

SUMMARY
RNA modifications that once escaped detection are now thought to be pivotal for governing RNA lifetimes in both prokaryotes and eukaryotes. For example, converting the 5′-terminal triphosphate of bacterial transcripts to a monophosphate triggers 5′-end-dependent degradation by RNase E. However, the existence of diphosphorylated RNA in bacteria has never been reported, and no biological role for such a modification has ever been proposed. By using a novel assay, we show here for representative Escherichia coli mRNAs that ~35–50% of each transcript is diphosphorylated. The remainder is primarily monophosphorylated, with surprisingly little triphosphorylated RNA evident. Furthermore, diphosphorylated RNA is the preferred substrate of the RNA pyrophosphohydrolase RppH, whose biological function was previously assumed to be pyrophosphate removal from triphosphorylated transcripts. We conclude that triphosphate-to-monophosphate conversion to induce 5′-end-dependent RNA degradation is a two-step process in E. coli involving γ-phosphate removal by an unidentified enzyme to enable subsequent β-phosphate removal by RppH.
Keywords: Diphosphate, RNA decay, pyrophosphatase, guanylyltransferase, Pce1, cap, PACO, PABLO, transcription initiation, RNA polymerase, E. coli, yeiP, metE
eTOC blurb
Luciano et al. show that diphosphorylated mRNA 5′ ends are abundant in E. coli and that the E. coli RNA pyrophosphohydrolase RppH prefers diphosphorylated substrates to their triphosphorylated counterparts. Their findings suggest that converting triphosphorylated to monophosphorylated 5′ ends before RNase E cleavage involves sequential phosphate removal by distinct enzymes.
INTRODUCTION
Of the various mechanisms by which genes are regulated, mRNA degradation is among the least well understood. In both eukaryotes and prokaryotes, the lifetimes of individual messages can differ by as much as two orders of magnitude, with proportionate effects on protein synthesis. However, in spite of the widespread regulatory impact of this process, the basis for most differences in mRNA longevity has yet to be explained.
Eukaryotic mRNAs are typically protected from unrestrained degradation by a 5′-terminal cap and a 3′-terminal poly(A) tail, the removal of which is tightly controlled and exposes messages to rapid exonucleolytic destruction (Garneau et al., 2007). By contrast, bacterial mRNAs are thought to be protected by a 5′-terminal triphosphate and a 3′-terminal stem-loop and to be degraded by a distinct set of ribonucleases (Hui et al., 2014). Despite progress toward elucidating mRNA decay in bacteria, many of the most important molecular mechanisms that govern mRNA longevity in such organisms have remained obscure, even in so well studied a species as Escherichia coli.
In E. coli, mRNA degradation usually proceeds via either of two pathways that both involve cleavage by RNase E, an endonuclease that cuts with only modest sequence specificity in single-stranded regions that are AU-rich (McDowall et al., 1994; Lin-Chao et al., 1994; Kaberdin, 2003; Del Campo et al., 2015; Chao et al., 2017). In one of these pathways, RNase E gains direct access to internal sites within unprocessed primary transcripts bearing a 5′-terminal triphosphate. Cleavage at these sites produces two fragments: (i) a 5′ fragment that lacks a protective stem-loop at its 3′ terminus and therefore undergoes rapid 3′-exonucleolytic degradation and (ii) a monophosphorylated 3′ fragment that is highly susceptible to further RNase E cleavage due to the preference of this enzyme for substrates that have a single phosphate at the 5′ end (Mackie, 1998). Degradation by the other pathway is 5′-end-dependent, as it is triggered by removal of two of the three phosphates from the 5′ terminus of primary transcripts so as to generate a full-length mRNA that is monophosphorylated and therefore vulnerable to attack by RNase E (Celesnik et al., 2007). Central to this initial triggering event is the RNA pyrophosphohydrolase RppH (Deana et al., 2008).
In vitro, E. coli RppH converts triphosphorylated RNA to monophosphorylated RNA in a single step by cutting between the two phosphates (α and β) adjacent to the first nucleoside, generating pyrophosphate as the principal by-product (Deana et al., 2008). This reaction requires at least two unpaired nucleotides at the 5′ end, helping to explain the protective effect of the 5′-terminal stem-loop structures that are present on some long-lived transcripts in E. coli (Foley et al., 2015). Apart from this strict requirement, E. coli RppH is able to remove pyrophosphate from RNAs that begin with any 5′-terminal sequence, although it has a modest preference for A over G at position 1 and for a purine at position 2 (Foley et al., 2015; Vasilyev and Serganov, 2015). The purified enzyme is also able to remove a single phosphate from the 5′ end of diphosphorylated transcripts synthesized in vitro (Vasilyev and Serganov, 2015), again by cutting between the α and β phosphates.
RppH plays a crucial role in 5′-end-dependent mRNA degradation in E. coli, where deletion of the rppH gene reduces the percentage of full-length transcripts that are monophosphorylated almost to zero and prolongs the lifetime of hundreds of mRNAs (Deana et al., 2008) while also impairing the ability of pathogenic E. coli to invade human cells (Badger et al., 2000). By contrast, overproducing RppH has little effect on the phosphorylation state or half-life of the mRNAs it targets, suggesting that the rate of formation of the full-length monophosphorylated intermediate may be limited by the cellular concentration of an ancillary factor or by a hypothetical step that precedes attack by RppH (Luciano et al., 2012). This observation led to the discovery that the specific activity of RppH is boosted 2–3 fold in E. coli by binding to the diaminopimelate epimerase DapF (Lee et al., 2014). However, the possibility of a prior step that triggers the action of RppH has not previously been explored.
We have now investigated whether conversion of a 5′-terminal triphosphate to a monophosphate in E. coli occurs in a single step by pyrophosphate removal, as has commonly been assumed. Our examination of representative triphosphorylated E. coli transcripts reveals that they are first transformed into diphosphorylated RNAs, which accumulate to a high cellular concentration, and that the latter RNAs, not their triphosphorylated precursors, are the preferred substrates of RppH. These results indicate that a previously unrecognized event enables 5′-end-dependent mRNA degradation in E. coli and raise the possibility of an important role for diphosphorylated 5′ ends in other RNA-related cellular processes.
RESULTS
Comparative reactivity of diphosphorylated and triphosphorylated RNA as RppH substrates
In principle, the conversion of a 5′-terminal triphosphate to a monophosphate could occur in one step by pyrophosphate removal or in two steps by a mechanism involving sequential loss of the γ and β phosphates and the transient production of a diphosphorylated intermediate. However, whether diphosphorylated RNA molecules exist in bacterial cells has not previously been investigated. To begin to address the possibility of a diphosphorylated intermediate in 5′-end-dependent mRNA degradation in E. coli, we first compared the reactivity of diphosphorylated and triphosphorylated RNA as substrates for RppH (Figure 1A). To conduct this test, we used a variant of the E. coli yeiP transcript, which encodes a paralog of the translation elongation factor EF-P and decays by an RppH-dependent mechanism in vivo (Deana et al., 2008; Richards et al., 2012). This variant (yeiP-U2G) contained a U-to-G substitution at the second nucleotide to facilitate its synthesis by T7 RNA polymerase.
Figure 1. Relative reactivity of diphosphorylated and triphosphorylated RNA as substrates of purified E. coli RppH.

(A) Substrates and product of the reaction of RppH with diphosphorylated and triphosphorylated yeiP-U2G RNA. The first six nucleotides of yeiP-U2G are shown. p, 5′-terminal α, β, or γ phosphate. (B, C) The reaction of diphosphorylated and triphosphorylated yeiP-U2G with RppH was compared in the presence of the ancillary factor DapF by using PABLO to detect the monophosphorylated reaction products. In each case, a diphosphorylated derivative of yeiP-U2G with the same 5′-terminal sequence but a distinct length served as an internal standard. The increase in the concentration of monophosphorylated reaction products is graphed beneath each blot. Representative experiments are shown. Bent arrows beside the blots lead from each RNA substrate to its PABLO ligation product. M, fully monophosphorylated yeiP-U2G generated by treating an equivalent substrate mixture with excess RppH for an extended period of time. Filled triangles, reaction of diphosphorylated yeiP-U2G (DiP). Gray circles, reaction of triphosphorylated yeiP-U2G (TriP). Empty triangles, reaction of the diphosphorylated internal standard (Std). In multiple experiments of this kind, the initial reaction rate of diphosphorylated yeiP-U2G was 3.2 ± 0.6 times that of its triphosphorylated counterpart; because the reactions slowed as a function of time, this value represents a lower limit on the difference in the initial rates.
Diphosphorylated and triphosphorylated yeiP-U2G RNAs were prepared by in vitro transcription along with another diphosphorylated transcript that was included as an internal standard in every experiment. The pairwise reaction of these substrates with purified E. coli RppH was examined by monitoring the conversion of each to a monophosphorylated product as a function of time. This was accomplished by PABLO, a splinted ligation assay in which the 5′ end of monophosphorylated RNA is joined to a DNA oligonucleotide (Celesnik et al., 2007, 2008). By contrast, diphosphorylated RNA undergoes ligation at a meager rate (Figure S1), and triphosphorylated RNA is completely unreactive (Celesnik et al., 2007). The ligation product and any remaining unligated RNA are then separated by gel electrophoresis and quantified by Northern blotting. Although the ligation reaction is not sufficiently fast to proceed all the way to completion, even with fully monophosphorylated RNA, the percentage of 5′ ends that are monophosphorylated can be accurately calculated from the ratio of the ligation yield for a sample of interest to that of a fully monophosphorylated control sample (Luciano et al., 2012).
Using this procedure, the reactivity of diphosphorylated and triphosphorylated yeiP-U2G RNA as substrates of E. coli RppH was compared (Figure 1B, C). These rate measurements demonstrated that diphosphorylated yeiP-U2G reacts with RppH at least three times faster than its triphosphorylated counterpart, while the reaction rate of the diphosphorylated internal standard did not vary.
Previous studies have demonstrated that the reactivity of triphosphorylated substrates with E. coli RppH is influenced by the identity of the nucleotide at the second position, where a purine (especially G) is preferred over a pyrimidine (Foley et al., 2015; Vasilyev and Serganov, 2015). To determine whether this sequence preference also applies to diphosphorylated substrates, we compared the reaction rates of four dinucleotide substrates (AG, AA, AU, and AC) bearing two or three phosphates at the 5′ end. The progress of each reaction was monitored by using anion-exchange chromatography to separate the substrate and product, which were then detected spectrophotometrically (Figure 2A). In every case, the diphosphorylated substrate reacted with E. coli RppH at least ten times faster than its triphosphorylated counterpart (Figure 2B, Table S1). Each substrate yielded a monophosphorylated dinucleotide as the sole reaction product; no diphosphorylated intermediate was detected for the triphosphorylated substrates. Moreover, as previously reported for triphosphorylated RNA, the order of reactivity of the diphosphorylated substrates with RppH was AG ≥ AA > AU ≥ AC.
Figure 2. Sequence-independent preference of purified E. coli RppH for diphosphorylated over triphosphorylated dinucleotide substrates.

(A) Chromatographic separation of diphosphorylated and triphosphorylated AG dinucleotide substrates from monophosphorylated reaction products. Representative samples of separate reactions were quenched before and 10 min after adding RppH and fractionated by anion-exchange chromatography. Substrates and products were detected spectrophotometrically as a function of elution volume, and the chromatographic traces were superimposed. Black traces, reaction of the diphosphorylated substrate (ppAG) to produce a monophosphorylated product (pAG). Gray traces, reaction of the triphosphorylated substrate (pppAG) to produce a monophosphorylated product (pAG).
(B) Relative reactivity of diphosphorylated versus triphosphorylated AG, AA, AU, and AC dinucleotides. Reaction samples were quenched at 1- to 2-min intervals after adding equal amounts of RppH and analyzed chromatographically to determine the percentage of substrate remaining. Black triangles, reaction of diphosphorylated substrates (DiP). Gray circles, reaction of triphosphorylated substrates (TriP). Mean values from multiple experiments are plotted. To avoid obscured error bars, the standard deviation of each measurement is reported in Table S1.
Assay for detecting diphosphorylated intermediates in cellular RNA
The much greater reactivity of E. coli RppH toward diphosphorylated versus triphosphorylated RNA raised the possibility that the former might be its principal biological substrate, a notion consistent with the hypothesis that the conversion of 5′-terminal triphosphates to monophosphates might often proceed via a diphosphorylated intermediate. To ascertain whether such decay intermediates exist in cells, we devised an assay – Phosphorylation Assay by Capping Outcome (PACO) – that makes it possible to determine the percentage of a particular RNA that is diphosphorylated at the 5′ end (Figure 3A). This method is based on the substrate specificity of the RNA guanylyltransferase that catalyzes the second step of RNA capping in eukaryotic cells: the reaction of GTP with a diphosphorylated 5′ end to add an unmethylated cap (Shuman, 2001). We reasoned that such capping would render the 5′ end of diphosphorylated RNA resistant to alkaline phosphatase, whereas the 5′-terminal phosphates of triphosphorylated or monophosphorylated RNA would remain susceptible to removal by alkaline phosphatase due to the inability of the guanylyltransferase to cap those RNAs. Subsequent pyrophosphatase treatment would release the cap from transcripts that had originally been diphosphorylated to produce a monophosphorylated product that could be detected and quantified by PABLO.
Figure 3. Detection of diphosphorylated yeiP-U2G mRNA in E. coli.

(A) Method for detecting diphosphorylated RNA (PACO). Pce1 preferentially guanylylates the 5′ end of diphosphorylated RNA. Subsequent treatment with alkaline phosphatase removes the exposed 5′-terminal phosphates of triphosphorylated and monophosphorylated RNA but not the protected phosphates of guanylylated RNA, which is then converted to monophosphorylated RNA by treatment with a pyrophosphatase and detected by PABLO.
(B) Specificity of PACO as determined by analysis of in vitro transcribed yeiP-U2G RNA bearing a 5′-terminal triphosphate (TriP), diphosphate (DiP), or monophosphate (MonoP). The analytical reactions were performed in the presence of total E. coli RNA from ΔyeiP cells to simulate the assay of an endogenous E. coli transcript. See also Tables S2 and S3.
(C) Relative abundance of diphosphorylated yeiP-U2G mRNA in wild-type (WT) and ΔrppH cells, as determined by PACO. A vertical line marks the location of superfluous lanes omitted from the image. See also Tables S4 and S5 and Figures S2 and S4.
(D) Relative abundance of monophosphorylated yeiP-U2G mRNA in wild-type (WT) and ΔrppH cells, as determined by PABLO. See also Tables S4 and S5 and Figures S1 and S4.
(E) Number of phosphates in the yeiP-U2G cap after guanylylation by Pce1. Equimolar amounts of triphosphorylated, diphosphorylated, or monophosphorylated yeiP-U2G standards synthesized by in vitro transcription and yeiP-U2G mRNA selectively purified from total RNA that had been extracted from wild-type or ΔrppH cells were capped by Pce1 in the presence of [α-32P]GTP, digested with nuclease S1 to release the capped 5′-terminal nucleotide, treated with alkaline phosphatase to hydrolyze the remaining GTP, and analyzed by thin layer chromatography on PEI-cellulose and autoradiography. The radiolabeled reaction products comigrated with unlabeled samples of GpppA and GppppA that were obtained commercially and detected by their ability to quench fluorescence (not shown). Capping and subsequent hydrolysis of the diphosphorylated standard produced GpppA. Capping and subsequent hydrolysis of the triphosphorylated standard produced primarily GppppA, along with a small quantity of GpppA whose mechanism of formation is unclear. *, GppppG produced by Pce1-catalyzed capping of GTP (Wang and Shatkin, 1984); Origin, denatured reaction intermediate comprising [α-32 P]guanylate covalently bonded to Pce1 (Shuman and Hurwitz, 1981).
For a detailed explanation of how the values in panels B, C, and D were calculated, see STAR Methods. Minor discrepancies versus the theoretical maximum (100%) or minimum (0%) resulted from the small mathematical correction factors that were used and are either within the margin of error or close to it. A bent arrow leads from each RNA substrate to its ligation product.
The specificity of PACO was tested by using the RNA guanylyltransferase of Schizosaccharomyces pombe (Pce1) (Pei and Shuman, 2002) to analyze in vitro transcribed yeiP- U2G RNA bearing one, two, or three phosphates at the 5′ end (Figure 3B). In each case, the in vitro transcript was mixed with total E. coli RNA from ΔyeiP cells to simulate the conditions of an actual assay of an endogenous E. coli transcript. As anticipated, diphosphorylated yeiP-U2G was capped and subsequently ligated with a high yield (69%). The failure to achieve a yield of 100% was due primarily to incomplete ligation of the monophosphorylated assay intermediate to the DNA oligonucleotide, as evidenced by the 80% ligation yield observed when all prior steps except pyrophosphatase treatment were omitted from the procedure, but was also due in part to incomplete capping by Pce1. From these values and some other minor corrections (see STAR Methods), we calculated a capping efficiency (ED) of 0.90 ± 0.06 for the diphosphorylated substrate on a scale from 0 (no capping) to 1 (complete capping) (Tables S2 and S3). Ligation was observed for this substrate only when it was capped by Pce1 prior to being treated with alkaline phosphatase. The triphosphorylated and monophosphorylated RNAs yielded significantly smaller amounts of ligation product in the assay (ET = 0.31 ± 0.09 and EM = 0.04 ± 0.03, respectively). This lack of strict specificity appears to be an intrinsic property of Pce1, as demonstrated by the complete absence of a ligation product when treatment with this enzyme was omitted from the assay, and it is a property shared by other RNA guanylyltransferases (Yu and Shuman, 1996; Chen et al., 1999). These imperfections can all be accommodated when analyzing biological RNA samples, as it is possible to compensate mathematically for both the incomplete reactivity of diphosphorylated RNA and partial reactivity of triphosphorylated and monophosphorylated RNA by using the following equation to calculate the fraction of a particular RNA that is diphosphorylated:
where D is the diphosphorylated fraction, M is the monophosphorylated fraction (as determined by PABLO), Y is the fractional PACO ligation yield, and EL is the ligation efficiency of fully monophosphorylated RNA. This equation is derived from two simpler ones: the fraction of RNA that becomes capped (Y/EL) = ET T + ED D + EM M and T + D + M = 1, where T is the triphosphorylated fraction.
Presence of diphosphorylated RNA in E. coli
We next used PACO to investigate whether diphosphorylated RNA could be detected in E. coli. If formed, such RNAs might have a fleeting existence in vivo due to their susceptibility to rapid phosphate removal by RppH. Therefore, we decided to test for their presence in cells containing or lacking this enzyme. By performing these tests on cellular yeiP-U2G mRNA in parallel with the in vitro transcribed yeiP-U2G standards shown in Figure 3B, whose length and sequence were identical to those of the cellular transcript, it was possible to accurately quantify the fraction of this transcript that was diphosphorylated in E. coli.
Diphosphorylated yeiP-U2G mRNA was detected at substantial levels in both the presence and absence of RppH. In log-phase rppH+ cells, 34 ± 6% of yeiP-U2G was diphosphorylated at steady state (Figure 3C, Tables S4 and S5). This value rose to 108 ± 13% in the absence of RppH. No ligation was observed without prior capping by Pce1, evidence that none of the cellular yeiP-U2G mRNA was intrinsically resistant to treatment with alkaline phosphatase. Parallel analysis of the same RNA samples by PABLO showed that nearly all the rest of the yeiP-U2G mRNA in the rppH+ strain was monophosphorylated (71 ± 3%), whereas little or none was monophosphorylated in ΔrppH cells (−3 ± 1%) (Figure 3D). Consequently, it appears that almost none of this transcript remained triphosphorylated at steady state in either cell type. We also compared PACO ligation yields for wild-type yeiP mRNA in cells containing or lacking RppH. Although the 5′-terminal sequence of this transcript (AU…) prevented its synthesis by T7 RNA polymerase and therefore precluded precise quantitative analysis, a substantial fraction of yeiP mRNA appeared to be diphosphorylated in rppH+ cells and even more was diphosphorylated in ΔrppH cells (Figure S2), results consistent with those for the U2G variant.
To begin to assess how common diphosphorylated mRNAs are in E. coli, we also examined the 5′ phosphorylation state of the metE transcript, which encodes a homocysteine transmethylase important for methionine biosynthesis. This message was chosen because its decay rate in E. coli is RppH-dependent (Figure S3) and its 5′-terminal nucleotide sequence (GUAAAC…) is completely different from that of yeiP-U2G (AGAUUU…). PACO analysis in conjunction with a set of in vitro transcribed metE standards (Figure 4A), whose length and sequence matched those of the cellular transcript, revealed that 51 ± 8% of metE mRNA is diphosphorylated in cells containing RppH, a value that rises to 96 ± 7% in ΔrppH cells (Figure 4B, Tables S2–S5). Because 27 ± 2% of metE transcripts are monophosphorylated when RppH is present (versus −5 ± 1% in ΔrppH cells) (Figure 4C), we calculate that only ~22% of metE mRNA is ordinarily triphosphorylated at steady state.
Figure 4. Detection of diphosphorylated metE mRNA in E. coli.

(A) Examination of in vitro transcribed metE standards bearing a 5′-terminal triphosphate (TriP), diphosphate (DiP), or monophosphate (MonoP) by PACO. The reactions were performed in the presence of total E. coli RNA from ΔmetE cells. See also Tables S2 and S3.
(B) Relative abundance of diphosphorylated metE mRNA in wild-type (WT) and ΔrppH cells, as determined by PACO. A vertical line marks the location of superfluous lanes omitted from the image. See also Tables S4 and S5.
(C) Relative abundance of monophosphorylated metE mRNA in wild-type (WT) and ΔrppH cells, as determined by PABLO. See also Tables S4 and S5 and Figures S1 and S3.
For a detailed explanation of how the values in panels A, B, and C were calculated, see STAR Methods. Minor discrepancies versus the theoretical maximum (100%) or minimum (0%) resulted from the small mathematical correction factors that were used and are either within the margin of error or close to it. A bent arrow leads from each RNA substrate to its ligation product.
To confirm the presence of diphosphorylated mRNA in E. coli, we compared the number of phosphates present in the capped 5′ ends produced by the reaction of Pce1 with either cellular yeiP-U2G mRNA or triphosphorylated, diphosphorylated, and monophosphorylated yeiP-U2G standards synthesized by in vitro transcription. Each was capped by prolonged reaction with Pce1 in the presence of [α-32P]GTP, and the capped 5′-terminal nucleotide was then released from the RNA by hydrolysis with nuclease S1 and examined by thin layer chromatography and autoradiography. As expected, Pce1 reacted with the diphosphorylated standard to generate a capped product whose digestion with S1 released radiolabeled P1-guanosine-P3-adenosine-5′,5′-triphosphate (GpppA) (Figure 3E). Capping an equal concentration of the triphosphorylated standard and subsequent hydrolysis produced primarily P1-guanosine-P4-adenosine-5′,5′-tetraphosphate (GppppA). Little if any capped reaction product was observed for the monophosphorylated standard. Parallel analysis of yeiP-U2G mRNA purified from total RNA that had been extracted from either wild-type or ΔrppH cells yielded GpppA as the sole radioactive product of RNA capping, verifying that diphosphorylated mRNA is abundant in E. coli. As expected, yeiP-U2G from ΔrppH cells, where virtually all of that transcript is diphosphorylated, yielded about three times as much GpppA as did a similar amount of the same transcript from wild-type cells, where 71% is monophosphorylated.
Finally, to rule out the possibility that the phosphorylation state of transcripts might be perturbed during RNA extraction from E. coli, fully triphosphorylated, diphosphorylated, or monophosphorylated yeiP-U2G mRNA that had been synthesized by in vitro transcription was added to intact E. coli cells lacking a yeiP gene, and total RNA was then extracted from those cells. The extraction procedure had no effect on the phosphorylation state of the spiked-in transcripts, as judged by PACO and PABLO (Figure S4).
Competitive 5′-terminal incorporation of ATP versus ADP by E. coli RNA polymerase
In principal, the great excess of diphosphorylated yeiP-U2G and metE mRNA over their triphosphorylated counterparts could be a consequence of the preferential synthesis of diphosphorylated transcripts by RNA polymerase or the swift removal of the γ phosphate of triphosphorylated primary transcripts shortly after they are made. To distinguish between these possibilities, we compared the incorporation of ATP and ADP at the 5′ end of a transcript during its synthesis by purified E. coli RNA polymerase. This RNA, EcIVTa, which bore a single, 5′-terminal adenylate nucleotide, was produced by in vitro transcription at various ATP : ADP ratios, and its 5′ phosphorylation state was examined by PACO. The ratio of triphosphorylated to diphosphorylated EcIVTa exceeded the ratio of ATP to ADP present during its synthesis by approximately five-fold (Figure 5). These results indicate that E. coli RNA polymerase favors ATP over ADP when initiating transcription. In view of the ratio of nucleotides available for transcription initiation in E. coli, which are predominantly triphosphorylated (ATP : ADP : AMP = 92 : 5 : 3 and GTP : GDP : GMP = 87 : 12 : 1 in minimal glucose medium (Bennett et al., 2009)), we conclude that both yeiP-U2G and metE mRNA are synthesized with a 5′-terminal triphosphate. Evidently, the triphosphate is then rapidly converted to a more stable diphosphate, which eventually undergoes β-phosphate removal by RppH as a prelude to 5′-end-dependent degradation.
Figure 5. Competitive incorporation of ADP and ATP at the 5′ end of transcripts synthesized by E. coli RNA polymerase.
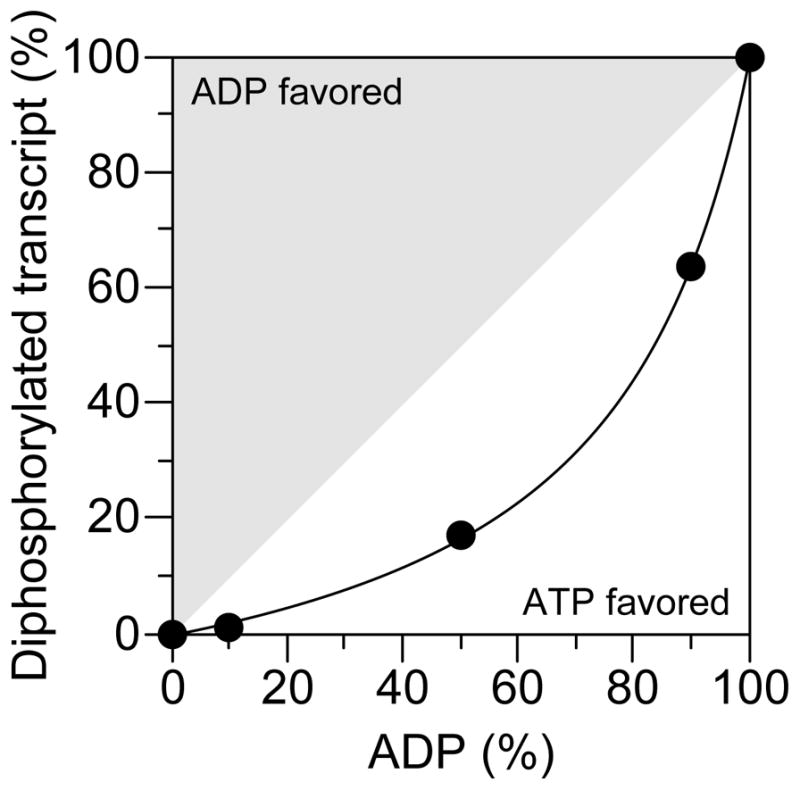
EcIVTa RNA (5′ AUCUUUCUCUGUUGUUCUUCUGUUGGUCUUUUUCCCUGCCCUGUGCGGUCGGCGUUGCGUUCGGU 3′) was synthesized by E. coli RNA polymerase at various ADP : ATP ratios, and the relative abundance of diphosphorylated versus triphosphorylated 5′ termini was determined by PACO (filled circles). The PACO ligation yields for RNA synthesized at ratios of 100 : 0 and 0 : 100 (100% diphosphorylated and 100% triphosphorylated EcIVTa, respectively) provided correction factors that allowed precise quantification of the data obtained at intermediate ratios. The curved line represents the relative yield of diphosphorylated EcIVTa that would be expected if ATP is incorporated 5 times more readily than ADP, and the gray and white zones encompass the range of experimental outcomes expected for preferential incorporation of ADP or ATP, respectively. A representative experiment is shown. The observed preference for 5′-terminal incorporation of ATP versus ADP was 5.0 ± 0.2 (ATP/ADP).
Relative reactivity of diphosphorylated RNA as an RNase E substrate
RNase E has a well documented preference for monophosphorylated over triphosphorylated substrates (Mackie, 1998; Jiang et al., 2000; Tock et al., 2000). The long lifetime of the diphosphorylated mRNAs that accumulate in E. coli when RppH is absent suggested that they, like their triphosphorylated precursors, are likely to be cleaved slowly by RNase E. To test this hypothesis, we examined the rate at which this enzyme cuts diphosphorylated yeiP-U2G RNA, whose the 5′ untranslated region is known to contain several RNase E cleavage sites (Richards et al., 2012; Richards and Belasco, 2016). Purified N-RNase E (the catalytic amino-terminal domain of RNase E) was added to in vitro transcribed yeiP-U2G RNA bearing one, two, or three phosphates at the 5′ end, and the reaction was monitored by Northern blotting (Figure 6). The monophosphorylated substrate was rapidly cut to produce multiple discrete cleavage products resembling decay intermediates observed in vivo. By contrast, the diphosphorylated and triphosphorylated substrates were both cut much more slowly, demonstrating that a 5′-terminal diphosphate is not able to accelerate RNase E cleavage significantly.
Figure 6. Relative reactivity of diphosphorylated RNA as an RNase E substrate.

(A) Reaction of monophosphorylated (MonoP), diphosphorylated (DiP), and triphosphorylated (TriP) yeiP-U2G with equal amounts of E. coli N-RNase E. Reaction samples were quenched at time intervals and analyzed by Northern blotting.
(B) Semilogarithmic plots of the amount of intact yeiP-U2G RNA remaining as a function of time after N-RNase E addition. Empty squares, monophosphorylated yeiP-U2G. Filled triangles, diphosphorylated yeiP-U2G. Gray circles, triphosphorylated yeiP-U2G. Mean values from multiple experiments are plotted. Error bars represent standard deviations.
DISCUSSION
Until now, it has commonly been assumed that the 5′ ends of bacterial RNAs are ordinarily either triphosphorylated as a result of transcription initiation with a nucleoside triphosphate (Maitra et al., 1965) or monophosphorylated as a consequence of subsequent pyrophosphate removal or RNA cleavage (Crouch, 1974; Kole and Altman, 1981; Szeberényi et al., 1983; Deana et al., 2008), with rare exceptions (Collins et al., 2007; Cook et al., 2013; Cahová et al., 2015). However, direct evidence for triphosphorylated RNA in bacterial cells is rather limited, as it is based largely on the detection of triphosphorylated fragments among the products of chemical or enzymatic digestion of E. coli RNA (Jorgensen et al., 1969; Ginsburg and Steitz, 1975; Bieger and Nierlich, 1989). Our finding that diphosphorylated yeiP-U2G and metE mRNA are each much more abundant than their triphosphorylated counterparts in E. coli (34–51% versus only 0–22%) refutes the putative predominance of triphosphorylated mRNA 5′ ends and suggests that the 5′-terminal triphosphate of both primary transcripts is quickly converted to a diphosphate as the initial event in their 5′-end-dependent degradation (Figure 7). This modification creates a better substrate for RppH, which is able to remove the β phosphate from the diphosphorylated species much faster than it could have removed pyrophosphate from the triphosphorylated primary transcript. Sequential loss of the γ and β phosphates generates a monophosphorylated intermediate susceptible to rapid cleavage by RNase E.
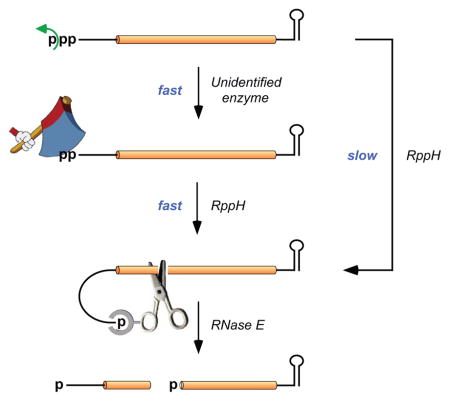
Figure 7. Principal mechanism of 5′-end-dependent RNA degradation in E. coli.

Swift removal of the γ phosphate of a triphosphorylated primary transcript by an unidentified enzyme generates a 5′-terminal diphosphate susceptible to rapid β phosphate hydrolysis by RppH. Less often, RppH may convert the 5′-terminal triphosphate to a monophosphate in a single step. The monophosphorylated 5′ end of the resulting intermediate binds to a pocket on the surface of RNase E, thereby facilitating endonucleolytic cleavage by this ribonuclease at various downstream locations. The cleavage products are subsequently degraded by the combined action of RNase E and 3′ exonucleases (not shown). Curved arrows represent phosphate removal.
The finding that a substantial fraction of each mRNA tested is diphosphorylated and that this fraction increases to ~100% in the absence of RppH suggests that stepwise conversion of the triphosphorylated primary transcript to a monophosphorylated product vulnerable to ribonuclease attack is the principal mechanism by which 5′-end-dependent RNA degradation is triggered in E. coli. This mechanism is also likely to predominate in other proteobacterial species, most of which contain orthologs of E. coli RppH (Foley et al., 2015) that are expected to share its preference for diphosphorylated substrates. Nevertheless, it is possible that in some cases γ phosphate removal may be sufficiently slow that RppH converts triphosphorylated 5′ ends to monophosphates in a single step without waiting for the γ phosphate to be removed.
Previously, the presence and functional significance of diphosphorylated 5′ ends had gone unrecognized due to the lack of an analytical method for distinguishing such ends from their triphosphorylated counterparts. To detect and quantify diphosphorylated mRNA in E. coli, we devised a novel assay (PACO) that is based on the specificity of the RNA guanylyltransferase important for mRNA capping in eukaryotic nuclei. Key to the utility of PACO is the quantitative nature of this assay, which makes it possible not only to detect diphosphorylated RNAs but also to determine their relative abundance. Like other enzymes of its kind (Yu and Shuman, 1996; Chen et al., 1999), the RNA guanylyltransferase employed in these assays (Pce1 of Schizosaccharomyces pombe) is not entirely specific for diphosphorylated RNA, making it impractical to drive the capping reaction to completion by using an excess of this enzyme. Instead, it was necessary to interrupt the reaction just as the capping of diphosphorylated RNA was nearing completion and to compare the reaction yield for cellular RNA to that for a set of triphosphorylated, diphosphorylated, monophosphorylated standards that had been synthesized by in vitro transcription and were identical in sequence and length to the cellular transcript under investigation. This knowledge, together with an independent measurement of the percentage of the cellular transcript that is monophosphorylated, provided all the information required to calculate how much of that transcript is diphosphorylated in vivo. The fact that no PACO ligation was observed without prior capping by Pce1, i.e. that none of the cellular yeiP-U2G or metE mRNA was intrinsically resistant to alkaline phosphatase, indicates that neither 5′ end is protected detectably by a cap-like structure, a finding consistent with previous evidence (Cahová et al., 2015). Though effective for determining the phosphorylation state of individual RNAs, PACO is not suitable for examining an entire transcriptome because the assay must be calibrated with a distinct set of cognate standards and a PABLO measurement for each transcript under investigation.
Our finding that the diphosphorylated and monophosphorylated forms of yeiP-U2G and metE together account for ~80–100% of each full-length transcript means that little triphosphorylated RNA remains for either of them. The great excess of diphosphorylated over triphosphorylated 5′ ends stands in stark contrast to the ratio of nucleotides available for transcription initiation in E. coli, which are predominantly triphosphorylated. Moreover, our data indicate that E. coli RNA polymerase preferentially incorporates NTPs rather than NDPs at the 5′ end of transcripts. Therefore, it appears that E. coli cells contain one or more enzymes that can rapidly remove the γ phosphate from primary transcripts. RppH does not appear to be the source of this triphosphatase activity, as no diphosphorylated intermediate was detected in vitro during the conversion of triphosphorylated 5′ ends to monophosphorylated 5′ ends by the purified E. coli enzyme (Figure 2A), a reaction in which pyrophosphate, not orthophosphate, is the principal by-product (Deana et al., 2008). Furthermore, contrary to the phenotype expected for a triphosphatase-deficient mutant, deleting the rppH gene does not diminish the abundance of diphosphorylated 5′ ends in E. coli (Figures 3C and 4B). For similar reasons (Lee et al., 2014 and PACO analysis of RNA from ΔdapF cells [data not shown]), it is also unlikely that DapF, which forms a complex with RppH, functions as an RNA triphosphatase in E. coli. Nor does E. coli contain a homolog of the eukaryotic RNA triphosphatases that catalyze the first step in RNA capping. Whatever the source of this activity may be, it apparently can act on RNAs with a variety of 5′-terminal sequences, including those that begin with either A or G. Nevertheless, it appears to favor the former, as judged from the markedly greater steady-state ratio of diphosphorylated to triphosphorylated yeiP-U2G versus metE mRNA despite the likely preference of RppH for the first two nucleotides of yeiP-U2G (AG versus GU (Foley et al., 2015; Vasilyev and Serganov, 2015)).
Irrespective of its 5′-terminal sequence, the diphosphorylated RNA generated by the putative RNA triphosphatase is a much better substrate for RppH than its triphosphorylated precursor. The ≥3–10-fold greater reactivity of diphosphorylated RNA explains the need for γphosphate removal despite the ability of E. coli RppH to convert 5′-terminal triphosphates directly to monophosphates. The basis for the faster reaction rate of diphosphorylated substrates is not apparent from the published structures of the liganded enzyme (Vasilyev and Serganov, 2015) or the relevant pKa of the leaving group, which is significantly higher for phosphate than for pyrophosphate (12.4 versus 9.3).
RNA modifications that previously went undetected are now known to play major roles in governing gene expression in both prokaryotic and eukaryotic organisms (Deana et al., 2008; Schwartz, 2016). For instance, recent studies have shown that adenosine methylation can stimulate translation and accelerate mRNA degradation in human cells (Wang et al., 2014; Wang et al., 2015; Meyer et al., 2015). The discovery that the formation of diphosphorylated RNA enables 5′-end-dependent degradation in E. coli provides a prime example of such a phenomenon in bacteria and a key insight into the mechanism of an important regulatory pathway that has been implicated in microbial pathogenesis. Nevertheless, it seems unlikely that bacteria evolved the ability to produce diphosphorylated RNA solely as a mechanism for initiating degradation, as RppH alone is able to convert 5′-terminal triphosphates directly to monophosphates, albeit more slowly than it acts on diphosphates. Instead, the remarkable abundance of diphosphorylated RNA in E. coli raises the possibility that this novel phosphorylation state may govern additional RNA-dependent processes, perhaps by enabling binding by regulatory proteins other than RppH or by preventing RNA 5′ ends from being mistaken for nucleoside triphosphates. However diverse its biological functions might be, the discovery of diphosphorylated RNA in E. coli demonstrates that modifying the phosphorylation state of bacterial mRNAs is a more complex process than previously realized and suggests that the 5′ end of bacterial transcripts may be a busier and more influential locale than heretofore imagined.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joel G. Belasco (joel.belasco@med.nyu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Growing cells for RNA extraction
Saturated cultures of E. coli grown in MOPS-glucose (Neidhardt et al., 1974) were diluted into fresh MOPS-glucose (without antibiotic) to an initial OD650 of less than 0.01 and shaken at 37ºC until the OD650 reached 0.28–0.32.
METHOD DETAILS
Strains and plasmids
Measurements of the 5′ phosphorylation state and half-life of mRNA were performed in the E. coli K-12 strain BW25113 (Datsenko and Wanner, 2000) and an isogenic derivative thereof bearing an in-frame deletion of the rppH coding region. In addition, the strains used to analyze yeiP and yeiP-U2G mRNA lacked the chromosomal copy of the yeiP gene and instead contained a plasmid (pYeiP1 or pYeiP1-U2G, respectively (Richards et al., 2012; Foley et al., 2015)) encoding the transcript of interest. Each chromosomal deletion mutant was constructed by P1 transduction of BW25113 with the corresponding kan-disrupted gene from the Keio collection (Baba et al., 2006) followed by excision of the kan gene.
Protein purification
E. coli RppH bearing an amino-terminal hexahistidine tag (MHHHHHHG) was produced in E. coli from plasmid pPlacRppH6, purified by affinity chromatography on TALON beads (Clontech), and stored at −80°C in a buffer containing 10 mM HEPES (pH 7.5), 0.3 M NaCl, and 50% (v/v) glycerol (Foley et al., 2015; Vasilyev and Serganov, 2015). E. coli DapF bearing six additional residues (SGSGSG) at the amino terminus was generated from a protein fusion in which hexahistidine and SUMO tags were joined in tandem to the amino terminus of DapF. This protein fusion was produced in E. coli from plasmid pSumo-DapF and purified from cell extracts by affinity chromatography on a HisTrap column (GE Healthcare). The tags were removed by cleavage with hexahistidine-tagged ULP1 protease, and the flow-through from a second HisTrap column (no imidazole) was then concentrated and further fractionated by gel filtration through Superdex 200 (GE Healthcare). The purified DapF was stored at −80°C in a buffer containing 20 mM Tris-HCl (pH 8.0), 0.25 M NaCl, and 1 mM dithiothreitol. ULP1 bearing an amino-terminal hexahistidine tag was produced in E. coli from plasmid pET28b-ULP1 (Mossessova and Lima, 2000), purified from cell extracts by affinity chromatography on a HisTrap column (GE Healthcare), concentrated/exchanged into a buffer containing 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 1 mM EDTA, and 0.05% 2-mercaptoethanol, and stored at −80°C.
Schizosaccharomyces pombe Pce1 bearing an amino-terminal His6Smt3 domain was produced in E. coli from plasmid pSMT3-Pce1, which was a generous gift from Christopher Lima (Memorial Sloan Kettering Cancer Center, New York) (Doamekpor et al., 2014), purified from cell extracts by elution from TALON beads (Clontech), concentrated/exchanged into a buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 1 mM 2-mercaptoethanol, and 20% (v/v) glycerol, and stored at −20°C.
T4 DNA ligase bearing an amino-terminal hexahistidine tag was produced from plasmid pPlacT4L in the RNase H-deficient E. coli strain MIC2067 (Itaya et al., 1999) and then purified from cell extracts by elution from TALON beads (Clontech) followed by anion-exchange chromatography on an UNO Q1 column (Bio-Rad). The purified protein was concentrated/exchanged into a buffer containing 10 mM Tris-HCl (pH 7.4), 50 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, and 50% (v/v) glycerol and stored at −80°C.
E. coli RNA polymerase holoenzyme containing σ70 was provided by Vitaly Epshtein and Evgeny Nudler (NYU School of Medicine, New York) (Nudler et al., 2003).
N-RNase E comprised amino acid residues 1-529 of E. coli RNase E joined to a carboxy-terminal extension containing hexahistidine and Myc-epitope tags (GGAAAHHHHHHVAAEQKLISEEDLNGAARSA). It was produced from plasmid pNRNE7529 in E. coli, purified from cell extracts by elution from TALON beads (Clontech), concentrated/exchanged into a buffer containing 20 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 10 mM dithiothreitol, and 20% (v/v) glycerol, and stored at −80°C (Richards and Belasco, 2016).
RNA synthesis by in vitro transcription
Triphosphorylated yeiP-U2G and metE RNA was synthesized in vitro by using T7 RNA polymerase to transcribe a PCR-amplified DNA template containing a T7 ϕ2.5 (yeiP-U2G) or ϕ6.5 (metE) promoter in the presence of ATP, GTP, CTP, and UTP. Their diphosphorylated and monophosphorylated counterparts were synthesized in the presence of an 18–30 fold molar excess of ADP (4.4 mM) or AMP (7.5 mM) over ATP (0.25 mM) in the case of yeiP-U2G or of GDP (7.5 mM) or GMP (7.5 mM) over GTP (0.25 mM) in the case of metE. These in vitro transcripts were identical to the corresponding cellular transcripts in their entirety (yeiP-U2G: 654 nt in length; metE: 2,464 nt in length). In addition, a diphosphorylated RNA (yeiP-std128) comprising the first 128 nucleotides of yeiP-U2G except for a GGU → CCC substitution at positions 68–70 was prepared by in vitro transcription for use as an internal standard in rate measurements with purified RppH. For details about nucleotide purity and oligonucleotide primers, see Tables S6 and S7.
Reactivity of RppH with polynucleotides
Either diphosphorylated or triphosphorylated yeiP-U2G RNA (1 pmol) was combined with a diphosphorylated internal standard (1 pmol of yeiP-std128, a yeiP-U2G variant resistant to cleavage by the 10–23 DNAzyme DZyeiP69) in a total volume of 50 μl. A 5-μl sample was removed as the 0-min timepoint, and another 5-μl sample was removed and processed as a ligation control by exhaustive treatment with excess RppH (200 nM at 37°C for 2 hr). The remainder (40 μl) was prewarmed to 37°C and combined with prewarmed RppH (40 μl) to initiate the reaction. The final reaction mixture (80 μl) contained the following: diphosphorylated or triphosphorylated yeiP-U2G RNA (10 nM), diphosphorylated internal standard RNA (10 nM), HEPES, pH 7.5 (20 mM), MgCl2 (10 mM), dithiothreitol (1 mM), glycerol (1%), rRNasin (2 units/μl; Promega), RppH (5 nM), and DapF (10 nM). To monitor the progress of the reaction, samples (10 μl) were removed at time intervals, quenched with excess EDTA, phenol extracted, and ethanol precipitated. Each sample was then examined by PABLO in conjunction with the selective cleavage of yeiP-U2G RNA and its ligation product by DZyeiP69 to increase their electrophoretic mobility. The extent of conversion of diphosphates or triphosphates to monophosphates at each timepoint was calculated by normalizing the measured ligation yield to that of the fully monophosphorylated ligation control.
Reactivity of RppH with dinucleotides
Dinucleotide substrates bearing a 5′-terminal diphosphate or triphosphate were prepared by in vitro transcription with T7 RNA polymerase (Vasilyev and Serganov, 2015; Vasilyev and Serganov, 2016). To measure RppH activity, reaction mixtures containing 50 mM HEPES, pH 7.5, 10 mM MgCl2, 0.1% Triton X-100, 100 μM di- or triphosphorylated dinucleotide, and 100 nM RppH were incubated at 37°C for 0–10 min, and 50-μl samples taken at time intervals were quenched by dilution with 50 mM sodium acetate, pH 5.0 (500 μl). The substrate and product in each sample were separated by anion-exchange chromatography on a 5×50 mm Mono Q column by applying a gradient of 0–0.5 M NaCl in 10 mM Tris-HCl, pH 8.0, while recording the elution profile spectrophotometrically at 260 nm. Peak areas corresponding to the substrate and product were integrated using UNICORN software (GE Healthcare). Each reaction was repeated two times.
RNA extraction from E. coli
Total cellular RNA was extracted from E. coli cultures that had been grown to mid-log phase at 37°C in MOPS-glucose medium. A culture sample (10–15 ml) was rapidly chilled by pipetting it into a 50-ml centrifuge tube filled with crushed ice, and the bacteria were pelleted by centrifugation at 6,000 × g for 10 min at 0°C. The cell pellet was resuspended at 0°C in 125 μl of ice-cold buffer A (0.3 M sucrose, 10 mM sodium acetate, pH 4.5, 10 mM EDTA), transferred to a chilled microfuge tube, mixed with 250 μl of room-temperature buffer B (2% SDS, 10 mM sodium acetate, pH 4.5), and heated to 75°C for 3 min to lyse the cells. The cell lysate was extracted three times with 250 μl of hot, unneutralized, water-saturated phenol by vigorously agitating the mixture and then heating it to 75°C for 3 min. After each phenol extraction, the mixture was chilled rapidly in a dry ice/ethanol bath and then centrifuged at 21,000 × g for 5 min before transferring the aqueous layer to a new microfuge tube. Finally, the RNA was precipitated by mixing the aqueous layer of the last phenol extraction (up to 450 μl) with 3 M sodium acetate, pH 4.8 (45 μl) and ethanol (1 ml), cooled to −20°C for 30 min, pelleted by centrifugation at 21,000 × g for 15 min, washed with 70% ethanol, air-dried, dissolved in water (30–70 μl) to a concentration of 0.5–2.0 μg/μl, and stored at −20°C.
PABLO
To determine the percentage of a particular E. coli transcript that was monophosphorylated, total cellular RNA was combined with oligonucleotide X (0.44 μM), oligonucleotide Y (0.09 μM), and a transcript-specific 10–23 DNAzyme (8.9 μM) (see Table S7) in a total volume of 45 μl, heated to 85°C for 5 min, slowly cooled to 30°C, and then placed on ice for at least 2 min. A solution (35 μl) containing Tris-HCl, pH 7.5 (114 mM), MgCl2 (23 mM), dithiothreitol (23 mM), ATP (2.7 mM), rRNasin (1 unit/μl; Promega), and T4 DNA ligase (1.3 μM) was added, and ligation was allowed to proceed for 2 or 4 hr at 37°C before being quenched with EDTA (150 μl of an 8.3 mM solution). The reaction products were phenol extracted, ethanol precipitated, separated by electrophoresis on a 5–6% polyacrylamide-urea gel, and transferred to a Hybond-XL membrane (GE Healthcare) by electroblotting. The blot was then UV-crosslinked and probed with a 5′-radiolabeled oligonucleotide complementary to the RNA of interest. Bands were visualized on a Typhoon Trio imager (GE Healthcare) and quantified by using ImageQuant TL software. As a ligation control, fully monophosphorylated RNA generated by prior treatment with excess RppH (200 nM for 2 hr at 37°C) was analyzed in parallel. When testing reactivity with RppH in vitro, the PABLO reactions were performed as above but with four times as much oligo X (1.8 μM), oligo Y (0.36 μM), and 10–23 DNAzyme (35.6 μM) in a total volume of 45 μl.
PACO
To determine the percentage of a particular E. coli transcript that was diphosphorylated, total cellular RNA (10 μl, prewarmed) was combined with Pce1 (10 μl, prewarmed) in a reaction mixture containing RNA (5 μg), Tris-HCl, pH 7.5 (50 mM), dithiothreitol (5 mM), GTP (1 mM), MgCl2 (1 mM), rRNasin (2 units/μl; Promega), and Pce1 (100 nM). In parallel, triphosphorylated, diphosphorylated, and monophosphorylated RNAs identical to the entire E. coli transcript of interest were synthesized by in vitro transcription, added to total RNA (5 μg) from E. coli cells devoid of that transcript, and treated identically. The capping reaction was allowed to proceed for 4 min at 37°C before being quenched with EDTA (195 μl of a 4 mM solution), and the reaction products were phenol extracted and ethanol precipitated. The 5′-terminal phosphates of uncapped RNA were next removed by treatment with calf intestine alkaline phosphatase (0.5 units/μl; New England Biolabs) in Tris-HCl, pH 7.9 (100 mM), NaCl (100 mM), MgCl2 (10 mM), dithiothreitol (1 mM), and rRNasin (2 units/μl; Promega) for 1 hr at 37°C, and the reaction products were again phenol extracted and ethanol precipitated. Subsequent treatment of the capped 5’ ends with excess RppH (200 nM for 2 hr at 37°C) converted them all to monophosphates, which were then detected by PABLO in conjunction with DNAzyme cleavage. As a ligation control, fully monophosphorylated RNA generated by prior treatment with excess RppH (200 nM for 2 hr at 37°C) was analyzed in parallel.
Characterization of caps added by Pce1
Total RNA (260 μg) extracted from E. coli cells that lacked the chromosomal yeiP gene and instead contained plasmid pYeiP1-U2G was fractionated by electrophoresis on a 4.5% polyacrylamide/7 M urea gel. A gel slice containing full-length yeiP-U2G mRNA was excised, crushed, and soaked overnight in EDTA (1 mM, pH 8.0), and the extracted RNA was ethanol precipitated. To selectively reduce the length of yeiP-U2G mRNA relative to other, co-purified RNAs of a similar size, it was then cleaved 444 nucleotides from the 5′ end by treatment with DZyeiP444 (20 μM) in Tris-HCl, pH 7.5 (50 mM), MgCl2 (10 mM), and dithiothreitol (10 mM) for 4 hr at 37°C. The cleavage products were separated by electrophoresis on a 4.5% polyacrylamide/7 M urea gel, and the fragment comprising the 5′-terminal 444 nucleotides of yeiP-U2G mRNA was recovered as described above. In addition, triphosphorylated, diphosphorylated, and monophosphorylated standards identical to the 5′-terminal 444 nucleotides of yeiP-U2G mRNA were synthesized in vitro by run-off transcription with T7 RNA polymerase. Each RNA was dissolved in 20 μl of a buffer containing Tris-HCl, pH 7.5 (50 mM), dithiothreitol (5 mM), [α-32P] GTP (0.65 μCi, 0.078 μM), MgCl2 (1 mM), and rRNasin (2 units/μl; Promega) and capped with Pce1 (3.5 μM) for 2 hr at 37°C. The capping reaction was quenched with EDTA (180 μl of a 4.2 mM solution), and the reaction products were phenol extracted and ethanol precipitated. The capped 5′-terminal nucleotide was then released by RNA digestion with S1 nuclease (50 units; Thermo Fisher Scientific) for 30 min at 37°C in 9 μl of a buffer containing sodium acetate, pH 4.5 (40 mM), NaCl (300 mM), and ZnSO4 (2 mM), and the remaining unreacted [α-32P] GTP was hydrolyzed to guanosine and orthophosphate by treatment with calf intestinal alkaline phosphatase (1 unit/μl; New England Biolabs) for 30 min at 37°C in a buffer containing Tris-HCl, pH 8.0 (50 mM) and MgCl2 (10 mM). Finally, the products were analyzed by thin layer chromatography beside GpppA (1 nmol; New England Biolabs) and GppppA (1 nmol; Jena Bioscience) markers on a fluorescent PEI-cellulose plate (J.T. Baker) developed with ammonium sulfate (0.4 M, pH 5.5). The radiolabeled products were detected with a Typhoon Trio imager (GE Healthcare), and the unlabeled GpppA and GppppA markers were detected by UV shadowing.
5 ′-terminal nucleotide incorporation by E. coli RNA polymerase
EcIVTa RNA was synthesized in vitro by run-off transcription with E. coli RNA polymerase of a 174-bp DNA template that contained a σ70 promoter (CTTAAGGTTAACCACCGAATTCATGAGCGCGCACAGTACCACCTTTTTTGCACCAGCAAAAGTGCGAATACCACTTGACAGAAAGGCCCGTCGCGAGTACTTTGTCGCGATCTTTCTCTGTTGTTCTTCTGTTGGTCTTTTTCCCTGCCCTGTGCGGTCGGCGTTGCGTTCGGT, where the region encoding RNA is underlined). Purified E. coli RNA polymerase holoenzyme (16 pmol) was combined with the purified DNA template (8 pmol) in buffer TB50 (40 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2) containing GTP (0.1 mM), CTP (0.1 mM), UTP (0.1 mM), and a mixture of ATP and ADP at various molar ratios (2.0 + 0.0 mM, 1.8 + 0.2 mM, 1.0 + 1.0 mM, 0.2 + 1.8 mM, or 0.0 + 2.0 mM). After 1 hr at 37°C, each reaction was quenched with an equal volume of stop solution (20 mM EDTA, 7 M urea), phenol extracted, and ethanol precipitated. The reaction products were then combined with total E. coli RNA (5 μg), cleaved 54 nt from the 5′ end by treatment with the 10–23 DNAzyme DZEcIVTa54 (11 μM; see Table S7) for 2 hr at 37°C in a buffer containing Tris-HCl, pH 7.5 (50 mM), MgCl2 (10 mM), and dithiothreitol (10 mM), and analyzed by PACO to determine the ratio of 5′-terminal ADP versus ATP incorporation.
RNA cleavage by purified N-RNase E
Monophosphorylated, diphosphorylated, and triphosphorylated in vitro transcripts comprising the first 80 nucleotides of yeiP-U2G RNA (37 nM) were digested with purified N-RNase E (175 nM enzyme subunits) at 30°C in a buffer containing 25 mM Bis-tris propane (pH 8.0), 100 mM NaCl, and 15 mM MgCl2. Reaction samples (0.33 pmol) were quenched with excess EDTA at time intervals. These samples were then mixed with total RNA (7.5 μg) from E. coli strain BW25113 ΔyeiP to facilitate precipitation, ethanol precipitated, subjected to electrophoresis on a 7.5% polyacrylamide-urea gel, and transferred to a Hybond-XL membrane (GE Healthcare) by electroblotting. The blot was UV-crosslinked and probed with a 5′-radiolabeled oligonucleotide (yeiP 80 rev) complementary to the 3′ end of the RNA. Bands were visualized on a Typhoon Trio imager (GE Healthcare) and quantified by using ImageQuant TL software.
Decay rate of metE mRNA in E. coli
At time intervals after inhibiting transcription with rifampicin (200 μg/ml), total RNA was extracted from isogenic E. coli strains containing or lacking RppH, and the half-life of the metE transcript was determined by quantitative RT-PCR analysis with SuperScript III reverse transcriptase (Thermo Fisher Scientific), FastStart Universal SYBR Green Master (Rox) (Roche), and primers specific for metE mRNA and 16S rRNA (see Table S7). The initial concentration of the two cDNAs in each sample was determined by fitting the qPCR data to a two-parameter MAK2 model (Boggy and Woolf, 2010; Ritz and Spiess, 2008; Luciano et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
Northern blot band intensities were quantified by using ImageQuant TL software (GE Healthcare). Peak areas for the substrates and products of the reaction of dinucleotides with RppH were determined by using UNICORN software (GE Healthcare).
Statistical analysis
Mean values and standard deviations are provided in Figures 6 and S3 and in the legends of Figures 1 and 5. Standard deviations of the data plotted in Figure 2 are listed in Table S1. Statistical analysis of the 5′ phosphorylation data shown in Figures 3 and 4 is presented in Tables S2, S3, S4, and S5.
Calculating capping efficiencies and phosphorylation states
The capping efficiency for each standard was calculated by using the following set of equations:
where ET, ED, and EM are the capping efficiencies of the triphosphorylated, diphosphorylated, and monophosphorylated standards, respectively. YTS, YDS, and YMS are the fractional PACO ligation yields obtained for each standard, DS and MS are the fraction of those standards that are diphosphorylated or monophosphorylated, as calculated from the molar ratio of NDP or NMP to the corresponding NTP during their synthesis by in vitro transcription, and ELS is the ligation efficiency of the fully monophosphorylated transcript. (Note that DS = MS = 0 for the in vitro transcribed triphosphorylated standards, MS = 0 for the in vitro transcribed diphosphorylated standards, and DS = 0 for the in vitro transcribed monophosphorylated standards and that the correction factors (YTS/ELS) (1 – DS) and (YTS/ELS) (1 – MS) are very small: 0.005 to 0.021 and 0.005 to 0.007, respectively, depending on the RNA.) These equations were derived from two simpler equations: (YS/ELS) = ET (TS) + ED (DS) + EM (MS) and TS + DS + MS = 1, where YS is the fractional PACO ligation yield for a given standard and TS is the fraction of a particular standard that is triphosphorylated. Mean values and standard deviations were calculated for YTS, YDS, YMS, and ELS from multiple measurements of each (Table S2) and subsequently used to generate random Gaussian sample sets of 100,000 values for each of these variables. ET, ED, and EM and their errors were then calculated by Monte Carlo simulation (ciCalc). For each simulation, a histogram was constructed and fit to a spline function, and the mode (peak value) was determined. Because of the asymmetry of the distribution, an error equivalent to one standard deviation was then calculated by integrating outward from the mode until a confidence level of 68.3%, equivalent to one standard deviation, was attained. The modes and errors are reported in Table S3.
The distributions of ET, ED, and EM were then combined with data from PACO and PABLO analysis of cellular RNA to calculate the fraction of diphosphorylated and monophosphorylated yeiP-U2G and metE mRNA in E. coli by Monte Carlo simulation (ciCalc) with the following equations:
where D is the diphosphorylated fraction, M is the monophosphorylated fraction, YD and YM are the fractional PACO and PABLO ligation yields, ELD and ELM are the ligation efficiencies of fully monophosphorylated RNA in PACO and PABLO experiments, and EPD is the small fraction of diphosphorylated RNA (0.045 for yeiP-U2G and 0.075 for metE (see Figure S1)) that undergoes ligation by T4 DNA ligase during PABLO. (Note that the correction factors EM (M) and EPD (D) are very small: −0.002 to 0.030 and 0.015 to 0.073, respectively, depending on the RNA and host genotype.) Random Gaussian sample sets (100,000 values each) were generated from the means and standard deviations of YD, ELD, YM, and ELM (Table S4) and then used to calculate D and M by iterative Monte Carlo simulation. For an initial estimate of D and its error, we performed a Monte Carlo simulation in which YM/ELM was used as an approximation of M. The resulting distribution of values for D and the complete equation for M were then used in a Monte Carlo simulation to calculate M more accurately. This distribution of M values was subsequently used to recalculate D, and so on. After three iterations, the modes of the distributions for D and M had each achieved convergence. These modes and their associated errors (confidence level of 68.3%, equivalent to one standard deviation) are reported in Table S5.
DATA AND SOFTWARE AVAILABILITY
ciCalc: https://www.dropbox.com/sh/tnmfozrubycxq56/AABDh2ex_wfMQ6204byQsYKXa?dl=0 ciCalc computes modes (peak values) and confidence intervals for variables of interest by the Monte Carlo method. These variables are defined by the user as arbitrary functions of other, input variables, which are assumed to have Gaussian distributions with known means and standard deviations that are entered by the user.
Supplementary Material
Highlights.
E. coli RppH prefers diphosphorylated RNAs to their triphosphorylated counterparts.
An assay was devised to detect and quantify diphosphorylated RNA 5′ ends.
Diphosphorylated 5′ ends were found to be abundant on mRNA in E. coli.
5′-end-dependent RNA decay begins with sequential removal of the γ and β phosphates.
Acknowledgments
We are grateful to Patricia Foley, Ang Gao, Vitaly Epshtein, and Evgeny Nudler for providing purified enzymes and to Kevin Belasco and Sisi Ma for help with statistical analysis. This research was supported by fellowships to D.J.L. from the National Institutes of Health (T32AI007180) and the Vilcek endowment and by grants to J.G.B. (R01GM035769) and A.S. (R01GM112940) from the National Institutes of Health.
Footnotes
AUTHOR CONTRIBUTIONS
D.J.L., N.V., J.R., A.S., and J.G.B. designed the experiments, D.J.L., N.V., and J.R. conducted the experiments, and D.J.L., N.V., J.R., A.S., and J.G.B. interpreted the data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006. doi: 10.1038/msb4100050. 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger JL, Wass CA, Kim KS. Identification of Escherichia coli K1 genes contributing to human brain microvascular endothelial cell invasion by differential fluorescence induction. Mol Microbiol. 2000;36:174–182. doi: 10.1046/j.1365-2958.2000.01840.x. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieger CD, Nierlich DP. Distribution of 5′-triphosphate termini on the mRNA of Escherichia coli. J Bacteriol. 1989;171:141–147. doi: 10.1128/jb.171.1.141-147.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggy GJ, Woolf PJ. A mechanistic model of PCR for accurate quantification of quantitative PCR data. PLoS One. 2010;5:e12355. doi: 10.1371/journal.pone.0012355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahová H, Winz ML, Höfer K, Nübel G, Jäschke A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature. 2015;519:374–377. doi: 10.1038/nature14020. [DOI] [PubMed] [Google Scholar]
- Celesnik H, Deana A, Belasco JG. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol Cell. 2007;27:79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celesnik H, Deana A, Belasco JG. PABLO analysis of RNA: 5′-phosphorylation state and 5′-end mapping. Methods Enzymol. 2008;447:83–98. doi: 10.1016/S0076-6879(08)02205-2. [DOI] [PubMed] [Google Scholar]
- Chao Y, Li L, Girodat D, Förstner KU, Said N, Corcoran C, Śmiga M, Papenfort K, Reinhardt R, Wieden HJ, et al. In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol Cell. 2017;65:39–51. doi: 10.1016/j.molcel.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Luongo CL, Nibert ML, Patton JT. Rotavirus open cores catalyze 5′-capping and methylation of exogenous RNA: evidence that VP3 is a methyltransferase. Virology. 1999;265:120–130. doi: 10.1006/viro.1999.0029. [DOI] [PubMed] [Google Scholar]
- Collins JA, Irnov I, Baker S, Winkler WC. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 2007;21:3356–3368. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook GM, Robson JR, Frampton RA, McKenzie J, Przybilski R, Fineran PC, Arcus VL. Ribonucleases in bacterial toxin-antitoxin systems. Biochim Biophys Acta. 2013;1829:523–531. doi: 10.1016/j.bbagrm.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Crouch RJ. Ribonuclease III does not degrade deoxyribonucleic acid-ribonucleic acid hybrids. J Biol Chem. 1974;249:1314–1316. [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008;451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- Del Campo C, Bartholomäus A, Fedyunin I, Ignatova Z. Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 2015;11:e1005613. doi: 10.1371/journal.pgen.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD. How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes. Genes Dev. 2014;28:1323–1336. doi: 10.1101/gad.242768.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley PL, Hsieh PK, Luciano DJ, Belasco JG. Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J Biol Chem. 2015;290:9478–9486. doi: 10.1074/jbc.M114.634659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Ginsburg D, Steitz JA. The 30 S ribosomal precursor RNA from Escherichia coli. A primary transcript containing 23 S, 16 S, and 5 S sequences. J Biol Chem. 1975;250:5647–5654. [PubMed] [Google Scholar]
- Hui MP, Foley PL, Belasco JG. Messenger RNA degradation in bacterial cells. Annu Rev Genet. 2014;48:537–559. doi: 10.1146/annurev-genet-120213-092340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M, Omori A, Kanaya S, Crouch RJ, Tanaka T, Kondo K. Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. Journal of bacteriology. 1999;181:2118–2123. doi: 10.1128/jb.181.7.2118-2123.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Diwa A, Belasco JG. Regions of RNase E important for 5′-end-dependent RNA cleavage and autoregulated synthesis. J Bacteriol. 2000;182:2468–2475. doi: 10.1128/jb.182.9.2468-2475.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SE, Buch LB, Nierlich DP. Nucleoside triphosphate termini from RNA synthesized in vivo by Escherichia coli. Science. 1969;164:1067–1070. doi: 10.1126/science.164.3883.1067. [DOI] [PubMed] [Google Scholar]
- Kaberdin VR. Probing the substrate specificity of Escherichia coli RNase E using a novel oligonucleotide-based assay. Nucleic Acids Res. 2003;31:4710–4716. doi: 10.1093/nar/gkg690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R, Altman S. Properties of purified ribonuclease P from Escherichia coli. Biochemistry. 1981;20:1902–1906. doi: 10.1021/bi00510a028. [DOI] [PubMed] [Google Scholar]
- Lee CR, Kim M, Park YH, Kim YR, Seok YJ. RppH-dependent pyrophosphohydrolysis of mRNAs is regulated by direct interaction with DapF in Escherichia coli. Nucleic Acids Res. 2014;42:12746–12757. doi: 10.1093/nar/gku926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Chao S, Wong TT, McDowall KJ, Cohen SN. Effects of nucleotide sequence on the specificity of rne-dependent and RNase E-mediated cleavages of RNA I encoded by the pBR322 plasmid. J Biol Chem. 1994;269:10797–10803. [PubMed] [Google Scholar]
- Luciano DJ, Hui MP, Deana A, Foley PL, Belasco KJ, Belasco JG. Differential control of the rate of 5′-end-dependent mRNA degradation in Escherichia coli. J Bacteriol. 2012;194:6233–6239. doi: 10.1128/JB.01223-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie GA. Ribonuclease E is a 5′-end-dependent endonuclease. Nature. 1998;395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- Maitra U, Novogrodsky A, Baltimore D, Hurwitz J. The identification of nucleoside triphosphate ends on RNA formed in the RNA polymerase reaction. Biochem Biophys Res Commun. 1965;18:801–811. [Google Scholar]
- McDowall KJ, Lin-Chao S, Cohen SN. A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J Biol Chem. 1994;269:10790–10796. [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E, Gusarov I, Bar-Nahum G. Methods of walking with the RNA polymerase. Methods Enzymol. 2003;371:160–169. doi: 10.1016/S0076-6879(03)71011-8. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J Biol Chem. 2002;277:19639–19648. doi: 10.1074/jbc.M200015200. [DOI] [PubMed] [Google Scholar]
- Richards J, Belasco JG. Distinct requirements for 5′-monophosphate-assisted RNA cleavage by Escherichia coli RNase E and RNase G. J. Biol. Chem. 2016;291:5038–5048. doi: 10.1074/jbc.M115.702555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J, Luciano DJ, Belasco JG. Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol Microbiol. 2012;86:1063–1072. doi: 10.1111/mmi.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Spiess AN. qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics. 2008;24:1549–1551. doi: 10.1093/bioinformatics/btn227. [DOI] [PubMed] [Google Scholar]
- Schwartz S. Cracking the epitranscriptome. RNA. 2016;22:169–174. doi: 10.1261/rna.054502.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog Nucleic Acid Res Mol Biol. 2001;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- Shuman S, Hurwitz J. Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme--guanylate intermediate. Proc Natl Acad Sci USA. 1981;78:187–191. doi: 10.1073/pnas.78.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeberényi J, Roy MK, Apirion D. 7 S RNA: a single site substrate for the RNA processing enzyme ribonuclease E of Escherichia coli. Biochim Biophys Acta. 1983;740:282–290. doi: 10.1016/0167-4781(83)90137-9. [DOI] [PubMed] [Google Scholar]
- Tock MR, Walsh AP, Carroll G, McDowall KJ. The CafA protein required for the 5′-maturation of 16 S rRNA is a 5′-end-dependent ribonuclease that has context-dependent broad sequence specificity. J Biol Chem. 2000;275:8726–8732. doi: 10.1074/jbc.275.12.8726. [DOI] [PubMed] [Google Scholar]
- Vasilyev N, Serganov A. Structures of RNA complexes with the Escherichia coli RNA pyrophosphohydrolase RppH unveil the basis for specific 5′-end-dependent mRNA decay. J Biol Chem. 2015;290:9487–9499. doi: 10.1074/jbc.M114.634824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev N, Serganov A. Preparation of short 5′-triphosphorylated oligoribonucleotides for crystallographic and biochemical studies. Methods Mol Biol. 2016;1320:11–20. doi: 10.1007/978-1-4939-2763-0_2. [DOI] [PubMed] [Google Scholar]
- Wang D, Shatkin AJ. Synthesis of Gp4N and Gp3N compounds by guanylyltransferase purified from yeast. Nucleic Acids Res. 1984;12:2303–2315. doi: 10.1093/nar/12.5.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Shuman S. Mutational analysis of the RNA triphosphatase component of vaccinia virus mRNA capping enzyme. J Virol. 1996;70:6162–6168. doi: 10.1128/jvi.70.9.6162-6168.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.