

Abstract
Human papillomaviruses (HPV) exhibit constitutive activation of ATM and ATR DNA damage response (DDR) pathways, which are required for productive viral replication. Expression of HPV31 E7 alone is sufficient to activate the DDR through an unknown mechanism. Here, we demonstrate that the E7 Rb binding domain is required to increase levels of many DDR proteins, including ATM, Chk2, Chk1, the MRN components MRE11, Rad50, and NBS1, as well as the homologous recombination repair proteins BRCA1 and Rad51. Interestingly, we have found that the increase in these DNA repair proteins does not occur solely at the level of transcription, but that E7 broadly increases the half-life of these DDR factors, a phenotype that is lost in the E7 Rb binding mutant. These data suggest that HPV-31 upregulates DNA repair factors necessary for replication by increasing protein half-life in a manner requiring the E7 Rb binding domain.
Keywords: Virus, Replication, DNA damage response, Human papillomavirus
1. Introduction
High-risk human papillomaviruses (HPV) are the etiological agents of cervical cancer, and are also associated with other genital malignancies, as well as an increasing number of head and neck cancers (zur Hausen, 2009). HPV has adapted its life cycle to be linked closely with epithelial differentiation, with late viral events being restricted to the uppermost layers of the epithelium (Longworth and Laimins, 2004b). HPV is thought to infect dividing, basal cells of the stratified epithelium through a microwound, where upon entry into the nucleus, the virus is maintained as a low copy episome. As an infected cell divides, one daughter cell migrates upward and initiates differentiation. Differentiation triggers the productive phase of the viral life cycle, which requires cellular factors. HPV maintains differentiating cells active in the cell cycle through viral gene expression, allowing for initiation of DNA synthesis and amplification of viral genomes to thousands of copies per cell. Late gene expression, as well as virion assembly and release occur concomitantly with productive replication (Moody and Laimins, 2010).
Previous studies demonstrated that the productive replication of high-risk HPV31 requires activation of an ATM-dependent DNA damage response (DDR) (Moody and Laimins, 2009). ATM is a serine/threonine kinase that is activated primarily in response to double strand DNA breaks (DSBs), resulting in phosphorylation of a variety of substrates important in activating cell cycle checkpoints, as well as DNA repair (Ciccia and Elledge, 2010). If left unrepaired, DSBs have the potential to generate chromosomal translocations, aneuploidy, and increased incidence of malignancy (Agarwal et al., 2006; Helleday et al., 2007). Though the DNA damage response plays a crucial role in the maintenance of genomic stability, many viruses have been shown to exploit repair pathways to facilitate replication (Hollingworth and Grand, 2015; Ryan et al., 2016). We have previously shown that DNA repair factors localize to sites of HPV replication, indicating a direct role for these factors in efficient viral replication (Gillespie et al., 2012). In support of this, we have found that the histone variant H2AX, one of the first targets of ATM, is bound to HPV DNA (Gillespie et al., 2012). In addition, we recently demonstrated that the MRN complex (Mre11, Rad50, Nbs1), which facilitates ATM activation in response to ionizing radiation (Lee and Paull, 2004, 2005; Paull and Lee, 2005), is localized to HPV31 genomes and required for productive replication (Anacker et al., 2014). In addition, we have found that Rad51 and BRCA1, two factors essential for repair of DSBs through homologous recombination (Ciccia and Elledge, 2010), are required for productive viral replication (Chappell et al., 2015). Although numerous studies support a role for the ATM pathway in productive replication of HPV (Anacker et al., 2014; Chappell et al., 2015; Gillespie et al., 2012; Hong et al., 2015b; Hong and Laimins, 2013; Moody and Laimins, 2009), the mechanism by which ATM is activated in HPV-infected cells remains unclear.
The E6 and E7 oncoproteins of high-risk HPV types contribute to carcinogenesis largely through their ability to target the tumor suppressors p53 and Rb for degradation, respectively (Howie et al., 2009; Roman and Munger, 2013). This is especially important upon differentiation, as E6 and E7 ensure virus production by promoting S-phase re-entry of a subset of differentiating cells (Moody and Laimins, 2010). The ability of E6 and E7 to target critical regulators of cell cycle progression results in the bypass of checkpoints normally involved in the elimination of abnormal cells (Munger et al., 2004). While this is necessary for viral replication, checkpoint abrogation can also result in genomic instability in HPV-immortalized cells that eventually leads to cancer (Solinas-Toldo et al., 1997). High-risk E7 expression has been shown to lead to DSB induction and genomic instability in a manner thought to be dependent on Rb inactivation and deregulation of E2F transcription factors (Bester et al., 2011; Duensing and Munger, 2002; Pickering and Kowalik, 2006). Inactivation of Rb outside the context of HPV infection is sufficient to induce DSBs, formation of γH2AX and MRE11 positive foci, and increase levels of ATM, Chk2, and Chk1 (Frame et al., 2006; Pickering and Kowalik, 2006; Shamma et al., 2013, 2009). Previous studies demonstrated that expression of high-risk HPV31 E7 alone is sufficient to induce ATM signaling (Moody and Laimins, 2009), raising the possibility that E7 contributes to the differentiation-dependent phase of the life cycle through modulation of the DDR. More recent studies by Hong et al. demonstrated that the phosphorylation of STAT5 is required for ATM activation in HPV31 positive cells, potentially through E7-mediated Rb inactivation (Hong et al., 2015b; Hong and Laimins, 2013). Studies by this same group have also provided a link between STAT5, activation of the ATR DNA damage kinase and productive replication of HPV31 (Hong et al., 2015a). Although Rb inactivation by E7 is important for providing an environment conducive to productive viral replication, whether a direct link exists between Rb binding and ATM/ATR activation has not been demonstrated. However, recent studies linked the development of female reproductive tract cancers, as well as head and neck cancers in HPV16 transgenic mice to E7-mediated inactivation of pocket proteins (Rb, p107, p130) and resultant DNA damage (Park et al., 2014, 2013).
In this study, we investigated whether the Rb binding domain of E7 is required for activation of ATM, as well as ATR signaling pathways in HPV31 positive cells. We have found that deletion of the Rb binding domain in the context of the HPV31 genome results in decreased levels of DNA repair factors compared to cells maintaining wild-type HPV31 genomes. A similar phenotype was observed in cells expressing wild-type HPV31 E7 alone compared to cells expressing an E7 Rb binding mutant. Interestingly, we found that E7 maintains high levels of DNA repair factors required for productive replication through increased protein stability, rather than exclusively through increased gene expression, with exceptions limited to the ATR target Chk1, and the homologous recombination repair factors BRCA1 and Rad51. Together, these data suggests that the E7 Rb binding domain is important for increased ATM/ATR activation by virtue of increasing total protein levels through increased protein stability, and that increases in DNA repair factor levels in HPV positive cells depends largely on the ability of E7 to bind and target Rb for degradation.
2. Materials and methods
2.1. Cell culture
Human foreskin keratinocytes (HFKs) were isolated from neonatal foreskins as previously described and were cultured in E medium supplemented with 5 ng/ml mouse epidermal growth factor (EGF; BD Biosciences) (Wilson and Laimins, 2005). HFK-31, HFK-31 ΔLHCYE, pLXSN-31 E7, pLXSN-31 E7 ΔLHCYE cells were also cultured in E medium supplemented with 5 ng/ml mouse epidermal growth factor. All lines were cultured in the presence of mitomycin C-treated J2 3T3 fibroblast feeder cells, as previously described (Wilson and Laimins, 2005). J2 feeder cells were removed from HPV-positive cells with 1 mM EDTA in phospho-buffered saline (PBS) as necessary.
2.2. Plasmids
The pBR322-HPV31 plasmids containing the wild-type HPV31, HPV31 E7 ΔLHCYE mutant genomes have been previously described (Hubert and Laimins, 2002; Longworth and Laimins, 2004a). Briefly, the E7 ΔLHCYE plasmid contains an in-frame deletion of the Rb binding site. The pLXSN retroviral vectors encoding wild-type HPV31 E7 and the E7-Rb binding mutant (ΔLHCYE) have been described previously (Longworth and Laimins, 2004a).
2.3. Generation of HFK-31 lines
HFKs maintaining wild type HPV31, as well as mutant HPV 31 genomes (HFK-31 E7 ΔLHCYE) were generated as previously described (Wilson and Laimins, 2005). Briefly, HPV genomes were digested with HindIII to release them from the pBR322 plasmid backbone. T4 DNA ligase (Life Technologies) was then used to religate the excised HPV genomes. HFKs were co-transfected with 2.5 μg of ligated HPV genomes and 2.5 μg of pSV2-neo using PolyJet transfection reagent as per the manufacturer’s instructions (Signagen Laboratories), followed by selection in G418 (Sigma). Surviving populations were expanded for further analysis.
2.4. Keratinocyte differentiation
Differentiation of keratinocytes was performed by suspending cells in 1.5% methylcellulose, as previously described (Wilson and Laimins, 2005). Cells were harvested as an undifferentiated sample (T0), as well as 24 and 48hrs after suspension in methylcellulose. At each time point, DNA, RNA and protein were harvested.
2.5. Western blot analysis
Whole cell lysates were harvested by lysing cell pellets in RIPA buffer supplemented with Complete Mini protease inhibitor (Thermo Scientific) and PhoSTOP phosphatase inhibitor tablets (Roche). Total protein concentrations were determined by Bio-Rad protein assay (Bio-Rad). Equal amounts of protein were separated by SDS-page and transferred to polyvinylidene difluoride (PVDF) membranes. (Immobilon-P; Millipore). The following primary antibodies were used: phospho-ATM Ser1981, Chk1, NBS1 (Abcam); ATM (Bethyl laboratories); phospho-Chk1 Ser345, phospho-Chk2 Ser68, Chk2, E2F1 (Cell Signaling Technology); E2F2, E2F3, GAPDH, Rad51 (Santa Cruz); MRE11, Rad50, and BRCA1 (GeneTex). Secondary antibodies used were horseradish peroxidase (HRP)-conjugated anti-rabbit (Cell Signaling Technology) and HRP-conjugated anti-mouse (GE Life Sciences). Western blots were developed using Enhanced Chemiluminescence Prime blotting substrate (GE Life Sciences). Images were captured with the Biorad ChemidocMP imaging system, and analyzed with Biorad Imagelab 5.0 software.
2.6. Southern Blot Analysis
DNA isolation and Southern blotting were performed as previously described (Fehrmann et al., 2003). Briefly, cells were harvested in DNA lysis buffer (400 mM NaCl, 10 mM Tis pH 7.5 and 10 mM EDTA), then lysed by the addition of 30uL 20% SDS. Samples were subsequently treated with 15 ul of 10 mg/ml proteinase K overnight at 37 °C. DNA was extracted using phenol chloroform, followed by ethanol precipitation in the presence of sodium acetate. 5 μg of DNA was digested with either BamHI (New England Biolabs) (does not cut the genome), or HindIII (New England Biolabs) (cuts the genome once). DNAs were resolved on a 0.8% agarose gel for 15 h at 40 V and were then transferred to a positively charged nylon membrane (Immobilon-Ny+; EMD Millipore). The DNA was fixed to the membrane via UV irradiation and then hybridized to a radioactive DNA probe consisting of 32P-labeled linearized HPV31 genome.
2.7. Real-time PCR
RNA was extracted from primary HFKs, HFK-31, HFK-31 ΔLHCYE, pLXSN, pLXSN-31 E7, and pLXSN-31 E7 ΔLHCYE cells using RNA STAT 60 (Tel-test). DNA was removed from samples via treatment with RQ1 DNAse (Promega) via the manufacturer’s protocol. cDNA was made using the iScript reverse transcription kit (Bio-Rad). Quantitative RT-PCR was performed in triplicate on 50 ng of cDNA using 375 nM primers and iTaq Universal SYBR Green Supermix (Bio-Rad) in a total reaction volume of 10 μl. Reactions were performed using an ABI QuantStudio 6 Flex thermal cycler and analyzed with version 1.0 of the QuantStudio 6 and 7 Flex software. The thermal profile used for PCR is as follows: 10 min denaturation at 95 °C followed by 40 cycles of 95 °C for 15 s, then 60 s at 60 °C (ATM, Chk2, Chk1, MRE11, Rad50, NBS1, E2F2, E7) or 63 °C (BRCA1, Rad51, E2F1), followed by 72 °C for 30 s. A melt curve was run to ensure primer annealing. Relative transcript levels were determined using the threshold cycle method (ΔΔCT) with GAPDH serving as an endogenous control gene. Values were normalized relative to transcript levels of primary HFKs or pLXSN control cells. The primer sequences are as follows: ATM Forward, 5′-TGTTCCAGGACACGAAGGGAGA-3′; ATM Reverse, 5′-CAGGGTTCTCAGCACTATGGGA-3′; BRCA1 Forward, 5′-CTGAAGACTGCTCAGGGCTATC-3′; BRCA1 Reverse, 5′-AGGGTAGCTGTTAGAAGGCTGG-3′; Chk1 Forward 5′-TGAGAATCCATCAGCAAGAATTACC-3′; Chk1 Reverse, 5′-ATCCACTGGGAGACTCTGACACA-3′; Chk2 Forward, 5′-GCAGCAGTGCCTGTTCACA-3′; Chk2 Reverse, 5′-TGGATATGCCCTGGGACTGT-3′; E2F1 Reverse 5′-ATCTGTGGTGAGGGATGAGG-3′ E2F2 Forward, 5′-CTCTCTGAGCTTCAAGCACCTG-3′; E2F2 Reverse, 5′-CTTGACGGCAATCACTGTCTGC-3′; HPV31 E7 Forward, 5′-ACACCTACGTTGCAAGACTATG-3′; GAPDH Forward, 5′-CTGTTGCTGTAGCCAAATTCGT-3′; GAPDH Reverse, 5′-ACCCACTCCTCCACCTTTGAC-3′; HPV31 E7 Reverse, 5′-CGAATATCTACTTGTGTGCTCTGT-3′, Mre11 Forward, 5′-GCCTTCCCGAAATGTCACTA-3′; Mre11 Reverse, 5′-TTCAAAATCAACCCCTTTCG-3′; NBS1 Forward, 5′-TCTGTCAGGACGGCAGGAAAGA-3′; NBS1 Reverse, 5′-CACCCCAAAGACAACTGCGGA-3′; Rad50 Forward, 5′-GGAAGAGCAGTTGTCCAGTTACG-3′; Rad50 Reverse, 5′-GAGTAAACTGCTGTGGCTCCAG-3′; Rad51 Forward, 5′-TCTCTGGCAGTGATGTCCTGGA-3′, Rad51 Reverse, 5′-TAAAGGGCGGTGGCACTGTCTA-3′.
2.8. Measurement of protein half-life
Primary HFKs, HFK-31, HFK-31 ΔLHCYE, pLXSN, pLXSN-31E7 and pLXSN-31E7 ΔLHCYE cells were grown in 10 cm dishes until ~80% confluency. Whole cell lysates were then harvested from one dish for the 0 h time point and at the indicated time points after treatment with 50 μg/ml cycloheximide. J2 fibroblasts were removed prior to harvest using Versene (1 mM EDTA in PBS). Western blot analysis was performed using 50 μg of total protein as described above. Westerns were digitally imaged using the Bio-Rad Chemidoc MP system, and densitometry was performed with the Biorad ImageLab 5.0 software.
3. Results
3.1. The Rb binding domain of E7 is required for ATM and ATR activation in HPV31 positive cells
To examine the importance of the E7 Rb binding domain to DDR activation and maintenance of high levels of DDR factors in HPV31 positive cells, we generated human foreskin keratinocyte (HFKs) lines that maintain either wild-type HPV31 genomes (HFK-31), or genomes containing a mutation in the E7 LXCXE Rb binding domain (HFK-31 ΔLHCYE). Previous studies demonstrated that this mutation does not alter the stability of E7, and that HFK-31 ΔLHCYE mutant genomes are maintained extrachromosomally, though copy number decreases over time compared to wild-type genomes (Longworth and Laimins, 2004a). In addition, these studies showed that the E7 Rb binding site is required for efficient amplification of viral genomes upon differentiation (Longworth and Laimins, 2004a), and we have found similar results in this study (Fig. 1A). To determine the effect of the ΔLHCYE mutation on DDR activation throughout the viral life cycle, cells were harvested both prior to and after the induction of epithelial differentiation in methylcellulose, a method commonly used to activate the productive phase of the viral life cycle. We first examined the importance of the E7 Rb binding domain on the activation of the ATM and ATR pathways. As shown in Fig. 1B, both the phosphorylated and total levels of ATM, the ATM target Chk2, and the ATR target Chk1 were increased in HFK-31 cells compared to uninfected HFKs, indicating activation of ATM/ATR signaling pathways, as published previously (Hong et al., 2015a; Moody and Laimins, 2009). While the levels of total ATM and Chk2 decreased in HFK-31 positive cells upon differentiation, the phosphorylated levels of ATM and Chk2 remained elevated, suggesting that DDR activation is increased during the productive phase of the viral life cycle. In contrast, both phosphorylated and total levels of Chk1 decreased upon differentiation, despite being required for productive replication (Hong et al., 2015a). Interestingly, we found that the levels of phosphorylated and total ATM and Chk2, as well as Chk1 dramatically decreased in the HFK-31 ΔLHCYE cells at all time points, exhibiting levels similar to that found in the uninfected HFKs (Fig. 1B). These data suggest that in the setting of an HPV infection, the Rb binding domain of E7 is necessary for ATM and ATR activation.
Fig. 1.
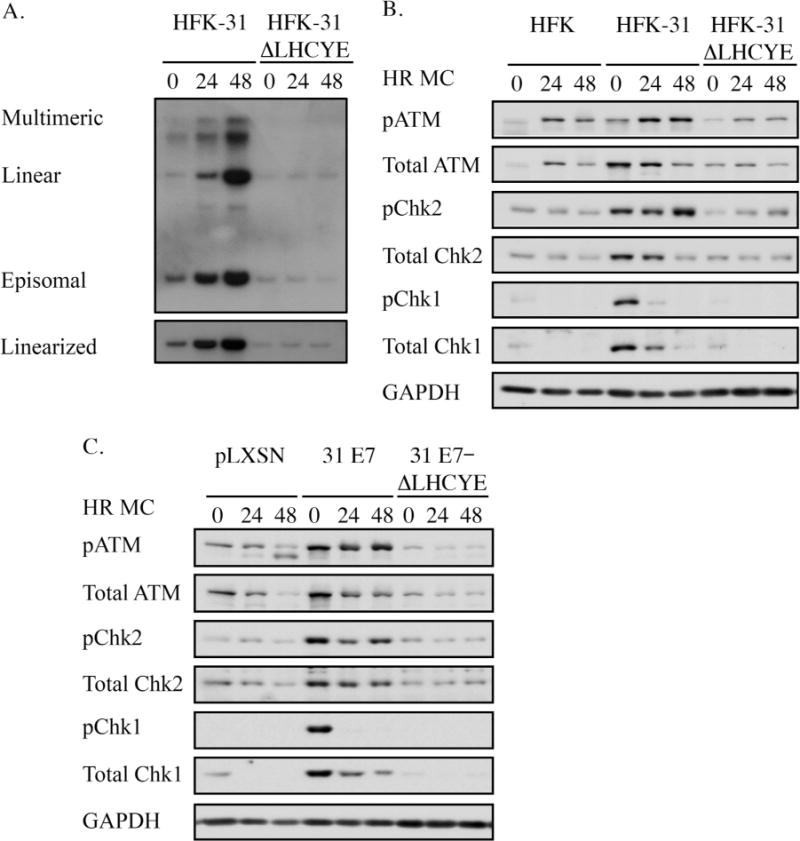
The Rb binding domain of E7 is required for ATM and ATR activation in HPV31 positive and E7-expressing keratinocytes. (A) Southern blot analysis performed on DNA harvested at the indicated time points from human foreskin keratinocytes maintaining wild-type HPV31 genomes (HFK-31), as well as HFK-31 cells containing a mutation in the E7 Rb binding site (HFK-31 ΔLHCYE). DNA was digested with either BamH1, which does not cut the viral genome, or HindIII, which cuts the genome once. The HPV31 genome was used as a probe. (B) Western blot analysis was performed on lysates harvested from HFKs, HFK-31 cells, or HFK-31 ΔLHCYE cells. Lysates were harvested from undifferentiated cells (T0), as well as after 24 and 48hr differentiation in methylcellulose (MC). Primary antibodies used were phosphorylated ATM on Ser1981 (pATM), total ATM, phosphorylated Chk2 on Ser68 (pChk2), total Chk2, phosphorylated Chk1 on Ser345 (pChk1), and total Chk1. GAPDH served as a loading control. Shown are blots representative of five independent experiments across four HFK backgrounds. (C) Whole cell lysates were harvested from undifferentiated (T0) as well as differentiated (24, 48hr MC) HFKs stably transduced with either a retroviral control construct (pLXSN) or retroviral constructs expressing wild-type HPV31 E7 (pLXSN-31 E7), or HPV31 E7 containing a mutation in the Rb binding domain (pLXSN-31 E7 ΔLHCYE). Immunoblotting was performed as described in panel B. Western blots shown are representative of three independent experiments across two HFK backgrounds.
Previous studies demonstrated that HPV31 E7 expression is alone sufficient to activate the ATM and ATR pathways in keratinocytes (Hong et al., 2015a; Moody and Laimins, 2009). Additionally, studies by other groups have shown that E7 expression alone induces DNA damage and activates the DDR (Duensing and Munger, 2002; Pickering and Kowalik, 2006). To determine if the E7 Rb binding domain is required for DDR activation outside the context of viral infection, we stably transduced HFKs with either a retroviral control vector (pLXSN), a vector encoding wild type HPV31 E7, or HPV31 E7 containing the ΔLHCYE mutation. We then analyzed ATM and ATR activation from both undifferentiated and methylcellulose-differentiated cells by Western blot analysis. As shown in Fig. 1C, HFKs expressing wild-type HPV31 E7 exhibited increased levels of both phosphorylated and total ATM, Chk2, and Chk1 relative to control cells, confirming that E7 expression alone is sufficient to activate ATM and ATR signaling pathways. However, this phenotype was lost in cells expressing the E7 ΔLHCYE mutant (Fig. 1C). Overall, these results indicate that maintenance of high levels of ATM, Chk2 and Chk1 in HPV31 positive cells requires the Rb binding domain of E7, which likely contributes to the ability of HPV to activate ATM and ATR signaling pathways.
3.2. Maintenance of high levels of DNA repair factors in HPV31 positive cells requires the Rb binding domain of E7
The MRN protein complex (consisting of MRE11, Rad50, and NBS1) is a DNA damage sensor that activates the DDR by recognizing DSBs and activating ATM (Lee and Paull, 2004, 2005; Paull and Lee, 2005). Previously, we showed that the levels of MRN components are increased in HPV31 positive cells, and that the maintenance of the MRN complex is necessary for productive viral replication (Anacker et al., 2014). We next wanted to determine if the E7 Rb binding domain is also required for the maintenance of high levels of MRN complex members in HPV31 positive cells. As shown in Fig. 2A, levels of all three MRN components were elevated in undifferentiated HFK-31 cells compared to primary HFKs, and this phenotype was maintained upon differentiation in methylcellulose. However, this phenotype was lost in cells containing E7 ΔLHCYE mutant genomes, with levels of MRN components resembling those found in HFKs (Fig. 2A). Similar results were observed in E7-expressing lines, with cells expressing the E7 ΔLHCYE mutant exhibiting levels of MRN components similar to that of HFKs (Fig. 2B). Together, these data suggest that the Rb binding site of E7 is necessary for maintenance of high levels of MRN components in HPV31 positive cells.
Fig. 2.
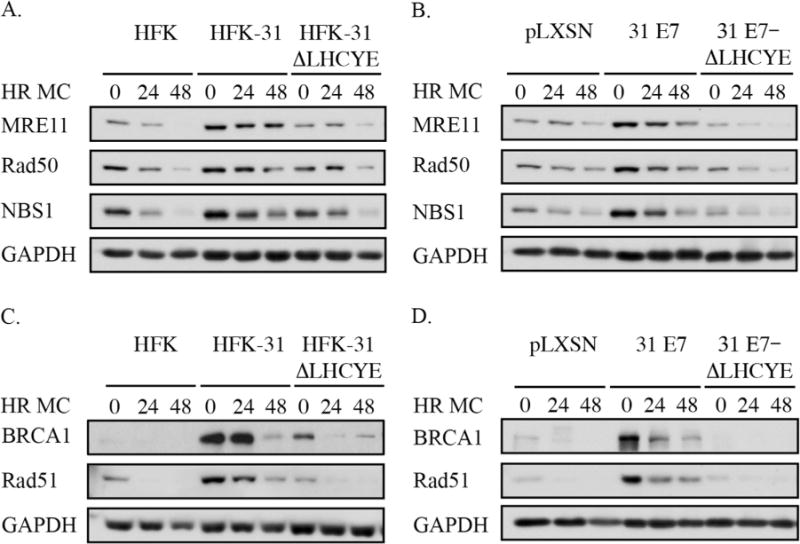
Levels of the MRN complex, as well as the homologous recombination proteins BRCA1 and Rad51, are maintained in HPV31 positive cells in a manner dependent on the E7 Rb binding domain. (A) Whole cell lysates were harvested from HFKs, HFKs stably transfected with wild-type HPV31 genomes (HFK-31), or HPV31 genomes with a mutation in the E7 Rb binding domain (HFK-31 ΔLHCYE) at T0 (undifferentiated), as well as after 24 and 48hr differentiation in methylcellulose (MC). Western blotting was performed using antibodies for MRE11, Rad50, and NBS1, with GAPDH serving as a loading control. Western blots shown are representative of five independent experiments across four HFK backgrounds. (B) Whole cell lysates were harvested from HFKs stably transduced with either the empty retroviral vector (pLXSN), wild type HPV31 E7 (pLXSN-31 E7), or an E7 containing a mutation in the Rb binding domain (pLXSN-31 E7-ΔLHCYE) at T0 (undifferentiated) as well as after 24 and 48hr differentiation in MC. Western blotting was performed with antibodies targeting MRE11, Rad50, and NBS1. The blots shown are representative of three independent experiments across two HFK backgrounds. (C) Whole cell lysates were harvested from HFKs, HFK-31, and HFK-31 ΔLHCYE cells, both at T0 and after 24 and 48hr differentiation in MC. Western blot analysis was performed for BRCA1 and Rad51, with GAPDH serving as a loading control. The data shown is a representative example of five independent experiments across four HFK backgrounds. (D) Whole cell lysates were harvested from pLXSN, pLXSN-31 E7, and pLXSN-31 E7-ΔLHYCE cells both prior to and 24 and 48 h post differentiation in MC, and Western blot analysis was performed using BRCA1 and Rad51 antibodies, with GAPDH serving as a loading control. Shown is a representative Western blot from three independent experiments across two HFK backgrounds.
BRCA1 and Rad51 are two proteins essential to homologous recombination, a DSB repair mechanism that also requires ATM activity (Ciccia and Elledge, 2010). Previously, we demonstrated that levels of BRCA1 and Rad51 are increased in HPV31 positive cells and are required for productive viral replication (Chappell et al., 2015). As shown in Fig. 2C, HFK-31 cells exhibited increased levels of BRCA1 and Rad51 compared to HFKs, as previously reported (Chappell et al., 2015; Gillespie et al., 2012). However, the E7 ΔLHCYE mutation resulted in decreased BRCA1 and Rad51 levels, similar to those found in HFKs, indicating the E7 Rb binding domain is required for increased levels of BRCA1 and Rad51 in HPV31 positive cells. These findings were recapitulated in cells expressing E7 alone, with the E7 Rb binding domain being required for maintenance of high levels of BRCA1 and Rad51 (Fig. 2D). Overall, these results indicate that E7 expression is necessary for HPV31 to increase the levels of DDR factors and that this increase requires the E7 Rb binding domain.
3.3. Elevated levels of DNA repair factors in HPV31 positive cells cannot solely be explained by increased transcription
The inactivation of Rb results in the constitutive activation of a subset of E2F transcription factors termed the activator E2Fs (E2F1-3) that drive transcription of a number of cellular genes not only involved in facilitating S-phase entry, but DNA repair as well, including ATM, Chk1, BRCA1, and Rad51 (Bracken et al., 2004). Previous studies by the Laimins lab showed that E2F2 protein levels are increased in HPV31 positive cells upon differentiation in a manner dependent on the E7 Rb binding domain (Longworth et al., 2005). E7 may therefore contribute to activation of the DDR, at least in part, by increasing transcription of key DNA repair genes in an E2F-dependent manner. To investigate this possibility, we first examined if the protein levels of the other activator E2Fs, E2F1 and E2F3, are also affected by the E7 ΔLHCYE mutation in HPV31 positive cells. As shown in Fig. 3A, the levels of E2F1 and E2F2, but not E2F3, were increased in HFK-31 cells compared to uninfected HFKs, and this phenotype was lost in cells containing E7 ΔLHCYE mutant genomes. We next confirmed these results in cells stably expressing E7 alone. As shown in Fig. 3B, while HFKs expressing wild-type E7 exhibited elevated E2F1 and E2F2 protein levels, this increase was not observed in the E7 ΔLHCYE mutant, similar to results found with the HFK-31 ΔLHCYE mutant genome lines (Fig. 3A). Additionally, E2F3 was regulated in a manner similar to E2F1 and E2F2 in E7-expressing cells (Fig. 3B), which was not observed within the context of the HPV genome (Fig. 3A). Given the relationship between the E7 Rb binding domain and maintenance of E2F proteins, we examined the possibility that E7 increases the levels of DNA repair factors at the level of gene expression.
Fig. 3.
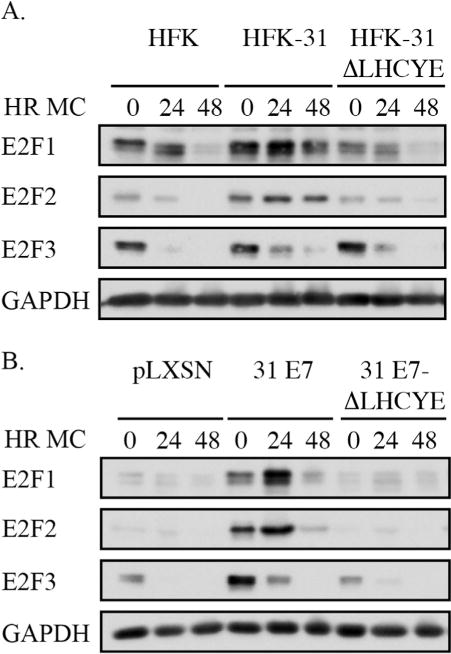
The Rb binding domain of E7 is necessary for increased levels of E2F1 and E2F2 in HPV positive and E7-expressing keratinocytes. (A) Whole cell lysates were harvested from HFKs, HFKs containing wild-type HPV31 genomes (HFK-31), or HPV31 genomes containing an E7 Rb deletion mutant (HFK-31 ΔLHCYE) that were undifferentiated (T0) or differentiated for 24 or 48 h in methylcellulose (MC). Western blot analysis was performed using antibodies to E2F1, E2F2, and E2F3. GAPDH served as a loading control. The Western blots shown are representative of five independent experiments across four HFK backgrounds. (B) Whole cell lysates were harvested from HFKs stably transduced with a retroviral control vector (pLXSN), or vector expressing wild-type HPV31 E7 (pLXSN-31 E7), or an E7 containing a mutation in the Rb (pLXSN-31 E7-ΔLHCYE) that were undifferentiated (T0) or differentiated in methylcellulose for 24 and 48 h. Western blot analysis was performed as described in Panel A. Shown are Western blots representative of three independent experiments across two HFK backgrounds.
We first examined the effect of mutating the E7 Rb binding domain on the transcript levels of ATM, Chk2, and Chk1 in HPV31 genome-containing HFKs (Fig. 4). As shown in Fig. 4A, HFK-31 cells exhibited no significant changes in transcript levels for ATM, Chk2, or Chk1 in comparison to uninfected HFKs, despite exhibiting substantially increased protein levels (Fig. 1A). While loss of the E7 Rb binding domain did slightly affect the levels of ATM, Chk2, and Chk1 transcripts, these changes were statistically insignificant (p > 0.05) (Fig. 4A). In E7-expressing HFKs, no significant changes were observed for ATM or Chk2 transcripts compared to control cells, however, mRNA levels of ATM and Chk2 were reduced in the E7 ΔLHCYE mutant (Fig. 4B). For Chk1, E7-expressing cells exhibited a minor, though significant increase in transcript levels compared to HFKs. Similar to ATM and Chk2, Chk1 transcript levels were decreased in the E7 ΔLHCYE mutant cells (Fig. 4B). These results indicate that while elevated transcription may contribute to the increased protein levels of Chk1, the moderate changes in ATM and Chk2 transcript levels in HFK-31 cells compared to HFKs cannot solely account for the high protein levels observed in HPV31 positive cells. In addition, the decrease in transcript levels observed in the ΔLHCYE mutant cannot fully account for the differences in protein levels observed in these cells and suggest that E7 may also regulate levels of these DDR factors in a post-transcriptional manner.
Fig. 4.
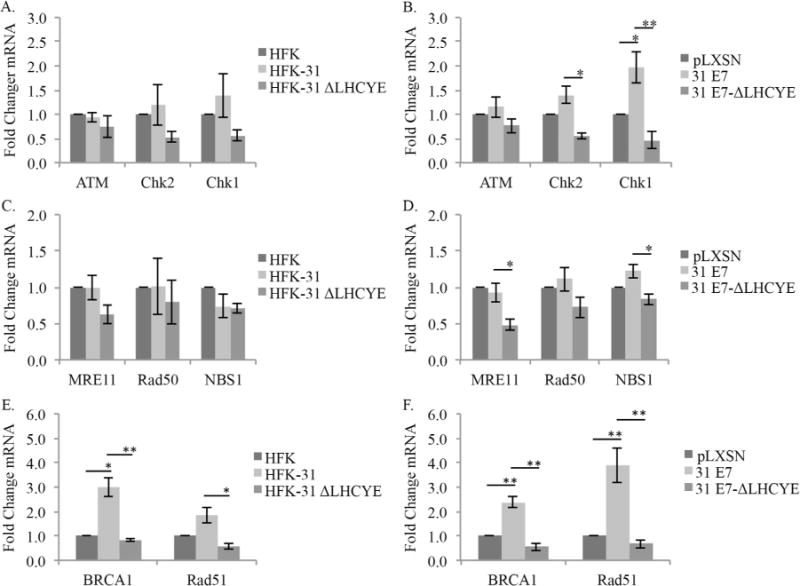
The E7 Rb binding domain is required for increased transcript levels of Chk1, BRCA1 and Rad51. RNA was extracted from undifferentiated (A, C, E) HFKs, HFKs maintaining wild type HPV-31 genomes (HFK-31), and HPV-31 genomes containing a deletion in the E7 Rb binding site (HFK-31 ΔLHCYE); (B, D, F) or undifferentiated HFKs retrovirally transduced with either a control vector (pLXSN), wild type HPV31 E7 (pLXSN-31 E7), or the E7-Rb binding mutant (pLXSN-31 E7-ΔLHCYE). Reverse transcription quantitative PCR (RQ-PCR) was performed using gene-specific primers for ATM, Chk2, and Chk1 (A, B), MRE11, Rad50, and NBS1 (C, D), or BRCA1 and Rad51 (E, F). Shown is fold change in transcripts calculated using the 2−ΔΔCT method. Values shown are transcript levels relative to either HFK (A, C, E) or pLXSN (B, D, F), which are set to 1. Error is indicated as +/− the standard error of the mean. Each panel represents the results from four independent experiments derived from two different HFK donors. For all panels, the student’s t-test was used to test significance. * P≤0.05, and ** P ≤0.01.
We next determined if loss of the Rb binding domain affects the mRNA levels of MRN complex components in HPV31 positive cells (Fig. 4C), as well as E7-expressing cells (Fig. 4D) Previously, we reported that in HPV31 E7-expressing cells, MRE11 gene expression was slightly, though significantly elevated compared to control cells, with no change in Rad50 and NBS1 transcripts (Anacker et al., 2014). To confirm these results in HPV31 positive cells and determine if loss of the Rb binding domain affects expression of MRE11, Rad50, and NBS1, we first examined mRNA levels in HFKs and HFK-31 cells, as well as in HFK-31 ΔLHCYE cells. As shown in Fig. 4C, we found no significant changes in transcript levels for MRE11, Rad50, or NBS1 between HFK and HFK-31 cells. Similarly, while small reductions in mRNA levels for MRE11, Rad50, and NBS1 were observed in HFK-31 ΔLHCYE cells when compared to HFK-31 cells, these changes were not significant. In HFKs expressing HPV31 E7 alone, we observed no significant changes between wild-type E7-expressing cells and the vector control for any of the MRN components (Fig. 4D). The disparity in these data for Mre11 and our previously published results likely owes to the very small changes observed in both cases (< 2 fold increase in MRE11). Similar to ATM, Chk2, and Chk1, we did observe a small but significant decrease in transcript levels for both MRE11 and NBS1, but not Rad50, in cells expressing the E7 ΔLHCYE mutant. Taken together, these data suggest that transcription may play a minor role in the increase in the MRN components observed in HPV31 positive cells, however, increased gene expression alone cannot fully explain the changes observed in MRN protein levels.
Previously, we demonstrated that HPV31 positive cells exhibit increased transcript levels of BRCA1 and Rad51, with E7 expression alone being sufficient for this increase (Chappell et al., 2015). To determine if the increase in gene expression requires the E7 Rb binding domain, we examined BRCA1 and Rad51 mRNA levels in uninfected HFKs, as well as HFKs containing wild-type HPV31 genomes, or E7 ΔLHCYE mutant genomes (Fig. 4E). As shown in Fig. 4E, transcript levels for BRCA1 and Rad51 were increased in HFK-31 cells compared to uninfected HFKs, and this increase was lost in cells expressing the E7 ΔLHYCE mutant. Similar results were observed for E7-expressing cells (Fig. 4F), with BRCA1 and Rad51 transcripts being present at a significantly increased level compared to control cells, as published previously (Chappell et al., 2015). This phenotype was again lost in the E7 ΔLHCYE mutant (Fig. 4F), indicating that in addition to maintenance of BRCA1 and Rad51 protein levels (Fig. 2C–D), the E7 Rb binding domain is also required for increased gene expression of BRCA1 and Rad51. Overall, these data suggest that E7 regulates the levels of a subset of DDR proteins in a transcription-independent manner (ATM, Chk2, MRN), while conferring a mechanism of transcriptional dependence, to a certain extent, on other DDR factors (Chk1, BRCA1, Rad51).
3.4. The E7 Rb binding domain is required for increased protein stability of a subset of DNA repair factors in HPV31 positive cells
Previous studies have demonstrated that the E7 Rb binding domain is required for maintenance of high levels of E2F2 in a post-transcriptional manner (Longworth et al., 2005). In addition, our lab has previously shown that BRCA1 and Rad51 exhibit an increased protein half-life in HPV31 positive CIN612 cells (Chappell et al., 2015). These studies, taken together with the observation that the transcript levels of ATM, Chk2, Chk1, and the MRN components were not substantially altered in HFK-31 ΔLHCYE cells raises the possibility that E7 may contribute to increased DDR protein levels in infected cells through influencing protein stability.
To determine the impact of protein stability on the maintenance of DDR factors in HPV31 positive cells, we first examined the half-life of ATM, Chk2, and Chk1 in HKFs compared to HFK-31 cells using cycloheximide to inhibit protein synthesis. As shown in Fig. 5A and B and summarized in Table 1, the protein half-lives for ATM, Chk2, and Chk1 in HFKs were 5.8+/−1.3, 7.0+/−0.02, and 4.5+/−0.3 h, respectively. In contrast, in HFK-31 cells the half-lives of ATM, and Chk2 increased to greater than 12 h (the longest time point measured). For Chk1, the half-life was also increased in HFK-31 cells (5.8+/−0.5 h vs. 4.5+/−0.3 in HFKs), though not to the same extent as ATM and Chk2. To determine if the E7 Rb binding domain was required for the increase in protein stability observed, we examined the half-lives of ATM, Chk2, and Chk1 in cells containing HPV31 genomes with the ΔLHCYE mutation. Interestingly, in HFK-31 ΔLHCYE cells, the half-lives of ATM, Chk2, and Chk1 were reduced to 4.5+/−0.8, 9.6+/−2.0, and 4.2+/−0.4 h, respectively, similar to the half-lives observed in HFKs for each protein (Fig. 5A–B, Table 1). These results suggest that high levels of ATM, Chk2, and Chk1 are maintained in HPV31 positive cells, at least in part, through an increase in protein stability.
Fig. 5.
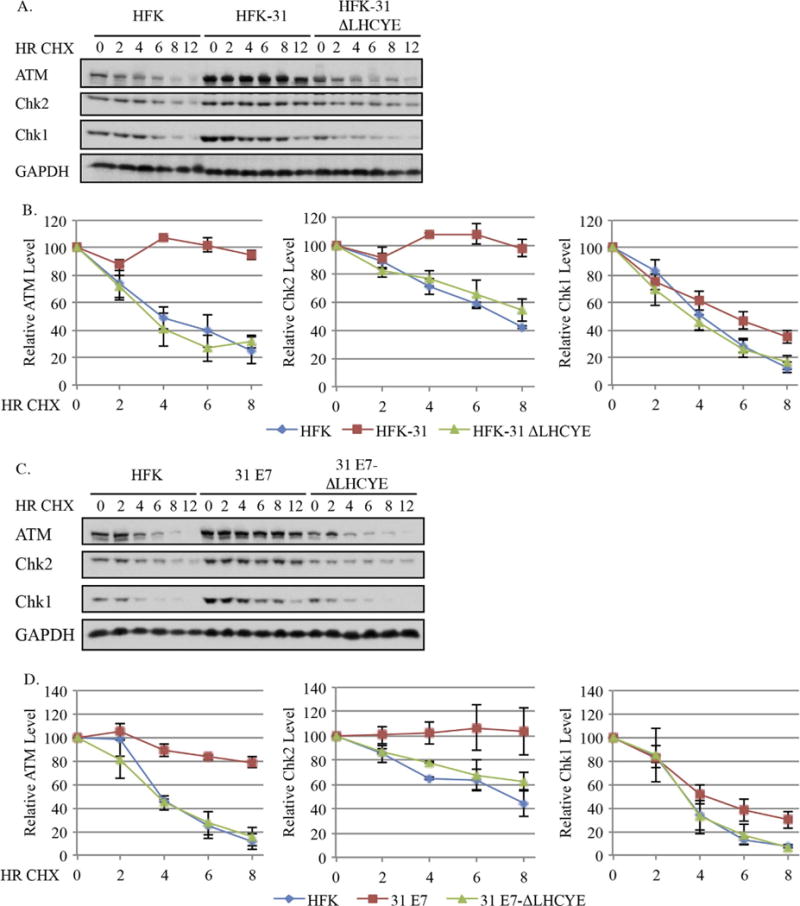
The half-lives of ATM, Chk2 and Chk1 are increased in HPV31 positives cells in an E7-dependent manner. (A, B) Uninfected HFKs, HFKs maintaining wild type HPV31 genomes (HFK-31), HPV31 genomes containing a mutation in the E7 Rb binding site (HFK-31 ΔLHYCE), as well as (C, D) HFKs stably transduced with a retroviral vector expressing wild type E7 (pLXSN-31 E7), or E7 with a mutation in the Rb binding domain (pLXSN-31 E7 ΔLHCYE) were treated with 50 μg/ml cycloheximide over 12 h time course, with whole cell lysates being harvested at the indicated time points. Western blot analysis was performed using antibodies targeting ATM, Chk2, and Chk1, as well as GAPDH, which served as a loading control. (A, B) Shown is representative Western blots from three independent experiments from two different HFK donors. (C, D) Data shown are representative Western blots from three independent experiments from one HFK donor. (B) Graphed are the average protein levels of ATM, Chk2, and Chk1 in HFK, HFK-31, and HFK-31 ΔLHCYE cells over three independent experiments +/− the standard error of the mean. Westerns were digitally imaged using the Bio-Rad Chemidoc MP system, and densitometry was performed with the Biorad ImageLab 5.0 software. Values are shown relative to each T0, which was set to100. (D) Graphed are the average protein levels of ATM, Chk2 and Chk1 in HFKs, pLXSN-E7 and pLXSN E7 ΔLHCYE cells over three independent experiments. Densitometry was performed as described above. Error bars represent +/− the standard error of the mean.
Table 1.
Protein half-lives for DDR factors in primary HFKs, HFK-31 cells, and HFK-31 ΔLHCYE cells. Half-lives of the indicated proteins were determined by performing linear regression on values obtained by densitometry from Fig. 5B, Fig. 6B, and Fig. 7B across three independent experiments from two HFK backgrounds. Error represents +/− standard error of the mean.
| Protein | HFK | HFK-31 | HFK-31 ΔLHCYE |
|---|---|---|---|
| ATM | 5.8+/−1.3 | > 12 | 4.5+/−0.8 |
| Chk2 | 7.0+/−0.02 | > 12 | 9.6+/−2.0 |
| Chk1 | 4.5+/−0.3 | 5.8+/−0.5 | 4.2+/−0.4 |
| MRE11 | 4.8+/−0.4 | > 12 | 4.2+/−0.2 |
| Rad50 | 8.3+/−0.9 | > 12 | > 12 |
| NBS1 | 7.1+/−1.1 | > 12 | 9.4+/−1.1 |
| BRCA1 | 2.9+/−0.3 | 6.0+/−0.5 | 3.0+/−0.3 |
| Rad51 | 5.4+/−0.2 | 10.2+/−0.3 | 4.6+/−0.2 |
To determine if E7 expression alone is sufficient for the increase in protein stability observed, we examined the half-life of ATM, Chk2, and Chk1 in HFKs expressing wild-type E7, as well as the E7 ΔLHCYE mutant (Fig. 5C–D). As shown in Fig. 5C and D and summarized in Table 2, similar to HPV31 genome-containing lines, the half-life of ATM increased from 4.5+/−0.3 h in HFKs to > 12 h in E7-expressing cells. In contrast, the half-life of ATM decreased to 4.9+/−0.9 h in the E7 ΔLHCYE mutant. Similar results were observed for Chk2, with the half-life increasing to > 12 h in HFK-31 cells from 7.5+/−0.5 h in HFKs. Similar to ATM, Chk2 exhibited a reduced half-life in cells expressing the E7 ΔLHCYE mutant (Fig. 5C–D, Table 1). Since the half-life of Chk2 in the E7 ΔLHCYE mutant cells extended past our time course, we were unable to determine the full effect of loss of the E7 Rb binding domain on Chk2 protein stability. However, the observation that the relative levels of Chk2 decrease in the E7 ΔLHCYE mutant at each time point compared to HFK-31 cells suggests that the E7 Rb binding domain is important for extending the half-life of the Chk2 protein. The half-life of Chk1 was also extended in E7-expressing cells, increasing to 5.6+/−0.8 h from 3.5+/−0.6 h in control cells. Similar to ATM and Chk2, the increase in protein stability was lost in cells expressing the E7 ΔLHYCE mutant (3.9+/−0.3 h). Taken together, these data indicate that HPV maintains high levels of ATM, as well as ATR pathway components by increasing protein stability. Furthermore, these data indicate that the increase in half-life observed in HPV31 positive cells occurs in an E7-dependent manner through its Rb binding domain.
Table 2.
Protein half-lives for DDR factors in primary HFKs, pLXSN 31-E7 cells, and pLXSN 31-E7 ΔLHCYE cells. Linear regression was performed on values obtained by densitometry in Fig. 5D, Fig. 6D, and Fig. 7D to determine the half-life of the indicated proteins. Shown are the average half-lives across three independent experiments from one HFK background with the error representing +/− standard error of the mean.
| Protein | HFK | 31-E7 | 31-E7 ΔLHCYE |
|---|---|---|---|
| ATM | 4.5+/−0.3 | > 12 | 4.9+/−0.9 |
| Chk2 | 7.5+/−0.5 | > 12 | 12.3+/−2.7 |
| Chk1 | 3.5+/−0.6 | 5.6+/−0.8 | 3.9+/−0.3 |
| MRE11 | 3.2+/−0.2 | > 12 | 3.6+/−0.8 |
| Rad50 | 8.4+/−0.3 | > 12 | > 12 |
| NBS1 | 5.0+/−0.3 | > 12 | 4.6+/−0.7 |
| BRCA1 | 3.3+/−0.4 | 6.9+/−0.2 | 2.6+/−0.1 |
| Rad51 | 5.0+/−1.4 | 12.1+/−1.4 | 5.0+/−0.9 |
We next determined if HPV31 also maintains components of the MRN complex by increasing protein stability. As shown in Fig. 6A and B and summarized in Table 1, the half-life of MRE11 was substantially increased from 4.8+/−0.4 h in HFKs to > 12 h in HFK-31 cells. In contrast, the half-life of MRE11 in HFK-31 ΔLHYCE cells (4.2+/−0.2 h) mirrored that found in HFKs. Similarly, the half-life of Rad50 was increased in HFK-31 cells (> 12 h) compared to HFKs (8.3+/−0.9 h). We were unable to determine the effect of the ΔLHCYE mutation on Rad50 protein stability as the half-life extended beyond our time course (> 12 h) (Fig. 6B). However, the observation that the relative levels of Rad50 decreased in the E7 ΔLHCYE mutant at each time point compared to HFK-31 cells suggests that protein stability is affected by loss of the Rb binding domain. For NBS1, the half-life increased from 7.1+/−1.1 h in HFKs to more than 12 h in HFK-31 cells, and was reduced to 9.4+/−1.1 h in HFK-31 ΔLHYCE cells. Similar results for the MRN complex components were observed in E7-expressing cells (Fig. 6C–D). As shown in Fig. 6C and D and summarized in Table 2, the half-lives of MRE11 and NBS1 increased from 3.2+/−0.2 and 5.0+/−0.4 h in HFKs, respectively, to > 12 h in cells expressing wild-type E7. In cells expressing the E7 ΔLHYCE mutant, the half-lives for MRE11 and NBS1 were reduced to 3.6+/−0.8 and 4.6+/−0.7 h, respectively, similar to that found in HFKs (Fig. 6C–D). Similar to the HPV31 genome containing lines, the half-life for Rad50 increased to > 12 h in E7-expressing cells compared to 8.4+/−0.3 h in HFKs. In cells expressing the E7 ΔLHCYE mutant, we were again unable to calculate the half-life of Rad50 based on the 12-h time-course utilized. However, at each time point the relative levels of Rad50 were lower in the E7 ΔLHCYE mutant compared to wild-type E7-expressing cells, again suggesting that loss of the Rb binding mutant affects Rad50 stability. Overall, these data suggest that protein stability is a contributing factor to the maintenance of MRN complex members in HPV31 positive cells and this regulation occurs through a mechanism requiring Rb binding domain of E7.
Fig. 6.
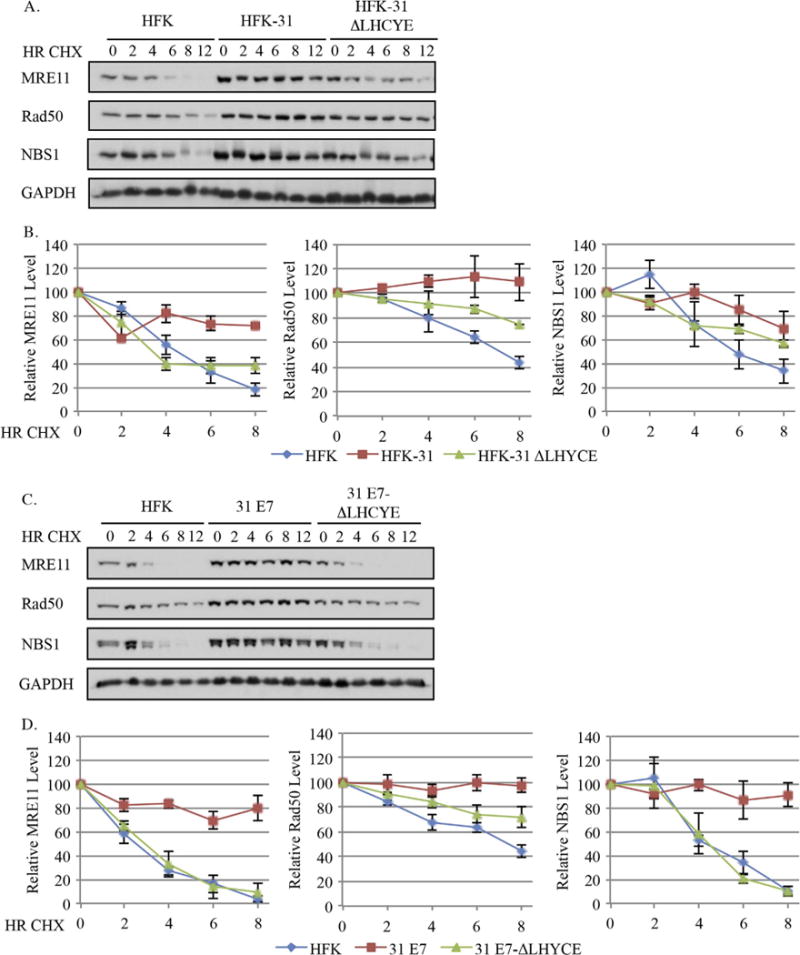
The E7 Rb binding domain is necessary for the increased half-lives of MRN complex components in HPV31 positive cells. (A, B) Uninfected HFKs, HFKs maintaining wild type HPV31 genomes (HFK-31), HPV31 genomes containing a mutation in the E7 Rb binding site (HFK-31 ΔLHYCE), as well as (C, D) HFKs stably transduced with a retroviral vector expressing wild type E7 (pLXSN-31 E7), or E7 with a mutation in the Rb binding domain (pLXSN-31 E7 ΔLHCYE) were treated with 50 μg/ml cycloheximide over 12 h time course, and whole cell lysates were harvested at the indicated time points. Western blot analysis was performed using antibodies to MRE11, Rad50, and NBS1, as well as GAPDH, which served as a loading control. (A, B) Data shown are representative blots from three independent experiments from two different HFK donors, and for (C, D) data shown are representative blots from three independent experiments from one HFK donor. (B, D) Graphed are the average protein levels of MRE11, Rad50, and NBS1 over three independent experiments. Westerns were digitally imaged using the Bio-Rad Chemidoc MP system, and densitometry was performed with the Biorad ImageLab 5.0 software. Values are shown relative to each T0, which was set to100. Error bars represent +/− the standard error of the mean.
As mentioned, we previously reported that BRCA1 and Rad51 exhibit increased protein stability in HPV31 positive CIN612 cells (Chappell et al., 2015). We next wanted to determine if this phenotype was affected by the E7 ΔLHCYE mutation. As shown in Fig. 7A and B and summarized in Table 1, the half-lives of BRCA1 and Rad51 were significantly elevated, increasing from 2.9+/−0.3 and 5.4+/−0.2 h in HFKs to 6.0+/−0.5 and 10.2+/−0.3 h in HFK-31 cells, respectively. Similar to the other DDR proteins examined, this increased stability was lost in HFK-31 ΔLHCYE cells for both BRCA1 (3.0+/−0.3 h) and Rad51 (4.6+/−0.2 h). Similar results were observed in the E7-expressing cells, as shown in Fig. 7C and D and summarized in Table 2. We found the half-lives of BRCA1 and Rad51 increased from 3.3+/−0.4 and 5.0+/−1.4 h in HFKs to 6.9+/−0.2 and 12.1+/−1.4 h in E7-expressing cells, respectively. In the E7 ΔLHCYE mutant cells, the half-lives decreased to levels similar to that found in HFKs for both BRCA1 (2.6+/−0.1 h) and Rad51 (5.0+/−0.9). This data, along with our analysis of BRCA1 and Rad51 gene expression (Fig. 4E–F), suggests that BRCA1 and Rad51 are regulated by HPV31 both transcriptionally, as well as post-transcriptionally via protein stability in an E7-dependent manner. Additionally, our data indicate that the Rb binding site of E7 contributes to both mechanisms.
Fig. 7.
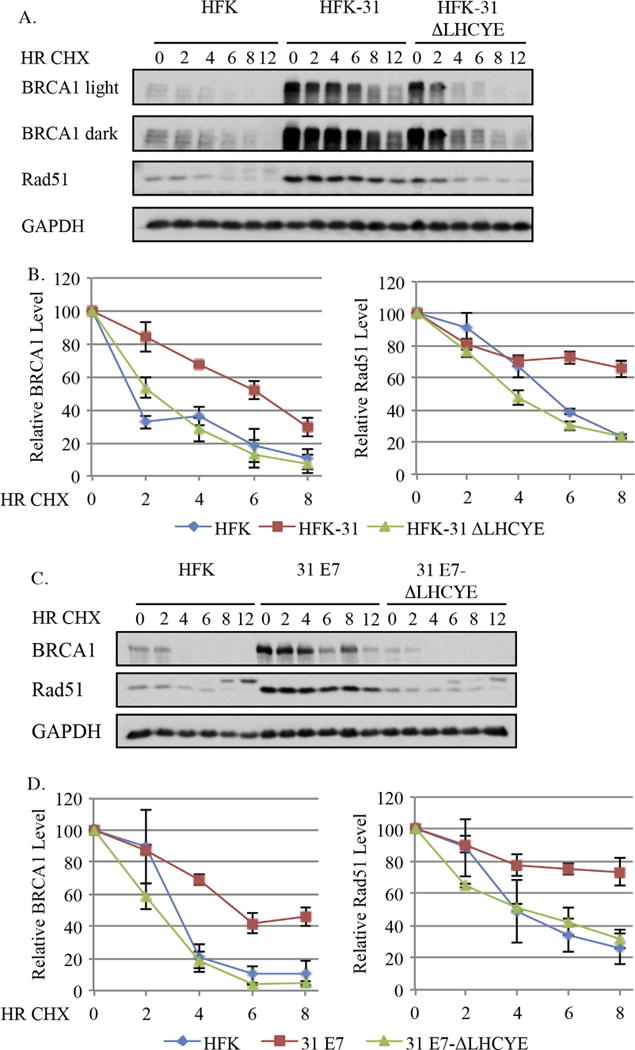
Increased protein stability in an E7-dependent manner contributes to the increased levels of BRCA1 and Rad51 in HPV31 positive cells. (A, B) Primary HFKs, HFKs maintaining wild type HPV31 genomes (HFK-31) or mutant HPV31 genomes with a mutation in the Rb binding site (HFK-31 ΔLHCYE), as well as (C, D) pLXSN-E7 and pLXSN E7 ΔLHCYE cells were treated with 50 μg/ml cycloheximide over a 12 h time course. Whole cell lysates were harvest at the indicated times using antibodies to BRCA1 and Rad51, with GAPDH serving as a loading control. (A, B) Data shown are representative blots from three independent experiments from two different HFK donors, and for (C, D) data shown are representative blots from three independent experiments from one HFK donor. (B, D) Graphed are the relative protein levels at each time point, with T0 for each cell line set to 100. Densitometry was performed across three independent experiments using Biorad ImageLab 5.0 software. Error bars represent means +/− standard error.
4. Discussion
Previous studies demonstrated that activation of both the ATM and ATR DNA damage response (DDR) pathways are required for the productive replication of HPV31 (Hong et al., 2015a; Moody and Laimins, 2009). Expression of E7 alone has been shown to be sufficient for activation of both ATM and ATR signaling (Hong et al., 2015a; Rogoff et al., 2004). It is well established that E7 plays a central role in facilitating viral replication by binding and targeting the degradation of Rb, resulting in S-phase re-entry by a subset of differentiating keratinocytes (Banerjee et al., 2006). The introduction of the HPV31 genome, or expression of HPV31 E7 alone is sufficient to increase the levels of a broad range of DNA repair factors in primary keratinocytes, including ATM, Chk2, Chk1, the MRN complex, BRCA1, and Rad51, all of which are required for productive replication (Anacker et al., 2014; Chappell et al., 2015; Moody and Laimins, 2009; Rogoff et al., 2004). Interestingly, loss of Rb activity has been shown to recapitulate many of these phenotypes (Frame et al., 2006; Pickering and Kowalik, 2006), raising the possibility that E7 activates the DDR and provides DNA repair factors for productive viral replication through its ability to bind and target Rb for degradation. Consistent with this idea, we have found that the E7 Rb binding domain is required for maintenance of ATM and ATR activation, as well as the increased levels of DNA repair factors observed in both HPV31 positive and E7-expressing cells.
We have found that deletion of the HPV31 E7 Rb binding domain results in decreased levels of both phosphorylated and total levels of ATM, as well as its downstream target Chk2. In addition, we have found that this domain is required for maintenance of total and phosphorylated levels of Chk1, a target of the ATR DNA damage kinase. The concomitant decrease in total levels along with the phosphorylated forms of ATM, Chk2, and Chk1 in E7 Rb binding deficient cells suggests that in addition to inducing DDR activation, E7 contributes to maintenance of the DDR through increasing total levels of DNA repair factors. Additionally, we show that while total levels of ATM and Chk2 decrease upon differentiation in HPV31 positive cells, the phosphorylated forms remain elevated, suggesting that the DDR is further activated during the productive phase of the viral life cycle. In contrast, loss of the Rb binding domain resulted in a minimal decrease in total levels of DDR factors upon differentiation, similar to that observed for uninfected HFKs. In cells expressing HPV31 E7 alone, these phenotypes were recapitulated, further highlighting the importance of the E7 Rb binding domain in the regulation of these DDR factors in HPV31 positive cells.
Our studies demonstrate that loss of the E7 Rb binding site results in reduced levels of all three components of the MRN complex (Mre11, Rad50, and Nbs1). The MRN complex is required for the activation of ATM in response to DNA damage (Lee and Paull, 2004, 2005; Paull and Lee, 2005). However, we previously published that the MRN complex is not required for ATM activation in HPV31 positive cells, but is required for productive viral replication (Anacker et al., 2014). These results suggest that the decrease observed in ATM phosphorylation upon the loss of the Rb binding domain is not due to decreased levels of MRN components. In addition, we have found that the levels of the homologous repair proteins BRCA1 and Rad51, both of which are also required for productive replication (Chappell et al., 2015), are maintained at high levels in HPV31 positive cells and E7-expressing cells in a manner dependent on the Rb binding domain. Together, these data further suggest that the Rb binding domain is required for HPV to maintain adequate levels of DNA repair factors required for viral DNA synthesis.
The most well known function of the E7 LXCXE domain is the binding of Rb, resulting in its targeted degradation and the constitutive activation of E2F transcription factors. Based on this function of E7, the simplest explanation of the reduction in total protein levels observed in Rb binding deficient cells is that transcription of DNA repair genes is decreased. In support of this, several of these factors are E2F responsive, including ATM, Chk1, BRCA1, and Rad51 (Bracken et al., 2004). However, while we found that E2F1 and E2F2 protein levels are increased in HPV positive cells in a manner dependent on the Rb binding domain, only Chk1, BRCA1, and Rad51 were significantly affected at the transcriptional level by the E7 ΔLHCYE mutation, suggesting that the other DNA repair factors examined (ATM, Chk2, and MRN) are regulated primarily in a post-transcriptional manner. In support of this, we found that all of the DDR proteins examined exhibited some level of regulation at the level of protein stability, with five proteins (ATM, Mre11, NBS1, BRCA1, and Rad51) at least doubling their half-lives in HPV31 positive and/or E7-expressing cells compared to uninfected HFKs. Importantly, this increase in half-life was lost in the ΔLHCYE mutant both in the context of the viral genome, as well as in cells expressing E7 alone. Together, these observations suggest that E7 uses a two-pronged approach to elevate levels of DNA repair factors: increasing the transcription of a subset of DNA repair factors and broadly increasing the stability of these proteins. Interestingly, our studies indicate that HPV ensures adequate levels of BRCA1 and Rad51 by targeting both the regulation of gene expression and protein stability.
The mechanism by which E7 increases the protein stability of DNA repair factors in HPV positive cells is currently unclear, although several possibilities exist. E7 has been shown to interact with multiple DDR components, including ATM, Rad50, NBS1, and BRCA1 (Anacker et al., 2014; Moody and Laimins, 2009; Zhang et al., 2005), which may influence the stability of these proteins. Importantly, the interaction of E7 with ATM and NBS1 is lost upon depletion of the Rb binding domain (Anacker et al., 2014; Moody and Laimins, 2009), and whether this affects the stability of these proteins will be the focus of future investigations. Another possibility is that E7 influences DDR factor protein stability through effects on protein degradation machinery. E7 has been shown to inhibit the anaphase promoting complex/cyclosome (APC/C) (Yu and Munger, 2013), a ubiquitin ligase complex involved in regulation of mitotic progression, as well as the DDR (de Boer et al., 2016). However, whether E7-mediated inhibition of this complex occurs in a manner dependent on the Rb binding domain is not known.
We have found that E7 requires the Rb binding domain to maintain ATM, as well as ATR activity. ATM has been shown to affect DDR factor stability both directly through phosphorylation, as well as indirectly through regulation of proteasomal degradation (Ciccia and Elledge, 2010). Therefore, loss of ATM activity in the E7 ΔLHCYE mutant may result in decreased stability of downstream targets through either loss of phosphorylation and/or increased degradation. The increase in pChk2 observed upon differentiation in HPV31 positive cells despite decreased total levels supports this possibility. ATM regulates the activity of ubiquitin ligases, including MDM2, through phosphorylation, in turn increasing the stability of p53, as well as Chk2 (Meek and Anderson, 2009; Kass et al., 2009). ATM also regulates the stability of Chk1 through phosphorylation and stabilization of the zinc-finger like protein ZEB1, which interacts with the deubiquitylase USP7 to prevent proteasomal degradation of Chk1 (Zhang et al., 2014). Additionally, p300 is phosphorylated and stabilized by ATM in response to DNA damage, and is in turn required for the stabilization NBS1 (Jang et al., 2010, 2011). Interestingly, p300 has also been shown to bind directly to the LXCXE domain of HPV16 E7 (Bernat et al., 2003), providing a potential direct link between the Rb binding site of E7 and the regulation of NBS1 stability. Furthermore, previous studies have linked Rb inactivation to ATM activation and DSB induction (Pickering and Kowalik, 2006; Shamma et al., 2013, 2009), as well as the control of Tip60-dependent acetylation of ATM (Shamma et al., 2013), which is required for ATM autophosphorylation and activation. Importantly, recent studies have shown that Tip60 is required for productive replication of HPV31 (Hong et al., 2015b), presumably through facilitating ATM activation. Determining if the Rb binding domain is required for ATM activation through Tip60, as well as whether ATM activity is required for maintenance of DDR factor stability in HPV31 positive cells will be important areas of future investigation.
In addition to increasing protein stability, our studies suggest that E7 elevates transcription of a subset of DNA repair genes (Chk1, Rad51, and BRCA1) in a manner dependent on the Rb binding domain, though the mechanism by which this occurs is unclear. Previous studies demonstrated a requirement for the STAT5 transcription factor in the activation of both the ATM and ATR pathways in HPV31 positive cells (Hong et al., 2015a; Hong and Laimins, 2013). The activation of STAT5 was shown to be E7-dependent and may require the Rb binding domain. STAT5 knockdown decreased the total levels of ATM, Chk2, BRCA1, and Rad51 in HPV31 positive cells, while only affecting the phosphorylated levels of ATR and Chk1 (Hong et al., 2015a; Hong and Laimins, 2013). However, whether the decrease in ATM, Chk2, BRCA1, and Rad51 is regulated transcriptionally or post-transcriptionally is unclear. Additionally, the expression of both BRCA1 and Rad51 is known to be regulated in manner dependent on E2F transcription factors (Bracken et al., 2004), opening the possibility that E7 increases their transcription through its ability to inactivate Rb or its related pocket proteins p107 and p130. Identifying the mechanism(s) by which E7 regulates the transcription of these DNA repair factors will be important to further understand how HPV manipulates DNA damage signaling to facilitate viral replication.
Acknowledgments
Funding: This project was supported by NIH grant 1R01CA181581 and American Cancer Society grant A14-0113 (to C.A.M).
References
- Agarwal S, Tafel AA, Kanaar R. DNA double-strand break repair and chromosome translocations. DNA Repair. 2006;5:1075–1081. doi: 10.1016/j.dnarep.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Anacker DC, Gautam D, Gillespie KA, Chappell WH, Moody CA. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J Virol. 2014;88:8528–8544. doi: 10.1128/JVI.00517-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee NS, Genovese NJ, Noya F, Chien WM, Broker TR, Chow LT. Conditionally activated E7 proteins of high-risk and low-risk human papillomaviruses induce S phase in postmitotic, differentiated human keratinocytes. J Virol. 2006;80:6517–6524. doi: 10.1128/JVI.02499-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Chappell WH, Gautam D, Ok ST, Johnson BA, Anacker DC, Moody CA. Homologous recombination repair factors Rad51 and BRCA1 are necessary for productive replication of human papillomavirus 31. J Virol. 2015;90:2639–2652. doi: 10.1128/JVI.02495-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer HR, Guerrero Llobet S, van Vugt MA. Controlling the response to DNA damage by the APC/C-Cdh1. Cell Mol Life Sci. 2016;73:949–960. doi: 10.1007/s00018-015-2096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol. 2003;77:2819–2831. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame FM, Rogoff HA, Pickering MT, Cress WD, Kowalik TF. E2F1 induces MRN foci formation and a cell cycle checkpoint response in human fibroblasts. Oncogene. 2006;25:3258–3266. doi: 10.1038/sj.onc.1209352. [DOI] [PubMed] [Google Scholar]
- Gillespie KA, Mehta KP, Laimins LA, Moody CA. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol. 2012;86:9520–9526. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair. 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses. 2015;7:2542–2591. doi: 10.3390/v7052542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Cheng S, Iovane A, Laimins LA. STAT-5 regulates transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene to activate the ATR pathway and promote human papillomavirus replication) MBio. 2015a;6:e02006–e02015. doi: 10.1128/mBio.02006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dutta A, Laimins LA. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J Virol. 2015b;89:4668–4675. doi: 10.1128/JVI.03455-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Laimins LA. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013;9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384:324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert WG, Laimins LA. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J Virol. 2002;76:2263–2273. doi: 10.1128/jvi.76.5.2263-2273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang ER, Choi JD, Jeong G, Lee JS. Phosphorylation of p300 by ATM controls the stability of NBS1. Biochem Biophys Res Commun. 2010;397:637–643. doi: 10.1016/j.bbrc.2010.05.060. [DOI] [PubMed] [Google Scholar]
- Jang ER, Choi JD, Lee JS. Acetyltransferase p300 regulates NBS1-mediated DNA damage response. FEBS Lett. 2011;585:47–52. doi: 10.1016/j.febslet.2010.11.034. [DOI] [PubMed] [Google Scholar]
- Kass EM, Poyurovsky MV, Zhu Y, Prives C. Mdm2 and PCAF increase Chk2 ubiquitination and degradation independently of their intrinsic E3 ligase activities. Cell Cycle. 2009;8:430–437. doi: 10.4161/cc.8.3.7624. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004a;78:3533–3541. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004b;68:362–372. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24:1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009;5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Shin MK, Lambert PF. High incidence of female reproductive tract cancers in FA-deficient HPV16-transgenic mice correlates with E7’s induction of DNA damage response, an activity mediated by E7′s inactivation of pocket proteins. Oncogene. 2014;33:3383–3391. doi: 10.1038/onc.2013.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Shin MK, Pitot HC, Lambert PF. High incidence of HPV-associated head and neck cancers in FA deficient mice is associated with E7′s induction of DNA damage through its inactivation of pocket proteins. PloS One. 2013;8:e75056. doi: 10.1371/journal.pone.0075056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle. 2005;4:737–740. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- Pickering MT, Kowalik TF. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene. 2006;25:746–755. doi: 10.1038/sj.onc.1209103. [DOI] [PubMed] [Google Scholar]
- Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, Kowalik TF. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24:2968–2977. doi: 10.1128/MCB.24.7.2968-2977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman A, Munger K. The papillomavirus E7 proteins. Virology. 2013;445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EL, Hollingworth R, Grand RJ. Activation of the DNA Damage Response by RNA Viruses. Biomolecules. 2016;6:2. doi: 10.3390/biom6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamma A, Suzuki M, Hayashi N, Kobayashi M, Sasaki N, Nishiuchi T, Doki Y, Okamoto T, Kohno S, Muranaka H, Kitajima S, Yamamoto K, Takahashi C. ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol Cell Biol. 2013;33:3113–3124. doi: 10.1128/MCB.01597-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, Takahashi C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell. 2009;15:255–269. doi: 10.1016/j.ccr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Solinas-Toldo S, Durst M, Lichter P. Specific chromosomal imbalances in human papillomavirus-transfected cells during progression toward immortality. Proc Natl Acad Sci USA. 1997;94:3854–3859. doi: 10.1073/pnas.94.8.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Laimins LA. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med. 2005;119:157–169. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- Yu Y, Munger K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology. 2013;446:251–259. doi: 10.1016/j.virol.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Y, Woodward WA, Liu Y, Dean DC, Liang H, Hu Y, Ang KK, Hung MC, Chen J, Ma L. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864–875. doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fan S, Meng Q, Ma Y, Katiyar P, Schlegel R, Rosen EM. BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem. 2005;280:33165–33177. doi: 10.1074/jbc.M505124200. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in the causation of human cancers – a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]