

Abstract
Mass spectrometry-based proteomic strategies can profile the expression level of proteins in response to external stimuli. Nicotine affects diverse cellular pathways, however, the nicotine-induced alterations on the global proteome across human cell lines have not been fully elucidated. We measured perturbations in protein levels resulting from nicotine treatment in four cell lines—HEK, HeLa, PaSC, and SH-SY5Y—in a single experiment using tandem mass tags (TMT10-plex) and high-resolution mass spectrometry. We quantified 8590 proteins across all cell lines. Of these, nicotine increased the abundance of 31 proteins 1.5-fold or greater in all cell lines. Likewise, considering proteins with altered levels in at least three of the four cell lines, 64 were up-regulated, while one was down-regulated. Gene ontology analysis revealed that ~40% of these proteins were membrane bound, and functioned in transmembrane signaling and receptor activity. We highlighted proteins, including APP, APLP2, LAPTM4B, and NCOA4, which were dysregulated by nicotine in all cell lines investigated and may have implications in downstream signaling pathways, particularly autophagy. Using the outlined methodology, studies in additional (including primary) cell lines will provide further evidence that alterations in the levels of these proteins are indeed a general response to nicotine and thereby merit further investigation.
Keywords: Cell biology, Nicotine, Orbitrap fusion, Pancreas, SPS-MS3, TMT
1 Introduction
Multiplexing strategies in mass spectrometry-based quantitative proteomics have significantly expanded the depth, efficiency, and throughput of comprehensive protein analyses. Isobaric tag-based methodologies, such as tandem mass tags (TMT), are well-established for relative quantification of peptides and associated proteins [1–3]. Using this technique, the relative abundances of several thousand proteins can be measured simultaneously in currently up to ten different samples—equivalent to performing thousands of quantitative Western blotting analyses in a single experiment [4]. Here, we use a strategy which employs TMT to enhance our ability to quantitatively analyze proteomic alterations resulting from nicotine treatment.
Cigarette smoke is a mixture of over 4000 compounds, including over 50 carcinogens [5]. The toxic compounds in tobacco smoke are absorbed into the bloodstream and several have been detected in other body fluids, such as urine [6] and pancreatic fluid [7]. Nicotine, a major component of tobacco, is directly absorbed by the lungs and by more distal organs via systemic circulation [8]. Although not considered carcinogenic, nicotine may act as a tumor promoter. Specifically, nicotine has been shown to enhance cell invasion, proliferation, migration and angiogenesis in many cell types [9]. Recent evidence suggests that nicotine may have a role in DNA damage and is potentially mutagenic [10, 11]. Although exposure to nicotine is known to be detrimental to cells, the full extent of its effects on the global cellular proteome has yet to be elucidated. Typically, nicotine has been studied in the context of lung and brain cell models, due in part to its oncological and psychological roles. However, once consumed, nicotine has a wide distribution in body systems presenting at high concentration in liver, kidney, and spleen [12]. Also, nicotine crosses the placental barrier and has been detected in fetal serum [13]. The extent of the systemic effects of nicotine are currently undetermined, but this drug may cause damage to the genome, disrupt metabolic processes, and facilitate the growth and spreading of cells, all of which have far-reaching pathological effects.
The extracellular targets of nicotine are predominantly nicotinic acetylcholine receptors (nAChR). Nicotine interacts with a broad population of these receptors that are composed of distinct subunits each having varying affinities and downstream effectors [14, 15]. Nicotine binding to these cell surface receptors can transduce extracellular signals to the intracellular space resulting in ion transport and/or initiation of phosphorylation cascades and other signaling pathways [16–18]. Although primarily associated with neuronal cells, these receptors, particularly the homopentameric α7 subtype, have been found also in non-neuronal cells, including mesenchymal, immune, endothelial, and epithelial cells, and are thought to be important regulators of cellular function [19–21].
Here we investigate the global proteomic effects of nicotine on four well-established human cell lines: HEK (embryonic kidney), HeLa (cervical adenocarcinoma), PaSC (pancreatic stellate cells) and SH-SY5Y (bone marrow-derived neuroblastoma). The HEK 293 cell line is used ubiquitously in cell biology research for protein expression-based studies, among many other applications [22]. Another common immortalized cell line, HeLa, is derived from adenocarcinoma of the cervix and is heavily investigated in cancer biology [23]. The PaSC cell line (RLT-PSC) is a myofibroblast-like cell line that has a role in the pathogenesis of pancreatitis and pancreatic cancer [24]. In contrast, SH-SY5Y is a human neuroblastoma cell line that is derived from bone marrow and is typically used in neuroscience studies, including those with nicotine and other nicotinic receptor ligands [25]. Although the effects of nicotine have been investigated in various other cell types, we present the first quantitative analysis of nicotine alterations across these four cell lines. In this study, we demonstrate that nicotine stimulates proteomic alterations across different cell lines. Here, we quantified the effect of nicotine on the abundance of over 8500 proteins in a single TMT-based experiment. Additionally, we classified proteins with altered abundances resulting from nicotine treatment and highlight those which may be indicative of a general response to nicotine.
2 Materials and methods
2.1 Materials
Tandem mass tag (TMT) isobaric reagents were from Thermo Fisher Scientific (Waltham, MA). Nicotine was purchased from Sigma (St. Louis, MO). Water and organic solvents were from J.T. Baker (Center Valley, PA). Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) were from LifeTechnologies (Waltham, MA). Unless otherwise noted, all other chemicals were from Sigma (St. Louis, MO).
2.2 Cell growth and harvesting
Methods of cell growth and propagation followed previously utilized techniques [26, 27]. In brief, cells were propagated in DMEM supplemented with 10% FBS. Upon achieving 80% confluency, the growth media was aspirated and the cells were washed thrice with ice-cold phosphate-buffered saline (PBS). Designated cell culture dishes were supplemented with 1 μM nicotine and control cell culture dishes were mock-treated with an equal volume of sterile deionized water, in buffer adjusted to pH 7.4. Twenty-four hours after the addition of the drug, the cells were dislodged with a non-enzymatic reagent, harvested by trituration following the addition of 10 mL PBS, pelleted by centrifugation at 3000 × g for 5 min at 4°C, and the supernatant was removed. One milliliter of HBSp (50 mM HEPES, 50 mM NaCl, pH 8.0 supplemented with 1X Roche Complete protease inhibitors), and 2% SDS were added per each cell pellet.
2.3 Cell lysis and protein digestion
Cells were homogenized by ten passes through a 21-gauge (1.25 inches long) needle and incubated at 4°C with gentle agitation for 30 min. The homogenate was sedimented by centrifugation at 21 000 × g for 5 min and the supernatant was transferred to a new tube. Protein concentrations were determined using the bicinchoninic acid (BCA) assay (ThermoFisher Scientific). Proteins were subjected to disulfide bond reduction with 5 mM tris (2-carboxyethyl)phosphine (room temperature, 30 min) and alkylation with 10 mM iodoacetamide (room temperature, 30 min in the dark). Excess iodoacetamide was quenched with 10 mM dithiotreitol (room temperature, 15 min in the dark). Methanol-chloroform precipitation was performed prior to protease digestion. In brief, four parts of neat methanol were added to each sample and vortexed, one part chloroform was added to the sample and vortexed, and three parts water was added to the sample and vortexed. The sample was centrifuged at 14 000 rpm for 2 min at room temperature and subsequently washed twice with 100% methanol. Samples were resuspended in 100 mM HEPES, pH 8.5 and digested at room temperature for 13 h with Lys-C protease at a 100:1 protein-to-protease ratio. Trypsin was then added at a 100:1 protein-to-protease ratio and the reaction was incubated for 6 h at 37°C.
2.4 Tandem mass tag labeling
Approximately, 50 μg of peptides from each sample were labeled with TMT reagent. A total of 5 μL of the 20 ng/μL stock of TMT reagent was added to the peptides along with 20 μL of acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.5% (v/v) for 15 min. The TMT-labeled samples were pooled at a 1:1:1:1:1:1:1:1:1:1 ratio. The sample was vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (SPE) (Sep-Pak, Waters).
2.5 Off-line basic pH reversed-phase (BPRP) fractionation
We fractionated the pooled TMT-labeled peptide sample using BPRP HPLC [28]. We used an Agilent 1100 pump equipped with a degasser and a photodiode array (PDA) detector (set at 220 and 280 nm wavelength) from Thermo Fisher Scientific (Waltham, MA). Peptides were subjected to a 50 min linear gradient from 5 to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 at a flow rate of 0.8 mL/min over an Agilent 300Extend C18 column (5 μm particles, 4.6 mm id and 220 mm in length). The peptide mixture was fractionated into a total of 96 fractions. Fractions were collected into a 96-well plate from left to right along a row. Samples were consolidated into 12 by combining the columns of the 96-well plate (outlined in Supporting Information Fig. 5 of [29]). Samples were subsequently acidified with 1% formic acid and vacuum centrifuged to near dryness. Each consolidated fraction was desalted via StageTip, dried again via vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
2.6 Liquid chromatography and tandem mass spectrometry
Our mass spectrometry data were collected using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC 1000 LC pump (Thermo Fisher Scientific). The capillary column was a 75 μm id microcapillary column packed with ~0.5 cm of Magic C4 resin (5 μm, 100 Å, Michrom Bioresources) followed by ~30 cm of GP-18 resin (1.8 μm, 200 Å, Sepax, Newark, DE). For each analysis, we loaded 2 μg onto the column.
Peptides were separated by using a 3 h gradient of 6 to 26% acetonitrile in 0.125% formic acid at a flow rate of ~450 nL/min. Each analysis used the Multi-Notch MS3-based TMT method [30]. The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution 120 000; mass range 400–1400 m/z; automatic gain control (AGC) target 2 × 105; maximum injection time 100 ms). Precursors for MS2/MS3 analysis were selected using a TopSpeed of 2 sec. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap analysis; AGC 4 × 103; normalized collision energy (NCE) 35; maximum injection time 150 ms). Following acquisition of each MS2 spectrum, we collected an MS3 spectrum using our recently described method in which multiple MS2 fragment ions were captured in the MS3 precursor population using isolation waveforms with multiple frequency notches [30]. MS3 precursors were fragmented by high energy collision-induced dissociation (HCD) and analyzed using the Orbitrap (NCE 55; AGC 5 × 104; maximum injection time 150 ms, resolution was 60 000 at 200 Th).
2.7 Data analysis
Mass spectra were processed using a Sequest-based in-house software pipeline [31]. Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the human UniProt database. This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 50 ppm precursor ion tolerance for total protein level analysis. The product ion tolerance was set to 0.9 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Sequest searches and linear discriminant analysis [31, 32]. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification.
Peptide-spectral matches (PSMs) were adjusted to a 1% false discovery rate (FDR) [33, 34]. PSM filtering was performed using a linear discriminant analysis, as described previously [31], while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. For TMT-based reporter ion quantitation, we extracted the summed signal-to-noise (S:N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. For protein-level comparisons, PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides.
Proteins were quantified by summing reporter ion counts across all matching PSMs using in-house software, as described previously [31]. PSMs with poor quality, MS3 spectra with more than eight TMT reporter ion channels missing, MS3 spectra with TMT reporter summed signal-to-noise ratio less than 100, or with no MS3 spectra were excluded from quantification [4]. Each reporter ion channel was summed across all quantified proteins and normalized assuming equal protein loading of all ten samples. Protein quantification values were exported for further analysis in Microsoft Excel or SAS JMP.
We used JMP Pro 12.2 to perform our Spearman correlation analysis. In our TMT data set, 17 zero measurements were present and these proteins were eliminated from the analysis. Spearman’s ρ correlation coefficient is computed on the ranks of the data, using the same formula as the Pearson correlation [35]. The p-value for significance was determined by the t-distribution, and significance level was set at p-value <0.05.
2.7.1 Data access
Supporting Information Tables 1 and 2 list all quantified proteins and peptides, respectively, as well as the associated TMT reporter ion intensities used for quantitative analysis. The mass spectrometry proteomics data (RAW files) have been deposited to the ProteomeXchange Consortium [36] via the PRIDE partner repository with the dataset identifier PXD005361.
3 Results and discussion
3.1 Our mass spectrometry workflow quantified over 8500 proteins across four cell lines in a single experiment
Our experimental design consisted of four cell types (HEK293, HeLa, PaSC, and SH-SY5Y) each of which were mock- or nicotine-treated for 24 h. As a control, we included a biological replicate of PaSC (for both mock- or nicotine-treated samples). These samples were processed via our TMT workflow and analyzed using Multi-Notch MS3 analysis on an Orbitrap Fusion mass spectrometer (Fig. 1).
Figure 1.
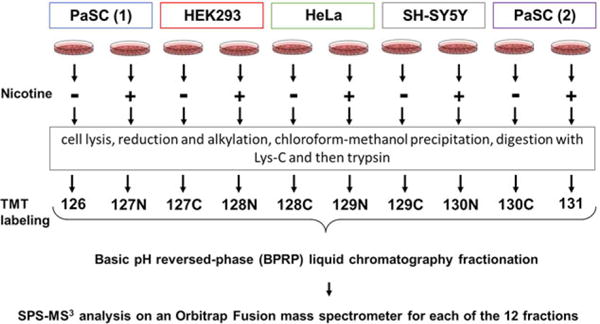
Experimental overview of the SPS-MS3 strategy. Four cell types (PaSC—in duplicate, HEK293, HeLa, and SH-SY5Y) were propagated and designated cultures were mock or nicotine treated. Proteins were extracted and then digested with Lys-C and trypsin. The resulting peptides were labeled with TMT, pooled, and fractionated via basic pH reversed-phase high performance liquid chromatography (BPRP-HPLC) prior to MS3 analysis on an Orbitrap Fusion mass spectrometer.
In total, we quantified 8590 proteins across all ten samples. These proteins were represented by 120 411 total peptides that corresponded to 83 043 unique peptides (Table 1). In Fig. 2A, a heat map illustrates the relative protein abundance across all samples and the associated dendrogram shows sample grouping. We noted that identical cell types clustered together closely and that the PaSC cells—which were analyzed in duplicate—did so by treatment. Clustering by cell type was expected as we did not anticipate that proteomic alterations resulting from the 24 h nicotine treatment would predominate over cell type-specific proteomes. Furthermore, we produced a Spearman correlation matrix comparing the relative protein abundance across the ten samples (Fig. 2B). As expected, protein abundance for the PaSC replicates correlated tightly (ρ>0.9), while correlation coefficients were lower between cell types. As such, we examined further the protein alterations resulting from nicotine stress across cell types having very diverse proteomes.
Table 1.
Summary of mass spectrometry data
| Unique peptides | Total peptides | Quantified proteinsa) |
|---|---|---|
| 83 043 | 120 411 | 8 590 |
a)Proteins quantified across all ten TMT channels, with an FDR <1%.
Figure 2.
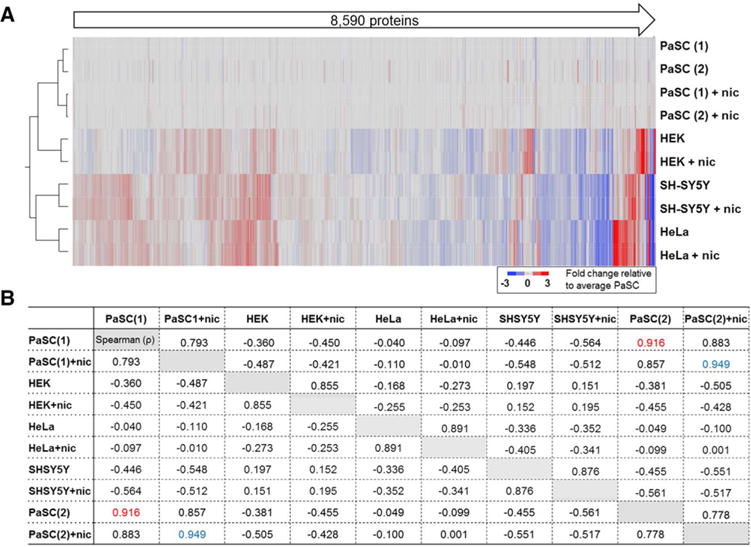
Total of 8590 proteins were quantified by using the SPS-MS3 strategy. (A) Heat map and associated dendrogram of samples analyzed in the TMT10-plex experiment. Data were normalized to the average untreated PaSC samples. (B) Spearman correlation (ρ) matrix of protein abundance for all ten samples. Highlighted in red and blue are the correlations of the replicates for PaSC and PaSC+nicotine, respectively, both of which demonstrated ρ>0.9. nic, nicotine.
We produced a biological replicate of this TMT10-plex workflow. Venn diagrams illustrated that approximately 75% of the identified proteins were in common between datasets (Supporting Information Fig. 1A). We also constructed scatter plots of the TMT relative abundance values to visualize the correlation between the two datasets for each cell line and treatment (Supporting Information Fig. 1B). In accordance with these plots, the Spearman coefficient, ρ, for the replicate sample pairs were >0.7 and the p-value < 0.001, showing strong correlation between the two datasets (Supporting Information Fig. 1C). These well-correlated datasets are encouraging, yet orthogonal validation of proteins meriting further investigation is recommended.
3.2 Nicotine altered the relative abundance of over 400 proteins in the cell lines investigated
Inclusively, we determined a total of 402 non-redundant proteins in our dataset as having increased levels greater than 1.5-fold when cells were treated with nicotine. Of these, 31 proteins were quantified in all four cell lines, while an additional 33 proteins showed such an increase in at least three of the four cell lines (Fig. 3A). In addition, we identified a total of 32 non-redundant proteins as having a greater than 1.5-fold decrease in abundance upon nicotine treatment in our dataset. Of these, none were determined to be down-regulated in all four cell lines, while only one was down-regulated in three of the four cell lines (Fig. 3B). In Fig. 4, we indicate the 31 proteins that demonstrated a 1.5-fold or greater increase in all four cell lines when treated with nicotine. The dot plot denotes the percentage of total signal contributed by each sample across the ten channels for a given protein. Similarly, we specify the 33 proteins with higher abundance in at least three of the four cell lines (Supporting Information Fig. 2A) and 1 protein that showed greater than 1.5-fold decrease when treated with nicotine (Supporting Information Fig. 2B) with a similar plot.
Figure 3.
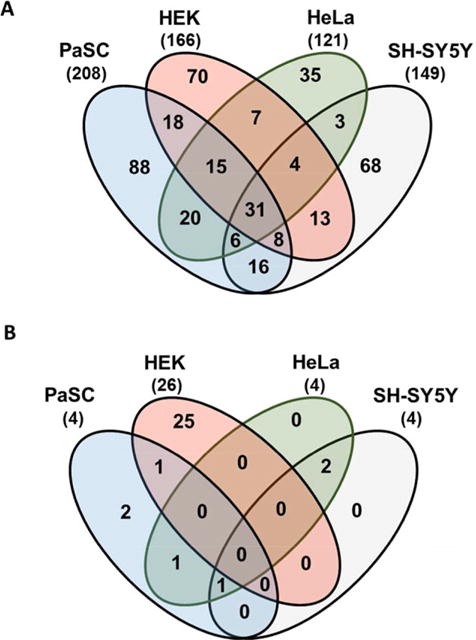
Venn diagrams of proteins with altered abundance following nicotine treatment. The Venn diagrams display the protein overlap for (A) 402 non-redundant proteins that exhibited an increase (≥1.5-fold) in abundance, and (B) the 32 that exhibited a decrease (≥1.5-fold) in abundance when the cell lines were treated with nicotine for 24 h.
Figure 4.
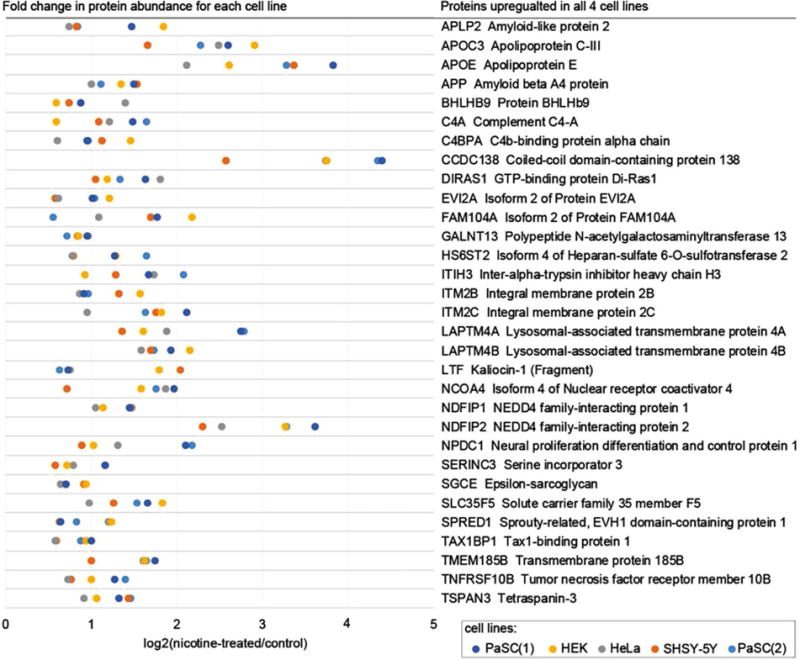
Proteins (n = 31) demonstrating a significant increase in abundance following nicotine treatment in all four cell lines. Heat map scale is the percentage of the total abundance of a protein across all ten channels.
3.3 Several proteins that participate in nAChR signaling, autophagy, and other signaling pathways demonstrated higher abundance across all cell lines
We highlighted eight proteins that demonstrated higher abundance when treated with nicotine across all cell lines. Amyloid beta A4 protein (APP, Fig. 5A) is well studied in the nervous system and is linked to the development of Alzheimer’s disease [37, 38]. APP has been identified previously in an interactome study of the α7 nAChR from mouse brain tissue [18] and several studies have shown APP to bind and modulate the activity of this receptor [39, 40]. APP has also been observed previously as up-regulated in nicotine-treated pancreatic cells in a cross-species analysis, which included PaSC from mouse, rat, and human species [41]. Moreover, an in vitro study using pancreatic ductal epithelial cells has indicated that increased APP expression in pancreatic cancer may influence cellular proliferation [42]. Like APP, Amyloid-like protein 2 (APLP2, Fig. 5B) was also among those proteins demonstrating higher abundance across cell lines when treated with nicotine [43]. Moreover, APLP2 may interact with proteins in cellular G-protein signaling pathways, which have been shown to associate with nAChR [44, 45], and likewise are members of a murine nAChR interactome [18].
Figure 5.
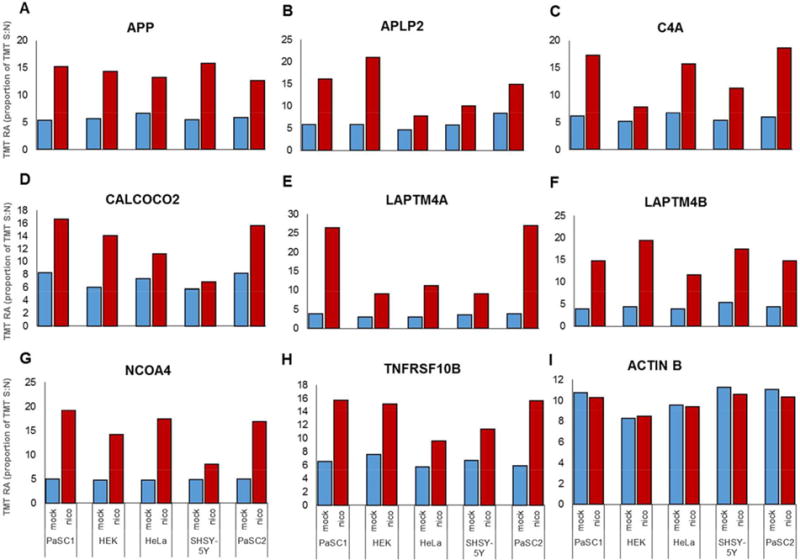
Bar graphs of proteins differentially expressed with statistical significance across cell lines. (A–H) Plots illustrate examples of proteins that demonstrate abundance greater than 1.5-fold in at least three of the four cell types investigated. (I) Actin is plotted as a control and does not show significant alteration in abundance in any cell line following nicotine treatment.
While the effects of nicotine have been associated with up-regulation of APP and APLP2 expression, we provide additional evidence that supports nicotine as having an effect on the expression of other proteins. For example, abundance differences in complement C4-A (C4A, Fig. 5C) have been shown previously to predict survival of patients with metastatic renal cell carcinoma [46] and members of the complement family have been also associated with nicotine exposure [47]. An unrelated protein, calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2, Fig. 5D) may be linked to the ionotropic role of nAChR. As α7 nAChR is a calcium channel, the influx of calcium in the presence of nicotine is likely to affect CALCOCO2 turnover [48]. CALCOCO2 has been observed previously to be elevated in PANC1 and HPNE pancreatic duct cells following treatment with nicotine [47]. Moreover, CALCOCO2 may have a role in actin cytoskeletal organization and is also involved in autophagy [49]. Potentially also linked to autophagy, lysosomal-associated transmembrane protein 4A (LAPTM4A, Fig. 5E) and 4B (LAPTM4B, Fig. 5F) are members of the mammalian four-transmembrane spanning protein superfamily that are of lysosomal and late endosomal origin [50]. While LAPTM4A confers multidrug resistance as a small molecule transporter, LAPTM4B promotes autophagy and tolerance to metabolic stress in cancer cells. LAPTM4B is overexpressed in human hepatocellular carcinoma [51] and is associated with colorectal and esophageal cancers [52]. Likewise, nuclear receptor coactivator 4 (NCOA4, Fig. 5G) has a potential role in thyroid carcinoma, papillary and prostate cancer [53, 54]. NCOA4 is a transcriptional co-regulator and is a coactivator of the peroxisome proliferator-activated receptor (PPAR) gamma [55]. NCOA4 also functions in ferritin degradation and autophagy [56]. Moreover, NCOA4 has been observed previously as elevated in PANC1 and HPNE pancreatic duct cells following nicotine treatment [47]. Another protein demonstrating higher abundance upon nicotine treatment is tumor necrosis factor receptor superfamily member 10B (TNFRSF10B, Fig. 5H). TNFRSF10B is a cell surface receptor of the TNF superfamily that mediates apoptosis [57]. Although no direct link has been established between TNFRSF10B and nicotine, nicotine can block TNF-mediated interaction with NMDA receptors [58].
In addition, we used actin (ACTB, Fig. 5I) as a housekeeping control. We show that no substantial effect on actin concentration was noted when the cells were exposed to nicotine. We have identified novel and previously-observed proteins as up-regulated when cells were treated with nicotine. In our replicate dataset we have quantified the same nine proteins highlighted in Fig. 5 (Supporting Information Fig. 3). These proteins show a very similar TMT relative abundance profiles in both datasets. However, further validation with orthogonal strategies will be needed to determine the role of these proteins in cells under nicotine stress.
3.4 Gene ontology and interaction analysis classification revealed enrichment of membrane protein-associated categories
We submitted differentially abundant proteins to the GOrilla server [59] to determine gene ontology enrichment (Table 2). Analysis of molecular function showed enrichment of proteins involved in transmembrane receptor and signaling function. It follows that the enriched terms for the cellular component category included plasma membrane-associated classifications. In addition, we examined the lists of differentially expressed proteins from each of the four cell lines individually, as nicotine may have cell line-specific effects. The proteins classified under a specific GO category were tallied and compared across the four cells lines (proteins are listed in Supporting Information Table 3). Generally, the percentage of differentially abundant proteins was similar across all cell lines. However, HeLa cells did not show enrichment for the categories: endosome, membrane raft, and receptor complex cellular component. This result may be expected as HeLa cells showed the least number of altered proteins and likewise these categories (i.e., endosome, membrane raft, and receptor complex cellular component) also represented the lowest percentage of altered proteins in the three other cell lines. Overall, these data provided supporting evidence that nicotine treatment promoted alterations in the abundance of membrane proteins and those involved in signaling pathways.
Table 2.
Gene ontology category enrichment of differentially abundant proteins
| GO term | Description | FDR q-valuea) | Fold enrichmentb) | No. of proteinsc) |
|---|---|---|---|---|
| Molecular function | ||||
| GO:0004888 | Transmembrane signaling receptor activity | 2.56E–08 | 6.82 | 20 |
| GO:0004872 | Receptor activity | 1.77E–08 | 4.74 | 27 |
| GO:0038023 | Signaling receptor activity | 8.73E–08 | 5.69 | 21 |
| GO:0060089 | Molecular transducer activity | 5.27E–06 | 3.2 | 31 |
| GO:0004871 | Signal transducer activity | 2.94E–05 | 3.36 | 26 |
| GO:0005102 | Receptor binding | 4.46E–05 | 2.64 | 35 |
| Cellular component | ||||
| GO:0005576 | Extracellular region | 3.47E–11 | 5.1 | 30 |
| GO:0005765 | Lysosomal membrane | 5.97E–03 | 3.5 | 13 |
| GO:0005768 | Endosome | 4.36E–03 | 2.69 | 20 |
| GO:0005886 | Plasma membrane | 7.61E–09 | 2.2 | 67 |
| GO:0010008 | Endosome membrane | 1.04E–04 | 3.52 | 20 |
| GO:0016021 | Integral component of membrane | 8.60E–31 | 3.26 | 105 |
| GO:0043235 | Receptor complex | 1.29E–05 | 5.86 | 14 |
Cut-off chosen was a false discovery rate <0.001.
Fold enrichment is with respect to all proteins quantified in the dataset.
Number of proteins with altered abundance (≥1.5-fold) in our dataset that were classified in a given category.
The 65 proteins that demonstrated changes in abundance ≥|1.5| in at least three of the four cell lines (as listed in Fig. 3 and Supporting Information Fig. 2) were subjected to String biological network analysis [60]. Figure 6 depicts the network diagram of these 65 proteins with respect to known and predicted protein-protein interactions. The degree of association is graphically illustrated by the thickness of the edges connecting nodes, which included the evidence parameters: experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence. The largest cluster consisted of 18 proteins and was anchored by APP and Notch1. The majority of these are membrane proteins and many have roles in metabolism and are involved in intramembrane cleavage of integral membrane proteins, such as Notch receptors, as well as in APP processing. To this end, previous evidence supports the potential effect of nicotine on Notch signaling [61,62] and APP expression [39,40]. A smaller, yet interesting, cluster was composed of the proteins MSMO1, FDFT1, and CYP51A1. These proteins are involved in steroid biosynthesis, a process which has recently been shown to be affected by nicotine exposure and may merit further targeted investigation [63]. This analysis has shown that some of the proteins altered by nicotine across cell lines fall into known biological networks. These data provide evidence supporting the merit of extending the methodologies outlined herein to investigate the effect of nicotine exposure in animal and/or primary cell models.
Figure 6.
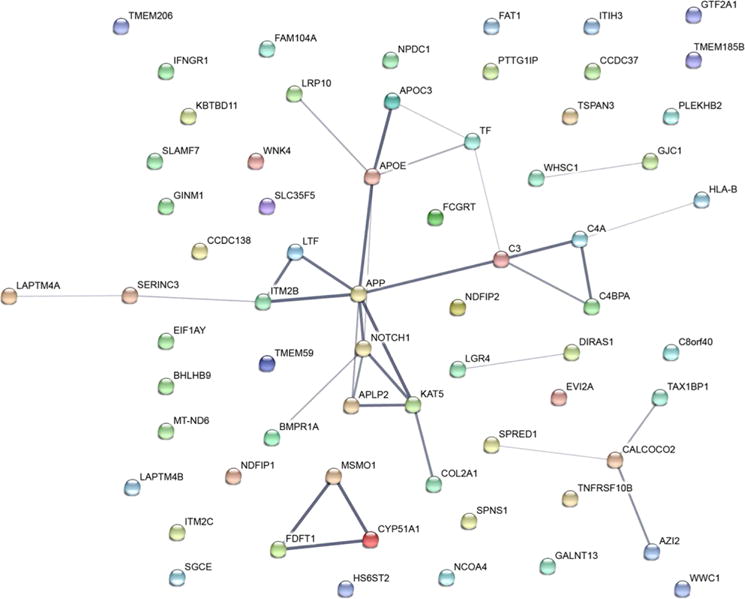
Network of 65 proteins that demonstrate differences in abundance (fold change ≥1.5). Line thickness of the edges indicates the strength of data supporting the interactions. Analysis was performed by using String [60] and evidence connecting nodes included experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence.
3.5 These data and the associated methodologies present a platform for further investigation of the cellular effects of nicotine
In this study, we measured perturbations in protein levels resulting from nicotine treatment in four cell lines using tandem mass tags (TMT10-plex). Our data show that several proteins increase in abundance across four cell lines. However, we recognize that this study is limited with respect to the effect of nicotine in a physiological environment. To understand better the true biological relevance of nicotine’s effect, a more rigorous experimental design should be applied. For example, the cellular dosage should be investigated further with nicotine concentrations spanning and extending beyond the physiological range. Alterations in protein abundance due to chemical treatments, such as nicotine, are typically concentration dependent. The data presented herein provide the protein profile of a single concentration of nicotine at a specific time point. The concentration used (1μM) and the time point chosen (24 h) mimics several previous studies [9, 64]. This concentration approximates the supposed upper limit in the blood of cigarette smokers, which can vary from 25 to 444 nM [65]. However, protein changes have also been noted at a range of nicotine concentrations. For example, one particular study investigated hepatic stellate cells and showed similar alterations at nicotine concentrations ranging from 100 pM to 10 μM [66]. As such, a wide range of nicotine concentrations may be investigated which would reveal a spectrum of proteomic alterations.
Studies may be designed to explore the consequences of nicotine treatment on cell lines resulting from dose-dependent effects. Particularly, lower concentrations should be used to show the minimum dosage needed for a significant alteration in abundance, as well as time-dependent protein abundance alterations elicited by nicotine treatment. Tracking the cellular proteomic response to these conditions may provide additional evidence for proteomic changes observed in the present study and result in further insights into the mechanisms of disease. These studies expand the scope of our investigation by considering the dosage and temporal effects of nicotine.
Moreover, the use of primary cell lines will produce a more physiologically relevant nicotine effect than do cell lines. Specifically, hepatic and hippocampal primary cells are known to be affected by nicotine and may serve as positive controls when examining other cells. Finally, determining a cellular read-out assay which is specific to nicotine treatment would better quantify nicotine’s impact on cellular function. For example, MTT cell proliferation assays, and wound healing (scratch) assays could complement well the proteomics data, thereby providing a phenotype which may be directly or indirectly associated with proteomic alterations.
In addition, future experiments may also extend the TMT10-plex strategy that we have outlined herein. For example, effects of different nAChR ligands could be investigated. Also, as the majority of proteins that are up-regulated with nicotine treatment have roles in cellular signaling events, the ubiquitylome and/or phosphoproteome may also be investigated using analogous TMT-based methods [67].
4 Concluding remarks
Here we highlight several proteins that are dysregulated in all four cell lines investigated when treated with nicotine. Future studies may use techniques similar to those herein to assay other cell lines or specifically target proteins with parallel reaction monitoring assays [68]. In addition to identifying individual candidate proteins for future studies, gene ontology analysis provides insight into the localization and functions of these altered proteins. This analysis reveals that several membrane-related processes may be altered. Accordingly, 58% of the dysregulated proteins are predicted as having transmembrane topology. The binding of nicotine to transmembrane nAChR modulates the internalization rate of these receptors, as such membrane protein landscape alterations are expected [69–71]. Additional studies using immunofluorescence or membrane-specific proteome analysis can examine further the induced differences in the membrane protein landscape. In summary, we have identified proteins and pathways that are altered following nicotine exposure in various cell types. The majority of the differentially abundant proteins are membrane-related and have roles in signal transduction and several are involved in cellular autophagy. Moreover, varying nicotine concentrations and incubation times may offer additional insight into potential time- and dose-dependent nicotine-induced changes in cellular proteomes. We conclude that nicotine exposure may cause widespread proteomic alterations and that this consequence merits further investigation in additional cell lines and model systems.
Supplementary Material
Significance of the study.
Nicotine is a stimulant drug found in tobacco plants and used as smoking prevention in forms of patches, gums, and popular e-cigarettes. However, nicotine can perturb cellular pathways and can induce alterations in many proteins across different cell types, yet the mechanisms thereof remain undetermined. We used a multiplexed Tandem Mass Tag (TMT10-plex)-based approach to study the proteomic alterations resulting from nicotine exposure in four common cell lines: HEK, HeLa, PaSC, and SH-SY5Y. We quantified 8590 proteins across the four cell lines. Of these proteins, 31 were altered by nicotine in all four cell lines, many of which were implicated in downstream signaling pathways, particularly autophagy. The outlined methodology along with studies in other cell lines will provide further evidence of proteomic alterations as a general response to nicotine exposure.
Acknowledgments
We would also like to thank members of the Gygi Lab at Harvard Medical School. This work was funded in part by the NIH/NIDDK grant K01 DK098285 (J.A.P.). The PaSC cell line (RLT-PSC) was a gift from Dr. Ralf Jesnowski (German Cancer Research Center).
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site
The authors have declared no conflict of interest.
References
- 1.Dayon L, Hainard A, Licker V, Turck N, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 2.Thompson A, Schafer J, Kuhn K, Kienle S, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 3.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.McAlister GC, Huttlin EL, Haas W, Ting L, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Toxicology, P. Tobacco-related exposures: tobacco smoking. Rep Carcinog. 2011;12:408–410. [PubMed] [Google Scholar]
- 6.McGuffey JE, Wei B, Bernert JT, Morrow JC, et al. Validation of a LC-MS/MS method for quantifying urinary nicotine, six nicotine metabolites and the minor tobacco alkaloids–anatabine and anabasine–in smokers’ urine. PLoS One. 2014;9:e101816. doi: 10.1371/journal.pone.0101816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prokopczyk B, Hoffmann D, Bologna M, Cunningham AJ, et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol. 2002;15:677–685. doi: 10.1021/tx0101088. [DOI] [PubMed] [Google Scholar]
- 8.Chowdhury P, Rayford PL. Smoking and pancreatic disorders. Eur J Gastroenterol Hepatol. 2000;12:869–877. doi: 10.1097/00042737-200012080-00006. [DOI] [PubMed] [Google Scholar]
- 9.Dasgupta P, Rizwani W, Pillai S, Kinkade R, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginzkey C, Kampfinger K, Friehs G, Kohler C, et al. Nicotine induces DNA damage in human salivary glands. Toxicol Lett. 2009;184:1–4. doi: 10.1016/j.toxlet.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Ginzkey C, Stueber T, Friehs G, Koehler C, et al. Analysis of nicotine-induced DNA damage in cells of the human respiratory tract. Toxicol Lett. 2012;208:23–29. doi: 10.1016/j.toxlet.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 12.Lindell O. Etiology, diagnosis and management of urinary lithiasis. Duodecim. 1996;112:1587–1592. [PubMed] [Google Scholar]
- 13.Dempsey DA, Benowitz NL. Risks and benefits of nicotine to aid smoking cessation in pregnancy. Drug Saf. 2001;24:277–322. doi: 10.2165/00002018-200124040-00005. [DOI] [PubMed] [Google Scholar]
- 14.Gotti C, Clementi F, Fornari A, Gaimarri A, et al. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 15.Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 16.Al-Wadei MH, Al-Wadei HA, Schuller HM. Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7. Mol Cancer Res. 2012;10:239–249. doi: 10.1158/1541-7786.MCR-11-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browne CJ, Sharma N, Waters KA, Machaalani R. The effects of nicotine on the alpha-7 and beta-2 nicotinic acetycholine receptor subunits in the developing piglet brainstem. Int J Develop Neurosci. 2010;28:1–7. doi: 10.1016/j.ijdevneu.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Paulo JA, Brucker WJ, Hawrot E. Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res. 2009;8:1849–1858. doi: 10.1021/pr800731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest. 2002;110:527–536. doi: 10.1172/JCI14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macklin KD, Maus AD, Pereira EF, Albuquerque EX, Conti-Fine BM. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 1998;287:435–439. [PubMed] [Google Scholar]
- 21.Sharma G, Vijayaraghavan S. Nicotinic receptor signaling in nonexcitable cells. J Neurobiol. 2002;53:524–534. doi: 10.1002/neu.10114. [DOI] [PubMed] [Google Scholar]
- 22.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 23.Scherer WF, Syverton JT, Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695–710. doi: 10.1084/jem.97.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jesnowski R, Furst D, Ringel J, Chen Y, et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and N-acetylcysteine. Lab Invest. 2005;85:1276–1291. doi: 10.1038/labinvest.3700329. [DOI] [PubMed] [Google Scholar]
- 25.Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973;33:2643–2652. [PubMed] [Google Scholar]
- 26.Paulo JA, Urrutia R, Banks PA, Conwell DL, Steen H. Proteomic analysis of a rat pancreatic stellate cell line using liquid chromatography tandem mass spectrometry (LC-MS/MS) J Proteomics. 2011;75:708–717. doi: 10.1016/j.jprot.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulo JA, Urrutia R, Banks PA, Conwell DL, Steen H. Proteomic analysis of an immortalized mouse pancreatic stellate cell line identifies differentially-expressed proteins in activated vs nonproliferating cell states. J Proteome Res. 2011;10:4835–4844. doi: 10.1021/pr2006318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Yang F, Gritsenko MA, Wang Y, et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulo JA, O’Connell JD, Everley RA, O’Brien J, et al. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotech-nol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 33.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 35.Myers JL, Well A. Research Design and Statistical Analysis. Lawrence Erlbaum Associates; Mahwah: 2003. [Google Scholar]
- 36.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 38.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York, NY) 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 39.Dineley KT, Westerman M, Bui D, Bell K, et al. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang HY, Lee DH, D’Andrea MR, Peterson PA, et al. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 41.Paulo JA, Urrutia R, Kadiyala V, Banks P, et al. Cross-species analysis of nicotine-induced proteomic alterations in pancreatic cells. Proteomics. 2013;13:1499–1512. doi: 10.1002/pmic.201200492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansel DE, Rahman A, Wehner S, Herzog V, et al. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 2003;63:7032–7037. [PubMed] [Google Scholar]
- 43.Gutala R, Wang J, Hwang YY, Haq R, Li MD. Nicotine modulates expression of amyloid precursor protein and amyloid precursor-like protein 2 in mouse brain and in SH-SY5Y neuroblastoma cells. Brain Res. 2006;1093:12–19. doi: 10.1016/j.brainres.2006.03.100. [DOI] [PubMed] [Google Scholar]
- 44.Inserra MC, Kompella SN, Vetter I, Brust A, et al. Isolation and characterization of alpha-conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem Pharmacol. 2013;86:791–799. doi: 10.1016/j.bcp.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 45.Adams DJ, Callaghan B, Berecki G. Analgesic conotoxins: block and G protein-coupled receptor modulation of N-type (Ca(V) 2.2) calcium channels. Br J Pharmacol. 2012;166:486–500. doi: 10.1111/j.1476-5381.2011.01781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zafar GI, Grimm EA, Wei W, Johnson MM, Ellerhorst JA. Genetic deficiency of complement isoforms C4A or C4B predicts improved survival of metastatic renal cell carcinoma. J Urol. 2009;181:1028–1034. doi: 10.1016/j.juro.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paulo JA. Nicotine alters the proteome of two human pancreatic duct cell lines. JOP. 2014;15:465–474. doi: 10.6092/1590-8577/2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang Y, Dai A, Zhou Y, Peng G, et al. Nicotine elevated intracellular Ca(2)(+) in rat airway smooth muscle cells via activating and up-regulating alpha7-nicotinic acetylcholine receptor. Cell Physiol Biochem. 2014;33:389–401. doi: 10.1159/000356678. [DOI] [PubMed] [Google Scholar]
- 49.Till A, Lipinski S, Ellinghaus D, Mayr G, et al. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy. 2013;9:1256–1257. doi: 10.4161/auto.25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kasper G, Vogel A, Klaman I, Grone J, et al. The human LAPTM4b transcript is upregulated in various types of solid tumours and seems to play a dual functional role during tumour progression. Cancer Lett. 2005;224:93–103. doi: 10.1016/j.canlet.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Zhou R, Zhang N, Rui J, Jin C. Biological function of a novel gene overexpressed in human hepatocellular carcinoma. Chin Med J (Engl) 2000;113:881–885. [PubMed] [Google Scholar]
- 52.Cheng XJ, Xu W, Zhang QY, Zhou RL. Relationship between LAPTM4B gene polymorphism and susceptibility of colorectal and esophageal cancers. Ann Oncol. 2008;19:527–532. doi: 10.1093/annonc/mdm469. [DOI] [PubMed] [Google Scholar]
- 53.Bongarzone I, Butti MG, Coronelli S, Borrello MG, et al. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res. 1994;54:2979–2985. [PubMed] [Google Scholar]
- 54.Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, et al. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9:509–516. [PubMed] [Google Scholar]
- 55.Heinlein CA, Ting HJ, Yeh S, Chang C. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor gamma. J Biol Chem. 1999;274:16147–16152. doi: 10.1074/jbc.274.23.16147. [DOI] [PubMed] [Google Scholar]
- 56.Dowdle WE, Nyfeler B, Nagel J, Elling RA, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 57.Chaudhary PM, Eby M, Jasmin A, Bookwalter A, et al. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 58.Carlson NG, Bacchi A, Rogers SW, Gahring LC. Nicotine blocks TNF-alpha-mediated neuroprotection to NMDA by an alpha-bungarotoxin-sensitive pathway. J Neurobiol. 1998;35:29–36. doi: 10.1002/(sici)1097-4695(199804)35:1<29::aid-neu3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 59.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48–55. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szklarczyk D, Franceschini A, Wyder S, Forslund K, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shang X, Shang Y, Fu J, Zhang T. Nicotine significantly improves chronic stress-induced impairments of cognition and synaptic plasticity in mice. Mol Neurobiol. 2016:1–15. doi: 10.1007/s12035-016-0012-2. [DOI] [PubMed] [Google Scholar]
- 62.Ng TK, Huang L, Cao D, Yip YW, et al. Cigarette smoking hinders human periodontal ligament-derived stem cell proliferation, migration and differentiation potentials. Sci Rep. 2015;5:7828–7834. doi: 10.1038/srep07828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu DM, He Z, Chen T, Liu Y, et al. DNA hypermethylation of acetoacetyl-CoA synthetase contributes to inhibited cholesterol supply and steroidogenesis in fetal rat adrenals under prenatal nicotine exposure. Toxicology. 2016;340:43–52. doi: 10.1016/j.tox.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 64.Wei PL, Kuo LJ, Huang MT, Ting WC, et al. Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann Surg Oncol. 2011;18:1782–1790. doi: 10.1245/s10434-010-1504-3. [DOI] [PubMed] [Google Scholar]
- 65.Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. doi: 10.1136/bmj.280.6219.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soeda J, Morgan M, McKee C, Mouralidarane A, et al. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun. 2012;417:17–22. doi: 10.1016/j.bbrc.2011.10.151. [DOI] [PubMed] [Google Scholar]
- 67.Paulo JA, McAllister FE, Everley RA, Beausoleil SA, et al. Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. Proteomics. 2015;15:462–473. doi: 10.1002/pmic.201400154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11:1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vallejo YF, Buisson B, Bertrand D, Green WN. Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci. 2005;25:5563–5572. doi: 10.1523/JNEUROSCI.5240-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hohnadel EJ, Hernandez CM, Gearhart DA, Terry AV., Jr Effect of repeated nicotine exposure on high-affinity nicotinic acetylcholine receptor density in spontaneously hypertensive rats. Neurosci Lett. 2005;382:158–163. doi: 10.1016/j.neulet.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 71.Gentry CL, Lukas RJ. Regulation of nicotinic acetylcholine receptor numbers and function by chronic nicotine exposure. Curr Drug Targets CNS Neurol Disord. 2002;1:359–385. doi: 10.2174/1568007023339184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.