

Abstract
Pathogenic variants in the gap junction protein beta-2 (GJB2) gene are the most common cause of hearing loss. Of these, the p.V37I variant of GJB2 has a high allele frequency (up to 10%) in East Asians. Characterization of the phenotypic spectrum associated with p.V37I, as well as the role of this variant in the onset of hearing loss could have a remarkable effect on future diagnostic strategies. Here, we performed a pedigree analysis of unrelated families exhibiting various hearing phenotypes, and then conducted a meta-analysis to comprehensively assess the association between the p.V37I and the risk of hearing loss. Pedigree analyses showed that both homozygous p.V37I variants, as well as compound heterozygous p.V37I with other GJB2 pathogenic variants, contributed to various phenotypes of hearing loss. Meanwhile, meta-analysis demonstrated that, compared with those in the wild type group, both p.V37I homozygotes and compound heterozygous p.V37I variants were at significantly higher risk of developing hearing loss (odds ratios = 7.14 and 3.63; 95% confidence intervals = 3.01-16.95 and 1.38–9.54, respectively). Conversely, heterozygous p.V37I variants alone did not increase the risk of hearing loss. Given the high allele carriage rate of p.V37I (up to 10%) within the general population, our work not only provides information that might influence future genetic screening policies, but also offers insight into clinical risk evaluation and genetic counseling regarding hearing loss.
Keywords: hearing loss (HL), GJB2, p.V37I, pedigree analysis, meta-analysis
INTRODUCTION
Hearing loss (HL) is the most sensory defect that affects 1-3 in every 1,000 newborns worldwide, and half of these cases are attributed to genetic factors [1]. Notably, while a large number of HL-related genes have been identified, the gap junction protein beta 2 (GJB2) gene accounts for nearly 20% of all cases of HL, as well as 50% of autosomal recessive non-syndromic HL, in many populations [2, 3]. GJB2 encodes the connexin 26 protein, which comprises a critical component of cochlear gap junctions, and is important to cell communication. To date, greater than 300 variants within GJB2 have been found to be associated with HL (http://www.hgmd.cf.ac.uk/ac/), including c.35delG, c.235delC, and c.176_191del16.
In particular, the p.V37I (c.109G>A) variant of GJB2 has a high allele frequency (up to 10%) among East Asian populations [4–6]. This variant, harboring a missense substitution from valine to isoleucine at codon 37, was first identified by Kelley et al. in 1998 [7]. Early studies regarded the p.V37I as a benign polymorphism, as it was observed in unaffected heterozygous controls [7–11]. However, the identification of increasing numbers of HL patients that are homozygous for p.V37I, or compound heterozygous for p.V37I and other GJB2 pathogenic variants, indicates that p.V37I likely increases the risk of HL, particularly for mild-to-moderate cases [6, 12–15]. Interestingly, a recent meta-analysis reported an insignificant association between the carriage rates of p.V37I and HL, which aroused wide concern regarding the pathogenicity of this variant [16].
Given the high allelic frequency of the p.V37I variant (up to 10%), it is estimated that greater than five million East Asians suffer from HL due to homozygous or compound heterozygous carriage of this allele [6]. It is therefore imperative to evaluate the risks associated with p.V37I for clinical genetic counseling and public health assessment purposes. In this study, we performed a pedigree analysis of families with probands exhibiting various HL phenotypes, and then conducted a meta-analysis of sporadic HL to comprehensively evaluate the role of p.V37I in the risk of HL.
RESULTS
Pedigree analyses
Seven families carrying the p.V37I variant were included for pedigree analyses (Table 1 and Figure 1). There were five male and two female probands. Six probands were children (7 months-9 years) and one proband was an adult (33 years). These probands exhibited both congenital and delayed-onset non-syndromic HL, and one member had sudden deafness. Bilateral and unilateral non-syndromic HL were also observed, with HL degrees of mild to moderate. Families 1–5 had probands that were homozygous for p.V37I (S1–S5), while the probands of Families 6 and 7 had compound heterozygous p.V37I variants (H1–H2). In this study, compound heterozygous p.V37I variants were defined as the p.V37I allele in a trans configuration with another pathogenic mutant allele of GJB2 gene. Pedigree analyses revealed that p.V37I was transmitted from heterozygous parents to their children, who suffered HL if he/she inherited two affected alleles (p.V37I homozygotes or compound heterozygous p.V37I variants). Meanwhile, siblings that inherited one affected allele retained normal hearing. These analyses strongly suggest that p.V37I increases the risk of HL through an autosomal recessive inheritance pattern.
Table 1. Clinical characteristics of probands carrying the p.V37I variant.
| ID | Sex | Age | p.V37I status | Onset | Site | Degree |
|---|---|---|---|---|---|---|
| S1 | Female | 7 months | Homozygote | Congenital | Bilateral | Moderate |
| S2 | Male | 8 years | Homozygote | Congenital | Bilateral | Mild |
| S3 | Male | 9 years | Homozygote | Congenital | Unilateral | Mild |
| S4 | Male | 33 years | Homozygote | Delayed-onset | Bilateral | Moderate |
| S5a | Male | 8 years | Homozygote | Sudden deafness | Bilateral | Mild |
| H1 | Female | 7 years | p.V37I/c.176_191del16 | Delayed-onset | Bilateral | Mild |
| H2 | Male | 3 years | p.V37I/c.235delC | Congenital | Bilateral | Moderate |
a The severity of deafness of proband S5 was 30 dB at the left ear, and 31.25 dB at the right ear, so his degree of HL was considered as mild.
Figure 1. Pedigree analyses for seven unrelated families carrying the p.V37I variant.
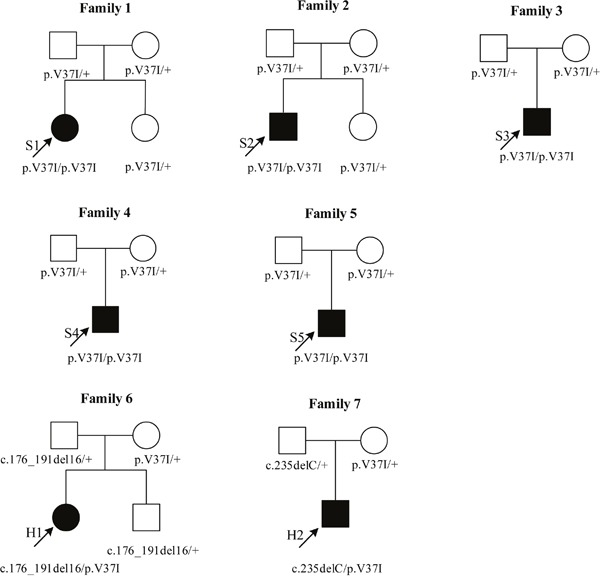
Five probands (S1-S5) carried two p.V37I alleles as homozygotes. Two probands (H1, H2) carried the p.V37I allele in a trans configuration with another well-known pathogenic mutant allele of GJB2, as compound heterozygotes. The hearing level of S1 was recorded by auditory brainstem responses (ABR) due to her very young age (7 months). The hearing level of H2 was recorded by the auditory steady-state responses (ASSR) because of his poor cooperation in pure-tone audiometry (PTA) test. Sanger sequencing results of pedigree analyses were shown in Supplementary Figure 4.
Meta-analysis on sporadic HL
To further determine the detrimental effects of p.V37I, we performed a meta-analysis. A flow chart of the literature search is shown in Supplementary Figure 1. A total of 1,085 potentially relevant records were initially identified in our search. Of these, 391 duplicates and 644 irrelevant records were removed upon reviewing titles and abstracts, yielding 50 full-text articles for further evaluation. Seventeen studies were subsequently excluded because they provided no genetic data regarding the p.V37I variant (n = 7), they lacked sufficient information to estimate odds ratio (OR) and 95% confidence interval (CI) (n = 7), they lacked a parallel control group (n = 2), or their data overlapped with that of another study (n = 1). Of the 33 remaining studies eligible for meta-analysis [5, 7–15, 17–39], 10 also provided genotypes of compound heterozygous p.V37I variants [8, 12, 13, 19, 21, 28, 33–35, 37].
As shown in Table 2, a total of 14,398 HL cases and 8,699 controls were included to evaluate the association between p.V37I and HL risk. Twenty of the studies were conducted in Asia (China, Japan, Malaysia and Indonesia) [5, 8, 9, 12–14, 18, 20, 22–24, 26, 30, 31, 34–37, 39], while six were conducted in North America (United States and Canada) [7, 11, 15, 19, 28, 33], five in Europe (Portugal, Italy, Finland, France and Spain) [10, 17, 21, 25, 38], one in Latin America (Argentina) [32], and one in Oceania (Australia) [27]. Overall, there was a significant association between the p.V37I variant and increased risk of developing HL (Figure 2). Specifically, the A allele of p.V37I was associated with a 2-fold higher risk of developing HL than the G allele (Figure 2A; OR = 1.91; 95% CI = 1.42–2.56; Pheterogeneity < 0.001; I2 = 74.9%). Moreover, compared with individuals with wild type, p.V37I homozygotes (Figure 2C; OR = 7.14; 95% CI = 3.01-16.95; Pheterogeneity < 0.001; I2 = 70.9%), but not p.V37I heterozygotes (Figure 2B; OR = 1.18; 95% CI = 0.92–1.52; Pheterogeneity = 0.034; I2 = 35.5%), had a significantly higher risk (7.14-fold greater) of developing HL. Similar results were obtained using the recessive model (Figure 2D; OR = 7.02; 95% CI = 2.95–16.66; Pheterogeneity < 0.001; I2 = 71.1%). Sensitivity analyses demonstrated that our results were quite stable (Supplementary Figure 2A–2D) and no obvious publication biases were found (all P > 0.10).
Table 2. Characteristics of studies included for meta-analysis.
| First author | Publication year | Country | Geological area | Population | Cases | Controls | PHWE |
|---|---|---|---|---|---|---|---|
| Chen [39] | 2016 | China | Asia | Children and adults | 50 | 53 | 0.399 |
| Caroça [38] | 2016 | Portugal | Europe | Children and adults | 134 | 177 | 0.970 |
| Huang [12] | 2015 | Shanghai, China | Asia | Infants | 300 | 484 | 0.747 |
| Huang [13] | 2015 | China | Asia | Infants and adults | 3,864 | 600 | 0.318 |
| Chai [14] | 2015 | China | Asia | Infants and adults | 945 | 1,550 | 0.557 |
| Chen [37] | 2014 | China | Asia | Infants and child | 107 | 61 | 0.486 |
| Zainal [36] | 2012 | Malaysia | Asia | Children | 32 | 37 | 0.479 |
| Zhang [35] | 2011 | China | Asia | Children and adults | 236 | 107 | 0.765 |
| Wu [34] | 2011 | China | Asia | Infants | 38 | 979 | 0.139 |
| Schimmenti [33] | 2011 | United States | North America | Infants | 1,177 | 1,177 | 0.884 |
| Tsukada [5] | 2010 | Japan | Asia | Infants and children | 1,343 | 252 | 0.924 |
| Dalamon [32] | 2010 | Argentina | Latin America | NR | 252 | 50 | 0.943 |
| Dai [31] | 2009 | China | Asia | Children and adults | 1,372 | 301 | NA |
| Chen [30] | 2009 | China | Asia | NR | 115 | 109 | NA |
| Yang [29] | 2007 | China | Asia | NR | 260 | 120 | <0.001 |
| Tang [28] | 2006 | United States | North America | NR | 610 | 294 | 0.004 |
| Huculak [15] | 2006 | Canada | North America | NR | 40 | 100 | 0.751 |
| Dahl [27] | 2006 | Australia | Oceania | Children | 48 | 90 | NA |
| Snoeckx [26] | 2005 | Indonesia | Asia | Patients: <20 years old; Control: adults | 120 | 100 | 0.879 |
| Ravecca [25] | 2005 | Italy | Europe | Children and adults | 39 | 40 | 0.936 |
| Xiao [24] | 2004 | China | Asia | NR | 131 | 100 | 0.100 |
| Wattanasirichaigoon [11] | 2004 | United States | North America | Children and adults | 166 | 205 | 0.181 |
| Shi [23] | 2004 | China | Asia | Patients: infants; Control: NR | 20 | 50 | 0.827 |
| Ohtsuka [22] | 2003 | Japan | Asia | NR | 1,227 | 147 | NA |
| Lopponen [21] | 2003 | Finland | Europe | Patients: children; Control: NR | 71 | 313 | NA |
| Hwa [20] | 2003 | China | Asia | NR | 324 | 432 | NA |
| Wu [19] | 2002 | United States | North America | Patients: children; Control: NR | 324 | 100 | NA |
| Liu [18] | 2002 | China | Asia | Patients: children and adults; Control: NR | 210 | 200 | NA |
| Marlin [17] | 2001 | France | Europe | Patients: children; Control: NR | 96 | 116 | 0.963 |
| Rabionet [10] | 2000 | Italy and Spain | Europe | Patients: children; Control: NR | 576 | 100 | NA |
| Kudo [9] | 2000 | Japan | Asia | Patients: children and adults; Control: NR | 78 | 63 | NA |
| Abe [8] | 2000 | Japan | Asia | NR | 35 | 96 | 0.918 |
| Kelley [7] | 1998 | United States | North America | NR | 58 | 96 | 0.959 |
NR: not reported; NA: not available; PHWE: P value for test of Hardy-Weinberg equilibrium.
Figure 2. Forest plots of the effects of p.V37I on HL risk under the (A) allelic, (B and C) codominant, and (D) recessive models.
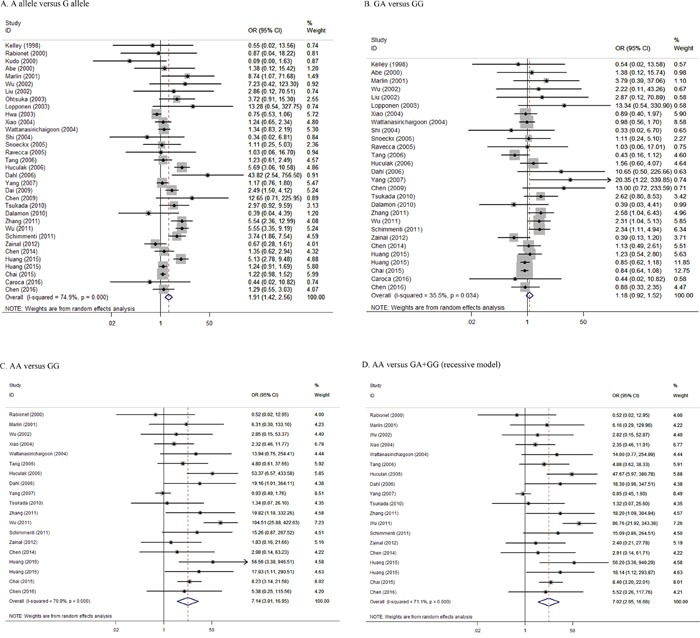
Allelic model referred to A allele versus G allele. Codominant model referred to GA genotype versus GG genotype (B), or AA genotype versus GG genotype (C). Recessive model referred to AA genotype versus GA+GG genotype.
Furthermore, we evaluated the association between compound heterozygous p.V37I variants on HL risk. Ten eligible studies comprising of 6,762 cases and 4,211 controls were included (Table 3). Notably, our results suggest that people harboring compound heterozygous p.V37I variants have a 3.63-fold higher risk of HL than those without these variants (Figure 3; OR = 3.63; 95% CI = 1.38–9.54; Pheterogeneity = 0.060; I2 = 44.9%). These results were strongly supported by sensitivity analyses (Supplementary Figure 3), and no publication bias was found (P = 0.152).
Table 3. Characteristics of studies for the association between compound heterozygous p.V37I variants and HL risk.
| First author | Publication year | Country | Population | Cases | Controls | Types |
|---|---|---|---|---|---|---|
| Huang [12] | 2015 | China | Infants | 300 | 484 | p.V37I/c.235delC, p.V37I/c.299_300delAT, p.V37I/c.79G>A, p.V37I/c.(79G>A+341A>G) |
| Huang [13] | 2015 | China | Infants and adults | 3,864 | 600 | p.V37I/c.235delC, p.V37I/c.299delAT, p.V37I/p.R143W, p.V37I/c.176del16, p.V37I/c.512insAACG, p.V37I/p.T86R, p.V37I/p.W77* |
| Chen [37] | 2014 | China | Infants and child | 107 | 61 | p.V37I/c.235delC, p.V37I/c.608T>C, p.V37I/c.(79G>A; 341A>G) |
| Zhang [35] | 2011 | China | Children and adults | 236 | 107 | p.V37I/c.235delC, p.V37I/c.427C>T, p.V27I/p.V37I/p.E114G, p.V27I/p.V37I, p.V37I/p.I203T, p.V27I/p.V37I/p.V84M |
| Wu [34] | 2011 | China | Infants | 38 | 979 | p.V37I/c.235delC |
| Schimmenti [33] | 2011 | United States | Infants | 1,177 | 1,177 | p.V37I/p.L90P, p.V37I/p.(V27I, E114G) |
| Tang [28] | 2006 | United States | NR | 610 | 294 | p.V37I/c.35delG, p.V37I/p.L90P, p.V37I/p.R216fsX232, p.V37I/p.V27I, p.V37I/(p.V27I+p.E114G), p.V37I/p.I203T |
| Lopponen [21] | 2003 | Finland | Patients: children; Control: NR | 71 | 313 | p.V37I/p.M34T |
| Wu [19] | 2002 | United States | Patients: children; Control: NR | 324 | 100 | p.V37I/p.M34T, p.V37I/c.167delT |
| Abe [8] | 2000 | Japan | NR | 35 | 96 | p.V37I/p.R143W, p.V37I/c.235delC |
NR: not reported.
Figure 3. Forest plots of the effects of compound heterozygous p.V37I variants on HL risk.
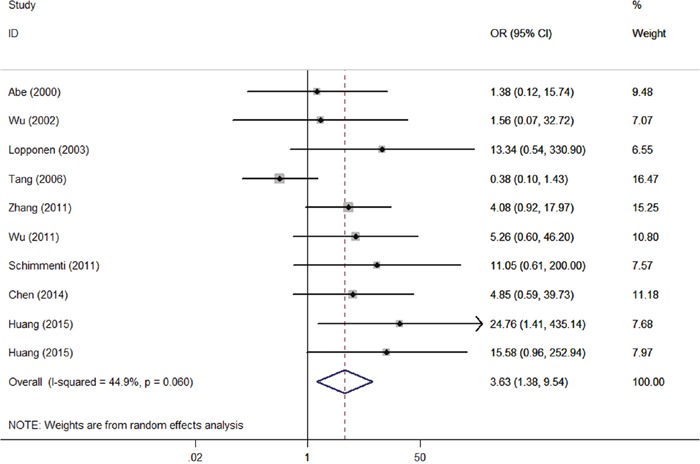
Compound heterozygous p.V37I variants referred to the heterozygous p.V37I allele in a trans configuration with another well-known pathogenic mutant allele of GJB2 gene.
DISCUSSION
In this study, we demonstrated that bi-allelic or compound heterozygous p.V37I variants are associated with increased risk of various HL phenotypes, and quantified the risk associated with these variants and development of HL.
Our pedigree analyses indicated that p.V37I can cause HL as either a homozygous variant or as compound heterozygous with other pathogenic variants in GJB2. Functional studies performed in cells and mouse models support this conclusion [40–42]. In consistent with previous reports [6, 12–14], we observed that the HL phenotypes associated with p.V37I varied by onset type, disease site, and degree of HL, implying that other causes, especially environmental factors, influence p.V37I-mediated onset of HL. Notably, we also found that homozygous p.V37I might give rise to sudden deafness in children. Indeed, similar results were reported in a recent study [39]. As such, p.V37I might be considered as a potential cause of sudden onset of deafness in future cases.
To quantify the pathogenic association between p.V37I and HL risk, we performed a meta-analysis. In keeping with previous cohort studies and functional experiments, our results supported the conclusion that the p.V37I variant significantly increases an individual's risk of developing HL. Compared with wild type individuals, the p.V37I homozygote group, but not the heterozygote group, showed a significantly greater likelihood of developing HL. This could explain why p.V37I heterozygotes are prevalent among the general healthy population, while bi-allelic p.V37I variants are found predominantly in patients with HL. Notably, our results also indicate that the compound heterozygous p.V37I variants are associated with a nearly four-fold greater risk of developing HL, compared to wild type individuals. In view of the high prevalence of p.V37I and the large number of other GJB2 pathogenic variants, our findings indicate that previous reports have likely underestimated HL risk in human populations. Compared with the previous meta-analysis [16], our work included more eligible studies and quantified the risky effects of p.V37I on HL in more detail.
The major strengths of our work were the variable phenotypic spectrum associated with p.V37I and that a large population (23,097 participants) was used to evaluate the association between this variant and HL risk. However, our results should be interpreted with caution. First, only commonly known HL-related variants (variants within the GJB2, SLC26A4, 12S rRNA, and GJB3 coding regions) were screened in our pedigree analysis. Second, the number of studies regarding compound heterozygous p.V37I variants evaluated herein was insufficient to further explore the concrete effects of particular compound variant types on HL risk. Third, although both our study and previous reports found that p.V37I is associated with various HL phenotypes, the specific mechanism by which this variant promotes HL, such as interactions between p.V37I and other genetic or environmental factors, remains unclear. Further studies are therefore needed to address these issues.
In summary, our work strongly suggests a pathogenic role for p.V37I in various HL phenotypes and provides a quantitative assessment of the risk associated with carriage of this variant and development of HL. Considering the high carriage rate of p.V37I within the general population, these findings provide compelling information that should influence future genetic screening policy and that offer insights into clinical risk evaluation and genetic counseling.
MATERIALS AND METHODS
Study participants and ethical statement
For this study, we recruited seven unrelated probands with non-syndromic, sensorineural hearing loss, and their family members. Each participant provided written informed consent. For underage participants, written informed consent was obtained from their parents. Our study was approved by the Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. All procedures were performed according to the ethical guidelines for human subjects research.
Clinical and audiometric evaluation
All participants were subjected to physical and neurological examinations to exclude syndromic deafness. Each participant's level of hearing was assessed via a comprehensive auditory evaluation, such as by otoscope examination, tympanometry, or pure-tone audiometry (PTA). For very young participants, auditory brainstem responses (ABR) and/or auditory steady-state responses (ASSR) were recorded. Degree of hearing loss was estimated as the average hearing levels at 0.5, 1.0, 2.0, and 4.0 kHz for the better ear. Severity of hearing loss was categorized as normal (<25 dB), mild (26-40 dB), moderate (41–70 dB), severe (71–95 dB), or profound ( >95 dB). In addition, we defined sudden deafness as over 30 dB of sensorineural hearing loss involving at least three frequencies occurring less than 72 hours [43].
DNA extraction and mutation analysis
Genomic DNA was extracted from anticoagulant peripheral blood by the QIAamp DNA blood mini kit (Qiagen, Germany). For mutation screening, the entire coding region and flanking sequences of four commonly mutated genes, GJB2, SLC26A4, 12S rRNA, and GJB3, were polymerase chain reaction (PCR) amplified and then subjected to bidirectional sequencing with an ABI 3500 DNA sequencer (Applied Biosystems). The primers and PCR conditions used for these analyses are described in detail in Supplementary Table 1. Sanger sequencing results of pedigree analyses were shown in Supplementary Figure 4.
Meta-analysis on sporadic HL
We further performed a meta-analysis to determine the detrimental effects of p.V37I on sporadic HL, according to the guidelines of Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [44]. A structured literature search was conducted of articles published through November 2016 using the PubMed, Web of Science, and EMBASE databases and the following search items: “GJB2 OR connexin 26”, “polymorphism OR variant OR mutation”, “hearing loss OR deafness”, and “case-control OR cohort OR population”. Only English language articles were included. The inclusion criteria were: (i) studies that investigated the association between p.V37I and HL under a case-control or cohort design; (ii) studies that reported genotypes or allelic data for p.V37I to estimate OR and 95%CI. In cases of overlapping populations, the study with the largest sample size was included. We used OR as the effect measure and combined data using a random-effects model. Hardy-Weinberg equilibrium (HWE) in the controls was checked by χ2 test. Meanwhile, Cochran χ2 test and I2 values were used to evaluate the heterogeneity between studies. Sensitivity analyses and publication bias assessment were also conducted. All statistical analyses were performed using Stata 12.1 software (StataCorp, College Station, TX, USA), and P ≤ 0.05 was considered significant for all tests.
SUPPLEMENTARY MATERIALS FIGURES AND TABLE
Footnotes
Author contributions
A.L. and Y.L. conceived and designed this study. N.S., A.L. and Y.L. recruited the study subjects and collected their characteristics and examination results. Y.Z. and W.L. extracted DNA. N.S. and J.P. conducted mutation analyses. J.P. and X.W. performed literature search, study selection and data extraction. X.W. and Y.Z. conducted statistical analysis. J.P. and N.S. prepared tables and figures. N.S., A.L. and Y.L. wrote the manuscript. All these authors completely consented with all the data in the study and approved the final manuscript. A.L. and Y.L. had the primary responsibility for final manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
FUNDING
This study was supported by the grants to Dr. Shen (81601839) from the National Natural Science Foundation of China.
REFERENCES
- 1.Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–64. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Zelante L, Gasparini P, Estivill X, Melchionda S, D'Agruma L, Govea N, Mila M, Monica MD, Lutfi J, Shohat M, Mansfield E, Delgrosso K, Rappaport E, et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet. 1997;6:1605–9. doi: 10.1093/hmg/6.9.1605. [DOI] [PubMed] [Google Scholar]
- 3.Minami SB, Mutai H, Nakano A, Arimoto Y, Taiji H, Morimoto N, Sakata H, Adachi N, Masuda S, Sakamoto H, Yoshida H, Tanaka F, Morita N, et al. GJB2-associated hearing loss undetected by hearing screening of newborns. Gene. 2013;532:41–5. doi: 10.1016/j.gene.2013.08.094. [DOI] [PubMed] [Google Scholar]
- 4.Han SH, Park HJ, Kang EJ, Ryu JS, Lee A, Yang YH, Lee KR. Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. J Hum Genet. 2008;53:1022–8. doi: 10.1007/s10038-008-0342-7. [DOI] [PubMed] [Google Scholar]
- 5.Tsukada K, Nishio S, Usami S. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet. 2010;78:464–70. doi: 10.1111/j.1399-0004.2010.01407.x. [DOI] [PubMed] [Google Scholar]
- 6.Wu CC, Tsai CH, Hung CC, Lin YH, Lin YH, Huang FL, Tsao PN, Su YN, Lee YL, Hsieh WS, Hsu CJ. Newborn genetic screening for hearing impairment: a population-based longitudinal study. Genet Med. 2017;19:6–12. doi: 10.1038/gim.2016.66. [DOI] [PubMed] [Google Scholar]
- 7.Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998;62:792–9. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:41–3. doi: 10.1136/jmg.37.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, Narisawa K, Takasaka T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet. 2000;90:141–5. doi: 10.1002/(SICI)1096-8628(20000117)90:2<141::AID-AJMG10>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 10.Rabionet R, Zelante L, Lopez-Bigas N, D'Agruma L, Melchionda S, Restagno G, Arbones ML, Gasparini P, Estivill X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106:40–4. doi: 10.1007/s004390051007. [DOI] [PubMed] [Google Scholar]
- 11.Wattanasirichaigoon D, Limwongse C, Jariengprasert C, Yenchitsomanus PT, Tocharoenthanaphol C, Thongnoppakhun W, Thawil C, Charoenpipop D, Pho-iam T, Thongpradit S, Duggal P. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin Genet. 2004;66:452–60. doi: 10.1111/j.1399-0004.2004.00325.x. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Yang XL, Chen WX, Duan B, Lu P, Wang Y, Xu ZM. Prevalence of p.V37I variant of GJB2 among Chinese infants with mild or moderate hearing loss. Int J Clin Exp Med. 2015;8:21674–8. [PMC free article] [PubMed] [Google Scholar]
- 13.Huang S, Huang B, Wang G, Yuan Y, Dai P. The relationship between the p.V37I mutation in GJB2 and hearing phenotypes in Chinese individuals. PLoS One. 2015;10:e0129662. doi: 10.1371/journal.pone.0129662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chai Y, Chen D, Sun L, Li L, Chen Y, Pang X, Zhang L, Wu H, Yang T. The homozygous p.V37I variant of GJB2 is associated with diverse hearing phenotypes. Clin Genet. 2015;87:350–5. doi: 10.1111/cge.12387. [DOI] [PubMed] [Google Scholar]
- 15.Huculak C, Bruyere H, Nelson TN, Kozak FK, Langlois S. V37I connexin 26 allele in patients with sensorineural hearing loss: evidence of its pathogenicity. Am J Med Genet A. 2006;140:2394–400. doi: 10.1002/ajmg.a.31486. [DOI] [PubMed] [Google Scholar]
- 16.Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope. 2014;124:E34–53. doi: 10.1002/lary.24332. [DOI] [PubMed] [Google Scholar]
- 17.Marlin S, Garabedian EN, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg. 2001;127:927–33. doi: 10.1001/archotol.127.8.927. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Ke X, Qi Y, Li W, Zhu P. Connexin26 gene ( GJB2): prevalence of mutations in the Chinese population. J Hum Genet. 2002;47:688–90. doi: 10.1007/s100380200106. [DOI] [PubMed] [Google Scholar]
- 19.Wu BL, Lindeman N, Lip V, Adams A, Amato RS, Cox G, Irons M, Kenna M, Korf B, Raisen J, Platt O. Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet Med. 2002;4:279–88. doi: 10.1097/00125817-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Hwa HL, Ko TM, Hsu CJ, Huang CH, Chiang YL, Oong JL, Chen CC, Hsu CK. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet Med. 2003;5:161–5. doi: 10.1097/01.gim.0000066796.11916.94. [DOI] [PubMed] [Google Scholar]
- 21.Lopponen T, Vaisanen ML, Luotonen M, Allinen M, Uusimaa J, Lindholm P, Maki-Torkko E, Vayrynen M, Lopponen H, Leisti J. Connexin 26 mutations and nonsyndromic hearing impairment in northern Finland. Laryngoscope. 2003;113:1758–63. doi: 10.1097/00005537-200310000-00018. [DOI] [PubMed] [Google Scholar]
- 22.Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, Van Camp G, Usami S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003;112:329–33. doi: 10.1007/s00439-002-0889-x. [DOI] [PubMed] [Google Scholar]
- 23.Shi GZ, Gong LX, Xu XH, Nie WY, Lin Q, Qi YS. GJB2 gene mutations in newborns with non-syndromic hearing impairment in Northern China. Hear Res. 2004;197:19–23. doi: 10.1016/j.heares.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Xiao ZA, Xie DH. GJB2 (Cx26) gene mutations in Chinese patients with congenital sensorineural deafness and a report of one novel mutation. Chin Med J (Engl) 2004;117:1797–801. [PubMed] [Google Scholar]
- 25.Ravecca F, Berrettini S, Forli F, Marcaccini M, Casani A, Baldinotti F, Fogli A, Siciliano G, Simi P. Cx26 gene mutations in idiopathic progressive hearing loss. J Otolaryngol. 2005;34:126–34. doi: 10.2310/7070.2005.04017. [DOI] [PubMed] [Google Scholar]
- 26.Snoeckx RL, Djelantik B, Van Laer L, Van de Heyning P, Van Camp G. GJB2 (connexin 26) mutations are not a major cause of hearing loss in the Indonesian population. Am J Med Genet A. 2005;135:126–9. doi: 10.1002/ajmg.a.30726. [DOI] [PubMed] [Google Scholar]
- 27.Dahl HH, Tobin SE, Poulakis Z, Rickards FW, Xu X, Gillam L, Williams J, Saunders K, Cone-Wesson B, Wake M. The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children. J Med Genet. 2006;43:850–5. doi: 10.1136/jmg.2006.042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang HY, Fang P, Ward PA, Schmitt E, Darilek S, Manolidis S, Oghalai JS, Roa BB, Alford RL. DNA sequence analysis of GJB2, encoding connexin 26: observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A. 2006;140:2401–15. doi: 10.1002/ajmg.a.31525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang JJ, Huang SH, Chou KH, Liao PJ, Su CC, Li SY. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol Neurootol. 2007;12:198–208. doi: 10.1159/000099024. [DOI] [PubMed] [Google Scholar]
- 30.Chen D, Chen X, Cao K, Zuo J, Jin X, Wei C, Fang F. High prevalence of the connexin 26 (GJB2) mutation in Chinese cochlear implant recipients. ORL J Otorhinolaryngol Relat Spec. 2009;71:212–5. doi: 10.1159/000229300. [DOI] [PubMed] [Google Scholar]
- 31.Dai P, Yu F, Han B, Liu X, Wang G, Li Q, Yuan Y, Liu X, Huang D, Kang D, Zhang X, Yuan H, Yao K, et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009;7:26. doi: 10.1186/1479-5876-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalamon V, Lotersztein V, Beheran A, Lipovsek M, Diamante F, Pallares N, Francipane L, Frechtel G, Paoli B, Mansilla E, Diamante V, Elgoyhen AB. GJB2 and GJB6 genes: molecular study and identification of novel GJB2 mutations in the hearing-impaired Argentinean population. Audiol Neurootol. 2010;15:194–202. doi: 10.1159/000254487. [DOI] [PubMed] [Google Scholar]
- 33.Schimmenti LA, Warman B, Schleiss MR, Daly KA, Ross JA, McCann M, Jurek AM, Berry SA. Evaluation of newborn screening bloodspot-based genetic testing as second tier screen for bedside newborn hearing screening. Genet Med. 2011;13:1006–10. doi: 10.1097/GIM.0b013e318226fc2e. [DOI] [PubMed] [Google Scholar]
- 34.Wu CC, Hung CC, Lin SY, Hsieh WS, Tsao PN, Lee CN, Su YN, Hsu CJ. Newborn genetic screening for hearing impairment: a preliminary study at a tertiary center. PLoS One. 2011;6:e22314. doi: 10.1371/journal.pone.0022314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Wang Z, Dai W, Zeng Y, Li H. GJB2 allele variants and the associated audiologic features identified in Chinese patients with less severe idiopathic hearing loss. Genet Test Mol Biomarkers. 2011;15:313–8. doi: 10.1089/gtmb.2010.0182. [DOI] [PubMed] [Google Scholar]
- 36.Zainal SA, Md Daud MK, Abd Rahman N, Zainuddin Z, Alwi Z. Mutation detection in GJB2 gene among Malays with non-syndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2012;76:1175–9. doi: 10.1016/j.ijporl.2012.04.027. [DOI] [PubMed] [Google Scholar]
- 37.Chen T, Jiang L, Liu C, Shan H, Chen J, Yang B, Ou Q. Update of the spectrum of GJB2 mutations in 107 patients with nonsyndromic hearing loss in the Fujian population of China. Ann Hum Genet. 2014;78:235–42. doi: 10.1111/ahg.12062. [DOI] [PubMed] [Google Scholar]
- 38.Caroça C, De Matos TM, Ribeiro D, Lourenço V, Martins T, Campelo P, Fialho G, Silva SN, Paço J, Caria H. Genetic basis of nonsyndromic sensorineural hearing loss in the sub-Saharan African island population of São Tomé and Príncipe: the role of the DFNB1 locus? OMICS. 2016;20:449–55. doi: 10.1089/omi.2016.0067. [DOI] [PubMed] [Google Scholar]
- 39.Chen K, Sun L, Zong L, Wu X, Zhan Y, Dong C, Cao H, Tang H, Jiang H. GJB2 and mitochondrial 12S rRNA susceptibility mutations in sudden deafness. Eur Arch Otorhinolaryngol. 2016;273:1393–8. doi: 10.1007/s00405-015-3693-7. [DOI] [PubMed] [Google Scholar]
- 40.Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D'Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/S0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- 41.Jara O, Acuna R, Garcia IE, Maripillan J, Figueroa V, Saez JC, Araya-Secchi R, Lagos CF, Perez-Acle T, Berthoud VM, Beyer EC, Martinez AD. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol Biol Cell. 2012;23:3299–311. doi: 10.1091/mbc.E11-12-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Hu L, Wang X, Sun C, Lin X, Li L, Mei L, Huang Z, Yang T, Wu H. Characterization of a knock-in mouse model of the homozygous p.V37I variant in Gjb2. Sci Rep. 2016;6:33279. doi: 10.1038/srep33279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stachler RJ, Chandrasekhar SS, Archer SM, Rosenfeld RM, Schwartz SR, Barrs DM, Brown SR, Fife TD, Ford P, Ganiats TG, Hollingsworth DB, Lewandowski CA, Montano JJ, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146:S1–35. doi: 10.1177/0194599812436449. [DOI] [PubMed] [Google Scholar]
- 44.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. Open Med. 2009;3:e123–30. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.