

Abstract
To investigate cytogenetic evolution after upfront autologous stem cell transplantation for newly diagnosed myeloma we retrospectively analyzed fluorescence in situ hybridization results of 128 patients with paired bone marrow samples from the time of primary diagnosis and at relapse. High-risk cytogenetic abnormalities (deletion 17p and/or gain 1q21) occurred more frequently after relapse (odds ratio: 6.33; 95% confidence interval: 1.86–33.42; P<0.001). No significant changes were observed for defined IGH translocations [t(4;14); t(11;14); t(14;16)] or hyperdiploid karyotypes between primary diagnosis and relapse. IGH translocations with unknown partners occurred more frequently at relapse. New deletion 17p and/or gain 1q21 were associated with cytogenetic heterogeneity, since some de novo lesions with different copy numbers were present only in subclones. No distinct baseline characteristics were associated with the occurrence of new high-risk cytogenetic abnormalities after progression. Patients who relapsed after novel agent-based induction therapy had an increased risk of developing high-risk aberrations (odds ratio 10.82; 95% confidence interval: 1.65–127.66; P=0.03) compared to those who were treated with conventional chemotherapy. Survival analysis revealed dismal outcomes regardless of whether high-risk aberrations were present at baseline (hazard ratio, 3.53; 95% confidence interval: 1.53–8.14; P=0.003) or developed at relapse only (hazard ratio, 3.06; 95% confidence interval: 1.09–8.59; P=0.03). Our results demonstrate cytogenetic evolution towards high-risk disease after autologous transplantation and underline the importance of repeated genetic testing in relapsed myeloma (EudraCT number of the HD4 trial: 2004-000944-26).
Introduction
Multiple myeloma (MM) is a genetically complex and heterogeneous disease.1,2 At the chromosomal level, MM can be subdivided according to the tumor-initiating event into hyperdiploid and non-hyperdiploid myeloma.3 While hyperdiploid MM is defined by gains of odd-numbered chromosomes, non-hyperdiploid myeloma mainly harbors IGH translocations.3 Patients with high-risk cytogenetic abnormalities at primary diagnosis, such as gain of chromosome 1q21, deletion 17p (del17p) and translocation t(4;14), show inferior outcome after high-dose therapy and autologous stem cell transplantation (ASCT), even in the era of novel agents.4,5 Recent studies have demonstrated that disease progression and refractoriness are caused by secondary genetic events.6,7 It has been discussed that systemic treatment might select pre-existing aggressive subclones or cause secondary genetic events and clonal evolution in patients with recurrent disease. Patients without the aforementioned abnormalities at primary diagnosis might, therefore, relapse with cytogenetically defined high-risk disease. However, there are only limited longitudinal data available elucidating cytogenetic changes in relapsed MM after primary therapy.6,8 We therefore performed a retrospective analysis of patients treated with upfront ASCT with an interphase fluorescence in situ hybridization (FISH) analysis of purified plasma cells at primary diagnosis and relapse. We tried to characterize cytogenetic evolution and studied which abnormalities occur upon relapse. We analyzed whether baseline characteristics, treatment or response affect cytogenetic evolution and investigated the prognostic significance of new high-risk abnormalities.
Methods
Patients and treatment
We identified 128 patients treated with ASCT for newly diagnosed MM with a FISH analysis at initial diagnosis (first FISH) and relapse (second FISH). Forty-four patients were initially enrolled in the prospective HD4 phase III trial of the German-Speaking Myeloma Multicenter Group (GMMG, EudraCT number: 2004-000944-26). Results of the GMMG HD4 trial have been published previously.9 In brief, patients were randomly assigned to either the control arm of three cycles of VAD (vincristine, doxorubicin, dexamethasone) followed by tandem-ASCT and thalidomide maintenance for 2 years, or the experimental arm with three cycles of PAD (bortezomib, doxorubicin, dexamethasone) followed by tandem-ASCT and bortezomib maintenance therapy for 2 years. Furthermore we identified 84 patients who were treated at our institution outside the HD4 trial with comparable induction and maintenance therapy regimens before and after ASCT (non-study patients, NSP). Table 1 summarizes baseline characteristics, treatment and response of both populations. The median time to second FISH analysis from progressive disease was 7.4 months (HD4 patients) and 4.4 months (NSP). The study was performed in accordance with the Declaration of Helsinki after informed consent had been obtained. Retrospective analysis was approved by the local ethics committee.
Table 1.
Baseline characteristics of patients in the HD4 trial and non-study patients.

Fluorescence in situ hybridization studies
FISH analyses were performed on CD138-purified plasma cells as described previously10 at the Institute of Human Genetics at Heidelberg University Hospital. The following probes were used: gain1q21, gain4p16, gain5q35, gain5p15, del6q21, del8p21, gain9q34, gain11q13, gain11q23, del13q14, gain14q32, gain15q22, del17p13, gain19q13, del22q11, t(4;14), t(11;14), t(14;16) and a probe for IGH splits. The threshold for all aberrations was 10%. If an aberration was found in 10–60% of cells it was defined as subclonal; if it was found in more than 60% it was defined as being the major clone. High-risk cytogenetic abnormalities were defined by the presence of del17p, t(4;14) or gain 1q21.
Statistical analysis
An exact McNemar test was used to assess changes between results of FISH assessments at baseline and relapse. A Fisher exact test was used to compare categorical parameters between groups. A multivariable logistic regression model was fitted accounting for baseline karyotype [hyperdiploid or t(11;14)], International Staging System (ISS) stage, time to first progression and to second FISH analysis, age, sex and type of induction therapy to investigate factors associated with the occurrence of high-risk cytogenetic abnormalities at relapse. A Wilcoxon test was used to compare time to progression, which was measured in months and defined as time from the start of chemotherapy to date of progression. Age in years was analyzed as a continuous parameter. The Kaplan-Meier method and log-rank test were used to analyze differences in overall survival times between patients with cytogenetic abnormalities at both FISH assessments and at the first or second FISH only. To analyze effects on overall survival, a multivariable Cox regression model accounting for the same variables as mentioned above was used. P-values <0.05 were considered statistically significant. The last follow-up evaluations were performed in June 2016 (NSP) and November 2015 (HD4). Analyses were carried out with R 3.3 statistical software.
Results
Differences between primary diagnosis and relapse
Table 2 summarizes the frequencies of cytogenetic abnormalities detected at first, second and both FISH examinations. The risk of developing high-risk cytogenetic abnormalities was significantly higher after relapse from ASCT [odds ratio (OR): 6.33; 95% confidence interval (CI): 1.86–33.42; P<0.001]. We found an increased risk of occurrence of del(17p) (OR: 3.4; 95% CI: 1.2–11.79; P=0.02) and gain 1q21 (OR: 16; 95% CI: 2.49–670.96; P<0.001). While none of the patients developed one of the defined IGH translocations [t(4;14), t(11;14) or t(14;16)] after relapse, IGH translocations involving an unknown partner occurred more frequently at second FISH only (OR: 8; 95% CI 1.07–354.98; P=0.039). Hyperdiploidy was observed before and after ASCT in 45 patients (44.6%). Only two patients developed a de novo hyperdiploid karyotype after relapse, while seven patients lost the hyperdiploid karyotype during followup. There were only two patients without detectable cytogenetic abnormalities at primary diagnosis who developed new cytogenetic abnormalities after progression. However, both patients carried a high-risk lesion at relapse (del17p and gain 1q21, respectively).
Table 2.
Number of patients with aberrations at first and/or second fluorescence in situ hybridization studies.

Clonal evolution in patients with high-risk cytogenetics
Analysis of patients with gain 1q21 at first and second FISH (n=48) revealed that most patients had subclones that harbored different copy numbers than the main clone (Figure 1). Five of the 16 patients with de novo gain 1q21 also had subclones with different copy numbers compared to the main clone (Figure 1). In seven of 17 patients with new del17p at relapse, the high-risk cytogenetic abnormality evolved as the major clone (>60% of analyzed cells). In ten patients del17p was detected only in a subclone of cells (10–60%). Figure 1 summarizes changes in copy numbers for patients with gain 1q21 and gives examples of clonal evolution of de novo del17p.
Figure 1.
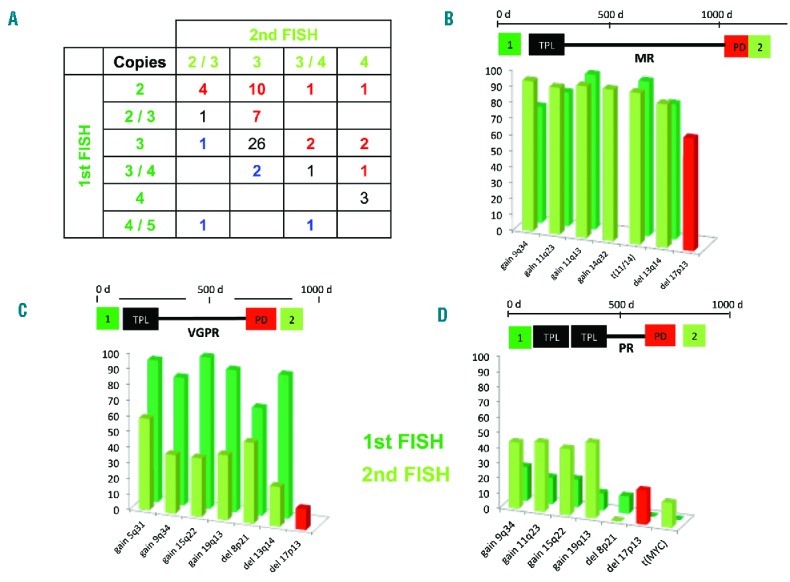
Clonal evolution in patients with gain of 1q21 and del17p. (A) Distribution of copy numbers of chromosome 1q21. Patients with increasing copy numbers after relapse are represented in red, patients with decreasing copy numbers in blue. (B–D) Examples of clonal evolution in three patients with de novo del17p. Panel B illustrates cause of the disease in a patient with minimal response (MR) after autologous transplantation (TPL). After relapse (PD), the second FISH analysis showed a new del17p present in 60% of analyzed plasma cells and no significant changes of the abnormalities already present at initial diagnosis. The patient represented in panel C achieved a very good partial remission (VGPR). The initially present hyperdiploid clone harboring a del13 and del8 was detected in a smaller subset of analyzed plasma cells with a new, subclonal del17p. In panel D, the patient relapsed after tandem TPL and partial remission with a new del17p and MYC translocation. Compared to the patient in panel C the maternal clone could be detected in a larger proportion of plasma cells after relapse.
Correlation with baseline characteristics and treatment response
We could not identify any baseline cytogenetic abnormalities associated with the occurrence of high-risk disease at relapse. Furthermore, there were no significant differences in high ISS stages: at baseline ISS stage 3 was detected in 29.6% of patients without high-risk cytogenetic abnormalities, in 31.2% of patients with high-risk cytogenetic abnormalities at both time points and in 33.3% of patient with high-risk cytogenetic abnormalities only at second FISH (P=0.75). We also observed no differences in rates of very good partial remission or better after ASCT between the three groups (no high-risk cytogenetic abnormalities: 53.1%, high-risk cytogenetic abnormalities at both FISH: 67.3%, high-risk cytogenetic abnormalities only at second FISH: 57.9%). Rates of very good partial remission or better after ASCT were significantly higher in patients with t(4;14) than in patients without this cytogenetic abnormality (92.3% versus 54.7%; P=0.01). Multivariate analysis revealed that patients who relapsed after ASCT and novel agent-based induction therapy had an increased risk of developing high-risk cytogenetic abnormalities compared to patients not treated with novel agents during induction (OR 10.82; 95% CI: 1.65–127.66; P=0.03).
Prognostic significance
When analyzing time to first progression based on cytogenetic abnormalities present at baseline and relapse, we did not observe significant differences. Patients who developed a de novo del17p or gain 1q21 after relapse had similar median times to progression (29.7 and 25.8 months, respectively) as patients with these high-risk cytogenetic abnormalities at both time points (35.5 and 23.7 months, respectively). We observed the same effect when analyzing overall survival in the different groups. Figure 2 shows Kaplan-Meier estimates for overall survival from different landmarks. Multivariate analysis confirmed the observed effects. Patients who developed de novo high-risk cytogenetic abnormalities had the same risk for shorter survival [hazard ratio (HR): 3.06; 95% CI: 1.09–8.59; P=0.03] as patients with high-risk cytogenetic abnormalities already present at baseline (HR: 3.53; 95% CI: 1.53–8.14; P=0.003). Other factors associated with poor outcome in the analyzed cohort were a short time to progression, higher age and higher ISS stage. Patients with a hyperdiploid karyotype at baseline had a better outcome (HR: 0.33; 95% CI: 0.17–0.66; P=0.002). No significant differences were observed for patients treated with novel agents during induction or between HD4 patients or NSP. Results from multivariate analysis from different landmarks are summarized in Figure 2.
Figure 2.
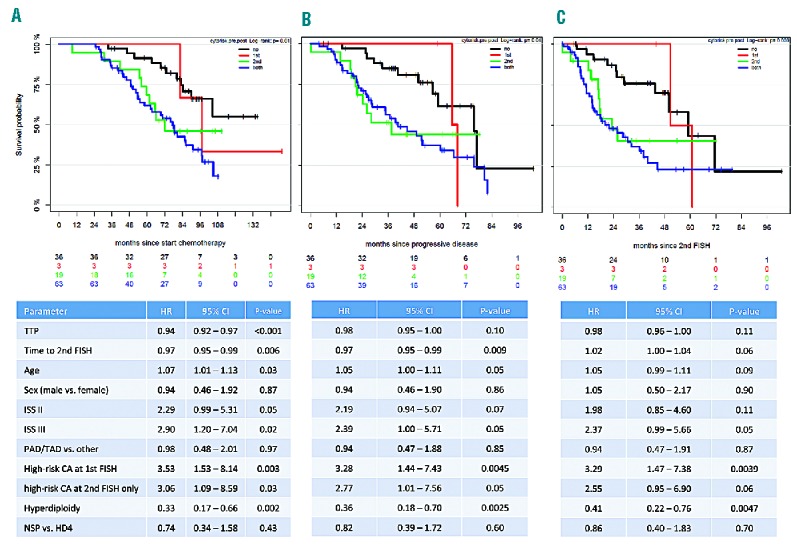
Multivariate survival analysis from different landmarks. Kaplan-Meier plots and multivariate analyses for overall survival in patients without high-risk cytogenetic abnormalities (CA) at both time points (black line), high-risk CA only at first (red line), only at second (green line) or at both (blue line) FISH analyses. Different landmarks were used: (A) from start of chemotherapy, (B) from progressive disease, (C) from second FISH analysis.
Discussion
The outcome of patients with MM has improved substantially during the last decades as a result of drug development and progress in the understanding of disease biology.11,12 However, even in the era of novel agents some patients with high-risk cytogenetic abnormalities or early relapse after first-line treatment have a dismal outcome.13,14 Clonal heterogeneity and evolution are contributors to disease progression and ultimately refractoriness in MM.6 So far, there are only limited data available that proved clonal evolution in patients relapsing after ASCT for newly diagnosed disease. With our current analysis of 128 patients with FISH data at primary diagnosis and relapse after ASCT we demonstrate that high-risk cytogenetic abnormalities occur more frequently at relapse. This observation was especially due to de novo gains of chromosome 1q and new deletions of chromosome 17p. No changes were observed between primary diagnosis and relapse for defined IGH translocations, including t(4;14).
A recent study demonstrated that chromosomal instability is a cornerstone of high-risk myeloma and propagated by bi-allelic inactivation of tumor suppressor genes, such as TP53 located on chromosome 17p.7 Furthermore, gain of chromosome 1q is associated with increased proliferation15 (e.g. compared to hyperdiploid MM) and genomic instability.16 Both cytogenetic abnormalities (del17p and gain 1q21) are considered secondary events in myelomatogenesis1 and associated with a high risk of progression from smoldering to symptomatic MM.17 Our current study confirmed this assumption for the first time in a large, longitudinally analyzed cohort of patients treated with ASCT.
In contrast to the increased rates of the high-risk cytogenetic abnormalities del17p and gain 1q21 after relapse, we did not observe the occurrence of new, defined IGH translocations, including t(4;14) with its adverse implications. This is most likely due to the fact that IGH translocations are considered primary, tumor-initiating events in MM.3 However, a study by the Intergroup Francophone Myélome found a de novo t(4;14) in 14 out of 268 patients, while in the same study 11 patients lost the t(4;14) after relapse.8 In accordance with our results, real time polymerase chain reaction analysis for IGH-MMSET confirmed the presence of minor t(4;14) subclones in selected cases.8 The authors concluded that t(4;14) can evolve from subclones, present at primary diagnosis, or can be silenced after chemotherapy until next relapse.8 This is in line with our finding, and those of recent studies,9 that patients with t(4;14) show a better response to primary therapy but still suffer from early relapse. To clarify whether clones with high-risk cytogenetic abnormalities remain present as minimal residual disease in patients with serological remission, future longitudinal genome sequencing studies in patients with relapse from minimal residual disease negativity as well as positivity are warranted.
Sublclonal evolution was also observed in patients with new del17p and gain 1q21. In a subset of patients, del17p occurred only in less than 60% of analyzed plasma cells, in line with the findings of a previous study.18 Furthermore, patients with gain 1q21 showed massive clonal heterogeneity after relapse, since many patients had subclones harboring different copy numbers compared to the main clone. Future studies will have to clarify whether the observed clonal heterogeneity in patients with del17p or gain 1q21 might be one of the reasons for the associated dismal outcome.
In contrast to the unchanged frequencies of the analyzed, defined IGH translocations [t(4;14), t(11;14) or t(14;16)] after relapse, we observed higher rates of IGH translocations with unknown partners with the IgH break-apart probe. Translocations involving the MYC locus have been associated with disease progression and adverse outcome.19,20 In our cohort, only a small group of patients was tested for MYC translocations at primary diagnosis and relapse, so that we were unable to perform a statistical analysis to confirm the hypothesis that MYC translocations are observed more frequently after relapse. Furthermore, sequencing studies are warranted not only to identify the translocation partners, but also to clarify, whether de novo translocations are caused by class switch recombination or by other mechanisms, as shown previously for t(11;14) and t(14;20).21
Patients with hyperdiploid MM had a favorable outcome in our current analysis and in the majority of patients a hyperdiploid karyotype proved to be stable after relapse. Remarkably, seven patients lost their hyperdiploid karyotype after relapse and gains of odd numbered chromosomes, especially of chromosome 5, were the only cytogenetic abnormalities occurring at lower frequencies after relapse. This might reflect chemosensitivity of the respective hyperdiploid clones. Since patients with hyperdiploid MM did not have a higher risk of developing high-risk cytogenetic abnormalities, other mechanisms might be responsible for disease progression, e.g. genomic mutations or epigenetic and microenvironmental modifications. However, it must be mentioned that we defined hyperdiploid status based on gains of the aforementioned odd-numbered chromosome loci. To rule out whether abnormalities other than the analyzed trisomies occurred after relapse, we would have had to include whole genome screening methods, such as single-nucleotide polymorphism assays.22
We could not identify other baseline characteristics or cytogenetic abnormalities associated with the occurrence of high-risk cytogenetic abnormalities at relapse and treatment response in patients with new del17p or gain 1q21 did not differ between patients with or without these cytogenetic abnormalities at both time points. Multivariate analysis revealed that patients who relapsed after novel agent-based induction therapy were at higher risk of developing the aforementioned cytogenetic abnormalities.
Since the first novel agents were combined with ASCT for the treatment of MM, physicians have debated whether relapses from primary therapy are becoming more aggressive. It has been suggested that the rates of extramedullary disease after novel agent-based treatment are higher23 and early relapse after primary therapy with novel agents is still associated with poor outcome.14 With our current analysis we provide the first evidence that relapse after treatment with novel agents might result in higher rates of high-risk cytogenetic abnormalities. One explanation for this finding might be that effective treatment selects pre-existing aggressive subclones that are below the level of sensitivity of FISH analyses, as proposed in the Intergroup Francophone Myélome study on t(4;14).8 Another explanation might be that chromosomal instability propagates disease progression and causes secondary genetic events.24
Lastly, we investigated the prognostic significance of new cytogenetic abnormalities after relapse. We found that patients with de novo del17p or gain 1q21 had the same dismal outcome as patients with the respective cytogenetic abnormalities detected at both time points. In fact, time to first progression was not significantly shorter whether the respective aberrations were already present at primary diagnosis or not. Again, this could imply that the evolving high-risk clone after relapse might have already been present at a subclonal level at primary diagnosis.
There are several potential criticisms of the current study that need to be addressed in the future. First of all, although patients were uniformly treated with ASCT, there were substantial differences in pre-transplant induction therapy in the cohort analyzed. Some patients were treated with proteasome inhibitors or immunomodulatory drugs before and/or after ASCT while others received only conventional chemotherapy. This might have caused different selection pressures on pre-existing clones and – more importantly – might have caused different secondary genetic events, since treatment with immunomodulatory drugs, in particular, has been associated with the occurrence of cytogenetic abnormalities typical of myelodysplastic syndromes.25 This hampers the comparison of study and non-study patients within our analysis.
Secondly, our retrospective data included only 128 patients and all patients had relapsed at the time of the second FISH. The findings for some subgroups are, therefore, based on very small numbers and we did not have a validation cohort of patients with a second FISH analysis in remission after ASCT. This weakness could be overcome in future studies including pre-planned bone marrow aspirates with FISH at certain time-points in a prospective clinical trial, e.g. at primary diagnosis, after ASCT before maintenance, at the end of maintenance and relapse. In this way, a longitudinal comparison of changes in the genetic profile of patients in remission and relapse would be possible. Lastly, as mentioned above, we will investigate the partners of the so far unknown IGH translocations observed after relapse in our current study.
In summary, our findings underline the importance of FISH analyses at relapse. We demonstrate that cytogenetic analyses need to be repeated, since the risk profile might be worse after treatment than at baseline, especially in the era of novel agents.3
Supplementary Material
Acknowledgments
The authors thank Maria Dörner, Ewelina Nickel, Hendrike Seidt, and Marie-Louise Brygider for technical assistance in the enrichment of CD138-positive plasma cells and Michaela Brough, Michelle Ebentheuer, Stephanie Pschowski-Zuck and Annekathrin Borowski for performing interphase FISH analyses.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/8/1432
Funding
This work was supported by grants from the German Federal Ministry of Education (BMBF) “CLIOMMICS” (01ZX1309) and “CAMPSIMM” (01ES1103), the Deutsche Forschungsgemeinschaft (SFB/TRR79), the Dietmar Hopp Stiftung “Heidelberger Konzept zur Optimierung der Diagnostik und Therapie des Multiplen Myeloms”, and the 7th EU-framework program “OverMyR”.
References
- 1.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100–113. [DOI] [PubMed] [Google Scholar]
- 2.Landgren O, Rajkumar SV. New developments in diagnosis, prognosis, and assessment of response in multiple myeloma. Clin Cancer Res. 2016;22(22):5428–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. [DOI] [PubMed] [Google Scholar]
- 4.Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression-related death. J Clin Oncol. 2014;32(20):2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raab MS, Lehners N, Xu J, et al. Spatially divergent clonal evolution in multiple myeloma: overcoming resistance to BRAF inhibition. Blood. 2016;127(17):2155–2157. [DOI] [PubMed] [Google Scholar]
- 7.Weinhold N, Ashby C, Rasche L, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016;128(13):1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hébraud B, Caillot D, Corre J, et al. The translocation t(4;14) can be present only in minor subclones in multiple myeloma. Clin Cancer Res. 2013;19(17):4634–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonneveld P, Schmidt-Wolf IGH, van der Holt B, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/GMMG-HD4 trial. J Clin Oncol. 2012;30(24):2946–2955. [DOI] [PubMed] [Google Scholar]
- 10.Neben K, Lokhorst HM, Jauch A, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119(4):940–948. [DOI] [PubMed] [Google Scholar]
- 11.Orlowski RZ, Lonial S. Integration of novel agents into the care of patients with multiple myeloma. Clin Cancer Res. 2016; 22(22):5443–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015; 385(9983):2197–2208. [DOI] [PubMed] [Google Scholar]
- 13.Kaufman GP, Gertz MA, Dispenzieri A, et al. Impact of cytogenetic classification on outcomes following early high-dose therapy in multiple myeloma. Leukemia 2016;30(3):633–639. [DOI] [PubMed] [Google Scholar]
- 14.Majithia N, Rajkumar SV, Lacy MQ, et al. Early relapse following initial therapy for multiple myeloma predicts poor outcomes in the era of novel agents. Leukemia. 2016; 30(11):2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hose D, Reme T, Hielscher T, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011;96(1):87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawyer JR, Tricot G, Lukacs JL, et al. Genomic instability in multiple myeloma: Evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer. 2005;42(1):95–106. [DOI] [PubMed] [Google Scholar]
- 17.Neben K, Jauch A, Hielscher T, Hillengass J, Lehners N, Seckinger A, et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 2013; 31(34):4325–4332. [DOI] [PubMed] [Google Scholar]
- 18.An G, Li Z, Tai Y-T, et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin Cancer Res. 2015;21(9):2148–2156. [DOI] [PubMed] [Google Scholar]
- 19.Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA. 2000; 97(1):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinhold N, Kirn D, Seckinger A, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17):3413–3419. [DOI] [PubMed] [Google Scholar]
- 22.Chretien M-L, Corre J, Lauwers-Cances V, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter¿ Blood. 2015;126(25): 2713–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Short KD, Rajkumar SV, Larson D, et al. Incidence of extramedullary disease in patients with multiple myeloma in the era of novel therapy, and the activity of pomalidomide on extramedullary myeloma. Leukemia. 2011;25(6):906–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji Z, Zhang L, Peng V, Ren X, McHale CM, Smith MT. A comparison of the cytogenetic alterations and global DNA hypomethylation induced by the benzene metabolite, hydroquinone, with those induced by melphalan and etoposide. Leukemia. 2010;24(5):986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Usmani SZ, Sawyer J, Rosenthal A, et al. Risk factors for MDS and acute leukemia following total therapy 2 and 3 for multiple myeloma. Blood. 2013;121(23):4753–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.