

Abstract
The incorporation of fluorine into organic scaffolds often improves the bioactivity of pharmaceutically relevant compounds. C–F/C–N/C–O stereotriad motifs are prevalent in antivirals, neuraminidase inhibitors, and modulators of androgen receptors, but are challenging to install. An oxidative allene amination strategy using Selectfluor rapidly delivers triply functionalized triads of the form C–F/C–N/C–O, exhibiting good scope and diastereoselectivity for all syn products. The resulting stereotriads are readily transformed into fluorinated pyrrolidines and protected α-, β-, and γ-amino acids.
Graphical abstract
The introduction of fluorine into organic compounds is a popular strategy to improve the physical properties of molecules that range from pharmaceuticals and agrochemicals to advanced materials, polymers, and ligands.1 Fluorine substitution often results in increased lipophilicity and bioavailability, promotes hydrophobic interactions between the drug and binding sites on receptors or enzymes, and imparts higher oxidative and thermal stability to a molecule as compared to the corresponding C–Hbonds.2 The recent burst of activity directed toward the development of reagents and methods to introduce fluorine into organic scaffolds points to the continued need for approaches that furnish complex, highly functionalized heterotriad motifs, especially ones that are found in useful bioactive molecules (Figure 1).3,4
Figure 1.
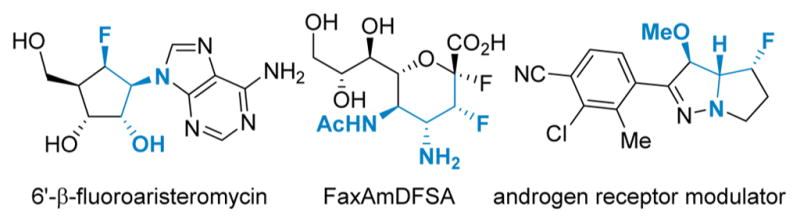
Bioactive molecules containing fluorinated amine heterotriads.
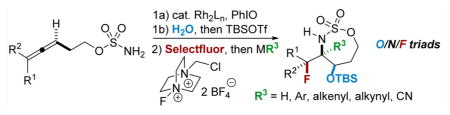
Our group has previously developed oxidative amination strategies that install a new heteroatom at each of the sites of unsaturation in an allene substrate such as 1, where a bicyclic methyleneaziridine 2 functions as the key intermediate (Scheme 1).5 These allenes are easily synthesized from the corresponding propargyl alcohols through a three-step sequence involving a Johnson–Claisen rearrangement using (EtO)3CMe, followed by LiAlH4 reduction and sulfamate formation.5a,d,f,i Employing an enantioenriched propargyl alcohol in the allene synthesis delivers efficient axial-to-point chirality transfer to furnish enantioenriched stereotriads.5a,i,j However, our early efforts to incorporate valuable fluorines into the triad products 3 delivered only moderate yields and poor dr, stimulating further efforts to improve upon these initial results. In this letter, we report methods to introduce fluorine into allenes in both an electrophilic and nucleophilic manner to provide novel building blocks not easily synthesized using alkene functionalization strategies. In particular, oxidative allene amination methodologies are enabling for providing access to quaternary fluorine-bearing stereocenters difficult to prepare through deoxyfluorination protocols that are best-suited for the synthesis of primary and secondary alkyl fluorides.6
Scheme 1.
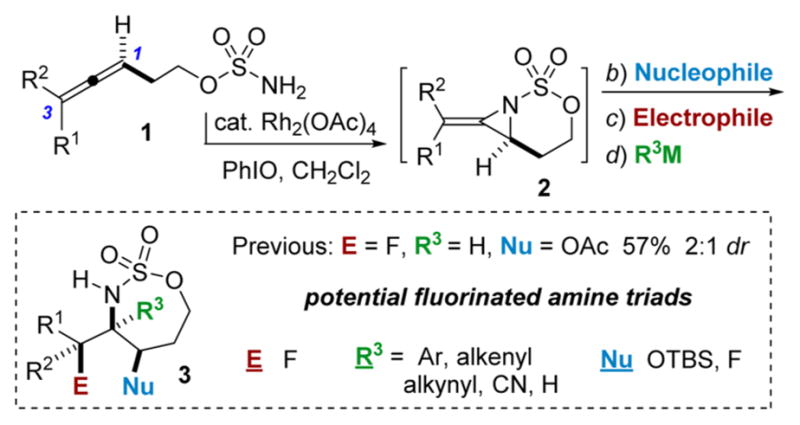
Strategy for Generating Fluorinated Amine ‘Triads’
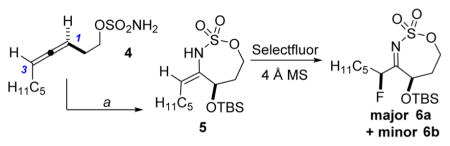
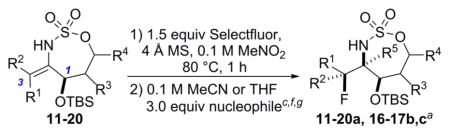
The first challenge was to identify the appropriate nucleophile/electrophile combinations capable of furnishing high dr in the oxidative allene amination to furnish the desired fluorinated stereotriads. A variety of nucleophiles are known to successfully open the intermediate bicyclic methyleneaziridine resulting from Rh-catalyzed aziridination of 4 (Scheme 1), including H2O, MeOH, PhSH, and PhNH2.5a Unfortunately, treatment of the corresponding enesulfamates with an array of electrophilic fluoride reagents, including Selectfluor, NFSI, 1-fluoropyridinium tetrafluoroborate, and 2,6-dichloro-1-fluoropyridinium tetrafluoroborate, showed variable dr or unproductive reaction in the imine formation. Eventually, we found that the combination of a bulky OTBS group at C1 of the enesulfamate 5 with Selectfluor enabled promising diastereocontrol in the addition of 5 (Table 1, entry 1) to the electrophile, furnishing 6a as the major stereoisomer. MeNO2 and MeCN proved to be the best solvents, but MeNO2 showed minimal loss in yield and dr when the temperature was increased from 60 to 90 °C (Table 1, compare entries 2 vs 4–6), in contrast to the drop in yield noted when reactions in MeCN were run at higher temperatures (compare entry 1 vs 3). Increasing the equivalents of Selectfluor (entry 7) resulted in no improvement in the yield or the dr; therefore, the reaction conditions in entry 5 were chosen for further study.
Table 1.
Optimization of the Electrophilic Fluorination Reaction in the Reaction of 5 with Selectfluor
| ||||||
|---|---|---|---|---|---|---|
| entry | solvent | F+ (equiv) | temp (°C) | time (h) | yieldb | dr (6a:6b)b |
| 1 | CD3CN | 1.5 | 60 | 4 | 69% | 8.6:1 |
| 2 | CD3NO2 | 1.5 | 60 | 4 | 77% | 8.7:1 |
| 3 | CD3CN | 1.5 | 80 | 2 | 56% | 7.9:1 |
| 4 | CD3NO2 | 1.5 | 80 | 2 | 72% | 8:1 |
| 5 | CD3NO2 | 1.5 | 80 | 1 | 76% | 8.3:1 |
| 6 | CD3NO2 | 1.5 | 90 | 0.5 | 77% | 7.7:1 |
| 7 | CD3NO2 | 2.5 | 80 | 1 | 75% | 8.4:1 |
0.75 mol % Rh2(OAc)4, PhlO, CH2Cl2, rt; H2O; then 4 Å MS, TBSOTf, Et3N.
Yields and dr (syn:anti) determined by 1H and 19F NMR using 1-Br-2,5-difluorobenzene as the internal standard.
The stereochemistry of the major imine product 6a was confirmed to be 1,3-syn diastereomer by subsequent reduction to 7a. The observed syn geometry is consistent with the model proposed in Figure 2, where the conformation A is less favored as compared to B, due to the A1,3 strain generated by interactions between the sterically bulky OTBS group and the C5H11 side chain that reside in the same plane. Relief of this A1,3 strain drives the enesulfamate to adopt a conformation where the Selectfluor favors approach from the face opposite to the OTBS group, as illustrated in B. This rationale explains why the installation of small groups at C1 in our preliminary studies resulted in low and variable diastereoselectivities.
Figure 2.
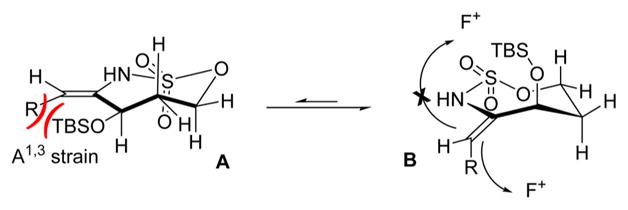
Model rationalizing the observed overall syn selectivity in electrophilic fluorinations of enesulfamates.
Reaction of 5 with Selectfluor, coupled with subsequent reduction of the imine stereoisomer 6a with Me4NHB(OAc)3, could be carried out in one pot to furnish the 1,2-syn:2,3-syn amine triad product 7a as the major diastereomer (Table 2); two minor diastereomers were also observed (see the Supporting Information (SI) for details). Again, the minimization of A1,3 strain in the imine 6a likely drives the observed stereochemical outcome in the reduction, where the relative all-syn configuration of the three new sp3 stereogenic centers of 7a was confirmed by single crystal X-ray crystallography (see the SI for details). Carbon nucleophiles, including Grignard reagents and Bu4NCN, could be employed instead of a hydride source to furnish the stereotriads 8a–10a in good yields and with dr of up to >20:1. The relative stereochemistry of 10a was confirmed by X-ray crystallography (see the SI for details) to ensure that carbon nucleophiles behave similarly to hydrides. The very different dipole moments of the products 7a–10a permitted ready separation of the diastereomers by column chromatography.
Table 2.
Scope of Nucleophiles in the Fluorination of 5
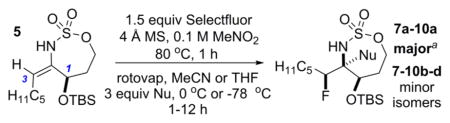
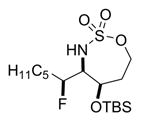
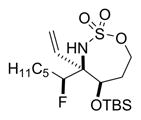
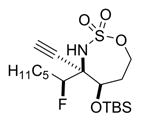
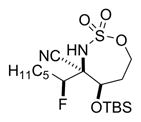
Yield of the isolated major diastereomer. After the fluorination and filtration were complete, the following reagents were added.
0.1 M MeCN/AcOH, 3 equiv of Me4N(OAc)3BH, 0 °C to rt, 12 h.
0.1 M THF, 3 equiv of HCCMgBr, 0 °C, 1 h.
0.1 M THF, 3 equiv of CH2=CHMgBr, −78 °C, 30 min.
0.1 M MeCN, 2.0 equiv of Bu4NCN, rt, 2.6 h.
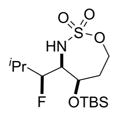
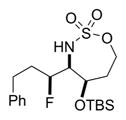
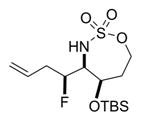
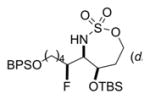
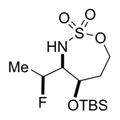
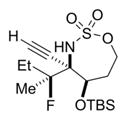
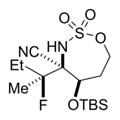
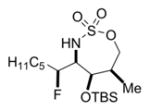
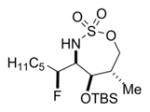
The scope of the reaction was explored using a series of 1,3-di- and 1,3,3-trisubstituted allenes (Table 3). Increasing the bulk of the C3 R group to an iPr in 11 was not well-tolerated and gave 11a in a low 19% yield and a 3:1 dr after reduction with Me4N(OAc)3BH. However, fluorination proceeded more smoothly when the allene side chains in 12–14 contained vinyl, phenyl, and silyl ether groups, giving moderate to good yields and dr of 12a–14a after the reduction. Decreasing the size of the C3 substituent had no detrimental impact, as 15a was obtained in 69% yield and 11:1 dr. Interestingly, even 1,3,3-trisubstituted allenes 16 and 17, which previously failed to give either good yields or dr in oxidative allene aminations, were successful using this fluorination protocol. Reduction of the intermediate imines with Me4N(OAc)3BH delivered 16a in 56% yield and >19:1 dr over the two steps. We were pleased to note that carbon nucleophiles, including HCCMgBr or Bu4NCN, were also effective and resulted in the production of stereotriads 16b and 16c, containing two adjacent and fully substituted stereocenters in excellent dr of >19:1. Even when the sterics of the two alkyl groups on C3 of the original allene substrate were similar, as in the Et and Me groups of 17, the triads 17a–c could be obtained in diastereoselectivities ranging from 4:1 to >19:1, albeit in lowered yields. These densely functionalized motifs are not readily accessible from conventional olefin oxidation methodologies or aziridine ring-opening.
Table 3.
Scope of 1,2-syn:2,3-syn Fluorinated Amine Triads
| |||
|---|---|---|---|
| product | yield dr |
product | yield dr |
|
19% 11a (dr 3:1)b,c |
|
68% 12a (dr 9:1)c,d |
|
58% 13a (dr 8:1)b,c |
|
69% 14a (dr 7:1.5:1)c,d |
|
69% 15a (dr 11:1)c,d |
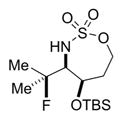
|
56% 16a (dr >19:1)c,d |
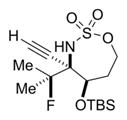
|
52% 16b (dr >19:1)d,e |
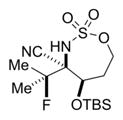
|
89% 16c (dr >19:1)d,e,g |
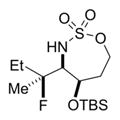
|
27% 17a (dr >19:1)d,e |
|
28% 17b (dr 4:1)d,f |
|
75% 17c (dr 5:1)d,e,g |
|
51% 18a (dr 3:1)c,d |
|
24% 19a (dr 3:1)c,d |
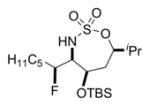
|
41% 20a (dr 6:3:2:1)c,d |
Listed yields are for the isolated major diastereomer.
Diastereomer ratios of the crude product are in parentheses.
0.1 M MeCN/AcOH, 3 equiv of Me4N (OAc)3BH, 0 °C to rt, 12 h.
Isolated dr.
Fluorination in MeCN, 40 0 °C, 14 h.
0.1 M THF, 3 equiv of HCCMgBr, 0 °C, 1 h.
0.1 M MeCN, 2.0 equiv of Bu4NCN, rt, 2.6 h.
Additional substitutions in the allene tethers of 18–20 would be expected to influence the conformation of the intermediate enesulfamate, which, in turn, would be likely to impact the stereochemical outcome of the electrophilic fluorination. The diastereoselectivities in the fluorination of substrates 18–20 were 3:1, 3:1, and 6:3:2:1, respectively. The models in Figure 3 help to explain the observed stereochemical outcomes. For substrate 18, there is a preference to avoid the eclipsed conformation in which OTBS and the methyl substituents are in close proximity. Thus, a gauche conformation is more favored, where the OTBS substituent is almost perpendicular to the ring. This results in effective blocking of the top face of the ring and increases the dr to 3:1. In terms of substrate 19, a conformation where the OTBS and methyl substituents are anti to each other minimizes both A1,3 strain between OTBS and the C5H11 side chain, as well as the gauche interaction between the OTBS and the adjacent methyl group. However, this conformation resulted in a dr of 3:1, due to the methyl group blocking the bottom face of the ring. In 20, the ring is forced into a conformation that avoids the syn-pentane interaction between the isopropyl and the OTBS groups. This orients the OTBS group a bit more in the plane of the ring; therefore, it is less effective at blocking the top face of the ring and the dr is lowered to 6:3:2:1.
Figure 3.
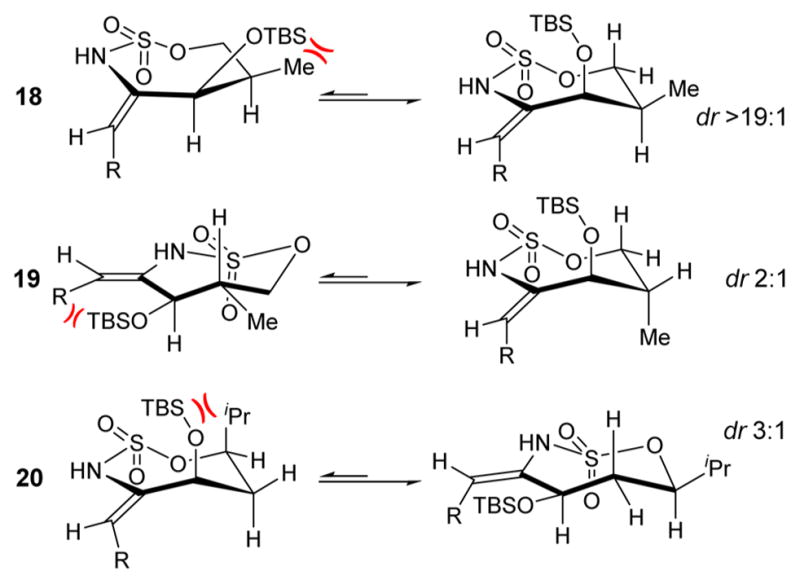
Model rationalizing the observed dr in electrophilic fluorination of tethered enesulfamates.
The fluorinated amine triads proved to be useful and flexible building blocks for the synthesis of novel pyrrolidines and unnatural amino acid scaffolds (Scheme 2). For example, N-Boc protection and a NaI-mediated ring contraction of 7a yielded the fluorinated pyrrolidine 28 in a good yield of 72% over the two steps and in a dr > 19:1. This motif appears in bioactive molecules that include the powerful glucosidase inhibitor 2,5-dideoxy-2,5-imino-D-mannitol (DMDP)7 and various inhibitors of purine nucleoside phosphorylase (PNP).8 Fluorinated amino acids are important building blocks in the design of hyperstable protein folds. They have also played roles in directing protein–protein interactions9 and can act as potential antitumor agents or antimicrobial agents.10 The use of fluorinated, unnatural amino acids in such applications could prove interesting and can be readily accessed in a few steps from either fluorinated triad 7a or 9a, as illustrated in the remainder of Scheme 2. For example, the ozonolysis of 9a was followed by a standard Pinnick oxidation to afford the α-amino acid precursor 29 with no loss in the stereochemical information contained in the precursor. An alternative approach could employ the Strecker product 10a (see Table 2), with hydrolysis of the nitrile delivering the same product 29. N-Boc protection/ring-opening/elimination of 7a, similar to that previously reported,5j furnished 30. Compound 30 could be further elaborated into the protected β-amino acid 31 through a simple sequence of ozonolysis, Pinnick oxidation, and esterification. The same 30 was transformed into the protected γ-amino acid 32 by first carrying out a hydroboration reaction, followed by a RuCl3-catalyzed oxidation of the alcohol to the carboxylic acid.
Scheme 2.
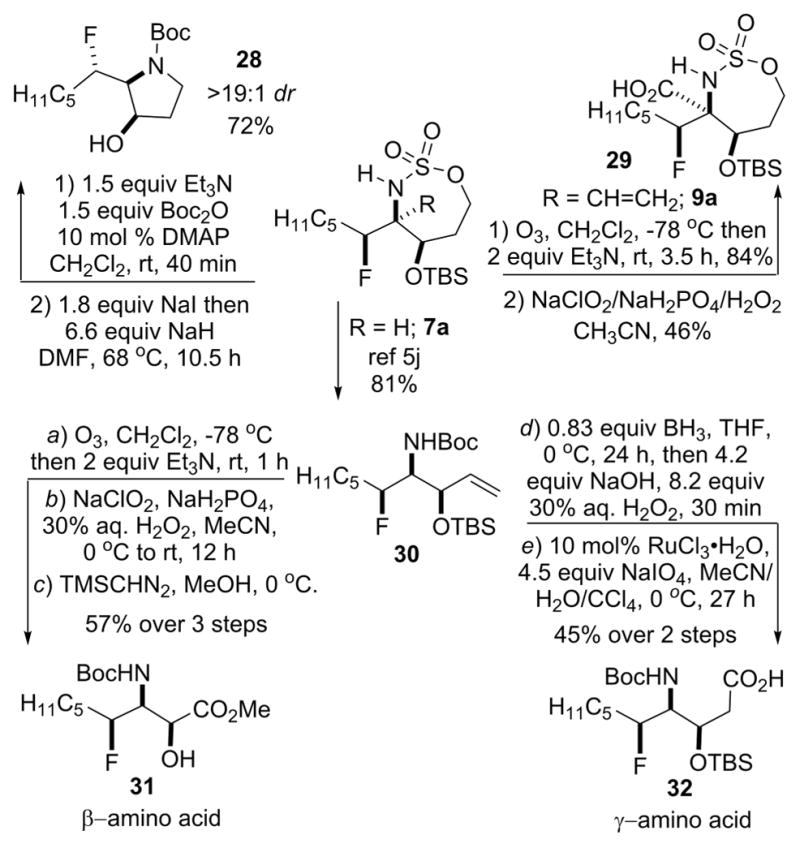
Motifs Available from Fluorinated Triads
In conclusion, we have demonstrated that allenes can be converted into stereochemically complex and densely functionalized C–F/C–N/C–O stereotriad motifs in a two-pot process. Allene aziridination, ring-opening, and protection of the alcohol with a bulky TBS group furnish enesulfamates with high regiocontrol and E:Z selectivity, even when differentiating between groups as similar as Me and Et. Selectfluor was the most effective electrophilic fluorination reagent; reduction of the intermediate imine with either hydride or carbon-based nucleophiles furnished the products with reasonable scope and diastereoselectivity. Especially noteworthy was the ability to generate motifs with adjacent fluorine- and amine-bearing quaternary stereocenters in excellent dr. The C–F/C–N/C–O motifs can be readily transformed into fluorinated pyrrolidines and protected unnatural α-, β-, and γ-amino acids.
Supplementary Material
Acknowledgments
J.M.S. thanks the NIH R01GM111412 for funding of this research. The NMR facilities at UW—Madison are funded by the NSF (CHE-1048642, CHE-0342998) and NIH S10 OD012245. The National Magnetic Resonance Facility at Madison is supported by the NIH (P41GM103399, S10RR08438, S10RR029220) and the NSF (BIR-0214394).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b01342.
Experimental procedures and characterization data for all new compounds (PDF)
X-ray crystallographic data for compound 7a (CIF)
X-ray crystallographic data for compound 10a (CIF)
References
- 1.(a) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (b) Vitale A, Bongiovanni R, Ameduri B. Chem Rev. 2015;115:8835–66. doi: 10.1021/acs.chemrev.5b00120. [DOI] [PubMed] [Google Scholar]; (c) Ilardi EA, Vitaku E, Njardarson JT. J Med Chem. 2014;57:2832–42. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]; (d) Jeschke P. ChemBioChem. 2004;5:570–89. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; (e) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (f) Isanbor C, O’Hagan D. J Fluorine Chem. 2006;127:303–319. [Google Scholar]
- 2.(a) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–43. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; (b) O’Hagan D. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]; (c) Müller K, Faeh C, Diederich F. Science. 2007;317:1881–86. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 3.(a) Liang T, Neumann CN, Ritter T. Angew Chem, Int Ed. 2013;52:8214–64. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]; (b) Kirk KL. Org Process Res Dev. 2008;12:305–321. [Google Scholar]
- 4.(a) Yin X, Schneller SW. Tetrahedron Lett. 2005;46:7535–38. [Google Scholar]; (b) Withers S, Watts AG, Kim J-H, Wennekes T. 2,3-Fluorinated Glycosides as Neuramidase Inhibitors and Their Use as Anti-virals. 8,815,941. US Patent. 2011; (c) Ullrich T, Sasmal S, Boorgu V, Pasagadi S, Cheera S, Rajagopalan S, Bhumireddy A, Shashikumar D, Chelur S, Belliappa C, Pandit C, Krishnamurthy N, Mukherjee S, Ramanathan A, Ghadiyaram C, Ramachandra M, Santos PG, Lagu B, Bock MG, Perrone MH, Weiler S, Keller H. J Med Chem. 2014;57:7396–7411. doi: 10.1021/jm5009049. [DOI] [PubMed] [Google Scholar]
- 5.(a) Adams CS, Boralsky LA, Guzei IA, Schomaker JM. J Am Chem Soc. 2012;134:10807–10. doi: 10.1021/ja304859w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boralsky LA, Marston D, Grigg RD, Hershberger JC, Schomaker JM. Org Lett. 2011;13:1924–27. doi: 10.1021/ol2002418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rigoli JW, Boralsky LA, Hershberger JC, Meis AR, Guzei IA, Schomaker JM. J Org Chem. 2012;77:2446–55. doi: 10.1021/jo3000282. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Weatherly CD, Rigoli JW, Schomaker JM. Org Lett. 2012;14:1704–7. doi: 10.1021/ol300269u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Weatherly CD, Schomaker JM. Eur J Org Chem. 2013;2013:3667–70. [Google Scholar]; (f) Rigoli JW, Guzei IA, Schomaker JM. Org Lett. 2014;16:1696–99. doi: 10.1021/ol5003576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Adams CS, Weatherly CD, Burke EG, Schomaker JM. Chem Soc Rev. 2014;43:3136–63. doi: 10.1039/c3cs60416k. [DOI] [PubMed] [Google Scholar]; (h) Adams CS, Grigg RD, Schomaker JM. Tetrahedron. 2014;70:4128–34. [Google Scholar]; (i) Adams CS, Grigg RD, Schomaker JM. Chem Sci. 2014;5:3046–56. [Google Scholar]; (j) Burke EG, Schomaker JM. Angew Chem, Int Ed. 2015;54:12097–12101. doi: 10.1002/anie.201504723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Gerstner NC, Adams CS, Grigg RD, Tretbar M, Rigoli JW, Schomaker JM. Org Lett. 2016;18:284–87. doi: 10.1021/acs.orglett.5b03453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected deoxyfluorination protocols, see: Goldberg NW, Shen X, Li J, Ritter T. Org Lett. 2016;18:6102–5. doi: 10.1021/acs.orglett.6b03086.Li L, Ni C, Wang F, Hu J. Nat Commun. 2016;7:13320. doi: 10.1038/ncomms13320. and references therein.
- 7.(a) Legler G. Adv Carbohydr Chem Biochem. 1990;48:319–84. doi: 10.1016/s0065-2318(08)60034-7. [DOI] [PubMed] [Google Scholar]; (b) Wrodnigg T. Monatsh Chem. 2002;133:393–426. [Google Scholar]; (c) Andersen SM, Ebner M, Ekhart CW, Gradnig G, Legler G, Lundt I, Stütz AE, Withers SG, Wrodnigg T. Carbohydr Res. 1997;301:155–66. [Google Scholar]; (d) Wrodnigg TM, Diness F, Gruber C, Häusler H, Lundt I, Rupitz K, Steiner AJ, Stütz AE, Tarling CA, Withers SG, Wölfler H. Bioorg Med Chem. 2004;12:3485–95. doi: 10.1016/j.bmc.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 8.(a) Krenitsky TA. Mol Pharmacol. 1967;3:526–36. [PubMed] [Google Scholar]; (b) Mason JM, Murkin AS, Li L, Schramm VL, Gainsford GJ, Skelton BW. J Med Chem. 2008;51:5880. doi: 10.1021/jm800792b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. J Med Chem. 2003;46:3412–23. doi: 10.1021/jm030145r. [DOI] [PubMed] [Google Scholar]
- 9.For reviews and selected examples of fluorinated amino acids used in protein design and engineering, see: Yoder NC, Kumar K. Chem Soc Rev. 2002;31:335–41. doi: 10.1039/b201097f.Odar C, Winkler M, Wiltschi B. Biotechnol J. 2015;10:427–46. doi: 10.1002/biot.201400587.Wang P, Tang Y, Tirrell DA. J Am Chem Soc. 2003;125:6900–06. doi: 10.1021/ja0298287.Niemz A, Tirrell DA. J Am Chem Soc. 2001;123:7407–13. doi: 10.1021/ja004351p.Bilgiçer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–99. doi: 10.1021/ja002961j.
- 10.For selected examples of antitumor and antimicrobial activities of fluorinated amino acids, see: Gottler LM, Lee HY, Shelburne CE, Ramamoorthy A, Marsh ENG. ChemBioChem. 2008;9:370–73. doi: 10.1002/cbic.200700643.Meng H, Kumar K. J Am Chem Soc. 2007;129:15615–22. doi: 10.1021/ja075373f.Tsushima T, Kawada K, Ishihara S, Uchida N, Shiratori O, Higaki J, Hirata M. Tetrahedron. 1988;44:5375–87.Tsushima T, Kawada K. Tetrahedron Lett. 1985;26:2445–48.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.