

The WRAMP structure is a Wnt5-induced association of a cell adhesion molecule with F-actin and myosin IIB at the rear of migrating cells. WRAMP structures control the speed and persistence of directional cell movement in melanoma and nonmelanoma cells.
Abstract
In contrast to events at the cell leading edge, rear-polarized mechanisms that control directional cell migration are poorly defined. Previous work described a new intracellular complex, the Wnt5a-receptor-actomyosin polarity (WRAMP) structure, which coordinates the polarized localization of MCAM, actin, and myosin IIB in a Wnt5a-induced manner. However, the polarity and function for the WRAMP structure during cell movement were not determined. Here we characterize WRAMP structures during extended cell migration using live-cell imaging. The results demonstrate that cells undergoing prolonged migration show WRAMP structures stably polarized at the rear, where they are strongly associated with enhanced speed and persistence of directional movement. Strikingly, WRAMP structures form transiently, with cells displaying directional persistence during periods when they are present and cells changing directions randomly when they are absent. Cells appear to pause locomotion when WRAMP structures disassemble and then migrate in new directions after reassembly at a different location, which forms the new rear. We conclude that WRAMP structures represent a rear-directed cellular mechanism to control directional migration and that their ability to form dynamically within cells may control changes in direction during extended migration.
INTRODUCTION
Cell movement requires the spatial control of signal transduction, including cell polarity mechanisms that move proteins to specific intracellular locations (Huttenlocher, 2005; McCaffrey and Macara, 2012). During cell locomotion, cells must coordinate the formation of membrane protrusions and attachments to extracellular matrix at the front, with membrane retraction and disassembly of attachments at the rear. Much is known about events at the leading edge, where actin polymerization via Rac, Cdc42, WASP/WAVE, and Arp2/3 form lamellipodia and membrane protrusions, which promote focal contact attachments to extracellular matrix and mediate forward movement (Ridley et al., 2003; Petrie et al., 2009; Parsons et al., 2010). These have been implicated in the maintenance and regulation of directional migration (Petrie et al., 2009; Krause and Gautreau, 2014). In contrast, mechanisms at the rear of migrating cells are less well defined. At the cell rear, localized F-actin and nonmuscle myosin create mechanical force for driving forward translocation of the cell body (Cramer, 2013). How this controls directional cell movement, however, is poorly understood. Directional cell migration underlies many processes, including development, angiogenesis, inflammation, and cancer. Thus novel signaling processes that control cellular migration may yield new therapeutic targets for many human diseases.
We have described a new cellular entity that polarizes to the rear in a manner that is induced by Wnt5a (Bowerman, 2008; Witze et al., 2008, 2013; Mladinich and Huttenlocher, 2013). Wnt5a is a signaling ligand that guides spatial organization in cells by moving proteins anisotropically to specific intracellular locations (Nishita et al., 2010; Kikuchi et al., 2012; Gomez-Orte et al., 2013). In metazoan development, Wnt5a controls cell polarity responses such as body axis formation and oriented cell division through planar cell polarity/convergent extension (PCP/CE) pathways (Gao, 2012). Wnt5a increases cytosolic Ca2+, although the mechanisms have not been clearly determined (Slusarski et al., 1997; Kohn and Moon, 2005). Wnt5a is also known to promote cell migration and invasion in melanoma, lung, gastric, pancreatic, and ovarian cancer cells (Bittner et al., 2000; Weeraratna et al., 2002; Kurayoshi et al., 2006; Ripka et al., 2007; O’Connell et al., 2010; Peng et al., 2011). Specifically, Wnt5a is elevated in aggressive, metastatic melanomas and at the invasive edge of primary tumors where cells metastasize toward lymph nodes (Bittner et al., 2000; Weeraratna et al., 2002). However, the mechanisms by which Wnt5a promotes migration and invasion of cancer cells are not known.
Previous findings in melanoma cells showed that Wnt5a induces the formation of a novel protein network that triggers rear-polarized actomyosin and Ca2+ signaling (Witze et al., 2013). This was named the Wnt5a-receptor-actomyosin polarity (WRAMP) structure and involves an anisotropic positioning of melanoma cell adhesion molecule (MCAM, also known as CD146 or MUC18). A proposed mechanism involves the internalization of transmembrane receptors into endosomes that interact with actomyosin cytoskeletal components to create the dynamic and transient WRAMP structure entity. Of importance, the WRAMP structure is connected temporally to the recruitment of F-actin and nonmuscle myosin-IIB and leads to a localized elevation of cytoplasmic Ca2+. Therefore the WRAMP structure is significant because it provides a mechanism for directing a highly localized second messenger signaling event that is needed for actomyosin contractility, membrane lifting, and retraction.
The characteristics of the WRAMP structure suggest that it may function in rear-directed events involved in cell migration. In response to isotropic Wnt5a and a gradient of the chemokine CXCL12/SDF1, WRAMP structures localized distally from the Golgi, consistent with rear polarity. In short-term movies, they localized to the rear with respect to the direction of nucleokinesis (Witze et al., 2008). However, the behavior of WRAMP structures during extended cell migration and its function in directional cell movement were not investigated. Here we perform a quantitative study using live-cell imaging to examine the importance of WRAMP structures for directional cell migration. The results demonstrate that WRAMP structures are maintained at the rear in migrating melanoma cells, as well as in nonmelanoma cells. Over extended times, individual cells are observed to fluctuate between periods when WRAMP structures are present and when they are absent. When WRAMP structures are present, cells translocate over longer distances with faster speed and greater persistence of directional movement. When WRAMP structures are absent, cells change direction through a process involving an intracellular relocalization of WRAMP structures to define the new rear position. Taken together, the results indicate that WRAMP structures are rear-polarized during cell migration and mediate speed and persistence of directional cell movement, fundamental processes required for cell locomotion. Our findings provide a deeper understanding of rear polarity mechanisms that control cellular migration and suggest a potential new target for intervening with cancer cell invasion.
RESULTS
WRAMP structures are characterized by Wnt5a-induced polarization of MCAM, F-actin, and myosin IIB
WRAMP structures were first observed in melanoma cells through the polarization of the cell surface receptor MCAM/CD146/MUC18, a member of the immunoglobulin G (IgG) superfamily of cell adhesion molecules. Formation of WRAMP structures can be measured by polarization of MCAM along the long axis of the cell, which is scored when it enriches with threefold or greater intensity over background. WRAMP structures form dynamically and are followed temporally by membrane retraction (Supplemental Movie S1).
Cytoskeletal proteins F-actin and myosin IIB associate with WRAMP structures such that they overlap but do not completely colocalize with MCAM (Figure 1A). Previous studies identified WRAMP structures in WM239a melanoma cells that show significant focal adhesions, stress fibers, and cell spreading on glass, as well as on two-dimensional collagen (Witze et al., 2008). Here we identify similar structures in A375 melanoma cells, which show greater cortical actin, lower stress fibers, and less adhesion (Figure 1B). A375 cells often show a stretched morphology, with spreading at one end and narrowing at the other end of the long axis, where the WRAMP structure is typically found. In each cell type, endogenous MCAM forms a polarized complex at one end of the cell, partially overlapping dense F-actin bundles with thick filaments extending to the plasma membrane. Myosin IIB forms a band-like configuration overlapping F-actin and MCAM, which is often located further from the cell edge and closer to the cell body and nucleus (Figure 1, A and B). Confocal microscopy reveals enrichment of MCAM, F-actin, and myosin IIB within the cell interior and not restricted to the plasma membrane (Figure 1, A and B).
FIGURE 1:

WRAMP structures are made up of polarized MCAM, F-actin, and myosin IIB. Monitoring of endogenous MCAM and myosin IIB by indirect immunofluorescence and F-actin by phalloidin staining in (A) WM239a cells and (B) A375 cells. Top, wide-field imaging (60×). Bottom, confocal imaging (100×). Scale bars, 10 µm. Merge, MCAM in green, F-actin in cyan, and myosin IIB in magenta. (C–F) Cells treated without or with 250 ng/ml Wnt5a for 30 min: percentages of cells with (C) polarized MCAM, (D) F-actin polarized along with MCAM, (E) myosin IIB polarized along with MCAM, and (F) both F-actin and myosin IIB polarized along with MCAM. Mean percentages and SEM from three or more slides (>750 cells), representing two or more biological replicates. **p < 0.01. The p values were calculated using standard two-tailed Student’s t test. The term “polarized” in this figure does not distinguish between rear and front polarity.
WRAMP structures were quantified by immunostaining of endogenous MCAM and myosin IIB and phalloidin staining of F-actin. Treatment of cells for 30 min with Wnt5a significantly enhanced the percentage of cells showing WRAMP structures, which increased by 2.5- to 3-fold as measured by polarized localization of MCAM (Figure 1C). Typically, WRAMP structures form within 15 min, but quantified at a single time point, they appear in only ∼24% of WM239A and 15% of A375 cells. This is explained by their transient nature; they assemble dynamically, followed by disassembly. When measured over a period of 0−2 h, >80% of cells formed WRAMP structures (unpublished data). Approximately 20% of WM239A cells and 12% of A375 cells showed F-actin polarized along with MCAM after Wnt5a treatment (Figure 1D). Therefore F-actin was present in 80% of WRAMP structures based on polarized MCAM. We also found myosin IIB polarized at WRAMP structures in >50% of cases (Figure 1E). F-actin was present in almost all of the WRAMP structures with myosin IIB (Figure 1F). Thus WRAMP structures were characterized by strong association between polarized MCAM, F-actin, and myosin IIB, forming with coordinately increased frequency in response to Wnt5a.
WRAMP structure formation involves coordinated movement of MCAM, F-actin, and myosin IIB
Confocal live cell imaging was used to examine the order of MCAM, F-actin, and myosin IIB recruitment into WRAMP structures. In both WM239a and A375 cells, MCAM–green fluorescent protein (GFP) polarized dynamically to form WRAMP structures and was always followed by membrane retraction (Figure 2, A and B). Cells were also cotransfected with MCAM-GFP and mCherry in controls, which confirmed that the polarized localization of MCAM-GFP was not an artifact caused by variations in cell volume or membrane thickness (Supplemental Figure S1). We then examined >100 cells cotransfected with MCAM-GFP and LifeAct-mCherry, a peptide that binds and labels F-actin. In each case, the accumulation of F-actin into WRAMP structures was synchronous with the polarization of MCAM-GFP (Figure 2, A and B). WM239a cells migrated in a manner that reflected spreading and adhesiveness reminiscent of mesenchymal cell movement, whereas A375 cells migrated with more-rounded morphologies. Nevertheless, the temporal dynamics of F-actin and MCAM-GFP in forming WRAMP structures were tightly coordinated in each cell type.
FIGURE 2:

Dynamic movement of WRAMP structures, followed by membrane retraction. Frames from confocal live-cell imaging experiments of (A) WM239a and (B) A375 cells cotransfected with MCAM-GFP and LifeAct-mCherry and (C) WM239a and (D) A375 cells cotransfected with MCAM-GFP and myosin IIB-N18-mCherry. Supplemental Movies S2–S5 (corresponding to A–D, respectively) show coordinated movement of MCAM, F-actin, and myosin IIB. White dot indicates starting position. Scale bars, 10 µm; times in hours:minutes. Controls for this experiment with MCAM-GFP plus mCherry in Supplemental Figure S1 and Supplemental Movies S6 and S7. Quantitation of MCAM-GFP and myosin IIB-mCherry (or LifeAct-mCherry) fluorescence vs. time is shown in Supplemental Figure S2.
In contrast, live imaging of cells coexpressing myosin IIB-mCherry and MCAM-GFP showed that the recruitment of myosin IIB to WRAMP structures was often delayed relative to MCAM. This was most obvious in WM239a cells with greater spreading, but could also be observed in A375 cells (Figure 2, C and D). Quantification of fluorescence intensity along the long axis of cells showed movement of myosin IIB to the cell edge in a manner often crossing MCAM (Supplemental Figure S2). In each case, the polarization of myosin IIB lagged behind that of MCAM, typically by 2–5 min. The delay in myosin IIB recruitment explains why myosin IIB is less strongly correlated with polarized MCAM than is F-actin (Figure 1, C–F). Membrane retraction then occurred after the appearance of myosin IIB at the cell edge (Figure 2, C and D, and Supplemental Figure S2). The results suggest that membrane retraction at WRAMP structures is initiated by the recruitment of myosin IIB to F-actin, which in turn promotes actomyosin contraction.
WRAMP structures also appear in nonmelanoma cells
We next asked whether WRAMP structures could be observed in nonmelanoma cells, which also express MCAM and in which Wnt5a signaling has been shown to promote cell migration. One such model is HT1080 fibrosarcoma cells, which are often used in studies of migration and invasion (Michl et al., 2005; Ripka et al., 2007; Schwartz et al., 2013; Wei et al., 2013; Tovari et al., 2014). HT1080 cells were treated with Wnt5a for 30 min and then fixed and immunostained for endogenous MCAM and myosin IIB, with phalloidin staining of F-actin. Like melanoma cells, HT1080 cells showed polarization of MCAM, overlapping with F-actin and myosin IIB (Figure 3A). Wnt5a treatment increased the percentage of cells with WRAMP structures by threefold or more, measured by polarized MCAM. F-actin overlapped with polarized MCAM in 85% of cases and with myosin IIB in 55% of cases, and all three markers overlapped in 55% of cases (Figure 3B). Thus HT1080 cells revealed WRAMP structures with characteristics nearly identical to those seen in melanoma cells, involving convergent polarization of MCAM, F-actin, and myosin IIB, in a manner stimulated by Wnt5a. Live imaging of HT1080 cells transiently transfected with MCAM-GFP and mCherry showed migration as rounded cells, with WRAMP structure formation followed by membrane retraction and forward movement of the cell body (Figure 3C). The polarized enrichment of MCAM was not accompanied by increased mCherry signal, confirming that it was not an artifact of increased cell volume or membrane retraction. Thus the appearance of WRAMP structures and their characteristics of actomyosin recruitment and coupling to membrane retraction are not limited to melanoma cells. We also observed MCAM polarized in a manner resembling WRAMP structures in WM1789, C2C12, and human umbilical vein endothelial cell lines (Witze et al., 2008, 2013).
FIGURE 3:

WRAMP structures in HT1080 cells. (A) Wide-field imaging (60×) of endogenous MCAM, F-actin, and myosin IIB in fixed HT1080 cells. Scale bars, 10 µm. (B) Cells treated without or with Wnt5a (250 ng/ml, 30 min) showing percentages of all cells with polarized MCAM, F-actin polarized along with MCAM, myosin IIB polarized along with MCAM, and F-actin plus myosin IIB polarized along with MCAM. Values show means and SEM from four or more slides (>800 cells) representing two or more biological replicates. *p < 0.05. The p values were calculated using standard two-tailed Student’s t test. (C) Live-cell imaging of HT1080 cells cotransfected with MCAM-GFP plus mCherry (also see Supplemental Movie S8) shows formation of WRAMP structures, followed by membrane retraction, not artifacts of cell volume or membrane thickness. Scale bars, 10 μm; times in hours:minutes.
Rear polarization of WRAMP structures in migrating cells
We next monitored WRAMP structures during cell migration over extended periods of time. WM239a or A375 cells expressing MCAM-GFP were plated on collagen-coated glass, and a scratch wound was introduced in the monolayer, followed by treatment with Wnt5a. Cells migrating into the wound by four cell lengths or more were then tracked by confocal time-lapse imaging. In both WM239a and A375 cells, WRAMP structures appeared stably maintained at the rear with respect to the direction of cell movement (Figure 4, A and B). This was measured quantitatively by tracking the trajectories of migrating cells from the x, y-coordinates of the cell center, and comparing them to the x, y-coordinates of the WRAMP structure in each frame. A polarity score was then calculated by the angle between the vector describing the location of the WRAMP structure relative to the cell center and a vector describing the path of cell migration measured by the cell position from the current frame to the next (Figure 4C). The localization of WRAMP structures relative to the path of cell migration was then binned into four quadrants based on angle. Angles between 0 and 45° reported rear-localized WRAMP structures, where an angle of 0° indicated two parallel vectors with the WRAMP structure located exactly at the rear with respect to the direction of migration. Angles between 135 and180° indicated front-localized WRAMP structures, where an angle of 180° indicated WRAMP structures at the leading edge. Angles between 45 and 135° indicated side localization of WRAMP structures. This metric allowed the polarity of WRAMP structures to be quantified over many frames in many cells.
FIGURE 4:
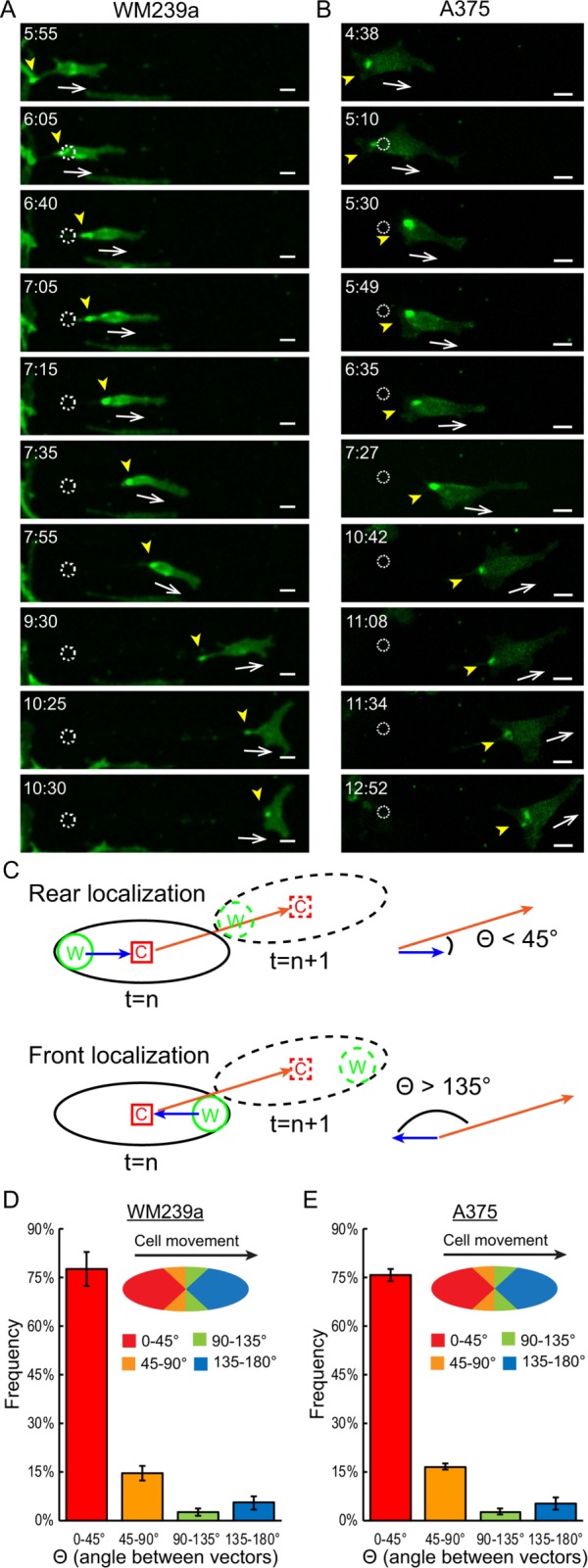
WRAMP structures are maintained at the rear during extended cell migration. Confocal live-cell imaging of MCAM-GFP in (A) WM239a cells and (B) A375 cells treated with recombinant Wnt5a, and monitored during extended migration by at least two cell lengths into scratch wounds. Yellow arrowheads mark WRAMP structures, dotted circles indicate starting position for the cell center, and white arrows indicate direction of cell movement. Supplemental Movies S9 and S10 correspond to A and B, respectively. Scale bars, 20 μm; times in hours:minutes. (C) Method for measuring WRAMP structure polarity. W indicates the position of WRAMP structures, measured by polarized MCAM; C indicates the position of the cell center; solid black line indicates the cell outline in frame n; dashed black line indicates the cell outline at frame n + 1. The red arrow depicts the movement vector, indicating the direction of cell movement from the cell center in frame n to frame n + 1. The blue arrow depicts the positioning vector, indicating the WRAMP structure position relative to the cell center. The polarity score measures the angle between these two vectors (Θ), where Θ ≤ 45° indicates rear polarity of WRAMP structures and Θ ≥ 135° indicates front polarity. (D, E) Histograms showing polarity scores for (D) WM239a and (E) A375 cells, measuring the mean for ≥30 cells (>2000 frames). Error bars indicate the range from biological replicates imaged in different experiments. All cells showed similar distributions, with primarily rear localization and only a small percentage of frames with side or front localization. Generally, WRAMP structures were found at the side or front only when cells were not migrating or before changing direction.
All cells showed similar behavior, with WRAMP structures primarily localized at the rear, but occasionally localized at the side or front, with respect to the direction of movement. Usually, the periods when WRAMP structures were present but not rear-localized were brief, lasting only one or two frames. The results showed WRAMP structures to be strongly biased to the rear measured in both WM239a and A375 cells in at least 1200 frames from 30 cells (Figure 4D). This confirmed that WRAMP structures are indeed rear-polarized during cell migration over long distances.
Inhibition of WRAMP structures slows wound healing and individual cell speed
We next examined whether WRAMP structures may be required for cell migration. Because WRAMP structures are induced by Wnt5a, it was reasonable to ask whether blocking signal transduction provided a way to inhibit WRAMP structures. We examined effects of Wnt5a–small interfering RNA (siRNA) knockdown as well as Box 5, a peptide derived from Wnt5a that antagonizes signaling (Jenei et al., 2009). In the presence of recombinant Wnt5a, Box 5 strongly inhibited the ability of both WM239A and A375 cells to form WRAMP structures, whereas Wnt5a-siRNA had a lesser effect, as expected (Supplemental Figure S3, A–C). In the absence of ligand, WRAMP structures in both cell types were significantly inhibited by either Box 5 or Wnt5a-siRNA alone and even further when the two treatments were combined (Supplemental Figure S3, A and B). Therefore we examined cells in which ligand was absent for the effects of Box 5, Wnt5a-siRNA, or their combination. We also examined cells treated with MCAM-siRNA. Knockdown of MCAM blocks WRAMP structures due to loss of the MCAM marker (Supplemental Figure S3D; Witze et al., 2008), although conceivably WRAMP structures lacking MCAM could still be present.
To quantify effects of inhibiting Wnt5a and WRAMP structures on cell migration, we transfected WM239A and A375 cells with H2B-mCherry and tracked individual cells migrating into scratch wounds after treatment with Box 5, Wnt5a-siRNA, or MCAM-siRNA. All individual cells that migrated into the wound were quantified for speed using a cell-tracking algorithm (described in Materials and Methods). The average speed of both cell types was significantly inhibited by each treatment compared with controls (Figure 5, A and B). Thus, inhibiting WRAMP structures by blocking Wnt5a or by knockdown of MCAM interfered with cell migration as measured by the rate of movement.
FIGURE 5:

Effect of blocking WRAMP structures on cell speed and wound healing. To measure cell speed, (A) WM239a and (B) A375 cells were transfected with PB-H2B-mCherry and seeded on glass-bottom 96-well plates, followed by scratch wounding. Every cell that migrated into the wound was tracked using a cell-tracking algorithm, calculating movement speed for individual cells. Bars show the mean and SEM from three wells (>360 cells/condition for WM239a cells and >180 cells/condition for A375 cells). *p < 0.01 and **p < 0.005. The p values were calculated using standard two-tailed Student’s t test. (C–F) Scratch wound assays were used to measure of the effects of suppressing Wnt5a signaling and MCAM expression on wound healing. Cells were imaged using bright-field microscopy in plastic 96-well dishes, and four wells were measured for each condition at various time points. We quantified (C) WM239a and (D) A375 cells in the presence of 10% FBS and (E) WM239a and (F) A375 cells in the absence of serum. Two-way analysis of variance was used to determine the significance between the treatments and nontargeting siRNA control across the time points. ‡Treatment is significantly different compared with nontargeting control (NTC) siRNA with p < 0.0001.
We then quantified effects of inhibiting WRAMP structures in scratch-wound healing assays, measuring the percentage of wound closure versus time. In both WM239a and A375 cells, the combination of Box 5 and Wnt5a-siRNA significantly suppressed the rate of wound healing in the presence of 10% fetal bovine serum (FBS; Figure 5, C and D). The suppression was even more pronounced under serum-free conditions (Figure 5, E and F). Knockdown of MCAM with siRNA also slowed wound healing under each condition but to a lesser extent than loss of Wnt5a signaling (Figure 5, C–F). Because the rate of wound closure can be enhanced by cell doubling as well as cell migration, controls were needed to examine effects of various treatments on cell proliferation. Neither WM239a nor A375 cells were affected by recombinant Wnt5a (rWnt5a), MCAM-siRNA, Box 5, Wnt5a-siRNA, or the Box 5+Wnt5a-siRNA combination in measurements of cell proliferation (Supplemental Figure S3, E and F). We conclude that inhibiting the ability of cells to form WRAMP structures by suppressing Wnt5a signaling or MCAM expression significantly interferes with cell migration as measured by movement speed and wound healing.
Cells move longer distances in the presence of WRAMP structures
Cells migrating for several hours revealed a periodicity in their formation of rear-directed WRAMP structures. Thus each cell could be segmented into periods when WRAMP structures were present in consecutive frames and periods when they were absent. Representative examples of WM239a and A375 cells are shown in Figure 6, A and B, where alternating periods when WRAMP structures are present (WRAMP+, orange) or absent (WRAMP–, blue) are indicated over time and plotted against the distance traveled from the origin. We then plotted individual trajectories describing the distance and direction of cell movement during WRAMP+ and WRAMP– segments in 30 cells, centering the start site for each segment at 0, 0 (Figure 6, C and D). Remarkably, cells migrated with significantly longer trajectories during WRAMP+ segments than with WRAMP- segments in both cell types. Histograms plotting segment period lengths over all cells revealed a median duration of 45 min for WM239a cells and 60 min for A375 cells but with little bias overall in the length of time when WRAMP structures were present versus when they were absent (Figure 6, E and F). Thus the longer trajectories for cell movement during WRAMP+ periods were not explained by differences in time duration between WRAMP+ versus WRAMP– segments.
FIGURE 6:

Migrating cells fluctuate between periods when WRAMP structures are present and periods when they are absent. (A, B) Trajectories for single representative (A) WM239a and (B) A375 cells, showing distance from origin vs. time. Periods when WRAMP structures are present (WRAMP+, orange) and absent (WRAMP–, blue) are indicated in trajectory plots and projections at the top, where each point represents one frame. (C, D) Cell trajectories for 250 WRAMP+ and 250 WRAMP– segments, each observed in (C) 28 WM239a and (D) 28 A375 cells. Cell position at the start of each segment starts at 0, 0 and follows trajectories to the end of the segment. (E, F) Lengths of periods in minutes for WRAMP+ and WRAMP– segments from (E) 30 WM239a and (F) 30 A375 cells. Histograms show similar period lengths between WRAMP+ and WRAMP– segments. All cells were treated with recombinant Wnt5a at the start of the experiment.
WRAMP structures coincide with faster cell speed and greater persistence of directional migration
The longer trajectories observed during WRAMP+ periods suggested potential effects on speed of movement. Cell speed was measured by live tracking of WRAMP+ and WRAMP– segments, measuring path distance over time for each segment. WM239a and A375 cells respectively showed 1.7- and 5-fold faster speeds during WRAMP+ segments than with WRAMP– segments (Figure 7, A and B). Therefore faster speed explains, at least in part, the longer trajectories that occurred during WRAMP+ segments (Figure 6, C and D).
FIGURE 7:

Quantitative measurements of cell movement. (A, B) Cell speed during WRAMP+ (WR+) and WRAMP– (WR–) segments were calculated in (A) WM239a (30 cells) and (B) A375 cells (30 cells). Values report mean and SEM, ***p < 0.0001. The p values were calculated using standard two-tailed Student’s t test. (C, D) Directionality ratio calculated at the final time point in WRAMP+ and WRAMP– segments; mean and SEM for 30 (C) WM239a and (D) A375 cells. The p values were calculated using standard two-tailed Student’s t test, ***p < 0.0001. (E, F) Directionality ratio calculated vs. time for WRAMP+ and WRAMP– segments in (E) WM239a and (F) A375 cells. (G, H) Direction autocorrelation of trajectories for WRAMP+ and WRAMP– segments at least 12 frames long in 30 (G) WM239a and (H) A375 cells. Error bars indicate SEM. All cells were treated with recombinant Wnt5a at the start of the experiment.
We also evaluated directional persistence of movement, measured in two ways. Directionality ratio is a metric commonly used to measure the “straightness” of a path. It is calculated as the ratio of the straight-line distance from the origin to the endpoint (d) divided by the total path distance traveled (D). The directionality ratio is equal to 1 for a cell that moves continuously in a straight line and approaches 0 for a cell that undergoes movement in random directions. Figure 7, C and D, shows that the average directionality ratio measured between the first and final time points in each period is greater for WRAMP+ than for WRAMP– segments in WM239a and A375 cells. We also calculated the average directionality ratio as a function of time during WRAMP+ and WRAMP– segments. Over time, the directionality ratio for movement typically decreases as cells change direction. However, during WRAMP+ segments, the directionality ratio decayed much more slowly than during WRAMP– segments (Figure 7, E and F). The results indicate that greater directional persistence of migration is associated with the presence of the WRAMP structure.
A second metric for directional persistence is the direction autocorrelation, which measures autocorrelation with varying time intervals between the cosines of the angle differences between vectors for directional movement. A direction autocorrelation coefficient equal to 1 reports parallel vectors for movement and indicates no change in angle or direction, whereas an autocorrelation coefficient equal to 0 indicates random movement (Gorelik and Gautreau, 2014). Typically, the direction autocorrelation coefficient starts at 1 at short time intervals and decays to 0 as cells undergo changes in direction with increasing time intervals. We observed significantly greater direction autocorrelation during WRAMP+ segments than with WRAMP– periods in WM239a cells (Figure 7G). Similar behavior was observed in A375 cells, which showed even greater differences between WRAMP+ and WRAMP– segments (Figure 7H).
The directionality ratio measures net path length over total path length without regard to angle, whereas direction autocorrelation measures the degree of variation in vector angle independently of how rapidly or slowly the cells move. Therefore, these two measurements quantify fundamentally different aspects of directional persistence. The fact that both showed significant differences between WRAMP+ and WRAMP– segments revealed an unambiguous enhancement of directional persistence in cell movement when WRAMP structures were present.
Cells lose directional persistence during periods when WRAMP structures are absent
Finally, we asked whether the total distance migrated from the origin correlated with the percentage of time in which cells displayed WRAMP structures. Examination of individual WM239a or A375 cells showed wide variations in WRAMP+ periods, ranging from 20 to 90% of total migration time (Figure 8, A and B). Unexpectedly, however, the distance traveled from the origin (d) was only weakly correlated with the percentage of time that cells formed WRAMP structures (Figure 8, A and B). Thus, despite the clear association of WRAMP structures with persistent directional movement, the overall degree of translocation could not be predicted from a cell’s propensity to form WRAMP structures.
FIGURE 8:

Cells change direction during WRAMP– segments. (A, B) Plots of distance from origin over the entire cell trajectory vs. fraction of time with WRAMP+ segments show little correlation in 30 (A) WM239a and (B) A375 cells. (C) Example of cell trajectory, showing changes in direction between consecutive WRAMP+ segments (arrows), measured by angle Φ. (D, E) Histogram of the angle Φ, showing the changes in direction between >600 consecutive WRAMP+ segments for (D) 30 WM239a and (E) 30 A375 cells. Bars represent the mean and range for two biological replicates each tracking 30 cells (>600 values). (F) Confocal live-cell imaging of MCAM-GFP in A375 and WM239a cells (also see Supplemental Movie S11) shows changes in cell direction during periods when WRAMP structures are absent. The dynamic movement of the WRAMP structure allows intracellular repositioning within the cell before migration in a new direction. Yellow arrowheads indicate WRAMP structures scored by MCAM-GFP; white arrows indicate direction of cell movement. Scale bar, 20 μm; times in hours:minutes. All cells were treated with recombinant Wnt5a at the start of the experiment. (G, H) Histograms showing the times at which WRAMP structures reappear relative to the start of migration in a new direction in (G) WM239a (n = 120 cases across 30 cells; frames acquired every 5.0 min) and (H) A375 cells (n = 123 cases across 30 cells; frames acquired every 6.5 min). Red indicates cells where WRAMP structures reappeared before the start of migration. Gray indicates WRAMP structures reappearing in the same frame in which cells begin migrating. Blue indicates WRAMP structures reappearing after cells begin migrating.
We asked whether this finding might signify that cells change direction during the WRAMP– periods, which occur between the WRAMP+ segments. This was consistent with observations in Figure 6, C and D, in which the directions taken in cell trajectories over many WRAMP+ segments appeared to be random, even though these WRAMP+ segments showed greater directionality ratio and direction autocorrelation than their intervening WRAMP– segments (Figure 7, C–H). We therefore asked whether cells change direction between consecutive WRAMP+ segments, by following their trajectories and measuring the vector angles between consecutive WRAMP+ segments, as illustrated by the representative cell in Figure 8C. We then plotted the histograms of angle changes between consecutive WRAMP+ segments for both WM239a and A375 cells (Figure 8, D and E). In each cell type, approximately half of the cases showed angles up to 90°, mostly between 0 and 45°, indicating similar trajectories from one WRAMP+ segment to the next. However, the other half of the cases showed angles >90°, mostly between 135 and 180°, indicating significant changes in direction between consecutive WRAMP+ segments. Therefore the frequency with which cells change directions is higher during WRAMP– periods. This explains the apparent random direction of cell movement over the full time course of migration.
The behavior of cells during WRAMP– segments is shown for representative WM239a and A375 cells. In each case, the cell changed directions (arrows) during the period when WRAMP structures were not observable (Figure 8F). In each case, the WRAMP structures appeared to disassemble and then reappear in a new location. In other words, dynamic movement of MCAM-GFP occurred that reflected intracellular movement of the WRAMP structures and accompanied each change in direction. Thus the cells did not appear to change direction by turning, but instead changed their polarization of WRAMP structures by up to 180°. After inspecting many cells, we noticed that the localization of MCAM-GFP generally shifted before or at the time of cell movement in a new direction, as if the newly formed WRAMP structure determined the new cell rear. This was quantified by monitoring cells during WRAMP– periods, followed by the reappearance of WRAMP structures and subsequent cell movement. We recorded the time at which WRAMP structures reappeared relative to the time that migration in a new direction began (Figure 8, G and H), taking a frame every 5 min (WM239A) or 6.5 min (A375). The results showed that in at least 90% of cases, WRAMP structures reappeared at least one frame before the start of directional migration. Therefore WRAMP structures precede cell movement by at least 5 min, 90% of the time. The findings are consistent with the hypothesis that the location at which WRAMP structures form may establish the new cell rear and, in doing so, determine the subsequent direction of cell migration.
DISCUSSION
Our study presents a quantitative characterization of the WRAMP structure, which coordinates the anisotropic localization of MCAM, F-actin, and myosin IIB in a Wnt5a-induced manner. WRAMP structures form dynamically and are associated with actomyosin contraction and membrane retraction (Witze et al., 2013). The results establish a function for the WRAMP structure in cells undergoing prolonged migration by showing that it polarizes stably at the rear and is strongly associated with enhanced speed and persistence of directional movement. Disrupting WRAMP structures by suppressing Wnt5a signaling or MCAM expression significantly interferes with cell migration, as shown by reductions in cell speed and the rate of wound healing. We propose that the polarization of the WRAMP structure promotes membrane retraction and nucleokinesis to help drive directional migration. We hypothesize that this could occur by promoting a sustained actomyosin-dependent force generation at the rear or alternatively through myosin IIB cross-linking functions (Vicente-Manzanares et al., 2009; Cramer, 2010).
Our findings reveal new insight into mechanisms controlling the persistence of directional migration. Current models involve the stabilization of lamellipodia and front-end protrusions to determine the orientation of migration (Petrie et al., 2009; Krause and Gautreau, 2014). Such mechanisms for “lamellipodia persistence” are driven by F-actin polymerization at the leading edge and occur in response to external gradients of chemotactic signaling molecules guiding the direction of chemotaxis (George et al., 2013; Loosley et al., 2015). By contrast, our characterization of the WRAMP structure shows a clear significance of the rear actomyosin machinery persistence of directional migration. Of importance, our results suggest an intrinsic signaling mechanism for cell directionality in response to Wnt5a, given that the cells in our experiments were plated on collagen and migrated in the absence of directionality cues. Nonetheless, cells display directional persistence when WRAMP structures form, and cells turn in random directions when WRAMP structures are absent. This supports a clear role of the WRAMP structure in the rear machinery for directional migration.
The periodicity of WRAMP structure formation provided us with a unique opportunity to characterize differences in cell movement when WRAMP structures were present and when they were absent. Strikingly, whereas cells displayed directional persistence in periods when WRAMP structures were present, they changed directions randomly when WRAMP structures were absent. Of importance, the cells did not change direction by slow turns in their trajectory. Instead, they appeared to pause their locomotion when WRAMP structures disassembled and, during this pause, reassembled the WRAMP structure at a different cellular location before migrating in a new direction with the WRAMP structure at the new rear. Thus the ability of WRAMP structures to form dynamically within cells may provide a mechanism for changing direction during extended migration. Further studies are needed to understand the mechanisms that guide the localization of WRAMP structures. For example, an intriguing possibility is that endosomal vesicles interacting with cytoskeletal components underlie the movement of WRAMP structures, influencing their rear positioning within cells. We also noted previously that in the presence of a gradient of CXCL12/SDF1, Wnt5a increases the positioning of WRAMP structures at the opposite pole from Golgi/ER–Golgi intermediate compartment markers, which report the anterior end of cells (Witze et al., 2008). This raises the possibility that external cues can control the positioning of WRAMP structures, possibly through leading-edge mechanisms. However, our findings in this study show consistent rear polarity of WRAMP structures in the absence of directional cues. Thus WRAMP structures may provide a mechanism allowing the orientation of cells and direction of cell movement to be guided from the rear.
Finally, our study shows clear evidence for WRAMP structures during cell migration in nonmelanoma as well as melanoma cell types. Our primary marker for WRAMP structures is MCAM, a protein known to promote cell invasion and metastasis in cancer cells (McGary et al., 2002; Iiizumi et al., 2008; Lei et al., 2015). However, we believe that WRAMP structures may not be limited to cells expressing MCAM, as unpublished observations show other cell adhesion molecules that endocytose in response to Wnt5a and localize at WRAMP structures. The cell types in which we do observe WRAMP structures in each case display a Wnt5a dependence of migration and invasion (Michl et al., 2005; Wang and Chen, 2010; Zhu et al., 2014a,b; Webster et al., 2015). We speculate that the force generation at WRAMP structures that leads to directional migration may also promote cell extravasation from primary tumors to metastatic sites. Indeed, immunohistochemical staining of human primary melanoma specimens show that whereas Wnt5a is weakly expressed within tumors, its levels increase at tumor margins (Weeraratna et al., 2002). This opens the possibility that the Wnt5a-stimulated WRAMP structures could provide a means to explain how Wnt5a promotes invasion and metastasis in several cancer types. Targeting metastasis at the level of Wnt5a is not simple, given that this ligand functions as a tumor suppressor in certain cancer types (McDonald and Silver, 2009). Thus the WRAMP structure mechanism may provide an alternative target for therapeutics against metastasis.
In summary, our findings provide compelling evidence that in addition to leading-edge lamellipodia formation and membrane protrusion, rear-directed mechanisms control the ability of cells to migrate directionally. The behavior of the WRAMP structure establishes a connection between rear-directed polarization of F-actin and myosin, which may lead to actomyosin cross-linking and/or force generation in a manner that enhances the speed, orientation, and persistence of directional migration through extracellular ligands that regulate cell polarity, such as Wnt5a. Further studies are needed to elucidate the details of WRAMP structure formation and function. We propose that the WRAMP structure provides a mechanism for a cell to maintain ongoing rear retraction as a cell moves forward. Integrating rear-directed mechanisms with events at the leading edge will lead to a more complete understanding of the mechanisms that determine the direction of cell migration.
MATERIALS AND METHODS
Materials
Plasmids for mammalian expression included MCAM-GFP, which was previously described (Witze et al., 2008); p-mCherry-N1, a gift of Esther Braselmann and Amy Palmer (University of Colorado, Boulder); and mCherry-MyosinIIB-N-18 and mCherry-LifeAct-7, from Michael Davidson’s lab (Florida State University; Addgene plasmids 55107 and 54491). PB-H2B-mCherry was a gift from Maria Lo and Amy Palmer (University of Colorado, Boulder) and was generated by restriction enzyme cloning of H2B-mCherry into the entry vector PB510-B1 from Systems Bio. Antibodies used included anti-MCAM (sc-18837; Santa Cruz Biotechnology) and anti-myosin IIB (M7939; Sigma-Aldrich); and Alexa Fluor 594 donkey anti-rabbit IgG (H+L) and Alexa Fluor 488 donkey anti-mouse IgG (H+L) (A21207 and A21202; Life Technologies). Coumarin-labeled phalloidin was obtained from Sigma-Aldrich (P2495); recombinant purified Wnt5A was obtained from R&D Systems (645-WN); and Box 5 was obtained from EMD-Millipore (Calbiochem 681673). ON-TARGETplus siRNA SMARTpool for human WNT5A (7474) and human MCAM (4162) and siGENOME Non-targeting siRNA pool #2 were purchased from Dharmacon. The antibodies used for Western blot analysis were anti-MCAM (13475S; Cell Signaling), anti-Wnt5a/b (253S; Cell Signaling), anti–glyceraldehyde-3-phosphate dehydrogenase (5174S; Cell Signaling), anti-nucleolin (sc-8031; Santa Cruz Biotechnology), anti-mouse IgG horseradish peroxidase (HRP) linked (7076; Cell Signaling), and anti-rabbit IgG HRP linked (7074S; Cell Signaling).
Cell culture
WM239a cells were a kind gift from Meenhard Herlyn (Wistar Institute). A375 and HT1080 cells were obtained from the American Type Culture Collection. All of our cell lines are routinely tested for mycoplasma infection using a PCR-based mycoplasma detection kit. WM239A cells and A375 cells were maintained in RPMI plus 10% (vol/vol) FBS at 37°C with 5% CO2, and HT1080 cells were maintained in DMEM plus 10% (vol/vol) FBS at 37°C with 5% CO2. Cells were transfected using the Neon electroporation device (Thermo Fisher Scientific) following the manufacturer’s standard protocol and 2 µg of each cDNA plasmid (1200 V × 20 ms × 2 pulses for WM239A cells; 1300 V × 10 ms × 3 pulses for A375 cells; and 950 V × 25 ms × 3 pulses for HT1080 cells). Cells were treated with 250 ng/ml Wnt5a for 30 min at 37°C with 5% CO2 or with 100 µM Box 5 (0.1% dimethyl sulfoxide [DMSO]) for at least 12 h before live-cell imaging or fixation and immunostaining. For knockdown experiments, cells were transfected with 100 nM siRNA either using DharmaFECT1 lipid transfection or by electroporation as described for cDNA. For lipid transfection, 16 µl of DharmaFECT1 reagent was incubated with siRNA in Opti-MEM for 30 min at room temperature and then added dropwise to 1 × 106 cells in suspension. After 8 h at 37°C and 5% CO2, the transfection mixture was replaced with full medium. Efficiency of protein knockdown was evaluated by Western blotting.
Cell proliferation assays
Cell proliferation was measured using the CellTiter-Glo luminescent cell viability assay (Promega) according to the manufacturer’s protocol. Cells were seeded in a 96-well plate (5000 cells/well for WM239a; 2500 cells/well for A375). Immediately before each time point, the cell culture medium was removed from each well and replaced with 100 µl of fresh medium. Then 100 µl of CellTiter-Glo reagent was added to each well, followed by shaking for 2 min, incubation in the dark at room temperature for 15 min, and luminescence recording using a BioTek Synergy H1 hybrid plate reader. Each sample was performed with six replicate wells, and standard curves were generated using 0, 250, 500, 1000, 2000, 4000, 8000, and 16,000 cells, which were seeded and allowed to adhere for 8 h before measurement. All values were normalized to an initial time point measured 8 h after seeding for each condition. Cells were either transfected with 100 nM siRNA or treated with 250 ng/ml Wnt5a, 100 µM Box 5, or 0.1% DMSO.
Microscopy
Cells analyzed by indirect immunofluorescence were seeded on glass coverslips for 18–24 h, followed by removal of medium, addition of fresh medium with or without 250 ng/ml Wnt5a, and further incubation at 37°C with 5% CO2 for 30 min. Cells were then washed with phosphate-buffered saline (PBS), fixed with PBS containing 4% paraformaldehyde (EM grade; Electron Microscopy Sciences), 5 mM MgCl2, 10 mM ethylene glycol tetraacetic acid, and 120 mM sucrose for 8 min at room temperature and then permeabilized with PBS plus 0.2% Triton X-100 for 5 min at room temperature. Cells were then blocked with PBS plus 5% bovine serum albumin (BSA) and then incubated with primary antibody for 1 h, secondary antibody (1:1000) for 1 h, and PBS plus phalloidin in 1% BSA for 40 min, all at room temperature. Samples were mounted using CitiFluor medium (Electron Microscopy Sciences) and sealed using clear nail polish. Images were collected using an Olympus IX-81 microscope at 60× or a Nikon A1R laser scanning confocal microscope at 100×.
Live-cell imaging was performed using a Nikon A1R laser scanning confocal microscope or a Nikon Ti-E spinning-disk confocal microscope, each equipped with an environmental chamber set to 37°C, 5% CO2, and 70% humidity. Cells were seeded on glass-bottom dishes for 12–18 h, washed with phenol red–free medium containing 10% (vol/vol) FBS, and incubated with phenol red–free medium plus 10% FBS with or without 250 ng/ml Wnt5a. For scratch-wound assays, cells were seeded in a confluent monolayer in glass-bottom 96-well plates coated with 55 µg/ml bovine collagen (Pure Col type I collagen, 5005-B; Advanced BioMatrix), allowed to adhere for 6–8 h at 37°C with 5% CO2, and then scratched with a 96-pin device to generate wounds in each well. Cells were washed three times with PBS and twice with phenol red–free medium plus 10% FBS and then incubated in phenol red–free medium plus 10% FBS with or without 250 ng/ml Wnt5a. The glass-bottom plates were treated with a very thin coating of collagen (not a collagen gel) according to the manufacturer’s protocol, with slight modification. Specifically, a collagen matrix was prepared by mixing 1 ml of 5× RPMI, 0.5 ml of FBS, and 3.5 ml of Pure Col and neutralized with 5 µl of 10 N NaOH. This collagen matrix was then further diluted to 55 µg/ml with cell culture medium and added to the glass-bottom plates with enough volume to coat the surface of the wells (50 µl/well in a 96-well plate) and incubated for 1–16 h. The coating matrix was aspirated from the wells before seeding of cells.
Quantitative measurements
Fixed cells were scored for the presence of WRAMP structures, as measured by the appearance of MCAM polarized at one end of the long axis of the cell with fluorescence intensity threefold or greater than the surrounding background. Quantification of F-actin and/or myosin IIB at WRAMP structures was scored by their elevated fluorescence at or proximal to polarized MCAM. Movements of live cells (for Figures 4 and 6–8) were tracked manually using the Manual Tracking plug-in for ImageJ, where the coordinates for cell position in each frame were placed at the cell center where long and short axes meet (using the mCherry channel). The tracked cells were then scored in each frame for the presence or absence of a WRAMP structure, as measured by MCAM-GFP polarized with intensity threefold or greater compared with surrounding background. When present, WRAMP structures were tracked for position by marking the approximate center of the structure using the Manual Tracking plug-in. The only criteria for selecting cells were that they 1) migrated into the wound and 2) traveled at least two cell lengths during the course of the experiment. Once these criteria were met, cells were chosen at random across multiple wells and multiple biological replicates. In 100% of cases, WRAMP structures were detectable.
The polarity of WRAMP structures was calculated from the angle between a vector reporting the positioning of the WRAMP structure and a vector reporting the direction of cell migration. The positioning vector was determined by the line drawn from the x, y-coordinates for the WRAMP structure in each frame n to the x, y-coordinates for the cell center in the same frame n. The migration vector was determined from the x, y-coordinates from the cell center in each frame n to the cell center in the next frame, n + 1. The angle between the two vectors was calculated as follows, where A is the positioning vector and B is the migration vector:
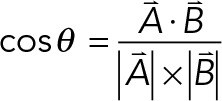 (1) (1)
|
Distances that each cell moved from the origin were calculated from the x, y-coordinates of the cell center at the start (frame 0) and each frame n. WRAMP+ and WRAMP– segments were scored over the full trajectory, based on consecutive frames when WRAMP structures were present and consecutive frames when they were absent. Sorting functions and other basic calculations were performed using MatLab software. The trajectories for these periods were plotted using the Plot_At_Origin function in DiPer software (Gorelik and Gautreau, 2014).
The directionality ratio, speed, and direction autocorrelation were also calculated using DiPer software (Gorelik and Gautreau, 2014). The directionality ratio was calculated from the distance from origin normalized to the full trajectory distance using the following equation, where d is the straight-line distance between the start and end points and D is the total path distance of the trajectory:
 (2) (2)
|
The direction autocorrelation coefficients were computed with overlapping time intervals using
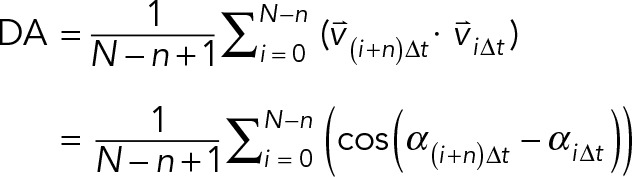 (3) (3)
|
where DA is the average direction autocorrelation coefficient for a given cell at step size n, N is the total number of displacements, and α is the angle at each time point of the trajectory. We plotted population averages per given time interval for C cell trajectories, calculating this DA from
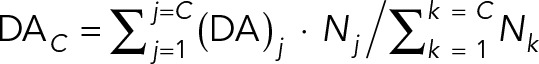 (4) (4)
|
Cell-tracking algorithm
An algorithm was developed to track cells and was used in Figure 5, A and B. These experiments reported all of the cells that were detected in the wound. Cell tracking was carried out in two steps: first, each frame of the movie was processed to segment (identify) the cells. The positions of the identified cells were then linked between consecutive frames to form tracks. During the segmentation step, each frame in the movie was first contrast enhanced by contrast-limited adaptive histogram equalization (Zuiderveld, 1994). The grayscale value corresponding to the upper 95th percentile of the enhanced intensity histogram was then used to threshold the image and generate the cell mask. The centroid of each identified cell in the mask was then calculated using a built-in algorithm in MATLAB, and a linear assignment algorithm (Jaqaman et al., 2008) was used to link cell positions between consecutive frames. In the linear assignment framework, a cost matrix was calculated for each frame of the movie. The total cost matrix consisted of the cost to link a cell in frame T to a new position in frame T + 1, a cost to link the cell in frame T to nothing (e.g., if the cell drifted out of the frame of view or was not correctly segmented), and the cost to start a new track. The linking costs were defined to equal the Euclidean distance between the cell position at frame T to all new positions at frame T + 1. To prevent physically impossible assignments, the maximum distance was set to 100 pixels, and the cost was set to infinity for distances above this point. The cost for the alternative outcomes was calculated as described (Jaqaman et al., 2008). Each cell was then assigned to one of these outcomes by solving the cost matrix for the global minimum cost using the Jonker–Volgenant algorithm (Jonker and Volgenant, 1987).
Wound-healing assays
Cells were seeded in a confluent monolayer in plastic-bottom 96-well plates coated with a thin layer of bovine collagen (55 µg/ml), allowed to adhere for 6–8 h at 37°C and 5% CO2, and then scratched with a 96-pin device (Essen Bioscience) to generate uniform wounds in each well. Cells were washed three times with PBS and twice with medium ± 10% FBS. Bright-field imaging was performed using a Nikon A1R microscope in wide-field mode equipped with an environmental chamber set to 37°C, 5% CO2, and 70% humidity. Four replicate wells were imaged for each condition. The wound area was measured at each time point using standard tools in ImageJ. The percentage of wound healing was calculated using
 (5) (5)
|
Supplementary Material
Acknowledgments
We are greatly indebted to Joseph Dragavon, Director of the BioFrontiers Institute Advanced Light Microscopy Core, for assistance with confocal microscopy. Instruments of the Core facility were obtained with the generous support of a NIST-CU Cooperative Agreement Award (70NANB15H226) and the Howard Hughes Medical Institute. We also thank Cassidy Thompson (BioFrontiers IT Core) for advice on computational analysis and Andrew Kavran for advice on statistical analysis. This work was supported by American Cancer Society Postdoctoral Fellowship Award PF-14-036-01-CSM (M.K.C.) and National Institutes of Health Grant R01GM105997 (N.G.A.).
Abbreviations used:
- MCAM
melanoma cell adhesion molecule
- WRAMP
Wnt5a-receptor-actin-myosin-polarity
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-12-0875) on June 7, 2017.
REFERENCES
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Bowerman B. Cell signaling. Wnt moves beyond the canon. Science. 2008;320:327–328. doi: 10.1126/science.1157590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer LP. Forming the cell rear first: breaking cell symmetry to trigger directed cell migration. Nat Cell Biol. 2010;12:628–632. doi: 10.1038/ncb0710-628. [DOI] [PubMed] [Google Scholar]
- Cramer LP. Mechanism of cell rear retraction in migrating cells. Curr Opin Cell Biol. 2013;25:591–599. doi: 10.1016/j.ceb.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Gao B. Wnt regulation of planar cell polarity (PCP) Curr Top Dev Biol. 2012;101:263–295. doi: 10.1016/B978-0-12-394592-1.00008-9. [DOI] [PubMed] [Google Scholar]
- George SP, Chen H, Conrad JC, Khurana S. Regulation of directional cell migration by membrane-induced actin bundling. J Cell Sci. 2013;126:312–326. doi: 10.1242/jcs.116244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Orte E, Saenz-Narciso B, Moreno S, Cabello J. Multiple functions of the noncanonical Wnt pathway. Trends Genet. 2013;29:545–553. doi: 10.1016/j.tig.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Gorelik R, Gautreau A. Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protoc. 2014;9:1931–1943. doi: 10.1038/nprot.2014.131. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A. Cell polarization mechanisms during directed cell migration. Nat Cell Biol. 2005;7:336–337. doi: 10.1038/ncb0405-336. [DOI] [PubMed] [Google Scholar]
- Iiizumi M, Liu W, Pai SK, Furuta E, Watabe K. Drug development against metastasis-related genes and their pathways: a rationale for cancer therapy. Biochim Biophys Acta. 2008;1786:87–104. doi: 10.1016/j.bbcan.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenei V, Sherwood V, Howlin J, Linnskog R, Safholm A, Axelsson L, Andersson T. A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion. Proc Natl Acad Sci USA. 2009;106:19473–19478. doi: 10.1073/pnas.0909409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker R, Volgenant A. A shortest augmenting path algorithm for dense and sparse linear assignment problems. Computing. 1987;38:325. [Google Scholar]
- Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Wnt5a: its signalling, functions and implication in diseases. Acta Physiol (Oxf) 2012;204:17–33. doi: 10.1111/j.1748-1716.2011.02294.x. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15:577–590. doi: 10.1038/nrm3861. [DOI] [PubMed] [Google Scholar]
- Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T, Yasui W, Kikuchi A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006;66:10439–10448. doi: 10.1158/0008-5472.CAN-06-2359. [DOI] [PubMed] [Google Scholar]
- Lei X, Guan CW, Song Y, Wang H. The multifaceted role of CD146/MCAM in the promotion of melanoma progression. Cancer Cell Int. 2015;15:3. doi: 10.1186/s12935-014-0147-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loosley AJ, O’Brien XM, Reichner JS, Tang JX. Describing directional cell migration with a characteristic directionality time. PLoS One. 2015;10:e0127425. doi: 10.1371/journal.pone.0127425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey LM, Macara IG. Signaling pathways in cell polarity. Cold Spring Harb Perspect Biol. 2012;4:a009654. doi: 10.1101/cshperspect.a009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SL, Silver A. The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009;101:209–214. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary EC, Lev DC, Bar-Eli M. Cellular adhesion pathways and metastatic potential of human melanoma. Cancer Biol Ther. 2002;1:459–465. doi: 10.4161/cbt.1.5.158. [DOI] [PubMed] [Google Scholar]
- Michl P, Ramjaun AR, Pardo OE, Warne PH, Wagner M, Poulsom R, D’Arrigo C, Ryder K, Menke A, Gress T, Downward J. CUTL1 is a target of TGF(beta) signaling that enhances cancer cell motility and invasiveness. Cancer Cell. 2005;7:521–532. doi: 10.1016/j.ccr.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Mladinich KM, Huttenlocher A. WRAMPing up calcium in migrating cells by localized ER transport. Dev Cell. 2013;26:560–561. doi: 10.1016/j.devcel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita M, Enomoto M, Yamagata K, Minami Y. Cell/tissue-tropic functions of Wnt5a signaling in normal and cancer cells. Trends Cell Biol. 2010;20:346–354. doi: 10.1016/j.tcb.2010.03.001. [DOI] [PubMed] [Google Scholar]
- O’Connell MP, Fiori JL, Xu M, Carter AD, Frank BP, Camilli TC, French AD, Dissanayake SK, Indig FE, Bernier M, et al. The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene. 2010;29:34–44. doi: 10.1038/onc.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Zhang X, Yu H, Wu D, Zheng J. Wnt5a as a predictor in poor clinical outcome of patients and a mediator in chemoresistance of ovarian cancer. Int J Gynecol Cancer. 2011;21:280–288. doi: 10.1097/IGC.0b013e31820aaadb. [DOI] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704, 1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Ripka S, Konig A, Buchholz M, Wagner M, Sipos B, Kloppel G, Downward J, Gress T, Michl P. WNT5A–target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis. 2007;28:1178–1187. doi: 10.1093/carcin/bgl255. [DOI] [PubMed] [Google Scholar]
- Schwartz MP, Rogers RE, Singh SP, Lee JY, Loveland SG, Koepsel JT, Witze ES, Montanez-Sauri SI, Sung KE, Tokuda EY, et al. A quantitative comparison of human HT-1080 fibrosarcoma cells and primary human dermal fibroblasts identifies a 3D migration mechanism with properties unique to the transformed phenotype. PLoS One. 2013;8:e81689. doi: 10.1371/journal.pone.0081689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarski DC, Yang-Snyder J, Busa WB, Moon RT. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev Biol. 1997;182:114–120. doi: 10.1006/dbio.1996.8463. [DOI] [PubMed] [Google Scholar]
- Tovari J, Futosi K, Bartal A, Tatrai E, Gacs A, Kenessey I, Paku S. Boyden chamber-based method for characterizing the distribution of adhesions and cytoskeletal structure in HT1080 fibrosarcoma cells. Cell Adh Migr. 2014;8:509–516. doi: 10.4161/cam.28734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chen H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res. 2010;30:4537–4545. [PubMed] [Google Scholar]
- Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 2015;28:184–195. doi: 10.1111/pcmr.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1:279–288. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- Wei W, Li H, Li N, Sun H, Li Q, Shen X. WNT5A/JNK signaling regulates pancreatic cancer cells migration by phosphorylating paxillin. Pancreatology. 2013;13:384–392. doi: 10.1016/j.pan.2013.05.008. [DOI] [PubMed] [Google Scholar]
- Witze ES, Connacher MK, Houel S, Schwartz MP, Morphew MK, Reid L, Sacks DB, Anseth KS, Ahn NG. Wnt5a directs polarized calcium gradients by recruiting cortical endoplasmic reticulum to the cell trailing edge. Dev Cell. 2013;26:645–657. doi: 10.1016/j.devcel.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. 2008;320:365–369. doi: 10.1126/science.1151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HH, Zhu XY, Zhou MH, Cheng GY, Lou WH. Effect of WNT5A on epithelial-mesenchymal transition and its correlation with tumor invasion and metastasis in nasopharyngeal carcinoma. Asian Pac J Trop Med. 2014a;7:488–491. doi: 10.1016/S1995-7645(14)60080-8. [DOI] [PubMed] [Google Scholar]
- Zhu N, Qin L, Luo Z, Guo Q, Yang L, Liao D. Challenging role of Wnt5a and its signaling pathway in cancer metastasis (Review) Exp Ther Med. 2014b;8:3–8. doi: 10.3892/etm.2014.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderveld KJ. Contrast limited adaptive histograph equalization. In: Heckbert PS, editor. Graphic Gems IV. San Diego, CA: Academic Press; 1994. pp. 474–485. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.