

Abstract
The XPF/Mus81 structure-specific endonucleases cleave double-stranded DNA (dsDNA) within asymmetric branched DNA substrates and play an essential role in nucleotide excision repair, recombination and genome integrity. We report the structure of an archaeal XPF homodimer alone and bound to dsDNA. Superposition of these structures reveals a large domain movement upon binding DNA, indicating how the (HhH)2 domain and the nuclease domain are coupled to allow the recognition of double-stranded/single-stranded DNA junctions. We identify two nonequivalent DNA-binding sites and propose a model in which XPF distorts the 3′ flap substrate in order to engage both binding sites and promote strand cleavage. The model rationalises published biochemical data and implies a novel role for the ERCC1 subunit of eukaryotic XPF complexes.
Keywords: archaea, DNA repair, endonuclease, HhH domain, XPF
Introduction
Nucleotide excision repair (NER) is a highly conserved DNA repair pathway able to detect and remove a variety of bulky DNA lesions caused by UV light and environmental mutagens and thereby contribute to the genomic integrity of an organism (Sancar, 1996; Lindahl and Wood, 1999). Defects in NER are associated with three inherited human diseases—xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and Cockayne syndrome (CS)—all of which have severe clinical consequences (reviewed in Lehmann, 2003). NER in higher eukaryotes involves the coordinated assembly of a large number of proteins, including the core NER factors XPC-HR23B, TFIIH, XPA, replication protein A (RPA) and two endonucleases XPG and ERCC1-XPF (Aboussekhra et al, 1995). Together these proteins cooperate to recognise, unravel and excise a 24–32-mer oligonucleotide bearing the DNA lesion prior to filling in the missing gap (Araujo and Wood, 1999). Although prokaryotes have a similar overall repair strategy, a much simpler and structurally unrelated multiprotein complex known as UvrABC carries out the same task (Petit and Sancar, 1999). The availability of fully sequenced archaeal genomes has revealed that many archaea have proteins related to eukaryotic NER factors, including the endonucleases XPF and XPG, rather than to the UvrABC bacterial repair system (White, 2003).
XPF (xeroderma pigmentosa complementation group F, also known as ERCC4) associates with a noncatalytic partner ERCC1 (excision repair cross complementarity group 1) to form a structure-specific endonuclease that preferentially cleaves DNA duplexes adjacent to a 3′ single-stranded flap (Figure 1A and B). Such double-stranded (ds)/single-stranded (ss) DNA junctions are found in bubbles, simple Y structures and hairpins and are generated on the 5′ side of bulky DNA lesions (Sijbers et al, 1996a, 1996b). All of these DNA structures can be cleaved by ERCC1-XPF (de Laat et al, 1998). The ERCC1-XPF 3′ flap substrate polarity complements the unrelated XPG endonuclease, which cleaves duplex DNA next to a 5′ single-stranded flap and together these endonucleases are responsible for the dual incision that eliminates the lesion-bearing oligonucleotide (Araujo et al, 2000). More recently, the XPF paralogue, Mus81, was shown to function as a 3′ flap endonuclease when associated with its noncatalytic partner Eme1/Mms4 (Boddy et al, 2001; Kaliraman et al, 2001). Mus81-Eme1 cleaves a different set of branched DNA substrates from eukaryotic XPF, including stalled replication forks, nicked Holliday junctions and D-loops (reviewed in Hollingsworth and Brill, 2004). It has a strict requirement for a 5′ DNA end near the flap junction and generates a nicked DNA product with a gap of five unpaired nucleotides (Bastin-Shanower et al, 2003; Osman et al, 2003). For both XPF and Mus81, the major cleavage site always lies within the upstream duplex present in all the branched DNA structures they recognise (Figure 1B).
Figure 1.
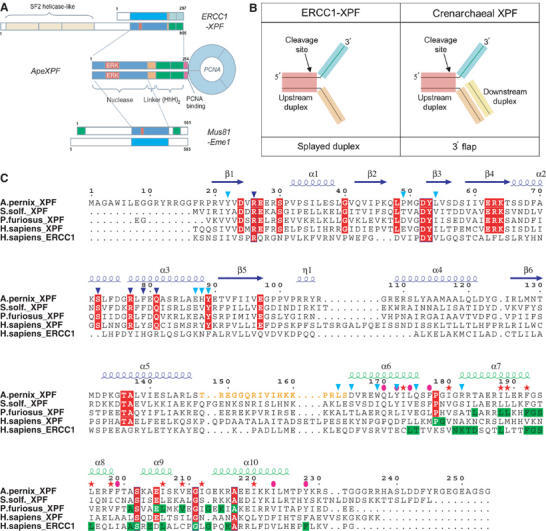
(A) Domain structures of the XPF superfamily. Sequence numbering corresponds to the human ERCC1-XPF heterodimer, ApeXPF and the human Mus81-Eme1 heterodimer. The red box indicates the catalytic ERK motif within the nuclease domain (blue). HhH motifs are shown in green. Cyan indicates a ‘nuclease-like' domain lacking catalytic residues. (B) Optimal substrates for crenarchaeal XPF and ERCC1-XPF. SsoXPF has a preference for substrates with dsDNA both upstream and downstream from the cleavage site (i.e. 3′ flap), while eukaryotic ERCC1-XPF prefers splayed duplex substrates. (C) Sequence alignment of selected XPF homologues. Representative sequences are shown for the nuclease and (HhH)2 domains from the euryarchaeal (18 sequences) and eukaryotic (18 sequences) XPFs, and for ERCC1 (18 sequences). Invariant residues within the XPF family are shown in red. Observed secondary structure of ApeXPF is indicated above the sequences. Residues involved in the nuclease–(HhH)2 interface are indicated by cyan triangles. Residues within the (HhH)2 dimer interface are indicated by pink ellipses, and those within the (HhH)2 domain hydrophobic core by pink stars. Mutated residues are indicated by dark blue triangles. Invariant residues conserved within the (HhH)2 domain of individual XPF subgroups are highlighted in green.
XPF family members have a catalytic domain followed by a DNA-binding domain containing two consecutive helix–hairpin–helix HhH motifs (Thayer et al, 1995;Aravind et al, 1999; Shao and Grishin, 2000). The catalytic domain contains the active site motif GDXnERKx3D related to prokaryotic endonucleases, while the two HhH motifs form a compact (HhH)2 domain that has been shown to contribute to XPF dimer formation and binds duplex DNA in a sequence-independent manner (Nishino et al, 2003). Mus81 also contains a similar nuclease domain, but is flanked by single HhH motifs. Eukaryotic XPFs have an N-terminal SF2-like helicase domain that apparently lacks essential catalytic residues for ATPase activity (Sgouros et al, 1999). A similar ‘long' form of XPF is present in most euryarchaea, one example being Hef (Helicase-associated endonuclease for fork-structured DNA) from Pyrococcus furiosus, which has an active helicase domain (Komori et al, 2002). Crenarchaea have a ‘short' form of XPF that lacks the helicase-like domain and whose catalytic activity is regulated by interaction with PCNA (Figure 1A) (Roberts et al, 2003). These differences in domain architecture may reflect differences in the recruitment of XPFs to branched DNA structures (Komori et al, 2002; Roberts et al, 2003). All XPF family proteins require divalent cations for nuclease activity (Sijbers et al, 1996a, 1996b; Nishino et al, 2003). They can either form heterodimers with much smaller but related partners, ERCC1 (eukaryotic XPF) or Eme1/Mms4 (Mus81), or form homodimers (archaea) (Sijbers et al, 1996a, 1996b; Nishino et al, 2003). This dimeric organisation is critical for stability and catalytic activity of XPF (Gaillard and Wood, 2001; Nishino et al, 2003). Additional functions for the noncatalytic partners of XPF have been proposed, for example, ERCC1 targets XPF to sites of DNA damage through its interaction with the DNA-binding protein XPA (Li et al, 1995).
To understand more about XPF architecture and the basis of ds/ssDNA substrate recognition, we have determined the structure of an essentially intact crenarchaeal XPF from Aeropyrum pernix (ApeXPF) in the presence and absence of dsDNA. Analysis of these structures has revealed for the first time a large domain movement on binding dsDNA and has led us to suggest a model for XPF substrate recognition involving distortion of branched and nicked DNA substrates.
Results
Structure determination
For crystallisation purposes, we used a truncated form of ApeXPF comprising residues 19–231 (denoted as ApeXPF throughout). In solution, ApeXPF forms homodimers as does the closely related Sulfolobus solfataricus XPF (SsoXPF) (Lally et al, 2004; Roberts et al, 2004). ApeXPF can be stimulated by a heterotrimeric PCNA to cleave 3′ flap structures; however, SsoXPF has a more robust activity in a nuclease assay and we therefore used SsoXPF for the mutational analysis (Supplementary Figure S1). We tried to crystallise ApeXPF in the presence of DNA with a wide range of 3′ flaps, hairpins and Y structures, and in some cases obtained crystals but none with good diffraction properties. Trigonal crystals were eventually obtained using a 15-mer DNA duplex; they diffracted to 2.8 Å resolution and contain a single ApeXPF dimer (Table I). The structure was solved by molecular replacement using as a search model the ApeXPF nuclease domain derived from a partially refined structure of residues 19–150 (Lally et al, 2004; Materials and methods). The initial electron density maps phased only on the nuclease domains showed good density for the (HhH)2 domain and the duplex DNA (Supplementary Figure S2) and allowed tracing of the entire molecule apart from the interdomain linker of one protomer, which is disordered. The final structure was refined to an R-factor of 23.0% and R-free of 29.0% at 2.8 Å resolution (Table I).
Table 1.
Data collection and refinement statistics
| Protein | ApeXPF | ApeXPF | ApeXPF |
| Form | Native | 1 mM K2OsO4 | Native+dsDNA |
| X-ray source | Station 14.2, SRS | Station 14.2, SRS | Station 9.6, SRS |
| Wavelength (Å) | 1.0000 | 1.0000 | 0.9780 |
| Space group | C2 | C2 | P3221 |
| Unit cell parameters (Å) | a=210, b=42.7, c=118.7 | a=209, b=42.8, c=119.9 | a=b=141.3, c=85.3 |
| β=121.4° | β=121.4° | ||
| Za | 4 | 4 | 2 |
| No. of measurements | 52 273 (7746) | 33 163 (4880) | 125 841 (8403) |
| No. of unique reflections | 14 887 (2159) | 9471 (1414) | 21 619 (2463) |
| Resolution limit (Å) | 3.2 (3.37–3.2) | 3.6 (3.79–3.6) | 2.8 (2.95–2.8) |
| Completeness | 99.9 (100) | 98.5 (99.9) | 97.5 (92.8) |
| Rsym | 6.2 (30.8) | 5.0 (32.1) | 8.3 (39.9) |
| Average I/σ(I) | 10.6 (2.4) | 13.1 (2.3) | 13.7 (2.8) |
| Ranomalous | — | 5.0 (23.1) | — |
| Rderivative | — | 26.0 (31.2) | — |
| R-factor | 0.24 | — | 0.23 |
| R-free | 0.31 | — | 0.29 |
| Model | 6516 protein atoms | — | 3357 protein atoms, 608 DNA atoms, 6 sulphate ions, 1 Mg ion, 7 H2O |
| R.m.s. bond lengths (Å) | 0.02 | — | 0.021 |
| R.m.s. bond angles (deg) | 1.9 | — | 2.2 |
| Ramachandran plota | 86.9%/12.6% | 90.0%/8.9% | |
| Values in parentheses refer to the highest resolution shell indicated in the row for resolution limit. | |||
| Percentage of residues located in the most favourable/additionally allowed regions of the Ramachandran plot. |
Monoclinic crystals of ApeXPF grown without DNA contain two ApeXPF dimers and diffracted to 3.2 Å resolution. This structure was solved by molecular replacement using the two domains of the DNA-bound structure separately as search models and guided by heavy atoms sites identified from an osmate derivative (see Materials and methods). Two of the four protomers had continuous density including the linker sequence, while the other two had disordered interdomain linkers. This structure was refined to an R-factor of 24.0% and R-free of 31.0% at 3.2 Å resolution (Table I).
Structure of apo-ApeXPF
ApeXPF 19–231 has two structured domains, an N-terminal nuclease domain (residues 19–148) and an (HhH)2 domain (residues 165–226) connected by a 15-residue linker (Figures 1C and 2A). As expected by analogy with Hef (Nishino et al, 2003), both the nuclease and (HhH)2 domains form independent, tightly associated dimers with an equivalent domain from the second protomer. In this arrangement, the domains are uncoupled. The α/β nuclease domain structure is closely related to the Hef nuclease domain and the protomers make similar dimer contacts via two helices (α4 and α5) and an edge β-strand (β6) (Nishino et al, 2003). The active site lies in a large cleft bounded by the structural elements α1′, α2, α3 and a loop between strands β2 and β3 (Figure 1C). This cleft is lined with the overwhelming majority of XPF conserved residues including the metal-binding and catalytic residues from the signature sequence E-R-K (E62-R63-K64) that runs along the base of the cleft as part of strand β4. There is density at a site equivalent to the metal site observed in Hef (Nishino et al, 2003). Similar density is present in the ApeXPF–DNA complex, which due to its higher resolution shows more precisely that the coordination geometry and distance to side chains of D52 and E62 are consistent with it being a metal ion, presumably magnesium.
Figure 2.
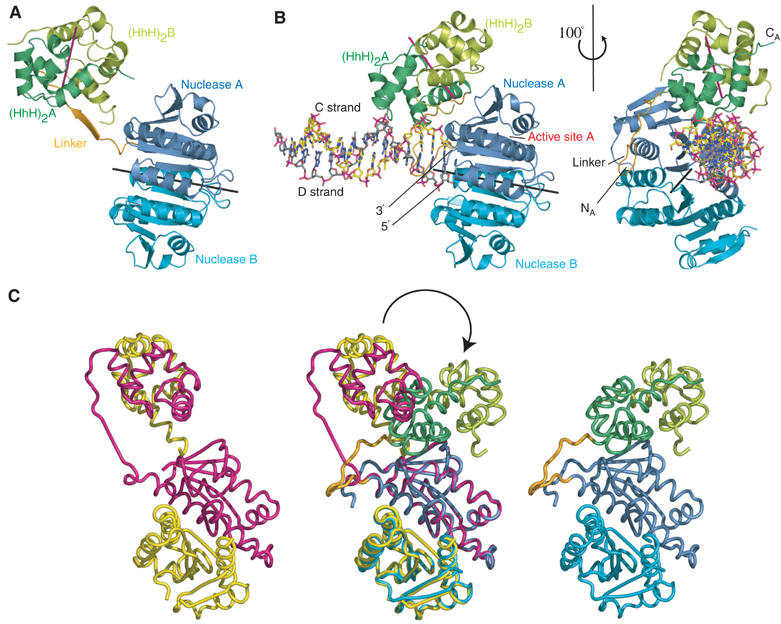
(A) Structure of the apo form of the ApeXPF dimer. Protein domains are coloured to correspond with Figure 1A. The local two-fold axes relating the nuclease and (HhH)2 domains are shown as black and magenta lines, respectively. (B) Two views of the ApeXPF–DNA complex. Protein domains are coloured to correspond with Figure 1A. The carbon atoms of the DNA C strand are yellow and those of the DNA D strand are grey. (C) Domain rearrangement between ApeXPF with and without DNA shown by superposing the nuclease domains of protomer A from the two structures. The ‘open' apo structure is on the left, the ‘closed' DNA-bound structure on the right and the two superposed in the middle.
Each (HhH)2 domain forms an integral five-helical domain bearing two functional helix–hairpin–helix motifs related by pseudo-two-fold symmetry (Thayer et al, 1995; Doherty et al, 1996; Shao and Grishin, 2000). A similar domain is present in RuvA (PDB code 1C7Y) (Ariyoshi et al, 2000). The RuvA (HhH)2 domain has an r.m.s. difference of 1.55 Å (54 C-alpha atoms) and 1.7 Å (53 C-alpha atoms) with the (HhH)2 domains from the two protomers of the ApeXPF–DNA structure. ApeXPF has two G-I-G hairpins at residues 179–181 and 211–213. Although the (HhH)2 domain fold has been observed previously (Thayer et al, 1995; Rafferty et al, 1996; Singh et al, 2002), this is the first structure of a dimer of (HhH)2 domains. There are three major contacts in this predominantly hydrophobic dimer interface (Figure 1C and Supplementary Figure S3). These centre around the connecting helix that links the two HhH motifs, residues from the first helix of the first HhH motif and residues C-terminal to the second HhH motif. A C-terminal extension to the domain contributes the highly conserved Y228 to the dimer interface.
The two independent dimers in the asymmetric unit of the apo ApeXPF crystals are very similar and both have the linker region of one protomer fully extended. The local two-fold axes relating the protomers of the nuclease and (HhH)2 domain dimers respectively are not coincident so that overall the ApeXPF dimer does not obey two-fold symmetry (Figure 2A). Each of the independent apo ApeXPF dimers is in close contact with another identical dimer related by operation of a crystallographic two-fold axis generating a tetramer. The contact involves two equivalent linker regions that form a two-stranded antiparallel sheet across the two-fold axis (Supplementary Figure S4). Comparison of both tetrameric arrangements indicates an r.m.s. difference of 3.4 Å on 802 C-alpha atoms.
Structure of an ApeXPF–dsDNA complex
The ApeXPF–dsDNA structure comprises two ApeXPF protomers (denoted A and B), together with one DNA duplex (strands denoted C and D) (Figure 2B). The DNA duplex is bound to the nuclease and (HhH)2 domains of protomer A, and this we define as site I. Overall, the ApeXPF in the complex forms an asymmetric dimer in which protomer A is more compact and has an ordered interdomain linker (Figure 2B). Comparing the A chains of the DNA-bound and DNA-free forms shows that when their nuclease domains are superimposed, the two orientations of the (HhH)2 domains are related by a rotation of 95°, which is primarily a closure rather than twist (Figure 2C). This shifts the (HhH)2 domains by over 30 Å. The linker forms a complex hinge region with the main flexures at residues 151–155 and 164. In the DNA-bound form, it contributes L163 to the interface between the nuclease and (HhH)2 domains (Figure 1C and Supplementary Figure S5).
The DNA-bound ApeXPF structure is dominated by the interaction of the (HhH)2 domain of protomer A with dsDNA. Interdomain interactions between the nuclease and (HhH)2 domains of the A protomer further stabilise the ApeXPF conformation and bury several aromatic residues at the interface close to W169 (Figure 1C and Supplementary Figure S5). Protomer B makes almost no contact with the DNA at site I, and its nuclease and (HhH)2 domain do not interact; its interdomain linker is not seen in the electron density maps, but must adopt an extended conformation because protomer B residues 148 and 160 are 29 Å apart. The interdomain linker has been suggested to be sensitive to proteolysis due to conformational flexibility (Nishino et al, 2003). We therefore dissolved ApeXPF–dsDNA crystals and confirmed by SDS–PAGE that no proteolytic cleavage had occurred, thus confirming the integrity of both linkers (data not shown).
Site I recognition involves binding of a six base-pair stretch of minor groove by the (HhH)2 domain of protomer A and the interaction of a blunt end (representing a ds/ss discontinuity) with the catalytic domain of protomer A. The stretch of minor groove recognised by the (HhH)2 domain is just two base pairs from the end of the duplex and this ensures that when the nuclease and (HhH)2 domains become coupled, the duplex DNA is oriented with the 3′ end of one strand adjacent to Y123 (Figure 3A and B). The 5′ end of strand D lies adjacent to a crystallisation-derived sulphate ion bound to R126 from both protomers A and B. Binding DNA with an opposite polarity would not be possible, as the HhH hairpin footprint could not accommodate the much wider major groove. The G-I-G hairpin from one HhH motif binds two backbone phosphates from T13 and G14 of DNA strand C, while the other hairpin binds phosphates from T7 and C8 of the opposite sense strand D (Figure 3A). Several positively charged residues immediately after the hairpins (R182, R183 and R187 of the first HhH motif and K215 and R216 of the second HhH motif) also contribute to the interaction with the minor groove. Of these, sequence-independent contacts include R187 interaction with the C12 phosphate backbone of the C strand, and R216 penetration into the minor groove to bind T7 (D strand) backbone phosphate. The unique orientation of dsDNA is assisted by a sequence-dependent contact from R182 of the first HhH motif to guanine-14 of strand C. Protomer A's nuclease domain contacts a blunt end of the DNA duplex through hydrophobic interactions (residues A86, Y123 and G124) with the two end bases A15 (strand C) and T1 (strand D) (Figure 3). The blunt end is not a feature of preferred XPF substrates but here represents a ds/ss discontinuity and lies adjacent to a shallow hydrophobic groove (Figure 3B and C) that may contact ssDNA (see below).
Figure 3.
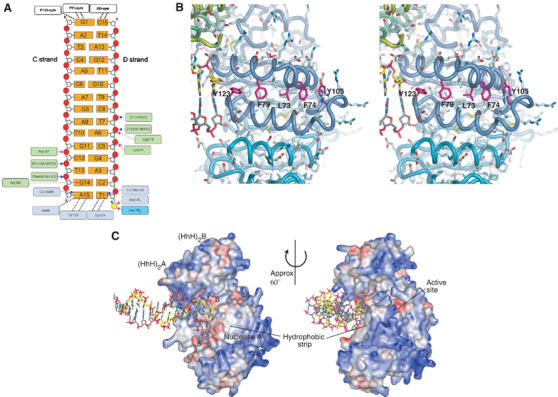
(A) Schematic of ApeXPF interaction with the 15-mer dsDNA. Hydrogen bonds to DNA involving protein side chains are shown as magenta arrows, and those involving protein backbone are shown as blue arrows. Hydrophobic interactions are represented by dashed lines. Blue and cyan boxes around residue numbers represent nuclease domains from protomers A and B, respectively, and green boxes indicate the (HhH)2 domain contacts. A small contact between DNA and a symmetry-related protein molecule is indicated for G1–C15. (B) The hydrophobic groove on the nuclease domain of protomer A. All side chains are shown as sticks. This is a possible ssDNA-binding site, and aromatic residues that might stack with or intercalate between DNA bases are labelled. The strip is bordered by positively charged side chains, some of which could interact with the phosphate backbone. (C) Protein electrostatic surface in the ApeXPF–DNA complex. Red indicates acidic regions and blue indicates basic regions. Only protein atoms were included in the calculation of the electrostatic potential with APBS (Baker et al, 2001).
A two-site model for ds/ssDNA junction recognition by XPF
The (HhH)2 domain of protomer A is a major component of site I, but the (HhH)2 domain of protomer B is also potentially capable of binding DNA. We note that the two-fold axis relating the (HhH)2 domains of protomers A and B makes an angle close to 45° with the helical axis of the dsDNA. Crystal contacts prevent the protomer B (HhH)2 domain from engaging dsDNA in the trigonal crystal form. Instead, a sulphate ion occupies a position equivalent to the phosphate backbone position of the DNA bound to protomer A's first HhH motif. We therefore modelled how protomer B's (HhH)2 domain might bind DNA. As the (HhH)2 domain dimer two-fold axis is at 45° to the helical axis of the dsDNA, rotating the dsDNA (strands C and D) about this two-fold axis generates a second putative dsDNA molecule (strands C′ and D′) at 90° to the experimentally observed dsDNA (Figure 4A). Inspection of the modelled DNA in the context of the crystallographically observed complex shows one end of the C′D′ duplex close to the active site of the nuclease domain of protomer A (Figure 4B). A second potential DNA-binding site (denoted site II) therefore exists, which comprises the (HhH)2 domain of protomer B and the active site of protomer A, although it is unoccupied in our structure.
Figure 4.
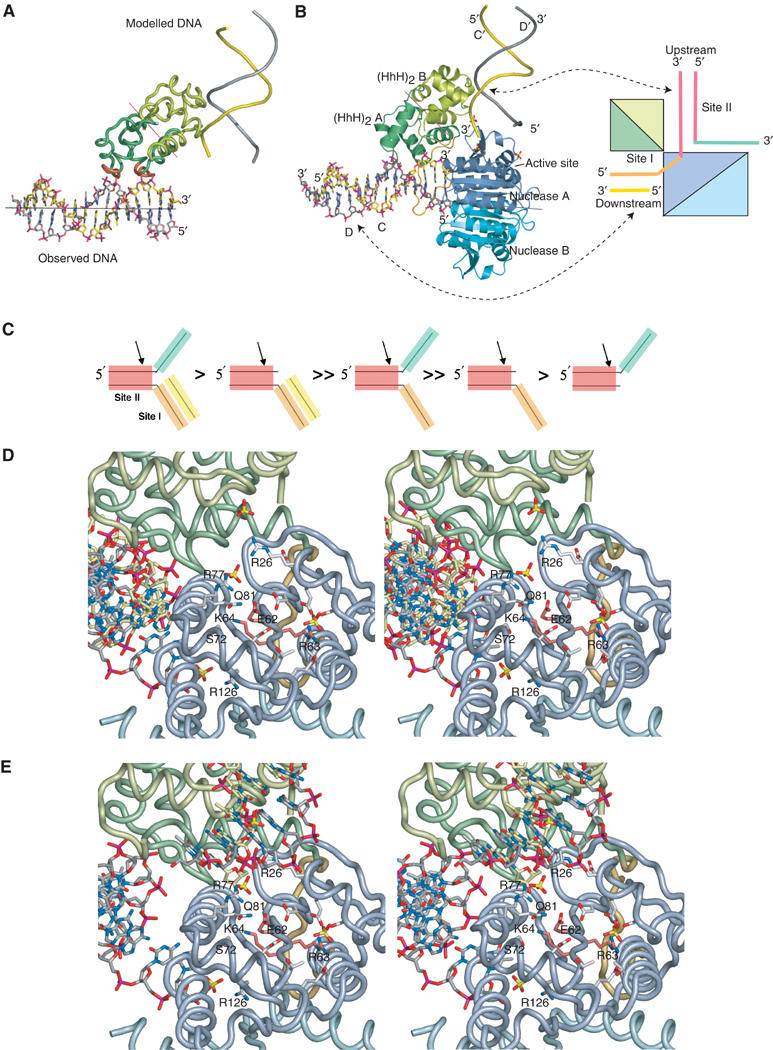
(A) Interaction of DNA with site I, and modelling of DNA bound to site II. Site I-bound dsDNA is shown in stick representation, and the GIG-containing hairpin regions of the protomer A (HhH)2 domain are highlighted in red. The two-fold axis between the (HhH)2 domains of protomers A and B is indicated by a magenta line, and the approximate direction of the DNA helical axis is shown by a black line. The modelled DNA generated by rotation of the observed DNA around the (HhH)2 two-fold axis (backbone representation) is in the correct orientation to interact with the two hairpin motifs of protomer B. (B) A structural model for DNA substrate binding to ApeXPF. (HhH)2 domains and DNA are drawn as in (A), but nuclease domains are also included. In our model, the observed DNA at site I would be downstream of the ds/ss junction and the modelled DNA at site II would be upstream. The sulphate ions found in the structure (shown in red/yellow) mimic the phosphate backbone of site II DNA. The binding sites are also shown schematically with DNA strands and protein chains coloured according to Figures 1B and 5C, and 2, respectively. (C) Substrate preferences for SsoXPF as determined by Roberts et al (2004). Distinct features of the preferred 3′ flap substrate are highlighted. A double greater than sign indicates at least a 10-fold improvement in the rate of cleavage by SsoXPF, whereas a single greater than sign indicates one- to three-fold improvement. (D) Stereo view of the protomer A active site. The ERK active site motif has light red side chains, and other key conserved side chains are labelled. Sulphate ions are shown where they may mimic phosphate backbone binding sites. Also shown are the side chain of R126 and the adjacent sulphate ion, which lies close to the 5′ end of strand D. (E) Identical stereo view of the protomer A active site to (A) but including the modelled second duplex DNA at site II generated by rotation about the (HhH)2 domain dimer dyad axis as described in the text.
We also assume that ApeXPF has similar substrate preferences to SsoXPF, which cleaves 3′ flaps and nicked duplexes preferentially over splayed duplexes (Figure 4C; Roberts et al, 2004). Both these preferred substrates have two segments of contiguous dsDNA and therefore the observed crystallographic DNA duplex at site I could represent either the upstream or downstream duplex of such substrates. Since XPF is known to cleave only within the upstream duplex of a 3′ flap (Sijbers et al, 1996a, 1996b; de Laat et al, 1998; Bastin-Shanower et al, 2003; Osman et al, 2003; Roberts et al, 2003), and the 3′ end of the C strand at site I is 17 Å from the active site of protomer A (and 35 Å from the active site of protomer B), we consider it more likely that dsDNA bound at site I represents the downstream part of such a substrate. Strand C would therefore be part of the continuous strand (or undamaged strand) (Figure 4B). This would imply that site II binds the upstream duplex, which would be processed by the active site of protomer A. This arrangement would allow for the XPF cleavage sites being upstream of the ds/ssDNA junction whether the duplex traverses the active site or the strands are separated. It is evident that in order for both (HhH)2 domains to engage DNA substrate simultaneously, a 3′ flap substrate would have to bend by almost 90° (Figure 4B).
The 3′ end of the modelled C′ strand lies close to the catalytic centre of protomer A and adjacent to the absolutely conserved residues solvent accessible R26 and R77, just outside of the catalytic cleft (Figure 4D and E). The C′ strand is therefore positioned with the expected polarity for the cleaved strand and with dsDNA adjacent to the active site, in agreement with experimental data. The polarity of the C′ strand (5′-3′) and the presence of two sulphate ions adjacent to the invariant Q81 and R61 suggest how the 3′ flap is led into the active site (Figure 4D and E). A closer view of how the cleaved strand is positioned within the active site cleft requires structures of XPF with longer and branched DNA substrates, a goal we are working towards.
The 3′ end of strand C at site I abuts a shallow hydrophobic groove in the nuclease domain extending between helices α3 and α4 (Figure 3B and C). The groove is lined with several conserved aromatic residues centred on F79 whose spacings approximate to base separation, and this is suggestive of an ssDNA-binding site. Variable length gapped products with a short single-stranded region in the continuous DNA strand are generated by SsoXPF, ERCC1-XPF and Mus81-Eme1 as the cleavage site is anywhere from 2 to 8 nucleotides 5′ of the ds/ssDNA junction (Figure 1B) (Sijbers et al, 1996a, 1996b; de Laat et al, 1998; Bastin-Shanower et al, 2003; Osman et al, 2003; Roberts et al, 2003). The hydrophobic groove may tether such a region extending from the 5′ end of the modelled strand D′ (and thus representing the continuous, uncleaved strand in all the substrates), by allowing stacking or intercalation with DNA bases from a stretch of ssDNA with electrostatic contacts from R104, R108 and R110 to the phosphodiester backbone (Figure 3B). Mutation of F63 of SsoXPF to alanine (equivalent to F79 of ApeXPF) central to this groove partially reduced the catalytic activity (by two- to three-fold) and may reflect the dominant role played by the duplex DNA rather than the single-stranded gap (Table II).
Table 2.
Catalytic rate constants for wild-type SsoXPF and selected mutants, in the presence of PCNA, towards a splayed duplex and a 3′ flap substrate (rates were calculated by linear regression, with standard errors (s.e.) derived from the line fit)
| SsoXPFa | Equivalent residue in ApeXPF | Splayed duplex kcat±s.e. (min−1 × 103) | 3′ flap kcat±s.e. (min−1 × 103) | kcat flap/kcat splayed |
|---|---|---|---|---|
| Wild type | — | 720±49 | 7200±880 | 10 |
| R61A | R77 | 3±0.1 | 840±23 | 280 |
| F63A | F79 | 277±27 | 4730±88 | 17 |
| Q65A | Q81 | 3.3±0.4 | 260±11 | 79 |
| We used SsoXPF for these assays as the enzyme was more robust than ApeXPF as shown in Supplementary Figure S4. |
A cluster of essential noncatalytic residues within site II
Previous experiments indicated that mutation of R678 in human XPF (equivalent to R26 of ApeXPF) resulted in a complete loss of activity (Enzlin and Scharer, 2002). To extend these results, we investigated the importance of R77 and Q81 by making the equivalent mutations R61A and Q65A in SsoXPF. We used SsoXPF for these assays due to its more robust activity and the availability of recombinant Sso heterotrimeric PCNA. Both mutations had large effects on the catalytic activity of the enzyme towards both a splayed duplex and a 3′ flap substrate (Table II). Q81 of ApeXPF separates R26/R77 from the metal-coordinating D52 and E62 of the active site. We interpret these data as showing that R26/R77/Q81 define a functionally important region bordering the catalytic cleft. These residues are not required for protein folding and they are too far from the active site to directly participate in catalysis. One possibility is that these residues play a role in substrate recognition by anchoring the backbone of strand C′, the model's candidate for the 3′ flap. Human ERCC1-XPF and SsoXPF have been shown to generate multiple cleavage sites near the ds/ss junction point, and several cleavage products accumulate over time (de Laat et al, 1998; Roberts et al, 2004). This observation can be explained using our model. Tethering the upstream duplex through protomer B (HhH)2 domain and R26/R77 could readily generate a heterogeneous set of anchored DNA positions by simple rotation of the duplex at site II about its helical axis. This would allow positioning of any of several phosphodiester bonds in the active site cleft but would retain the minor groove binding to the (HhH)2 domain.
Structural conservation within the XPF (HhH)2 domain
Our structure identifies the (HhH)2 domain as having a central role in DNA binding in agreement with recent biochemical work on an isolated (HhH)2 domain from SsoXPF and from UvrC (Singh et al, 2002; Roberts et al, 2004). We report the first example of how (HhH)2 domains dimerise and therefore we analysed (HhH)2 domain sequence conservation within the four distinct subgroups of XPF sequences: (1) eukaryotic XPFs (18 sequences), (2) ERCC1-like (18 sequences), (3) euryarchaeal XPFs (20 sequences) and (4) crenarchaeal XPFs (four sequences). Alignments were optimised to match core residues within the five helical bundle (Figure 1C; Aravind et al, 1999; Shao and Grishin, 2000). The hydrophobic character of the (HhH)2 dimer interface residues is conserved throughout the four groups, indicating that they are all likely to form the same dimer. Surprisingly, the catalytically inactive ERCC1-like group shows the greatest conservation of (HhH)2 domains, particularly within the two hairpin motifs S-V-N-K-T-D (hairpin 1) and G-L-G-P-Q-K (hairpin 2, absolutely conserved residues are in bold). Conservation of positively charged residues immediately after each hairpin is typical of many functional (HhH)2 domains as these residues lie spatially close to the DNA backbone. The euryarchaeal and crenarchaeal XPFs show the closest similarity between the (HhH)2 domain of both groups and each have conserved motifs within and adjacent to both hairpins. The (HhH)2 domains within eukaryotic XPFs are poorly conserved, lack any basic residues in their second hairpins and have almost no invariant surface residues. Hydrophobic core residues are generally conserved among eukaryotic XPFs, suggesting that these (HhH)2 domains retain their structural integrity but their function may have diverged or even been lost.
Discussion
We describe here the structures of both an apo- and a dsDNA-bound form of the crenarchaeal XPF from A. pernix (ApeXPF), which provide some insight into how XPF recognises ds/ssDNA junctions. ApeXPF cleaves 3′ flap substrates similarly to the closely related XPF from Sulfolobus solfataricus and both are dependent on PCNA for catalytic activity. Apo-ApeXPF has an open conformation and is likely to be inactive, while the dsDNA-bound form adopts a more compact conformation that is likely to resemble an activated form of the enzyme. Comparison of these two structures of ApeXPF reveals the following (1) the (HhH)2 domain plays a major role in interacting with DNA in agreement with studies on the isolated (HhH)2 domain from SsoXPF (Roberts et al, 2004); (2) the flexibility of the connecting linker sequence between the (HhH)2 and nuclease domain permits the domain movement on binding DNA thereby coupling the (HhH)2 domain and the nuclease domain to recognise and cleave DNA; (3) the linker's flexibility also enables the two functional domains to dimerise independently and permits formation of an overall asymmetric conformation for ApeXPF. This is likely to be important as the substrate itself is asymmetric and is cleaved by XPF in only one of the strands.
In contrast to eukaryotic ERCC1-XPF heterodimers, ApeXPF is homodimeric and therefore has two potential active sites. There are many catalogued examples of eukaryotic hetero-oligomers whose functional archaeal equivalents are homo-oligomeric allowing archaea to have a smaller, less complex genome. However, this also poses a problem for archaea, as such homo-oligomers often have to form stable, asymmetric conformations to bind substrate properly and for efficient catalysis. Our structural data suggest a mechanism that allows only one (operative) protomer to be catalytically active at a time, thereby mimicking the eukaryotic ERCC1-XPF heterodimers, which have only a single functional nuclease domain. With dimerised (HhH)2 domains that are symmetric and separated from the catalytic domain, a branched DNA substrate could bind the (HhH)2 domain dimer in two possible orientations. We suggest that binding in either orientation triggers the linker region to flex bringing one of the nuclease domains into position to produce a closed (active) conformation. In this way, the 3′ overhanging strand can enter a single active site and precludes the second protomer from having an active catalytic role (Figure 5B). Consistent with this idea, the interdomain linker of the apo-ApeXPF structure apparently blocks the nuclease domain from contacting the (HhH)2 domain while in its extended state (not shown).
Figure 5.
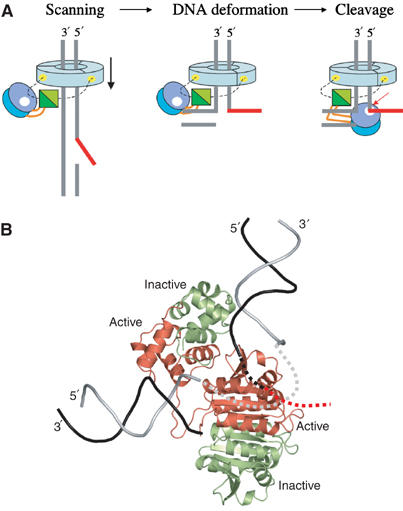
(A) Schematic of a possible mechanism for the recognition of ds/ssDNA junctions by crenarchaeal XPFs bound to heterotrimeric PCNA (pale blue). The active site is highlighted by a white dot, and the ApeXPF binding on PCNA by a yellow spot. The linker sequence connecting the nuclease and (HhH)2 domain is shown in orange and the PCNA-binding motif as a dashed line. (B) Catalytically active and inactive subunits in the complex. The view is the same as in Figure 4B, but protein subunits have been coloured according to the protomer and assigned as active (red) and inactive (light green); in eukaryotes, XPF and ERCC1 are the active and inactive subunits, respectively. The DNA strands have been coloured so that the black strand is the one cleaved by XPF, with its continuation into the active site and beyond indicated by black and red dashed lines. The continuous DNA strand is light grey, with the segment between sites I and II indicated by a dashed line.
The ApeXPF structure extends to Y228 but lacks the final 19 amino acids of which at least nine are homologous to the PCNA-binding site of SsoXPF and of FEN1 (Figure 1C). The intervening glycine-rich sequence after Y228 is likely to be flexible, and examination of the FEN1 C-terminal peptide/PCNA structure (PDB code 1RXM) suggests that the C-terminus for both ApeXPF protomers can be aligned to be roughly 17 Å from the N-terminus of the FEN1 PCNA-binding peptide. PCNA may therefore position ApeXPF via its C-terminal PCNA-binding motif close to the DNA being scanned for discontinuities and, once a ds/ssDNA junction is found, it could assist in domain realignment to activate ApeXPF (Figure 5B). Similar models have been proposed for PCNA-dependent enzymes DNA polymerase PolIV and FEN1/XPG whereby binding to PCNA switches these enzymes between inactive and active functional states (Bunting et al, 2003; Chapados et al, 2004; Sakurai et al, 2005).
The existence and relative position of the two minor-groove tracking (HhH)2 domains within the dimer leads us to propose a model in which the upstream and downstream duplex regions of a 3′ flap substrate are recognised separately by two nonequivalent DNA-binding sites on ApeXPF. Their nonequivalence is due to different contacts made by the catalytic domain at sites I and II. The two-site model rationalises the observed substrate preferences of SsoXPF, a close relative of ApeXPF, for substrates containing a downstream duplex. It also provides a framework for understanding why residues equivalent to R26/R77/Q81 of ApeXPF bordering the active site cleft are critical for catalytic activity. They would contribute to site II and act as an anchor point for the DNA strand that is ultimately to be cleaved.
An important implication of our two-site model is that the orthogonal positions of sites I and II require that a branched substrate has to be bent by around 90° in order to occupy both sites simultaneously. Stated another way, XPFs may recognise ds/ssDNA junctions by their susceptibility to distortion, which in turn helps to assist formation of a productive XPF endonuclease conformation. Even though our ApeXPF–DNA structure has only one occupied DNA-binding position, at site I, the conformational alterations induced in the enzyme are sufficient to prime site II ready for DNA engagement, suggesting that some cooperativity may exist between the two sites. As DNA duplexes cannot readily be distorted in this manner, this compressibility of branched or nicked XPF substrates provides a unique means for detecting ds/ssDNA junctions. Both SsoXPF and Mus81-Eme/Mms4 bind to and cleave efficiently nicked DNA duplexes, suggesting that the 3′ overhanging tail is not part of the recognition process, consistent with our model (Gaillard et al, 2003; Roberts et al, 2004). Both enzymes also cleave nicked Holliday junctions but are very inefficient at cleaving intact Holliday junctions; again this is consistent with a requirement for DNA distortion (Gaillard et al, 2003; Osman et al, 2003; Roberts et al, 2004). Although distortion of DNA substrates is energetically unfavourable, this may be offset by gains in enthalpy from binding both (HhH)2 domains at sites I and II of ApeXPF. An alternative possibility is that stabilisation of a kinked DNA junction is enhanced by other factors bound to ApeXPF such as the heterotrimeric PCNA (Daimon et al, 2002) or, for the euryarchaeal XPFs, by their intrinsic helicase activity (Komori et al, 2002). Severe DNA kinks have been observed by electron microscopy for the preincision UvrB helicase–DNA complex prior to binding the UvrC endonuclease (Shi et al, 1992). A similar 90° kink in DNA substrates has been proposed for FEN1/XPG that is also PCNA-dependent and this may be a common theme among PCNA-interacting repair enzymes (Chapados et al, 2004).
The DNA-bound ApeXPF structure provides the first template for understanding ERCC1-XPF heteromeric complexes, as there are clear similarities between each type of domain; for example, the respective nuclease domain and the (HhH)2 domain dimer interfaces are highly conserved between homo- and heterodimeric XPFs (Nishino et al, 2003) (Figure 1A and C).
However, there are significant differences: ApeXPF and SsoXPF prefer the same substrates as Mus81 rather than XPF despite being closer in domain organisation to the latter (Figure 1). Archaeal XPFs may thus represent a common ancestral form of both XPF and Mus81, since they exhibit properties of each nuclease. If we assume that protomer A of ApeXPF represents the active XPF subunit, then protomer B would represent the noncatalytic subunit of eukaryotic XPFs, namely ERCC1 (Figure 5B). Protomer B's (HhH)2 domain forms a major part of site II, which would imply that the ERCC1 hairpins perform the same role in heterodimeric XPFs (Figure 5B). Site II recognition of the upstream duplex is an invariant feature for all branched ds/ssDNA junctions and is especially important for the splayed duplexes preferred by heterodimeric XPFs. Therefore, if our model is correct, the ERCC1 (HhH)2 domain would play a crucial role in substrate recognition. The (HhH)2 domain of ERCC1 has almost complete conservation among the 18 aligned ERCC1 sequences and has been shown to have an essential function by the severity of the phenotype of a transgenic mouse bearing a truncated form of ERCC1 (Weeda et al, 1997). Removal of just seven C-terminal residues of ERCC1 to residue 291 in these mice eliminates F293, whose equivalent residue Y228 in ApeXPF contributes to the (HhH)2 dimer interface. In contrast, removal of the poorly conserved N-terminal 91 amino acids of mouse ERCC1 has no effect on ERCC1-XPF DNA repair function (Sijbers et al, 1996a, 1996b). This novel role for ERCC1 in site II substrate recognition would extend considerably its previously proposed roles in stabilising XPF and mediating protein interactions with XPA to recruit the endonuclease to the site of the DNA lesion (Li et al, 1995).
In contrast to ERCC1, eukaryotic XPFs have poorly conserved HhH motifs. Our model would suggest that their HhH motifs have a preference for ssDNA at site I (de Laat et al, 1998; Bastin-Shanower et al, 2003) (Figure 4A). Bubble and splayed duplex substrates, thought to be the relevant in vivo substrates of XPF, would indeed present a single-stranded region to site I. Mus81-Eme1 has a yet another variation with two HhH motifs present only in the Mus81 subunit and arranged such that they flank the nuclease domain close to either terminus (Figure 1A). Although the two motifs may be brought together in a similar arrangement to that of ApeXPF, through either intra- or intermolecular association, structural studies on Mus81-Eme1 are required to explain the basis for its substrate preference and recognition.
Eukaryotic XPF complexes are not PCNA dependent but are thought to be recruited to sites of DNA damage through interaction with other core NER factors such as XPA and RPA. One possibility is that opening up the DNA around the lesion by TFIIH sets up appropriate sharply kinked DNA structures, which are recognised and stabilised by XPA-RPA subcomplexes prior to recruitment of the XPF or XPG endonucleases (Tapias et al, 2004). Other regulatory mechanisms controlling the ERCC1-XPF incision have been suggested from recent work on XPB phosphorylation, indicating that there are further complexities to ERCC1-XPF positioning within the NER complex (Coin et al, 2004).
Materials and methods
Expression and nuclease assay
A. pernix XPF (1–254) was amplified from a plasmid derived from the A. pernix genome sequencing project, accession code C72622 and APE1436. A construct lacking the N-terminal 18 and C-terminal 23 residues was used for this study and herein denoted as ApeXPF was prepared and crystallised as described previously (Lally et al, 2004). Full-length ApeXPF was subcloned into the NcoI/BamHI site pET28c using a partial restriction digest to avoid cleavage at an internal NcoI site and purified as for SsoXPF (Roberts et al, 2003). SsoXPF and PCNA heterotrimer were expressed and purified as previously described (Roberts et al, 2003). Site-directed mutants of SsoXPF were constructed using the Quikchange method (Stratagene), verified by sequencing the complete gene and purified as for the wild-type enzyme. Wild-type and mutant SsoXPF activities were assayed in the presence of PCNA under single turnover conditions, as described previously (Roberts et al, 2003), using a splayed duplex or a 3′ flap substrate at 55°C. The splayed duplex and 3′ flap substrates were prepared as described by Roberts et al (2004). The assay for ApeXPF activity was carried out as described by Roberts et al (2004) with the modifications described in the caption to Supplementary Figure S1.
Crystallisation, data collection and structure determination
Crystals of ApeXPF, SeMet-substituted ApeXPF and ApeXPF bound to a 15-mer dsDNA were prepared and data collected as described previously (Lally et al, 2004) (Table I). CCP4 programs were used for all general crystallographic calculations (Collaborative Computational Project, 1994). A structure for the isolated ApeXPF nuclease domain was solved by molecular replacement using the Hef nuclease domain (PDB code IJ23) as a search model with a selenomethionyl-SAD data set collected from a triclinic crystal form (Lally et al, 2004). An anomalous difference Fourier map confirmed the molecular replacement solution by indicating the position of the selenium sites. The partially refined ApeXPF nuclease model (R-free 41.5% and R-factor 35.2%) was used to solve the ApeXPF–DNA complex in the trigonal crystal form by molecular replacement. An electron density map calculated using these molecular replacement phases showed the position of the (HhH)2 domains and duplex DNA. Phases were improved with RESOLVE prime-and-switch density modification (Terwilliger, 2000) aided by the high solvent content (72%). This allowed one complete protomer to be traced and all but the interdomain linker in the second protomer. This structure was refined and built using REFMAC5 (Murshudov et al, 1999) and O (Jones et al, 1991).
Of the numerous heavy atom derivatives screened for the monoclinic apo-ApeXPF crystals, an osmate derivative gave four sites found by SHELX (Sheldrick, 2002), but this had insufficient phasing power to give interpretable electron density maps. The separate domains of the refined ApeXPF model from the trigonal crystal form were therefore used to solve the monoclinic apo-ApeXPF crystals by molecular replacement and this solution was confirmed by crossphasing of the osmate derivative data set; typical density is shown in Supplementary Figure S2B. Each osmium site was found to lie close to His88 of one of the four nuclease domains in the asymmetric unit. The structure was refined with REFMAC5 using TLS refinement (Winn et al, 2001). There is a theoretical ambiguity in connecting the two domains from protomers with disordered linkers. However, inspection of symmetry equivalents for these protomers indicates that our assignment is the only possible one in view of the number of disordered residues in the linker and the distance to other symmetry equivalents of the two domains. Comparisons of the A chains of the DNA-bound and DNA-free forms were made using DynDom (see www.cmp.uea.ac.uk/dyndom/dyndomRef.do). The figures were made using PyMol (DeLano, 2002). Coordinates have been deposited at the PDB with codes 2bgw (DNA complex) and 2bhn (apo).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Acknowledgments
This work was supported by Cancer Research UK. We thank Rick Wood, Dale Wigley and Maureen Biggerstaff for helpful comments and Jen Roberts for unpublished data. MFW was funded by the AICR and is a Royal Society URF.
References
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80: 859–868 [DOI] [PubMed] [Google Scholar]
- Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev 14: 349–359 [PMC free article] [PubMed] [Google Scholar]
- Araujo SJ, Wood RD (1999) Protein complexes in nucleotide excision repair. Mutat Res 435: 23–33 [DOI] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res 27: 1223–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyoshi M, Nishino T, Iwasaki H, Shinagawa H, Morikawa K (2000) Crystal structure of the holliday junction DNA in complex with a single RuvA tetramer. Proc Natl Acad Sci USA 97: 8257–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ (2003) The mechanism of Mus81–Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1–Rad10. Mol Cell Biol 23: 3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR III, Russell P (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107: 537–548 [DOI] [PubMed] [Google Scholar]
- Bunting KA, Roe SM, Pearl LH (2003) Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J 22: 5883–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA (2004) Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116: 39–50 [DOI] [PubMed] [Google Scholar]
- Coin F, Auriol J, Tapias A, Clivio P, Vermeulen W, Egly JM (2004) Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. EMBO J 23: 4835–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project N (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Daimon K, Kawarabayasi Y, Kikuchi H, Sako Y, Ishino Y (2002) Three proliferating cell nuclear antigen-like proteins found in the hyperthermophilic archaeon Aeropyrum pernix: interactions with the two DNA polymerases. J Bacteriol 184: 687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JH (1998) DNA structural elements required for ERCC1-XPF endonuclease activity. J Biol Chem 273: 7835–7842 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL molecular graphics system. http://pymol.sourceforge.net/
- Doherty AJ, Serpell LC, Ponting CP (1996) The helix–hairpin–helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res 24: 2488–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enzlin JH, Scharer OD (2002) The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J 21: 2045–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard PH, Noguchi E, Shanahan P, Russell P (2003) The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell 12: 747–759 [DOI] [PubMed] [Google Scholar]
- Gaillard PH, Wood RD (2001) Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res 29: 872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth NM, Brill SJ (2004) The Mus81 solution to resolution: generating meiotic crossovers without Holliday junctions. Genes Dev 18: 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SWSW, Kjeldgaard M (1991) Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ (2001) Functional overlap between Sgs1–Top3 and the Mms4–Mus81 endonuclease. Genes Dev 15: 2730–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori K, Fujikane R, Shinagawa H, Ishino Y (2002) Novel endonuclease in Archaea cleaving DNA with various branched structure. Genes Genet Syst 77: 227–241 [DOI] [PubMed] [Google Scholar]
- Lally J, Newman M, Murray-Rust J, Fadden A, Kawayabashi Y, McDonald NQ (2004) Crystallisation of a xeroderma pigmentosum group F endonuclease from Aeropyrum pernix. Acta Crystallogr D 60: 1658–1661 [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Li L, Peterson CA, Lu X, Legerski RJ (1995) Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol Cell Biol 15: 1993–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Wood RD (1999) Quality control by DNA repair. Science 286: 1897–1905 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D 55 (Part 1): 247–255 [DOI] [PubMed] [Google Scholar]
- Nishino T, Komori K, Ishino Y, Morikawa K (2003) X-ray and biochemical anatomy of an archaeal XPF/Rad1/Mus81 family nuclease: similarity between its endonuclease domain and restriction enzymes. Structure (Camb) 11: 445–457 [DOI] [PubMed] [Google Scholar]
- Osman F, Dixon J, Doe CL, Whitby MC (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol Cell 12: 761–774 [DOI] [PubMed] [Google Scholar]
- Petit C, Sancar A (1999) Nucleotide excision repair: from E. coli to man. Biochimie 81: 15–25 [DOI] [PubMed] [Google Scholar]
- Rafferty JB, Sedelnikova SE, Hargreaves D, Artymiuk PJ, Baker PJ, Sharples GJ, Mahdi AA, Lloyd RG, Rice DW (1996) Crystal structure of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science 274: 415–421 [DOI] [PubMed] [Google Scholar]
- Roberts JA, Bell SD, White MF (2003) An archaeal XPF repair endonuclease dependent on a heterotrimeric PCNA. Mol Microbiol 48: 361–371 [DOI] [PubMed] [Google Scholar]
- Roberts JA, Rudolf J, White MF (2004) Evolution, specificity and domain organisation of the XPF/Mus81 endonuclease family. J Biol Chem, 10.1074/jbc.M412766200 [Google Scholar]
- Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T (2005) Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J 24: 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A (1996) DNA excision repair. Annu Rev Biochem 65: 43–81 [DOI] [PubMed] [Google Scholar]
- Sgouros J, Gaillard PH, Wood RD (1999) A relationship between DNA-repair/recombination nuclease family and archaeal helicases. Trends Biochem Sci 24: 95–97 [DOI] [PubMed] [Google Scholar]
- Shao X, Grishin NV (2000) Common fold in helix–hairpin–helix proteins. Nucleic Acids Res 28: 2643–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM (2002) Macromolecular phasing with SHELXE. Z Kristallogr 217: 644–650 [Google Scholar]
- Shi Q, Thresher R, Sancar A, Griffith J (1992) Electron microscopic study of (A)BC excinuclease. DNA is sharply bent in the UvrB–DNA complex. J Mol Biol 226: 425–432 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD (1996a) Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86: 811–822 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, van der Spek PJ, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers NG, Bootsma D, Hoeijmakers JH (1996b) Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res 24: 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Folkers GE, Bonvin AM, Boelens R, Wechselberger R, Niztayev A, Kaptein R (2002) Solution structure and DNA-binding properties of the C-terminal domain of UvrC from E. coli. EMBO J 21: 6257–6266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B, Egly JM (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem 279: 19074–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56 (Part 8): 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J 14: 4108–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol 7: 427–439 [DOI] [PubMed] [Google Scholar]
- White MF (2003) Archaeal DNA repair: paradigms and puzzles. Biochem Soc Trans 31 (Part 3): 690–693 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57 (Part 1): 122–133 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5