (GPVI binding and PpL recognition), while none of the recombinant Fab carrying the humanized VH1, VH4 or VL7b preserved structural features required for GPVI-binding. In turn, the humanized VL3b did not interact with PpL, which disqualified ACT010.
Figure 3.
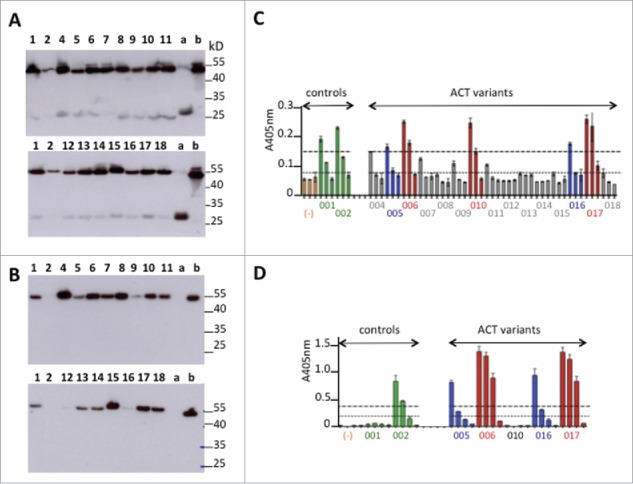
Primary screening of humanized anti-GPVI Fabs. (A-B) Western blot analysis under non-reducing conditions of light chain expression using anti-human kappa chain as primary antibody (A) or Fd expression using anti-human IgG Fd region as primary antibody (B). Deposit of 10 μL of supernatant. Numbers refer to ACT0xx variants. (a) trastuzumab Fab transiently expressed in similar conditions. (b) abciximab (Reopro®) 0.5μg. (C-D) ELISA selection of the best GPVI binders after developing with anti-human IgG (Fab specific)-peroxidase conjugate (C) or PpL-Peroxidase conjugate (D). Culture supernatants were analyzed after successive dilutions (1:10, 1:100, 1:1000) in C or (1:10, 1:50, 1:100, 1:1000 in D). Orange bars (−): culture supernatant with no Fab.
ACT005, ACT006, ACT016 and ACT017 were PpL-affinity purified before being analyzed in detail for GPVI-binding by surface plasmon resonance (SPR) in comparison to the mouse Fab 9O12 (Fig. 4). The results indicated a high affinity (KD< 25 nM) for all of them, ACT006 and ACT017 being the best 2 (KD< 8 nM).
Figure 4.
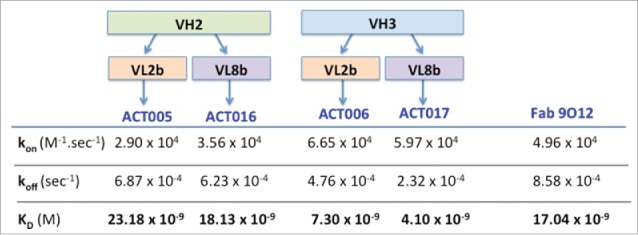
Kinetic parameters of the interaction between humanized Fab variants or Fab9O12 with immobilized GPVI-Fc as deduced from SPR analysis.
Based on these observations, ACT017 was selected as a lead because it was efficiently produced under non-optimized transient expression conditions (35 mg/L) with specificity and affinity (KD = 4.1 nM) equivalent to, or even better than, the parental Fab 9O12 (KD = 17 nM) analyzed under similar experimental conditions. Finally, ACT017 acquired the PpL recognition pattern required for affinity purification.
Stable expression of ACT017, structural and analytical characterization
ACT017 was produced from a pool of stably transfected GPEx GCHO cells at Catalent in a cGMP environment under standard conditions. ACT017 was purified from the cell culture supernatant on CaptoL affinity resin. No precipitation was observed in the elution peak. After buffer exchange and viral filtration, 0.42 g of purified ACT017 were recovered from one liter of clarified media at a concentration of 0.8 mg/mL.
We performed extensive analytical characterization of PpL-purified ACT017 (Fig. 5). We observed a major band close to 45 kDa under native conditions and 2 bands at 25 kDa, in equimolar amount after reduction (Fig. 5A). Western blot confirmed the Fab identity of the purified protein since it was recognized by both anti-kappa and anti-Fd chain antibodies under non-reducing conditions and by the anti-kappa chain antibody under reducing conditions (anti-Fd antibody did not function under reducing conditions). N-terminal sequencing up to the 6th residue of both chains was identical to the theoretical sequence DIQMTQ for kappa chain and QVQLVQ for Fd chain (data not shown). This confirmed the correct processing and cleavage of the signal sequence of ACT017. In addition, qualitative mass spectrometry analysis indicated an experimental Mr (M + H+) of 48 283 Da in the expected range of theoretical molecular mass ± 100 ppm (Fig. 5B). Size-exclusion chromatography revealed the presence of a single peak with an apparent Mr of 48 kDa ((Fig. 5C). Thus, the PpL-purified ACT017 reached a high level of homogeneity, the majority of the kappa and Fd chains being implied in the formation of the heterodimeric structure of the Fab.
Figure 5.
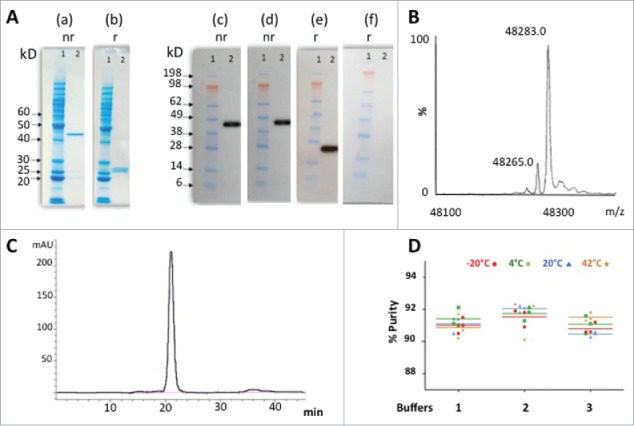
Purification and stability of ACT017. (A) SDS- PAGE analysis of PpL-purified ACT017 (500ng) after Coomassie staining (10% gel) (a-b) or Western-blotting (4–12% gel) with anti-kappa chain (c,e) or anti-Fd chain (d,f) antibody under non-reducing (nr) or reducing (r) conditions. (1) Molecular weight marker (kDa) (2) 500 ng of PpL-purified ACT017. (B) Mass spectrometry analysis of ACT017 by ESI-Q-ToF-MS at a protein concentration of 10 μM. (C) Typical elution profile of PpL purified ACT017 after gel filtration on a calibrated Superdex 75 10/300 GL column and stability profile under 4 temperatures in 3 different buffers. (D) Evolution of the purity of the main peak throughout the stability study for 3 buffers at 4 temperatures (−20°C to 42°C) as deduced from the gel filtration elution profile. Buffer 1: 50 mM Sodium acetate, pH 4.9. Buffer 2: 50 mM Sodium succinate, pH 5.1. Buffer 3: PBS pH 7.0.
Since Fab aggregation and conformational stability are of major concern for functional analysis and in vivo investigations, we performed an accelerated short-term stability study of ACT017 with the aim to monitor the potential appearance of aggregates and precipitates. The data collected during that stability study showed that the Fab was particularly stable in the 3 buffers and the 4 temperatures tested during the 7-day incubations, as well as after the 3 cycles of freezing / thawing (Fig. 5C-D). The purity of the whole Fab molecule remained between 90 and 92% throughout all the stability study for the 3 buffers and the 4 temperatures tested. We did not observe any change in A280nm between centrifuged versus non-centrifuged samples, indicating no loss of protein due to removal of precipitated material by centrifugation. A320nm values (between 0.000 and 0.005) remained very low, reflecting the absence of any aggregation or precipitation phenomenon. The apparent Mr calculated from the retention time after gel filtration did not show any shift or change in relative percentage of the peak areas or any loss of protein due to removal of protein by centrifugation.
NanoDSF analysis confirmed high conformational stability of ACT017 in phosphate-buffered saline (PBS) even at high concentration (10 mg/mL), with 2 unfolding transition points indicating that 2 domains unfold during the thermal denaturation (data not shown). The observed melting Tm1 and Tm2 temperature were 58.5°C and 82.5°C, respectively, and the aggregation onset temperature was 71°C without any significant change under pH variation. Therefore, PBS buffer was selected as the storage buffer for the subsequent analyses.
Residual immunogenicity
In addition to the humanization criteria used for selection of ACT017, we performed an extended analysis of potential immunogenicity using the computer algorithm EpiMatrix to identify epitope cluster and to fine map those individual residues that contribute most to the immunogenic potential of the cluster.
A total of 216 9-mer frames were analyzed and we performed 1728 allele-specific assessment of predicted binding affinity. Fig. 6 presents all the putative T cell epitope clusters found within the amino acid sequence of ACT017 variable domains. Two were identified in ACT017 VH; one (33–48) contains a known Tregitope and the other (76–94) is fully human, but occurring just in 3 human germlines. Three epitope clusters were identified in ACT017 VL domain, 2 of them ((1–15) and (73–91)) being fully human and containing known Tregitopes. The third cluster (48–71), whose sequence does not occur within the human germline, is derived from CDR L2. Therefore, it may be the only one that embodies some potential for immunogenicity. Finally, the immunogenicity of the complete ACT017 Fab was predicted to fall within the range of licensed, non-immunogenic antibodies. Compared to other antibodies, using the Epivax method for calculating immunogenicity, ACT017 should engender an anti-therapeutic response in approximately 2.00% ( ± 5%) of exposed subjects (Fig. 6B).
Figure 6.
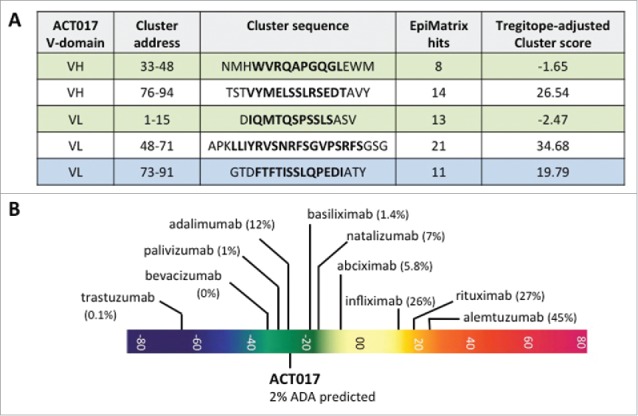
Overview of immunogenic potential of ACT017. (A) Putative T-cell epitope cluster found in ACT017 V-domains and main features. Clusters highlighted in green contain known Tregitope sequences. Cluster highlighted in blue contains homolog to known Tregitopes sequences. (B) Immunogenicity scale (Epimatrix protein score). Upper pane showcases: observed ADA responses in known antibodies. Lower pane showcases: predicted ADA responses in ACT017. All predictions are adjusted for the presence of Tregitopes.
ACT017 functional characterization
We first showed by ELISA that ACT017 binds recombinant GPVI-Fc in a dose-dependent and saturable manner (Fig. 7A). ACT017 was also able to inhibit GPVI-Fc binding to collagen immobilized on a microtitration plate with an IC50 of 1.37 ± 0.16 μg/mL (Fig. 7B).
Figure 7.
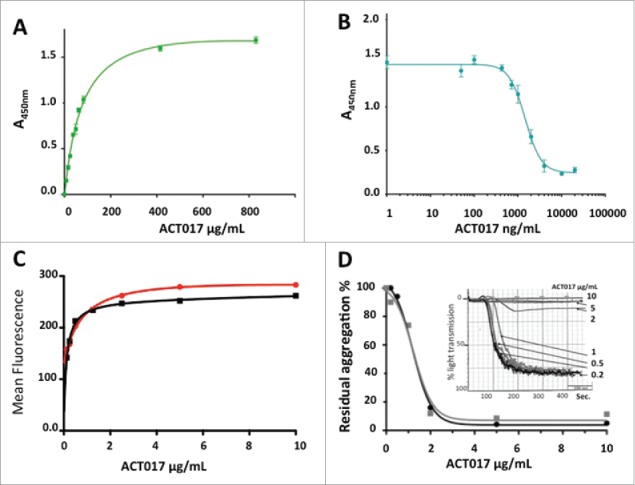
Functional characterization of ACT017 in vitro. (A) Isotherm binding curve of ACT017 to immobilized GPVI-Fc. (B) Inhibition of GPVI-Fc binding to immobilized collagen by increasing concentrations of ACT017. (C) In vitro binding of ACT017 conjugated to Alexa488 to human platelets analyzed by flow cytometry (red: whole blood; black: PRP). (D) Typical residual platelet aggregation as the ratio of the response in the presence of ACT017 to the response without ACT017 as a function of ACT017 concentration (Black: residual intensity, Gray: residual velocity). The insert shows a typical aggregation curve obtained after pre-incubation of human PRP with increasing concentrations of ACT017 (0–10 μg/mL).
The ability of ACT017 to bind GPVI expressed at the surface of human platelets was demonstrated by flow cytometry (Fig. 7C). Saturation of platelets was obtained in whole blood and platelet-rich plasma (PRP) for an ACT017 concentration of 1 to 2 μg/mL (20 to 40 nM). Inhibition of collagen-induced platelet aggregation was also analyzed after pre-incubation of human PRP with increasing concentrations of ACT017 (1 to 10 μg/mL) (Fig. 7D). The capacity of ACT017 to inhibit collagen-induced platelet aggregation was quantified on the velocity and intensity of the response. Using platelets from 3 different donors, the mean IC50 was of 3.2 ± 2.5 and 2 ± 0.7 μg/mL for the intensity and velocity respectively, while a total inhibition was observed for a concentration of 6.7 ± 2.9 μg/mL. These observations are in good agreement with the results of ACT017 binding to platelets and clearly demonstrate that ACT017 is a potent inhibitor of collagen-induced human platelet aggregation.
Pharmacology studies in non-human primates
To analyze ex vivo effects of ACT017 on platelet aggregation, platelet count, GPVI expression and bleeding time, ACT017 (1–8 mg/kg) or vehicle was intravenously injected to cynomolgus monkeys (n = 4) over 15 minutes. 0.5 hours, 2 hours and 24 hours after injection, blood was collected for analysis.
Ex vivo, ACT017 reversibly inhibited collagen-induced platelet aggregation (Fig. 8A). For ACT017 at 2 to 8 mg/kg doses, collagen-induced platelet aggregation measured 0.5 hour after the end of the administration was fully inhibited for all cynomolgus. At 2 hours, the inhibitory effect regressed for all tested doses. The regression of the effect was dose-dependent with mean aggregation intensity reaching 56% and 24% at 2 hours for 1 and 8 mg/kg, respectively. For the 8 mg/kg dose, collagen-induced platelet aggregation was normalized 24 hours post injection with the intensity of aggregation reaching 80% (data not shown).
Figure 8.
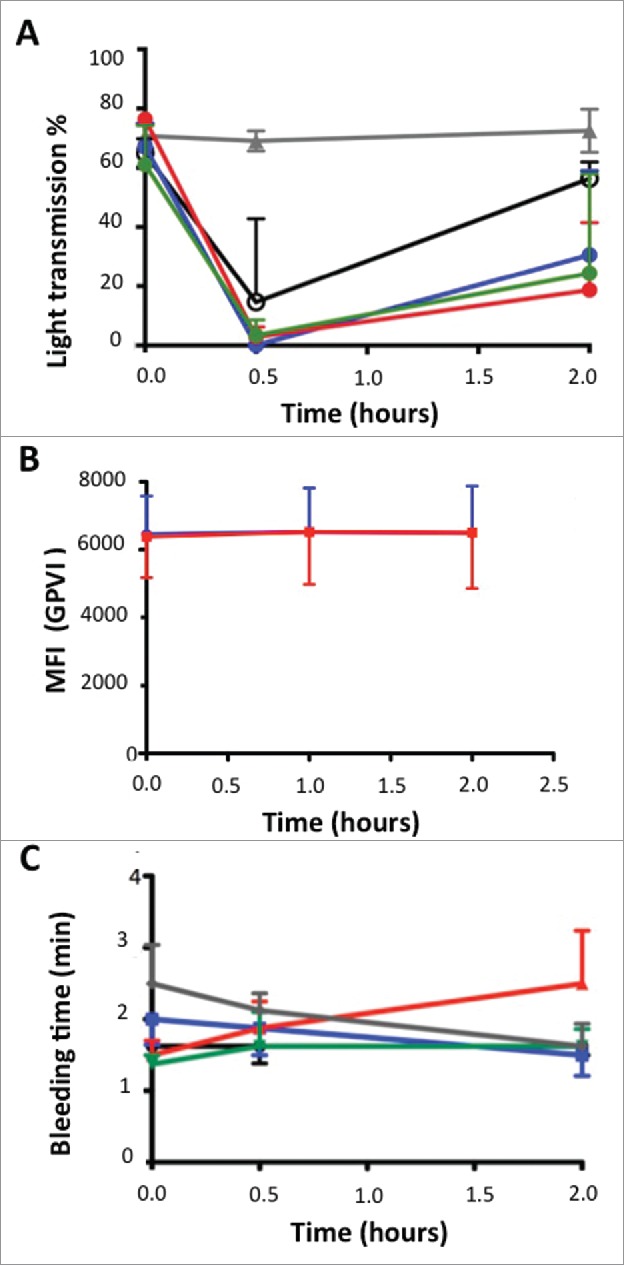
Effects of escalating doses of ACT017 on ex vivo collagen-induced blood platelet aggregation, GPVI expression level and bleeding time in 4 cynomolgus monkeys. (A) Mean intensity of collagen-induced platelet aggregation is expressed as a percent of light transmission. (B) GPVI expression level analyzed after immunolabeling with FITC-conjugated anti-human GPVI monoclonal antibody of blood samples and fluorescence measurement. (C) Average bleeding time evolution during the study. Average bleeding time at 30 min after injection of abciximab is out of the scale (> 30 min). Animals were treated with vehicle (gray triangle), ACT017 at 1 mg/kg (black circle), 2 mg/kg (blue), 4 mg/kg (red) and 8 mg/kg (green).
Platelet count remained stable and within the normal range (200–350 × 103/mm3) throughout the study in predose and 24 hours after each treatment. We did not observe any variation of GPVI expression level within each animal during the study regardless of the time and the dose that was administered (Fig. 8B). All other hematological parameters stayed normal, and we did not observe any adverse clinical signs or animal distress related to ACT017 administration. Concerning the bleeding time test, slight variations were observed without any statistical differences between the vehicle and each treatment dose tested (1–8 mg/kg) or between predose and post-treatment time-points (bleeding time 1.63 ± 0.82 min) (Fig. 8C). Under similar conditions, the anti-GPIIbIIIa abciximab Fab (0.4mg/kg) that is considered as a reference anti-aggregant molecule in human induced a significant increase of bleeding time (> 30 minutes) and large hematomas at the injection and bleeding time sites.
Together, these results demonstrated, in non-human primates, that administration of ACT017 efficiently and reversibly inhibits GPVI function without effects on platelet count or bleeding time.
Discussion
We designed humanized monovalent antibody fragments, selected and extensively characterized the lead (ACT017) that neutralizes collagen-induced platelet aggregation with the aim of developing a new safe and efficient antithrombotic drug. We focused on the Fab format that possesses some advantageous properties compared with single-chain antibody fragment (scFv), including a longer residence time in blood.32 This choice was also supported by previous data that demonstrated the capacity of mouse Fab 9O12 to inhibit in vivo thrombus formation in animal models.23,33
Since the first approval of a therapeutic humanized antibody (daclizumab), several strategies have been proposed for reducing potential immunogenicity of antibodies from animal sources while preserving their antigen-binding activity.24,34 Here, we selected the human framework acceptor region based on several criteria. Among the 15 humanized Fabs generated, 5 retained the parental antigen-binding activity, out of which 4 acquired the motif required for PpL-purification. These 4 Fabs simply resulted from the pairing of 2 humanized VH (VH2 and VH3) with 2 humanized VL (VL2b and VL8b). We also observed that the 2 best GPVI-binders (ACT006 and ACT017) were both derived from VH3. Interestingly, 2 substitutions with residues having very dissimilar physico-chemical properties compared with the parental 9O12 antibody were introduced in CDR H2 (F59>Y and K64Q). These substitutions were performed to improve the “humanness,” and because these residues did not belong to the structural loop that forms the basis for antigen binding (Fig. 2D). The improved antigen-binding affinity we observed clearly demonstrates that residues of the CDR are not all necessary to interact with an antigen in a particular antibody. It might be worthwhile to define more restricted regions than Kabat CDRs to generate fewer immunogenic non-human epitopes. However, this has not always proven true, and humanization procedures around CDRs still remain to be examined on a case-by-case basis.
The 2 best binders (ACT006 and ACT017) kept exactly the same predicted VH/VL packing angle as the parental mouse Fab. This feature is likely to mainly contribute to the stability of the quaternary structure as it was experimentally observed for ACT017. This point underlines how important it is to consider biophysical properties beside amino acid substitutions that reduce immunogenicity. Indeed, stability of the quaternary structure and aggregation propensity can critically affect the pharmacokinetics and immunogenicity of therapeutic antibody fragments.35,36
None of the variants containing VH1 whose template was the human germline sequence closest to mouse 9O12VH retained significant GPVI-binding activity. We noticed 6 very dissimilar residues between VH1 and the parental 9O12 VH. In particular, K73>E could affect the antigen-binding as a result of an opposite net charge in the vicinity of the hypervariable loop H1, as deduced from the 3D model. In the same way, VH4 and VL7b did not preserve structural features required for GPVI-binding. The reasons for these discrepancies have not yet been explored in detail, but a careful reassessment should certainly help in identifying residues critical for correct folding of the antigen-binding site and stability of the molecule. It should be noted that the FR3 region of VH4 is highly mutated with several dissimilar residues. In addition, the dipeptide sequence (P40P41) at the C-terminal extremity of β strand C is rarely observed and could introduce some instability and conformational conflicts (P41 is buried in VL-VH interfaces).
We selected CHO cells and the GPEx® technology for stable expression of ACT017 not only because this system, as some other mammalian cell systems, has been used with success for production of biopharmaceuticals at an industrial scale, but also because we observed unspecific glycosylation or partial proteolysis when ACT017 was produced and secreted from yeast (Pichia pastoris) or bacteria (E. coli), respectively (data not shown).
The PpL binding site we engineered onto the ACT017 VL domain, according to the criteria we reported above, not only preserved the Fab “humanness” and the canonical structures of the parental VL domain, but also allowed us to reach 90% purity with a yield of 0.42 g/L of media. This was achieved in single step affinity purification without optimization or additional cation exchange chromatography or polishing step. Therefore, the strategy consisting of grafting a PpL binding motif is very promising for industrial-scale downstream purification of antibody fragments. It could even be considered as an alternative to the current IgG purification gold standard (Protein A), while other strategies such as non-affinity processes and multimodal chromatography are still associated to disappointing outcomes.37-39
Extensive physico-chemical characterization using a large panel of technologies demonstrated that ACT017 reached the quality standard required for a biopharmaceutical candidate, i.e., correct processing, no unexpected post-translational modification, high solubility and thermal stability of the quaternary and secondary structures in various formulations. As a consequence, we did not observe any tendency to aggregate and this is a necessary pre-requisite for the intended therapeutic applications. The Food and Drug Administration (FDA)-recognized algorithm EpiMatrix that we used for immunogenicity prediction allowed us to demonstrate that ACT017 falls within the range of licensed non-immunogenic antibodies. The EpiMatrix score was well below that observed for abciximab (Reopro®) and in the same range as the human anti-tumor necrosis factor adalimumab (Humira®), which elicit anti-drug antibodies in 5.8% and 12% of treated patients, respectively.40 Taken together, these observations confirmed the efficiency of the humanization process undertaken and paved the way to preclinical pharmacological analysis.
Today, despite intensive researches, there is still a limited number of therapeutic options for the treatment of acute arterial thrombosis due to hematological disorders or bleeding risks associated to drugs directed against current pharmacological targets.4 This study clearly demonstrates that inhibition of GPVI with ACT017 does not affect normal hemostasis in animal models. ACT017 does not induce adverse clinical effects when injected at doses sufficient for ex vivo reversible inhibition of collagen-induced platelet aggregation. No transient thrombocytopenia or GPVI depletion is observed, unlike with other anti-GPVI antibodies such as JAQ1.41 In addition, ACT017 has no effect on bleeding time, a finding which is opposite to that seen with the FDA-approved abciximab (Reopro®), which is an antibody fragment (Fab) as well, but targeting GPIIb/IIIa.42 Therefore, ACT017 safety, associated to its inhibitory effect on collagen-induced platelet aggregation, is a promising feature that must be considered to develop new treatment options in the therapeutic landscape of arterial thrombosis. Further pharmacological and preclinical regulatory toxicology studies are now in progress. An interesting trial would be the early treatment (within 12 hours post-event) of acute ischemic stroke patients who received thrombolytic therapy or not. This time window is considered essential for recovery, but no suitable candidates are now available because of increased risks of intracranial hemorrhages associated with early administration of conventional drugs, including COX1 inhibitor aspirin and antagonists of the platelet ADP-receptor P2Y12.43
Materials and methods
Materials
The hybridoma cell line 9O12.2 secreting a mouse monoclonal IgG1,k (9012) directed against the GPVI of human platelets, and IgG and Fab fragment preparation and purification, have been reported previously.18 9O12 cross-reacts with non-human primate GPVI, but not with murine GPVI. 9O12 Fv sequences are registered in the EMBL data bank (AM 887763 and AM 887764)
Recombinant GPVI-Fc made of the extracellular domain of human GPVI fused to human IgG1 Fc domain44 was produced and purified at Syngene International Ltd, after transient expression in HEK 293–6E cells and affinity chromatography using MAbselect matrix (GE Healthcare, 17519901), followed by a polishing chromatography on Nuvia˜ HR-S cation exchange resin (BioRad, 156051).
Design of humanized V-domains
Protein physico-chemical properties were evaluated using the Swiss Institute of Bioinformatics software (PROTPARAM tool).45 Amino acid residues were identified according Kabat numbering as updated by Abhinandan and Martin.46 We used the online tool at UCL (www.bioinf.org.uk/abs/abnum).
The 3D-structure models were generated using the automatic modeling of antibody V-domains based on the canonical structure method.47
We used the EMBL-EBI facilities to identify human or humanized FR having high sequence identity with the 9O12 V-domains. We also identified the human germline genes most similar to 9O12 V-domains using the IMGT/DomainGapAlign online tool from the International ImmunoGeneTics information system (IMGT).48
For humanization, we considered several structural features of antibodies. These included not only sequence similarities, but also secondary structure characteristics and the main-chain conformations of 9O12 V-domains also called “canonical structures.”28,49,50 These conformations are determined first by the length of the loops and second by the presence of key residues not only in the loops, but also in the FRs that determine the conformation through their packing, hydrogen bonding or the ability to assume unusual main-chain conformations (http://www.bioinf.org.uk/abs/chothia.html). We paid special attention to residues buried in VH-VL interfaces and to the VH/VL “packing angle,” which affect the quaternary structure stability and antigen-binding site conformation.29,51
We grafted the CDRs as defined by Kabat et al.27,49 onto human FRs and each amino acid substitution was inspected individually, based on the physico-chemical classes of the amino acid differences.52 Before and after each backmutation, we considered the humanness score (Z score), which compares the sequence with a set of known human sequences assigned to germ-line derived families with the aim of achieving a score in the range of [−1.0; +1.0] (http://bioinf.org.uk/abs/shab).53 The residual immunogenicity was also analyzed using the BIMAS website (https://www-bimas.cit.nih.gov/molbio/hla_bind/). This allowed the location and ranking of 9-mer peptides that contain peptide-binding motifs for HLA class I molecules.54
To design a PpL binding site, which is highly dependent on several residues mainly belonging to V-kappa FR 1, some refinements were performed as previously reported, without altering predicted humanization and residual immunogenicity.30,31 Then, the designed sequences (4 humanized VH and 4 humanized VL) were compared with human germline genes using the Web interface IMGT tools and databases (IMGT/DomainGapAlign).48 Finally, the humanized VH and VL were paired together to generate 15 Fab variants (ACT004 to ACT018) in addition to the chimeric Fab9O12 (ACT001), which contains the mouse 9O12 V-domains and the Fab (ACT002) deduced from the first generation “humanized” scFv, previously reported.22
Generation of humanized Fab variants
cDNA encoding each humanized VL fused with Homo sapiens Ig kappa constant (IGKC*01) and cDNA encoding each humanized VH fused with Homo sapiens Ig heavy constant gamma 1 (IGHG1) followed by 9 residues (EPKSCDKTH) of the hinge region were synthesized at GeneArt after codon optimization for expression in mammalian cells and then inserted into vector pBIC-PS (NVH-Medicinal, F) downstream of the leader sequences encoding MDMRVPAQLLGLLLLWLRGARC and MDWTWRILFLVAAATGAH, respectively. Transient expression was performed at NVH-Medicinal in CHO-S cells under standard conditions (FreeStyle™ Max Reagent, Life Technology, 12651–014). Supernatant were collected 6 d post-transfection when cell viability fell down below 80%.
For stable expression of ACT017, Fd (heavy chain fragment) gene and light chain gene were cloned into GPEx® expression retrovector using GPEx® technology (Catalent, WI). Stably transfected GPEx® CHO (GCHO) cells were prepared at Catalent after multiple rounds of transduction. Cells were subcloned using the ClonePix˜2 system (Molecular Devices). The most viable clones were assayed by ELISA for GPVI-Fc binding and antibody titer determination, and then expanded for fed-batch overgrowth production.
Fabs purification
Fab variants were purified from transiently transfected CHO-S cells pool supernatants using Capto L affinity chromatography (GE Healthcare, 17–5478–01) according to the manufacturer instruction. ACT017 was also produced from stable transfected GCHO cells pool (Fedbatch, 2.8L flask) harvested on day 19 at 50% viability. It was purified from culture supernatant clarified by 0.5 μm glass fiber filtration after Capto L chromatography and elution with 0.1 M glycine pH 3.5. Viral filtration was performed using Virosart CPV, 10” capsule (Sartorius, 5452528V1). Then, ACT017 was formulated at 0.8 mg/mL in 10 mM sodium phosphate, 150 mM sodium chloride (PBS), pH 7.2.
Protein expression and purity were assayed by SDS-PAGE under reducing and non-reducing conditions followed by Coomassie blue staining or Western blot using either mouse anti-human IgG Fd region (ABIN, 952843) or mouse anti-human kappa chain (Tebu-bio, GTX21050) as primary antibodies and probed using peroxidase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, 715–035–151) as a secondary antibody. Protein transferred onto PVDF membrane after SDS-PAGE were also excised and loaded on a 494 Procise sequencer (Applied Biosystems) for N-terminal sequencing.
Protein integrity was investigated by ESI-Q-ToF mass spectrometry (Waters, Synapt G2 HDMS) in positive ion mode at 10 μM in ammonium bicarbonate 25 mM, 30% acetonitrile, 0.5% formic acid. Protein concentration was determined after micro BCA titration (ThermoFisher, 23235).
Thermal stability under various buffer conditions
Purified ACT017 was conditioned in 3 buffers (sodium acetate 50 mM, pH 4.9; sodium succinate 50 mM, pH 5.1; PBS, pH 7.0) by dialysis and then concentrated to 5 mg/mL by centrifugation using ultra-centrifugation devices with a cut-off of 10 kDa. Samples were aliquoted and stored at −20°C, 4°C, 20°C, 42°C and analyzed at 0, 4 and 7 d. In addition, 3 samples were submitted to 3 cycles of freezing (30 min) / thawing (30 min). UV-Vis spectra (220–350 nm) and SE-HPLC on a calibrated Superdex 75 10/300 GL column (GE) were performed on samples before and after centrifugation to characterize the aggregation state.
For nano differential scanning fluorimetry (nanoDSF) analysis, the protein concentration was set between 9.2 and 10 mg/mL. Samples were loaded in standard nanoDSF capillaries and measured using the Prometheus NT.48 instrument containing aggregation optics (Nanotemper-Technologies, PR001). The LED intensity was set to 10%, whereas the temperature ramp was set from 20 to 95°C with 1°C per min. As a control, one duplicate containing heat denatured protein was integrated. The onset temperatures were determined according to Menzen and Fries.55
Immunochemical characterization of Fab fragments
For GPVI-Fc binding assay in ELISA, immunoplates were coated with GPVI-Fc (0.2 μg/100μL in PBS). After blocking with BSA 1%, Fab preparations diluted in PBS, BSA 0.1%, Tween 20 0.05% in a range of protein concentration from 0 to 800 μg/mL were incubated on the coated antigen for 30 min at 37°C. Bound Fab were detected by adding anti-human IgG (Fab specific)-peroxidase conjugate (Sigma, A0293) or peroxidase conjugated PpL (Pierce, 32420), using 1-StepTM Ultra TMB-ELISA (ThermoFisher, 34028) for developing. The reaction was stopped with H2SO4 2M (50 μL/well). Absorbance was read at 450 nm. At least 3 washings with 200 µL of the dilution buffer were performed between each intermediate step. Each point was measured in triplicate.
GPVI-Fc binding kinetics of the purified Fabs was evaluated by SPR using BIAcore® 2000 (GE Healthcare). GPVI-Fc was immobilized (∼1000 RU) on a carboxy-Methyl Dextran CM5 sensor chip using the amine coupling method. All binding experiments were performed in triplicate, at 25°C, in PBS at flow rate of 20 μL/min and pulses of 10 mM HCl were used to regenerate the chip surface. Rate constants were calculated using the BIAevaluation 3.0 software.
Residual immunogenicity
The immunogenicity prediction software EpiMatrix developed at Epivax (Providence, RI) was applied to ACT017 V-domains sequences to identify putative T-cell epitopes and rate the immunogenic potential on a normalized scale. First, heavy chain and light chain sequences were parsed into overlapping 9-mer frames corresponding to the minimal length of an HLA-binding peptide and each frame was then evaluated with respect to a panel of 8 common class II HLA alleles that cover over 95% of the human population.56 This led to the identification of “hits” considered to have a significant chance of binding to HLA molecules, and therefore of being presented on the surface of antigen-presenting cells. The EpiMatrix Protein Score of an “average” protein is zero. Scores above zero indicate the presence of excess MHC ligands and denote a higher potential for immunogenicity while scores below zero indicate the presence of fewer potential MHC ligands than expected and a lower potential for immunogenicity. Proteins scoring above +20 are considered to have a significant immunogenic potential.
Any putative regulatory T-cell epitopes that may help to dampen the immune potential of ACT017 sequences were screened for homology with the non-redundant human proteome databases and sequences of published epitopes that have been cataloged in the immune epitope database at the La Jolla Institute for Allergy and Immunology. The Tregitope-adjusted scores deduced from such analysis have been shown to be correlated with clinical immune response for a set of 23 commercial antibodies.40
Potency test
In vitro quantification of ACT017 inhibitory activity was determined as its IC50 on the binding of GPVI-Fc to collagen. The microtiter plate wells were coated with Horm collagen (type I fibrils from equine tendon) (Takeda, batch 1130630) (2 μg/100μL in PBS) and incubated at 4°C overnight. Non-specific sites were saturated with Superblock blocking buffer (ThermoScientific, 37518). ACT017 dilutions (0 to 40 μg/mL in PBS, Superblock 10% and Tween 20 0.1%) and GPVI-Fc (4 μg/mL) were mixed in a one-to-one volume ratio and pre-incubated for 10 min at 37°C. 100 μL of each mix were loaded in wells, and incubated at 37°C for 30 min. The detection and revelation were performed using peroxidase-conjugated AffiniPure F(ab’)2 fragment goat anti-human IgG, Fcγ fragment-specific antibody (Jackson ImmuoResearch, 109–036–098) and 1-StepTM Ultra TMB-ELISA (ThermoFisher, 34028). The reaction was stopped with H2SO4 2M (50 µL per well). Absorbance was read at 450 nm. At least 5 washings were performed between each intermediate step. Each point was measured in triplicates. Results were expressed as A450nm as a function of ACT017 concentration and the inhibition curve was obtained by nonlinear regression, dose-response inhibition analysis using GraphPad Prism 7 software.
Human blood samples
Human blood samples were obtained after informed consent by venipuncture at the forearm of healthy volunteers free from medication for at least 10 d into vacutainers containing sodium citrate 3.2%. PRP and platelet-poor plasma (PPP) were successively separated after centrifugation at 120 g for 15 min at 20°C (PRP) followed by 13 000 g for 3 min at 20°C (PPP).
Binding to human platelets analysis
For flow cytometry analysis, ACT017 was coupled to Alexa488 (Molecular Probes, Z25402) according to the manufacturer recommendation. It was then incubated at different concentrations with human platelets in whole blood or PRP for 20 min at room temperature in the dark. After dilution in PBS, the cells were analyzed in flow cytometry (FACS LSR II BD, BD Biosciences). Events in the predetermined platelet gate were analyzed for their fluorescence intensity.
Platelet aggregation was analyzed by light transmission aggregometry: human PRP was incubated for 10 min at 37 °C with increasing quantities of ACT017 (0–10 μg/mL), without stirring. Then, platelet aggregation was initiated by adding type I collagen (Horm collagen, Nycomed, DE) (1 μg/mL) in the stirred platelet suspension at 37°C. Changes in light transmission were continuously recorded using an aggregometer (APACT 4004, 90.000.1020, Elitech). PRP and PPP were used to set the 0% and 100% light transmission, respectively corresponding to 0% and 100% aggregation.
Animals and pharmacology studies
All animal studies were reviewed by the animal welfare body of Cynbiose and the Ethics Committee of VetAgro Sup (Marcy l'Étoile, France) and approved under number 1611-V2 (MESR number: 2016030115166132). All experiments were conducted in accordance with the European Directive 2010/63/UE as published in the French Official Journal of February 7th, 2013.
The 4 cynomolgus monkeys (Macaca fascicularis) included in the study (2 males and 2 females) were housed at Cynbiose (Marcy l'Etoile, F). Complete examination was conducted before the study by a veterinarian and included blood pressure, heart rate, hematology and biochemical constants. The vehicle or each ACT017 dose (1, 2, 4, 8 mg/kg) were administered intravenously during 15 min (5 mL/kg) in conscious, trained and restrained animals.
Animal blood samples were obtained by femoral vessel puncture on conscious and restrained cynomolgus monkeys, into vacutainers containing sodium citrate 3.2% or EDTA. Blood samples were obtained at day 0 before the injection and at time 30 min, 2 hours and, in some cases, 24 hours post injection, each animal being its own control. PRP and PPP were prepared by successive centrifugation at 2300 g for 3 min and 1400 g for 3 min. Samples quality was checked by measuring the platelet count in the PRP.
Ex vivo platelet aggregation was measured on blood samples collected before injection, 30 min and 2 hours post-injection. The assay was performed within 30 minutes after blood collection. Light transmission platelet aggregometry was performed as described for human samples, but using collagen at a concentration of 2.5 μg/mL. Ex vivo platelet count was performed within 1 hour after blood collection on a platelet counter device (Scil Vet abc automatic cell counter) set to monkey parameters. To determine platelet surface GPVI expression levels, blood samples collected on EDTA were incubated with commercial FITC-coupled anti-human GPVI monoclonal antibody (Biocytex, clone 1G5) that cross-reacts with cynomolgus GPVI. After dilution, fluorescence was measured on events in the predetermined gate for cynomolgus platelets on a Beckman Coulter Gallios Flow cytometer.
The bleeding time was measured on vigil monkeys at the surface of the forearm, according to standard clinical procedure (Ivy's procedure) and using 0.5 cm Surgicutt™ bleeding time device (Fisher-scientific, 22–211–506). The bleeding time was recorded after the first blood drop until blood flow stops and clot formation. Local reactions were observed until day 3 post-injection. Blood samples collected into EDTA-containing vacutainers, before ACT017 administration and 24 hours after each treatment, were analyzed for the following hematological parameters: neutrophils, eosinophils, basophils, lymphocytes, monocytes, red blood cells, white blood cells count, hemoglobin concentration, hematocrit.
Disclosure of potential conflicts of interest
OFB and KL are employees of Acticor Biotech. MJP, GA and PB are cofounder of Acticor Biotech, own shares of Acticor Biotech. PB is Professor at the University of Paris-Sud.
Acknowledgements
We are indebted to Dr Nicolas Aubrey for stimulating discussions. We also acknowledge the valuable technical assistance provided by Betty Goudet and Stéphane Loyau.
References
- 1.Widimský P, Kožnar B, Vaško P, Peisker T, Štětkářová I. Acute myocardial infarction and acute stroke: What are the differences? Focus on reperfusion therapy. Cor Vasa 2013; 55:e111-6; https://doi.org/ 10.1016/j.crvasa.2013.02.002 [DOI] [Google Scholar]
- 2.Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke: Platelet adhesion and activation mechanisms. J Thromb Haemost 2011; 9:92-104; PMID:21781245; https://doi.org/ 10.1111/j.1538-7836.2011.04361.x [DOI] [PubMed] [Google Scholar]
- 3.McFadyen JD, Jackson SP. Differentiating haemostasis from thrombosis for therapeutic benefit. Thromb Haemost 2013; 110:859-67; PMID:23945664; https://doi.org/ 10.1160/TH13-05-0379 [DOI] [PubMed] [Google Scholar]
- 4.Gachet C. Antiplatelet drugs: Which targets for which treatments? J Thromb Haemost 2015; 13:S313-22; PMID:26149041; https://doi.org/ 10.1111/jth.12947 [DOI] [PubMed] [Google Scholar]
- 5.Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2014; 12:30-47; PMID:25286881; https://doi.org/ 10.1038/nrcardio.2014.156 [DOI] [PubMed] [Google Scholar]
- 6.Thachil J. Antiplatelet therapy - a summary for the general physicians. Clin Med Lond Engl 2016; 16:152-60; PMID:27037385; https://doi.org/ 10.7861/clinmedicine.16-2-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coutinho JM, Liebeskind DS, Slater L-A, Nogueira RG, Clark W, Dávalos A, Bonafé A, Jahan R, Fischer U, Gralla J, et al.. Combined Intravenous Thrombolysis and Thrombectomy vs Thrombectomy Alone for Acute Ischemic Stroke: A Pooled Analysis of the SWIFT and STAR Studies. JAMA Neurol 2017; 74:268-74; PMID:28097310; https://doi.org/ 10.1001/jamaneurol.2016.5374 [DOI] [PubMed] [Google Scholar]
- 8.Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor? Blood 2003; 102:449-61; PMID:12649139; https://doi.org/ 10.1182/blood-2002-12-3882 [DOI] [PubMed] [Google Scholar]
- 9.Jiang P, Jandrot-Perrus M. New advances in treating thrombotic diseases: GPVI as a platelet drug target. Drug Discov Today 2014; 19:1471-5; PMID:24931218; https://doi.org/ 10.1016/j.drudis.2014.06.005 [DOI] [PubMed] [Google Scholar]
- 10.Induruwa I, Jung SM, Warburton EA. Beyond antiplatelets: The role of glycoprotein VI in ischemic stroke. Int J Stroke Off J Int Stroke Soc 2016; 11:618-25; PMID:27312676; https://doi.org/ 10.1177/1747493016654532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, et al.. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015; 126:683-91; PMID:25977585; https://doi.org/ 10.1182/blood-2015-02-629717 [DOI] [PubMed] [Google Scholar]
- 12.Dumont B, Lasne D, Rothschild C, Bouabdelli M, Ollivier V, Oudin C, Ajzenberg N, Grandchamp B, Jandrot-Perrus M. Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood 2009; 114:1900-3; PMID:19549989; https://doi.org/ 10.1182/blood-2009-03-213504 [DOI] [PubMed] [Google Scholar]
- 13.Takahashi H, Moroi M. Antibody against platelet membrane glycoprotein VI in a patient with systemic lupus erythematosus. Am J Hematol 2001; 67:262-7; PMID:11443641; https://doi.org/ 10.1002/ajh.1128 [DOI] [PubMed] [Google Scholar]
- 14.Bhunia SS, Misra A, Khan IA, Gaur S, Jain M, Singh S, Saxena A, Hohlfield T, Dikshit M, Saxena AK. Novel glycoprotein VI antagonists as antithrombotics: synthesis, biological evaluation, and molecular modeling studies on 2,3-disubstituted tetrahydropyrido(3,4-b)indoles. J Med Chem 2017; 60:322-37; PMID:27996269; https://doi.org/ 10.1021/acs.jmedchem.6b01360 [DOI] [PubMed] [Google Scholar]
- 15.Reimann A, Li Z, Goebel S, Fassbender J, Holthoff H-P, Gawaz M, Münch G, Ungerer M. Combined administration of the GPVI-Fc fusion protein Revacept with low-dose thrombolysis in the treatment of stroke. Heart Int 2016; 11:e10-6; PMID:27924212; https://doi.org/ 10.5301/heartint.5000229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Lockyer S, Concepcion A, Gong X, Takizawa H, Guertin M, Matsumoto Y, Kambayashi J, Tandon NN, Liu Y. The fab fragment of a novel Anti-GPVI monoclonal antibody, OM4, reduces in vivo thrombosis without bleeding risk in rats. Arterioscler Thromb Vasc Biol 2007; 27:1199-205; PMID:17322098; https://doi.org/ 10.1161/ATVBAHA.107.140590 [DOI] [PubMed] [Google Scholar]
- 17.Takayama H, Hosaka Y, Nakayama K, Shirakawa K, Naitoh K, Matsusue T, Shinozaki M, Honda M, Yatagai Y, Kawahara T, et al.. A novel antiplatelet antibody therapy that induces cAMP-dependent endocytosis of the GPVI/Fc receptor gamma-chain complex. J Clin Invest 2008; 118:1785-95; PMID:18382762; https://doi.org/ 10.1172/JCI32513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lecut C, Feeney LA, Kingsbury G, Hopkins J, Lanza F, Gachet C, Villeval J-L, Jandrot-Perrus M. Human platelet glycoprotein VI function is antagonized by monoclonal antibody-derived Fab fragments. J Thromb Haemost JTH 2003; 1:2653-62; PMID:14675102; https://doi.org/ 10.1111/j.1538-7836.2003.00495.x [DOI] [PubMed] [Google Scholar]
- 19.Jamasbi J, Megens RTA, Bianchini M, Münch G, Ungerer M, Faussner A, Sherman S, Walker A, Goyal P, Jung S, et al.. Differential inhibition of human atherosclerotic plaque-induced platelet activation by dimeric GPVI-Fc and Anti-GPVI antibodies: Functional and imaging studies. J Am Coll Cardiol 2015; 65:2404-15; PMID:26046734; https://doi.org/ 10.1016/j.jacc.2015.03.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto Y, Takizawa H, Gong X, Le S, Lockyer S, Okuyama K, Tanaka M, Yoshitake M, Tandon NN, Kambayashi J. Highly potent anti-human GPVI monoclonal antibodies derived from GPVI knockout mouse immunization. Thromb Res 2007; 119:319-29; PMID:16566959; https://doi.org/ 10.1016/j.thromres.2006.01.023 [DOI] [PubMed] [Google Scholar]
- 21.Smethurst PA, Joutsi-Korhonen L, O'Connor MN, Wilson E, Jennings NS, Garner SF, Zhang Y, Knight CG, Dafforn TR, Buckle A, et al.. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood 2004; 103:903-11; PMID:14504096; https://doi.org/ 10.1182/blood-2003-01-0308 [DOI] [PubMed] [Google Scholar]
- 22.Muzard J, Bouabdelli M, Zahid M, Ollivier V, Lacapère JJ, Jandrot-Perrus M, Billiald P. Design and humanization of a murine scFv that blocks human platelet glycoprotein VI in vitro. FEBS J 2009; 276:4207-22; PMID:19558491; https://doi.org/ 10.1111/j.1742-4658.2009.07129.x [DOI] [PubMed] [Google Scholar]
- 23.Mangin PH, Tang C, Bourdon C, Loyau S, Freund M, Hechler B, Gachet C, Jandrot-Perrus M. A humanized glycoprotein VI (GPVI) mouse model to assess the antithrombotic efficacies of anti-GPVI agents. J Pharmacol Exp Ther 2012; 341:156-63; PMID:22238212; https://doi.org/ 10.1124/jpet.111.189050 [DOI] [PubMed] [Google Scholar]
- 24.Léger O, Saldanha JW. Humanization of antibodies In: Antibody drug discovery. Wood C.R; Imperial College Press 2011; 492. [Google Scholar]
- 25.Kabat EA. The structural basis of antibody complementarity. Adv Protein Chem 1978; 32:1-75. PMID:362869; https://doi.org/ 10.1186/1471-2164-5-12 [DOI] [PubMed] [Google Scholar]
- 26.Kabat EA, Wu TT, Bilofsky H. Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J Biol Chem 1977; 252:6609-16. PMID:408353; https://doi.org/ 10.1172/jc15118 [DOI] [PubMed] [Google Scholar]
- 27.Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol Baltim Md 1950 1991; 147:1709-19; PMID:1908882 [PubMed] [Google Scholar]
- 28.Chothia C, Lesk AM, Gherardi E, Tomlinson IM, Walter G, Marks JD, Llewelyn MB, Winter G. Structural repertoire of the human VH segments. J Mol Biol 1992; 227:799-817; PMID:1404389; https://doi.org/ 10.1016/0022-2836(92)90224-8 [DOI] [PubMed] [Google Scholar]
- 29.Chothia C, Novotný J, Bruccoleri R, Karplus M. Domain association in immunoglobulin molecules. The packing of variable domains. J Mol Biol 1985; 186:651-63; PMID:4093982 [DOI] [PubMed] [Google Scholar]
- 30.Muzard J, Adi-Bessalem S, Juste M, Laraba-Djebari F, Aubrey N, Billiald P. Grafting of protein L-binding activity onto recombinant antibody fragments. Anal Biochem 2009; 388:331-8; PMID:19268418; https://doi.org/ 10.1016/j.ab.2009.02.035 [DOI] [PubMed] [Google Scholar]
- 31.Lakhrif Z, Pugnière M, Henriquet C, di Tommaso A, Dimier-Poisson I, Billiald P, Juste MO, Aubrey N. A method to confer Protein L binding ability to any antibody fragment. mAbs 2016; 8:379-88; PMID:26683650; https://doi.org/ 10.1080/19420862.2015.1116657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23:1126-36; PMID:16151406; https://doi.org/ 10.1038/nbt1142 [DOI] [PubMed] [Google Scholar]
- 33.Ohlmann P, Hechler B, Ravanat C, Loyau S, Herrenschmidt N, Wanert F, Jandrot-Perrus M, Gachet C. Ex vivo inhibition of thrombus formation by an anti-glycoprotein VI Fab fragment in non-human primates without modification of glycoprotein VI expression. J Thromb Haemost JTH 2008; 6:1003-11; PMID:18419749; https://doi.org/ 10.1111/j.1538-7836.2008.02976.x [DOI] [PubMed] [Google Scholar]
- 34.Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature 1988; 332:323-7; PMID:3127726; https://doi.org/ 10.1038/332323a0 [DOI] [PubMed] [Google Scholar]
- 35.Mahler H-C, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci 2009; 98:2909-34; PMID:18823031; https://doi.org/ 10.1002/jps.21566 [DOI] [PubMed] [Google Scholar]
- 36.Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM. Immunogenicity of therapeutic protein aggregates. J Pharm Sci 2016; 105:417-30; PMID:26869409; https://doi.org/ 10.1016/j.xphs.2015.11.002 [DOI] [PubMed] [Google Scholar]
- 37.Kaleas KA, Tripodi M, Revelli S, Sharma V, Pizarro SA. Evaluation of a multimodal resin for selective capture of CHO-derived monoclonal antibodies directly from harvested cell culture fluid. J Chromatogr B Analyt Technol Biomed Life Sci 2014; 969:256-63; PMID:25203722; https://doi.org/ 10.1016/j.jchromb.2014.08.026 [DOI] [PubMed] [Google Scholar]
- 38.Rodrigo G, Gruvegård M, Van Alstine J. Antibody fragments and their purification by protein L affinity chromatography. Antibodies 2015; 4:259-77; https://doi.org/ 10.3390/antib4030259 [DOI] [Google Scholar]
- 39.Arora S, Saxena V, Ayyar BV. Affinity chromatography: A versatile technique for antibody purification. Methods 2017; 116:84-94; PMID:28012937; https://doi.org/ 10.1016/j.ymeth.2016.12.010 [DOI] [PubMed] [Google Scholar]
- 40.De Groot AS, Martin W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin Immunol 2009; 131:189-201; PMID:19269256; https://doi.org/ 10.1016/j.clim.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 41.Nieswandt B, Schulte V, Bergmeier W, Mokhtari-Nejad R, Rackebrandt K, Cazenave JP, Ohlmann P, Gachet C, Zirngibl H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med 2001; 193:459-69; PMID:11181698; https://doi.org/ 10.1084/jem.193.4.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simoons ML, GUSTO IV-ACS Investigators . Effect of glycoprotein IIb/IIIa receptor blocker abciximab on outcome in patients with acute coronary syndromes without early coronary revascularisation: the GUSTO IV-ACS randomised trial. Lancet Lond Engl 2001; 357:1915-24; PMID:11425411; https://doi.org/ 10.1016/S0140-6736(00)05060-1 [DOI] [PubMed] [Google Scholar]
- 43.Harris D, Hall C, Lobay K, McRae A, Monroe T, Perry JJ, Shearing A, Wollam G, Goddard T, Lang E. Canadian association of emergency physicians position statement on acute ischemic stroke. CJEM 2015; 17:217-26; PMID:26120643; https://doi.org/ 10.1017/cem.2015.26 [DOI] [PubMed] [Google Scholar]
- 44.Jandrot-Perrus M, Busfield S, Lagrue AH, Xiong X, Debili N, Chickering T, Le Couedic JP, Goodearl A, Dussault B, Fraser C, et al.. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: A platelet-specific collagen receptor from the immunoglobulin superfamily. Blood 2000; 96:1798-807. PMID:10961879 [PubMed] [Google Scholar]
- 45.Appel RD, Bairoch A, Hochstrasser DF. A new generation of information retrieval tools for biologists: The example of the ExPASy WWW server. Trends Biochem Sci 1994; 19:258-60; PMID:8073505; https://doi.org/ 10.1016/0968-0004(94)90153-8 [DOI] [PubMed] [Google Scholar]
- 46.Abhinandan KR, Martin ACR. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol 2008; 45:3832-9; PMID:18614234; https://doi.org/ 10.1016/j.molimm.2008.05.022 [DOI] [PubMed] [Google Scholar]
- 47.Marcatili P, Rosi A, Tramontano A. PIGS: Automatic prediction of antibody structures. Bioinforma Oxf Engl 2008; 24:1953-4; PMID:18641403; https://doi.org/ 10.1093/bioinformatics/btn341 [DOI] [PubMed] [Google Scholar]
- 48.Ehrenmann F. Lefranc M-P. IMGT/DomainGapAlign: IMGT standardized analysis of amino acid sequences of variable, constant, and groove domains (IG, TR, MH, IgSF, MhSF). Cold Spring Harb Protoc 2011; 2011:737-49. PMID:21632775 [DOI] [PubMed] [Google Scholar]
- 49.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; https://doi.org/ 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
- 50.North B, Lehmann A, Dunbrack RL. A new clustering of antibody CDR loop conformations. J Mol Biol 2011; 406:228-56; PMID:21035459; https://doi.org/ 10.1016/j.jmb.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abhinandan KR, Martin ACR. Analysis and prediction of VH/VL packing in antibodies. Protein Eng Des Sel PEDS 2010; 23:689-97; PMID:20591902; https://doi.org/ 10.1093/protein/gzq043 [DOI] [PubMed] [Google Scholar]
- 52.Honegger A, Plückthun A. Yet another numbering scheme for immunoglobulin variable domains: An automatic modeling and analysis tool. J Mol Biol 2001; 309:657-70; PMID:11397087; https://doi.org/ 10.1006/jmbi.2001.4662 [DOI] [PubMed] [Google Scholar]
- 53.Abhinandan KR, Martin ACR. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol 2007; 369:852-62; PMID:17442342; https://doi.org/ 10.1016/j.jmb.2007.02.100 [DOI] [PubMed] [Google Scholar]
- 54.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol Baltim Md 1950 1994; 152:163-75; PMID:8254189 [PubMed] [Google Scholar]
- 55.Menzen T, Friess W. High-throughput melting-temperature analysis of a monoclonal antibody by differential scanning fluorimetry in the presence of surfactants. J Pharm Sci 2013; 102:415-28; PMID:23212746; https://doi.org/ 10.1002/jps.23405 [DOI] [PubMed] [Google Scholar]
- 56.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol Baltim Md 1950 1998; 160:3363-73; PMID:9531296 [PubMed] [Google Scholar]