

Abstract
Cancer cells are exposed to various intrinsic and extrinsic factors that disrupt protein homeostasis, producing endoplasmic reticulum (ER) stress. To cope with these situations, cancer cells evoke a highly conserved adaptive mechanism called the unfolded protein response (UPR) to restore the ER homeostasis. Recently, several pharmacological agents have been found to exhibit anti-tumor activity by targeting the UPR components. The development of potent and specific compounds that target the UPR components has not only shed light on the regulation of the UPR in cancer cells, but also brought the field closer to clinical drug candidates. Here we present an overview of the milestones in the field of UPR biology in cancer with a focus on new strategies for pharmacological inhibition.
Keywords: Endoplasmic reticulum stress, unfolded protein response, cancer, anticancer therapy
Graphical abstract
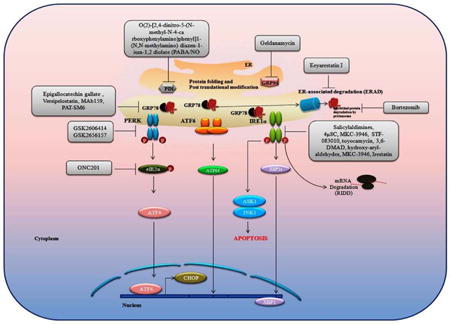
Introduction
The endoplasmic reticulum (ER) is a central cellular organelle that has crucial roles in various cellular processes like protein folding, post translational modifications, intracellular/extracellular shuttling of proteins, and lipid biosynthesis (1). The folding capacity of the ER is limited by tightly regulated expression of protein chaperones such as glucose related protein 78 (GRP78), GRP94 and calreticulin (CRT). The ER lumen has an oxidative environment which is crucial for formation of disulfide bonds via protein disulfide isomerase (PDI). Physiological or pathological insults which overwhelm the folding capacity of the ER activate an evolutionary conserved cascade of signaling events known as the ER stress response, or unfolded protein response (UPR)(2). The UPR is mediated by three molecular sensors present on the membrane of endoplasmic reticulum; PKR-like ER kinase (PERK), activated transcription factor 6 (ATF6) and inositol-requiring enzyme 1 alpha (IRE1α) (1, 3) (Figure 1). The UPR can exert both protective and deleterious effects on cell survival in cancer cells. Here we review what is known about the nature of the stimulus and duration of stress that tips the balance in UPR-dependent cell fate, agents that can target the UPR, and the connection of the UPR to other cell survival and death pathways.
Figure 1. The unfolded Protein Response (UPR).
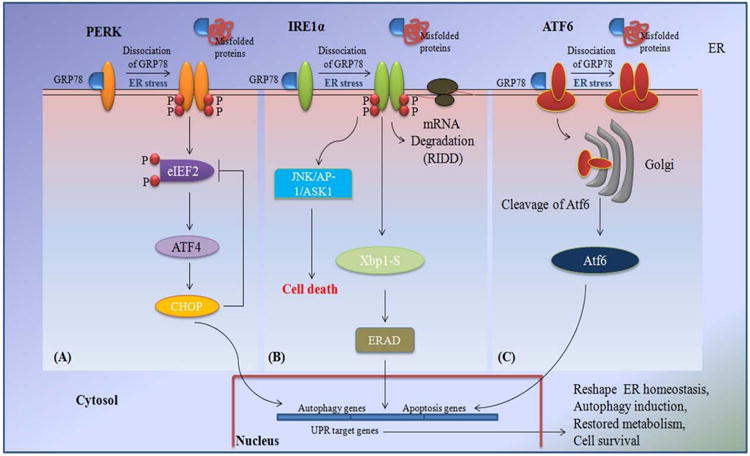
A. ER stress induces Inositol-requiring protein lα (IRE1α) dimerization and autotransphosphorylation, which triggers its RNase activity to processed the mRNA encoding unspliced X box-binding protein 1 (XBP1) to produce an active transcription factor, spliced XBP1. XBP1 controls the transcription of genes encoding proteins involved in protein folding, ER-associated- degradation (ERAD), protein quality control and phospholipid synthesis. During the adaptive response, IRElα conducts IRE 1-dependent decay RIDD on mRNAs encoding ER-translocating proteins to prevent further increases in protein-folding demand in the ER. IRE1α also induces cell death pathways including those driven by JUN N-terminal kinase (JNK) and AP1/ASK1, through binding to adaptor proteins. B. Following ER stress, GRP78 is released from PERK, thereby permitting PERK oligomerization and activation. PERK phosphorylates the initiation factor eukaryotic translation initiator factor 2α (eIF2α) to attenuate global protein synthesis. Phosphorylation of eIF2α allows the translation of ATF4 mRNA, which encodes a transcription factor controlling the transcription of genes involved in autophagy, apoptosis, amino acid metabolism and antioxidant responses. C. Activating transcription factor 6 (ATF6) exists as a dimer in association with GRP78 under physiological conditions. In response to ER stress, when GRP7S dissociates from ATF6, ATF6 translocates from the ER to the Golgi, where it is processed by site-1 protease (SIP) and site-2 protease (S2P) to generate an N-terminal cytosolic domain. Thereafter cleaved ATF6 translocates to the nucleus and binds to ER stress response element, resulting in induction of the UPR target genes.
Dual roles for the Unfolded Protein Response in cancer progression
The UPR is often upregulated in cancer, suggesting that this response is important for cell survival and should be considered a therapeutic target. Although activation of the UPR has been reported in a variety of human cancers, the role of the UPR in different forms of cancer is not well characterized (2, 4-7). The UPR may serve a pro-tumorigenic role by increasing the protein folding capacity, and prolonging resistance to anticancer drugs (5). However, when ER stress is too severe or prolonged, a persistent UPR can engage and activate cell death pathways including mitochondrial apoptosis. A mounting number of studies have revealed the dual role of the UPR in cancer, but without a clear understanding of molecular determinants of the switch between pro-survival and pro-death effects. Furthermore, the components of the ER stress pathway may play multiple roles and therefore, the effects of targeting ER stress and the UPR arms may be difficult to predict and difficult to control. Constitutive activation of the cytoprotective UPR signaling can promote tumorigenesis and resistance to various stresses, such as oxidative and therapeutic stress (8). Provided that the UPR can trigger pro-survival and pro-apoptotic signals, it is imperative to comprehend how modulation of the UPR alters the equilibrium between these processes and contributes to tumorigenesis, and therapy resistance in different cancer cell types. In this section, we will discuss the role of different UPR components in cancer progression.
Dual roles of PERK in cancer pose a challenge to drug development
PERK is a serine/threonine transmembrane ER kinase. Under homeostatic conditions, PERK exists as an inactive monomer associated with GRP78. Following ER stress, GRP78 disassociates from PERK in order to serve its protein chaperone function within the ER, thereby permitting PERK oligomerization and activation. The cytoplasmic domain of PERK is thereby activated resulting in signaling cascade that ends with nuclear transcription. The best-characterized PERK substrates are the eukaryotic initiation factor 2 (eIF2α) and the nuclear factor erythroid 2 relate factor 2 (Nrf2) (9). PERK-dependent phosphorylation of eIF2α is the best characterized consequence of PERK activation. Phosphorylation of eIF2α increases the affinity of eIF2α for the eIF2β guanine nucleotide exchange factor, thereby reducing global translation initiation of mRNA. However, activated eIF2α also increases the translational efficiency of some specific genes of the UPR such as activating transcription factor 4 (ATF4). Sustained activation of PERK-eIF2α-ATF4 pathway (but not with other branches of UPR) has also been implicated in the epithelial-mesenchymal transition (EMT), a process that contributes to tumor progression and metastasis (10). PERK deletion has been shown to reduce the Neu-dependent mammary tumor development and reduces lung metastasis, however long term PERK inactivation increases susceptibility to mammary tumorigenesis owing to increased genomic instability (11). PERK signaling plays a role in promoting tumor cell survival in response to reactive oxygen species (ROS) and hypoxia. PERK-dependent phosphorylation of Nrf2 promotes Nrf2 dissociation from Kelch-like ECH-associated protein 1 (Keap1), and nuclear translocation (12). Nrf2 is a master regulator of antioxidant enzymes that are associated with chemoresistance. The PERK-eIF2α-ATF4 signaling axis has been demonstrated to be critical for the adaptation of tumor cells to hypoxia (13) (14). Dominant negative PERK or eIF2α ser51A impairs the tumor growth, and induces apoptosis in hypoxic regions of tumor (15). Transformed MEFs derived from PERK-knockout mice undergo apoptosis when exposed to hypoxia. K-Ras- transformed PERK−/− xenografts demonstrated significant growth impairment compared to K-Ras-transformed PERK wild type tumors (16). The master oncogenic transcription factor Myc can activate the PERK-ATF4 arm of the UPR in both a mouse model and human lymphoma cells, suggesting that the UPR may play an important role in Myc driven cell survival and tumor progression (17).
In contrast to the pro-survival roles of PERK signaling described above, during chronic ER stress, constitutive PERK-mediated phosphorylation of eIF2α can also lead to apoptosis (18-20). ATF4 drives expression of the pro-apoptotic protein, C/EBP homologous protein (CHOP) during prolonged stress, thereby triggering cell death (20, 21). In addition, loss of PERK may increase resistance to cell death in pathological conditions linked to reactive oxygen species (ROS)-mediated ER stress (22). PERK-mediated translational arrest via eIF2α phosphorylation causes down-regulation of cyclin D1, resulting in subsequent cell cycle arrest (23). Therefore, PERK activation provides both pro-apoptotic and anti-apoptotic response depending on the severity of stress, genetic makeup and the type of cancer cell. Altogether, PERK activation seems to play a dual role in tumor promotion and suppression. Further research is needed to investigate which PERK-mediated signaling pathways affect tumor progression. Therefore, these pathways could potentially serve as candidates for development of targeted cancer therapies.
The dual roles of IRE1 in cancer could potentially be harnessed therapeutically
IRE1 is a type-I single pass transmembrane protein that undergoes oligomerization and trans-autophosphorylation during the ER stress response (24). The mammalian genome encodes two isoforms of IRE1; IRE1α and IRE1β. While IRE1α is expressed ubiquitously, the expression of IRE1β is restricted (25). The transcription factor XBP1 is the major RNA substrate of IRE1 (8). IRE1α performs splicing of the XBP1 mRNA, creating an active form of XBP1 (spliced XBP1). This activated form of XBP1 induces the expression of ER quality-control genes, thus enhancing the protein folding capacity of ER. Spliced XBP1 modulates expression of genes involved in protein folding, secretion, and endoplasmic reticulum-associated degradation (ERAD), a process by which ER localized proteins are exported to the proteasome for degradation in regulated fashion. Hypoxia has also been shown to activate IRE1α as a survival signaling axis in cancer cells. Inhibition of IRE1 in XBP1-knockout mice produced increased apoptosis and impaired tumor growth in response to hypoxia. The IRE1/XBP1 branch of the UPR may also be important in providing resistance against chemotherapy (26). Recently, NCOA3 (Nuclear receptor coactivator 3) has been shown important transcriptional target of spliced XBP1 in estrogen receptor positive breast cancer (27). Spliced XBP1 has been shown to regulate angiogenesis independent of vascular endothelial growth factor (VEGF) in pancreatic adenocarcinoma (28). Overexpression of spliced XBP1 plays important role in driving plasma cells towards a premalignant state (29). Interestingly, XBP1 deficiency inhibits tumor growth as well as blood vessel formation, and expression of spliced XBP1 restores angiogenesis in transgenic mice expressing a dominant negative form of IRE1 (29).
If attempts to reshape ER homeostasis fail, IRE1α curtails XBP-1 splicing, and activates apoptosis through IRE1 dependent decay of mRNA (RIDD) (30). During the transition from the IRE1-dependent adaptive response to the initiation of the apoptotic cascade, RIDD is thought to increases ER stress intensity through degradation of selective UPR target genes including the ER protein chaperone GRP78 (31, 32). Interestingly, many mRNAs that are degraded via RIDD encode proteins that have a crucial role in the negative regulation of apoptosis, cell growth, proliferation, and differentiation (33). Interestingly, RIDD substrates encode proteins of a diverse nature that modulate physiological processes that are distinct from ER homeostasis control. Of these various roles include production of inflammatory double-stranded RNA species through activation of an intracellular nucleic acid sensor (33). Therefore, prolonged ER stress is the tipping point that initiates IREα to act as a molecular switch that induces cell death instead of the adaptive response. Further research is needed to understand the molecular determinants which regulate the switch between pro- and anti-survival roles of IRE1.
ATF6: the least well understood arm of the UPR
ATF6 is a type-II transmembrane protein with an N-terminal cytoplasmic domain containing a DNA binding motif and a C-terminal ER luminal domain that binds GRP78. ATF6 regulates the expression of endoplasmic-reticulum-associated protein degradation (ERAD) pathway genes. In mammals, there are two homologs of ATF6: ATF6α (90 kDa) and ATF6β (110 kDa) (34). Both ATF6α and ATF6β are constitutively synthesized in the ER and activated by regulated intramembrane proteolysis during the ER stress response (35). In response to ER stress, ATF6 translocates from the ER to the Golgi, where it is processed by site-1 protease (S1P) and site-2 protease (S2P) to generate an N-terminal cytosolic domain, p50ATF6. (36). When the ER-membrane-bound precursor forms of ATF6α and ATF6β are cleaved during ER stress, their N-terminal halves become soluble transcription factors. These activated transcription factors then regulate the expression of UPR target genes via direct binding to the ER stress response element in gene promoters, in collaboration with the transcription factor nuclear factor-Y (‘NF-Y’) (37). Mounting evidence suggests that both ATF6α and ATF6β are crucial transcriptional regulators of the mammalian UPR. Overexpression of the active nuclear forms of ATF6α and ATF6β is sufficient for transcriptional upregulation of GRP78, CHOP and XBP1. ATF6α is essential for the adaptation of dormant cells to chemotherapy and nutrient stress. ATF6α provides a cell survival response through the up-regulation of Rheb and Akt-independent activation of mTOR (34). Down-regulation of ATF6α or Rheb increases the sensitivity of dormant tumor cells towards rapamycin thereby increasing antitumor activity of rapamycin. The role of ATF6 and the repertoire of ATF6 target genes have not been fully characterized, therefore, ATF6-mediated signaling remains the least understood of the three arms of the UPR. Development of specific chemical inhibitors of S1P, S2P or ATF6 itself will help to decipher the function of ATF6 in tumor cells under various stress conditions.
GRP78: a surprising target for therapeutics
GRP78 belongs to the heat shock protein 70 (HSP70) family of chaperones and resides primarily in the ER. GRP78 prevents aggregation and targeting of misfolded proteins for proteasomal degradation. GRP78 also binds Ca2+ and serves as an ER stress signaling regulator as described above (4, 38). Recent evidence indicates many additional non-canonical roles for GRP78. In prostate cancer cells, a cell surface form of GRP78 promotes tumor cell survival through modulation of the MAPK and PI3K/Akt-pathways (39). Targeting the carboxyl-terminal domain of cell surface GRP78 with a monoclonal antibody suppresses transcriptional activation of c-MYC-induced genes and impairs cell survival in glioma cell lines (40). ER stress has been shown to induce an alternatively spliced form of GRP78 that results in a cytosolic isoform that can also induce PERK signaling to increase the leukemic cell survival (41). GRP78 localization to the mitochondria has also been shown to increase during ER stress. Mitochondrial GRP78 maintains mitochondrial membrane integrity, thereby protecting against ER stress-induced apoptosis (42). Conversely, in response to TRAIL-mediated ER stress, GRP78 binds with Prostate apoptosis response-4 (Par4), translocates to the cell surface and leads to the activation of extrinsic apoptotic pathway (43). Additionally, cell surface GRP78 may also serve as a receptor for the angiogenesis inhibitor Kringle 5 (K5) and may act as a pro-apoptotic factor in stressed tumor cells (44). GRP78 and Bcl2 have been shown to form a complex which binds with separate domain of BIK, a pro-apoptotic protein. Sequestration of Bcl2 by BIK, reduces the localization of Bcl2 on ER and thereby leading to ER Ca2+ (release)-mediated mitochondrial apoptosis (45). These previously unrecognized roles for GRP78 offer new chances to therapeutically modulate the UPR.
The potential clinical relevance of the UPR in cancer
It has become increasingly evident that upregulation of UPR signaling components can be detected in clinical samples. XBP1 overexpression has been demonstrated in various human cancers including breast cancer and hepatocellular carcinoma (26). Furthermore, increased expression of NCOA3 was associated with poor prognosis, and higher levels of XBP1 in breast cancer tissues. High expression of spliced XBP1 protein correlates with poor prognosis in glioblastoma, triple negative breast cancer, and pre-B acute lymphoblastic leukemia (46). ATF6 expression has been shown to be positively associated with enhanced survival of differentiated thyroid cancer (47, 48). In accordance with previous work, a multi-cancer study showed higher ATF6 expression in metastatic lesions as compared to primary lesions (49). Colon cancer patients with increased expression of ATF6 in their primary tumors showed a higher propensity of the relapse (50). In squamous cell head and neck cancer, primary tumors which expressed higher ATF6 mRNA levels were more likely to be lymph node positive (48). The increased expression of GRP78 protein has been observed in melanoma, breast, colon and adenocarcinoma cancer cell lines, as well as in ex vivo human primary and animal model tissues. Several lines of evidence indicate that GRP78 expression is associated with poor prognosis and resistance to chemotherapeutics (51). GRP78 has been observed to be elevated in hepatocellular carcinoma, prostate, glioma, oral cancer, breast and gastric cancers. In many cancers, high GRP78 levels have been found to correlate with higher pathological grade and aggressive phenotype (52, 53, 54). Furthermore, autoantibodies against GRP78 have been observed in sera of prostate cancer patients and showed positive correlation with the aggressiveness of the disease (55). In solid tumors, GRP78 expression was associated with poor survival (56). In contrast, increased expression of GRP78 in neuroblastoma patients correlated with longer survival (57). Recently study analyzed the level of GRP78 expression that is associated with esophageal adenocarcinomas tumor growth. Results show that GRP78 was significantly up-regulated in early stages of cancer development but decreased in advanced stages of tumor growth (58). Notably, esophageal adenocarcinomas patients with higher GRP78 levels showed a survival advantage (58). These seemingly contradictory findings in human clinical samples demonstrate the need for further research into the precise mechanistic role of GRP78 in different contexts of tumorigenesis. Altogether, the above studies reflect the emerging evidence that variable expression of the components of the UPR may have important roles in human cancer. These observations can be paired with emerging preclinical studies indicate that UPR is intricately linked to transformation (16), genomic instability (59), tumorigenesis (60), metastasis (61), dormancy and resistance to anti-cancer therapy (62). Further studies are needed in both spheres to determine the context dependent roles of the UPR in such a manner that measurements of UPR components can one day serve as biomarkers for UPR targeted therapy.
The UPR presents multiple potential drug targets
The UPR could be exploited as a means of anticancer therapeutic strategy via two alternative approaches (Figure 2). The first approach is to block the UPR that has been activated as a cell survival response due to ER stress. Certain tumors such as myeloma and pancreatic cancer are believed to be reliant on the UPR, so impeding this process would sensitize these cells towards apoptosis (63). Furthermore, in certain tumors such as melanoma, the most effective strategy for exploiting ER stress would most likely involve the combination of agents that inhibit cytoprotective function of UPR arms along with those that actively induce ER stress/response (64)(64). The second approach is to pharmacologically increase ER stress above a certain threshold in cells that already have a high dependency on the UPR, thereby triggering cell death (65). For instance, Cerezo et al reported the development of the small molecule HA15 belonging to the thiazole benzensulfonamides. HA15 displayed antitumor activity on melanoma cells in vitro and in vivo. Interaction of HA15 with GRP78 leads to dissociation of GRP78 from the ER transmembrane UPR effectors thus activating the UPR with phosphorylation of PERK, eIF2α and ATF4 and CHOP expression. Interestingly, the authors showed that autophagy and apoptosis act simultaneously in HA15-induced cell death (66). In addition there is interest in pursuing both approaches simultaneously, i.e. pharmacologically increasing ER stress while simultaneously inhibiting the UPR. In the next section we will discuss UPR-suppressing and UPR-inducing agents (Table 1).
Figure 2.
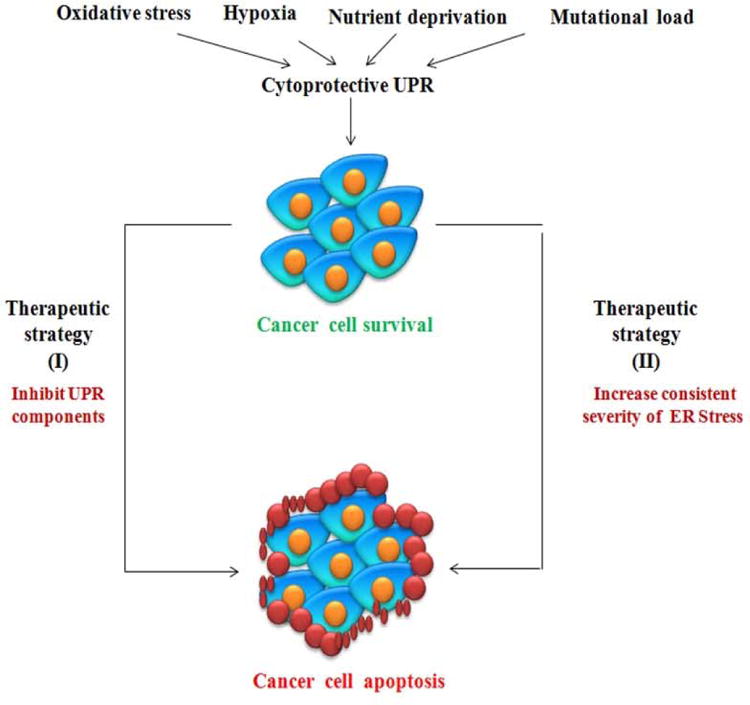
UPR can be exploited into two approaches for the purposes of cancer drug development. Targeting the unfolded protein response as an anticancer strategy attempts to divert cells from the survival pathway to the death pathway. [1] One approach is to inhibit components of the unfolded protein response so tumor cells can no longer deal with the stressful environment thereby leading to cell death. [2] Second approach is to increase the stress on the tumor cells, so the already activated unfolded protein response is overloaded and is unable to resolve the stress, thereby driving the cells towards the death pathway. Both strategies theoretically increase signaling to the death pathway and reduce survival signaling.
Table 1. Unfolded protein response-targeted drugs that inhibit cancer development.
| Therapeutic drugs | Therapeutic effect related to ER stress | Reference |
|---|---|---|
| GSK2606414, GSK2656157 (Second generation PERK inhibitor) | Inhibits PERK and eIF2α phosphorylation, ATF4 translation and CHOP mRNA expression | (67, 68) |
| ONC201 | Inhibits eIF2α | (114) |
| Salicylaldimines, 3-methoxy-6-bromosalicylaldehyde | Inhibits IRE1α activity | (71) |
| 4μ8C | Inhibits IRE1α activity | (72) |
| MKC-3946 | Inhibits IRE1α activity | (73) |
| STF-083010 | Inhibits IRE1α activity | (74) |
| Toyocamycin | Inhibits IRE1α activity | (75) |
| N9-(3-(dimethylamino) propyl)-N3,N3,N6,N6-tetramethylacridine-3,6,9-triamine (3,6-DMAD) | Inhibits IRE1α activity | (76) |
| Hydroxy-aryl-aldehydes (HAA) | Inhibits IRE1α activity | (77) |
| MKC-3946 | Inhibits IRE1α activity | (73) |
| STF-083010 | Inhibits IRE1α activity | (78) |
| Irestatin | Inhibits IRE1α activity | (60) |
| Eeyarestatin I (EerI) | ERAD inhibitor | (80) |
| Versipelostatin; Epigallocatechin gallate | Inhibition of GRP78 chaperone function compromises ER protein folding | (82, 83) |
| MAb159, PAT-SM6 | GRP78 inhibitors | (88, 115) |
PERK inhibitors
GSK2606414 is an ATP mimetic first generation PERK inhibitor (67). A second generation inhibitor GSK2656157 was developed with improved pharmacological properties (68). While PERK inhibitors remain an exciting anti-tumor strategy, there are likely dose limiting toxicities associated with pancreatic damage (69). The combination of a PERK inhibitor and proteasome inhibitor has been shown effective for multiple myeloma cells (70). It is thus interesting to speculate that the combined use of PERK and proteasome inhibitors may sensitize tumors, reducing the dose of the drug and decreasing the potential side effects. However, further studies are needed to investigate the side effects of PERK inhibition and how to limit them for the development of a new anticancer therapy.
IRE1α inhibitor
Numerous groups have identified small molecule inhibitors that selectively block IRE1α-XBP1 pathway. IRE1α inhibitors inhibit IRE1α activity either by binding to the catalytic core of the RNase domain, or to the ATP-binding pocket of the kinase domain. Salicylaldimines and their hydrolytic products can inhibit the endoribonuclease activity of IRE1α (71). Preventing XBP1 splicing, blocking transcriptional up-regulation of XBP1 targets and also inhibiting the degradation of mRNAs targeted by IRE1. Moreover, the salicylaldehyde analog 3-methoxy-6-bromosalicylaldehyde effectively inhibited the XBP1 splicing in an in vivo model of acute ER stress (71). Similarly, 4μ8C (72), MKC-3946 (73), STF-083010 (74), and toyocamycin (75) have potential inhibitory effects against IRE1α RNase activity. N9-(3-(dimethylamino) propyl)-N3,N3,N6,N6-tetramethylacridine-3,6,9-triamine (3,6-DMAD) inhibits both IRE1α oligomerization and its endoribonuclease activity. Inhibition of IRE1α-mediated XBP1 splicing by 3, 6-DMAD was shown to be cytotoxic in multiple myeloma cell lines (76). Another study showed that hydroxy-aryl-aldehydes (HAA) selectively inhibit IRE1α RNase activity and thus may represent a novel class of anti-cancer agents (77). MKC-3946, in combination with a proteosome inhibitor (bortezomib), enhanced ER stress by inhibiting XBP1 mRNA splicing (73). STF-083010 exerts an inhibitory effect in multiple myeloma xenografts (78). Irestatin, another inhibitor of IRE1α, also impairs the growth of malignant myeloma cells (60). Taken together, above studies indicate the clinical potential of IRE1α inhibitors; however, detailed mechanistic insights of these drugs are not explored yet. Though, there is a need for the development of more potent and specific compound to target IRE1α activity. Available compounds that target IRE1α activity have shown potential for anti-cancer treatment in combination with other conventional or existing chemotherapy. Therefore, the studies using small molecule inhibitors of IRE1α provide a fundamental background for understanding the cellular functions of this protein under stress, which may be also quite useful in designing the anti-cancer therapies using IRE1 inhibitors.
ERAD inhibitors
Molecular chaperones and lectin-like proteins are involved in the identification of misfolded proteins. These misfolded proteins are retained in the ER and subsequently degraded by the ERAD pathway. A defect in ERAD has been shown to cause accumulation of misfolded proteins in the ER and thus trigger the UPR. Eeyarestatin I (EerI) targets p97 (a cytosolic ATPase involved in polyubiquitinated protein transportation) inhibiting de-ubiquitination of p97-associated ERAD substrates (79). EerI has been shown to have antitumor activity in malignant myeloma cells (80). EerI treatment activates the transcription factors ATF3 and ATF4, which together activate NOXA expression. Interestingly, EerI blocks ubiquitination of histone H2A, thereby suppressing its inhibitory effect on NOXA transcription (81). These studies suggested that this example of an ERAD inhibitor may represent a novel class of anticancer drugs that concurrently targets the UPR and epigenetic signalling to limit the cancer cell progression.
GRP78 inhibitors
The tea-based polyphenol epigallocatechin gallate (EGCG) was shown to bind and inhibit the ATP-binding domain of GRP78, sensitizing glioma cells to chemotherapy (82). Versipelostatin inhibits GRP78 at transcriptional levels in combination with cisplatin in stomach cancer xenograft model (83). GRP78 silenced malignant glioma cells have shown increased sensitivity to temozomolide (84), etopside, and cisplatin (85). (83). Additionally, GRP78 inhibition by the GRP78 specific subtilase toxin and ER stress inducing agents, fenretinide or bortezomib, induced apoptosis in metastatic melanoma cells (86). GRP78 has been shown to function as an intracellular target for melanoma differentiation-associated gene-7/interleukin-24 (mda-7/IL-24), an IL-10 family cytokine which localizes to the ER and induces cancer cell specific growth suppression (87). Targeting GRP78 with a high affinity GRP78-specific mouse monoclonal IgG antibody (MAb159), was shown to enhance the apoptosis by degradation of cell surface GRP78 (88). Mab159 has been documented to suppress tumor growth in various mouse models such as colon, lung, metastatic breast cancer, melanoma, prostate cancer and leukemia (88). PAT-SM6, a human monoclonal IgM antibody, isolated from gastric cancer patients, was also shown to induce apoptosis by binding with cell surface GRP78 in human melanoma cells (89). Moreover, auto-antibodies against GRP78 in ovarian cancer patients decrease the metastatic properties of ovarian cancer cells (43). Altogether these studies suggest that strategies to inhibit GRP78 may represent promising therapeutic avenues for improving the sensitivity of apoptotic resistant cancer cells to various therapeutic drugs.
The extended reach of the UPR, and clinical translation
The UPR intricately interfaces with other stress response pathways including autophagy, and the integrated stress response. In addition, a new field of research focused on organelle membrane contact points is directly regulated by the UPR. The crosstalk between UPR and these stress pathways may finally determine the fates of cells under stress, and may offer indirect approaches to pharmacologically targeted the UPR, if the canonical targets of the UPR prove too difficult to drug. In this section, we will discuss the crosstalk of the UPR with autophagy, the membrane contact site regulatory network, and the integrated stress response.
UPR inhibitors and autophagy in cancer
The canonical branches of the UPR have been shown to regulate the autophagy in different ways during ER stress. Autophagy is a conserved pathway involving lysosomal degradation of long-lived proteins or damaged organelles such as mitochondria, ribosomes, peroxisome, and ER. Accumulating evidences show that the three arms of UPR differentially regulate the induction of autophagy (90). Cyclosporine A induces UPR-associated autophagy via IRE1 and PERK activation in glioma cells. Inhibition autophagy by rapamycin (mTOR/p70S6K1 pathway inhibitor) increases cell death significantly in this context (91). Resistance to BRAF inhibition in BRAF mutant melanoma was found to be mediated by UPR-induced autophagy. Surprisingly BRAF inhibitors can promote an unexpected binding of mutant BRAF to cytoplasmic GRP78, resulting in translocation of BRAF into the ER and enhanced ER stress. Subsequent activation of PERK leads to an upregulation of autophagy in melanoma (92). Inhibition of PERK or autophagy sensitized melanoma cells to BRAF inhibitor-induced cell death whereas inhibition of autophagy augmented BRAF inhibitor antitumor activity in a xenograft model (92, 93). ATF4 has been shown to be responsible for upregulation of autophagy genes such as ATG12 (94) and LC3 (95). The link between IRE1α and upregulation of autophagy is mediated by activation of JNK. (96). There are various drugs currently in preclinical and clinical trials that target processes that have a direct impact on the UPR and autophagy. Academic and industry researchers are aggressively developing agents to specifically target autophagy including inhibitors that target ULK1/ULK2 (97), ATG4 (98), and vps34 (99). However the only drug that has been used in clinical trials that targets autophagy is the lysosomal inhibitor hydroxychloroquine. Phase I clinical trials have been conducted that demonstrates the safety and preliminary activity of targeting autophagy to overcome therapeutic resistance (100-105). Collectively, these studies show that autophagy and UPR are tightly intertwined and together decides the fate of a cell under the stress. At this point it is unclear if in the context of therapy induced activation of UPR-associated autophagy, is targeting autophagy or upstream components of the UPR more effective at overcoming resistance to targeted therapy.
Role of ER-membrane contact sites in UPR and cancer
Contact sites between the ER and other organelles are now being extensively studied as a novel signaling hubs regulating cellular physiology (106). The ER must coordinate each of its specialized functions with mitochondria, plasma membrane and endosomes. Upon loss of ER membrane contact site signaling, cells may become dependent on other cell signaling and stress response systems, such as the UPR, to maintain organelle homeostasis and integrity (107). A growing number of reports suggest that these dynamic connections between ER and mitochondria are critical in fine tuning the UPR. PERK has been found to be uniquely enriched at the mitochondria-associated ER membranes (MAMs). An expending number of proteins such as inositol 1,4-5 triphosphate receptor, voltage dependent anion channel 1, calnexin, dynamin-1-like protein1, Ero1, phosphatidylserine synthase and mitofusin 2, have been shown to be enriched at the ER-mitochondria contact sites (108). PERK-/- cells display altered ER morphology, modified Ca2+ signaling, and weakened ER-mitochondria contact sites. Re-expression of a kinase-dead PERK mutant restores the ER-mitochondria contact sites which sensitize mitochondria to ROS-mediated stress (109). In addition, the ER has been known to form membrane contact sites with the Golgi complex to enable vesicular trafficking. In this regard, the crosstalk of these ER-Golgi contacts points with the UPR components has not been fully explored. Future studies defining the UPR interactome and its dynamics under ER stress condition will allow us to gain further insights into the role of ER stress sensors in organelle contact sites and tumor progression.
Integrated stress response and UPR
The integrated stress response (ISR) is an alternative route to activation of the UPR. EIF2α can be phosphorylated independently of the canonical UPR arms by three additional kinases: general control nonderepressible 2 (GCN2), heme regulated inhibitory kinase (HRI), and protein kinase R (PKR) (110). During the ISR, GCN2 phosphorylates eIF2α to conserve amino acid metabolism and induces ATF4 to promote recovery from nutrient deprivation. Loss of GCN2 causes a decrease in cancer cell survival during amino acid deprivation, and attenuated tumor growth in xenograft tumor models (111). Altogether, the additional mechanism of eIF2α phosphorylation enables the cells to integrate multiple stress stimuli into one common node, that being general control of protein synthesis via eIF2α. ONC201 (also called TIC10) is a novel small molecule anti-cancer agent that has been shown to elicit the ISR through the eIF2α kinases, resulting in activation of the eIF2α-ATF4 pathway (112). Recently, ONC201 has been shown to impair tumor growth by inhibiting survival-promoting kinases and inducing cell death through apoptosis (113). ONC201 is currently being tested in early phase human clinical trials in solid malignancies (114).
Conclusions
The UPR is an evolutionary conserved process which is linked to protein folding stress under physiological and pathological condition. The components of the UPR regulate numerous processes including lipid/cholesterol metabolism, energy homeostasis, inflammation, and cell differentiation. The UPR should not be viewed as three isolated signaling cascades, but a network that provides a strong crosstalk with autophagy, membrane contact sites, and the integrated stress response system. Altogether, three arms of UPR are a dynamic signaling framework integrated with crucial cellular process to maintain cellular homeostasis. Advances in the recent findings have made UPR a valid target for the development of new anticancer strategies. These strategies may offer future possibilities in personalized therapy and give hopes to the millions of patients suffering from cancer. However, more studies will be needed to investigate the oncogenic and tumor suppressive function of UPR in which the small molecule inhibitors of UPR components may play a vital role.
Footnotes
Conflict of interest statement: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Frontiers in bioscience (Scholar edition) 2012;4:412–431. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 4.Lee AS, Hendershot LM. ER stress and cancer. Cancer biology & therapy. 2006;5(7):721–722. doi: 10.4161/cbt.5.7.3120. [DOI] [PubMed] [Google Scholar]
- 5.Hazari YM, Bashir A, Haq EU, Fazili KM. Emerging tale of UPR and cancer: an essentiality for malignancy. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016 doi: 10.1007/s13277-016-5343-0. [DOI] [PubMed] [Google Scholar]
- 6.Mahdi AA, Rizvi SH, Parveen A. Role of Endoplasmic Reticulum Stress and Unfolded Protein Responses in Health and Diseases. Indian journal of clinical biochemistry : IJCB. 2016;31(2):127–137. doi: 10.1007/s12291-015-0502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mori K. The unfolded protein response: the dawn of a new field. Proceedings of the Japan Academy Series B, Physical and biological sciences. 2015;91(9):469–480. doi: 10.2183/pjab.91.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandewynckel YP, Laukens D, Geerts A, Bogaerts E, Paridaens A, Verhelst X, Janssens S, Heindryckx F, Van Vlierberghe H. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013;33(11):4683–4694. [PubMed] [Google Scholar]
- 9.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Molecular and cellular biology. 2003;23(20):7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q, Gupta PB. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer discovery. 2014;4(6):702–715. doi: 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- 11.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D, Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29(27):3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bu Y, Diehl JA. PERK Integrates Oncogenic Signaling and Cell Survival During Cancer Development. Journal of cellular physiology. 2016;231(10):2088–2096. doi: 10.1002/jcp.25336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Current molecular medicine. 2016;16(6):533–544. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Molecular and cellular biology. 2002;22(21):7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. The EMBO journal. 2005;24(19):3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, Li Y, Gao Y, Liu H, Li C, Maity A, Thomas-Tikhonenko A, Perl AE, Koong A, Fuchs SY, Diehl JA, Mills IG, Ruggero D, Koumenis C. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. The Journal of clinical investigation. 2012;122(12):4621–4634. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dey S, Tameire F, Koumenis C. PERK-ing up autophagy during MYC-induced tumorigenesis. Autophagy. 2013;9(4):612–614. doi: 10.4161/auto.23486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(37):16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishiwata-Kimata Y, Promlek T, Kohno K, Kimata Y. BiP-bound and nonclustered mode of Ire1 evokes a weak but sustained unfolded protein response. Genes to cells : devoted to molecular & cellular mechanisms. 2013;18(4):288–301. doi: 10.1111/gtc.12035. [DOI] [PubMed] [Google Scholar]
- 20.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro- apoptotic mRNAs and proteins. PLoS biology. 2006;4(11):e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature cell biology. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 22.Bonora M, Wieckowsk MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene. 2015;34(12):1608. doi: 10.1038/onc.2014.462. [DOI] [PubMed] [Google Scholar]
- 23.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(23):12625–12630. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends in cell biology. 2013;23(11):547–555. doi: 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imagawa Y, Hosoda A, Sasaka S, Tsuru A, Kohno K. RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 2008;582(5):656–660. doi: 10.1016/j.febslet.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 26.Koong AC, Chauhan V, Romero-Ramirez L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer biology & therapy. 2006;5(7):756–759. doi: 10.4161/cbt.5.7.2973. [DOI] [PubMed] [Google Scholar]
- 27.Gupta A, Hossain MM, Miller N, Kerin M, Callagy G, Gupta S. NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK-eIF2alpha-ATF4 signalling in breast cancer. Oncogene. 2016 doi: 10.1038/onc.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero-Ramirez L, Cao H, Regalado MP, Kambham N, Siemann D, Kim JJ, Le QT, Koong AC. X box- binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Translational oncology. 2009;2(1):31–38. doi: 10.1593/tlo.08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, Zheng M, Mani M, Henderson J, Pinkus GS, Munshi N, Horner J, Ivanova EV, Protopopov A, Anderson KC, Tonon G, DePinho RA. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer cell. 2007;11(4):349–360. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 31.Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132(1):24–26. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 32.Coelho DS, Domingos PM. Physiological roles of regulated Ire1 dependent decay. Front Genet. 2014;5:76. doi: 10.3389/fgene.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. The Journal of cell biology. 2009;186(3):323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(30):10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose- regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273(50):33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 36.Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer research. 2007;67(22):10631–10634. doi: 10.1158/0008-5472.CAN-07-1705. [DOI] [PubMed] [Google Scholar]
- 37.Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999;27(6):1437–1443. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer research. 2007;67(8):3496–3499. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 39.Misra UK, Pizzo SV. Activated alpha2-macroglobulin binding to cell surface GRP78 induces T-loop phosphorylation of Akt1 by PDK1 in association with Raptor. PloS one. 2014;9(2):e88373. doi: 10.1371/journal.pone.0088373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Gopal U, Gonzalez-Gronow M, Pizzo SV. Activated alpha2-Macroglobulin Regulates Transcriptional Activation of c-MYC Target Genes through Cell Surface GRP78 Protein. J Biol Chem. 2016;291(20):10904–10915. doi: 10.1074/jbc.M115.708131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ni M, Zhou H, Wey S, Baumeister P, Lee AS. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PloS one. 2009;4(8):e6868. doi: 10.1371/journal.pone.0006868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shu CW, Sun FC, Cho JH, Lin CC, Liu PF, Chen PY, Chang MD, Fu HW, Lai YK. GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum stress-induced apoptosis. Journal of cellular physiology. 2008;215(3):627–635. doi: 10.1002/jcp.21340. [DOI] [PubMed] [Google Scholar]
- 43.Cohen M, Ribaux P, Epiney M, Irion O. Role of prostate apoptosis response 4 in translocation of GRP78 from the endoplasmic reticulum to the cell surface of trophoblastic cells. PloS one. 2013;8(11):e80231. doi: 10.1371/journal.pone.0080231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen M, Zhang Y, Yu VC, Chong YS, Yoshioka T, Ge R. Isthmin targets cell-surface GRP78 and triggers apoptosis via induction of mitochondrial dysfunction. Cell Death Differ. 2014;21(5):797–810. doi: 10.1038/cdd.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou H, Zhang Y, Fu Y, Chan L, Lee AS. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK) J Biol Chem. 2011;286(29):25687–25696. doi: 10.1074/jbc.M110.212944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, Huang C, Swaminathan S, Sun H, Paietta E, Melnick AM, Koeffler P, Muschen M. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(21):E2219–2228. doi: 10.1073/pnas.1400958111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York, NY) 2004;6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer research. 2004;64(1):55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 49.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, Poggio T, Gerald W, Loda M, Lander ES, Golub TR. Multiclass cancer diagnosis using tumor gene expression signatures. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, Guilford PJ, Phillips V, Thompson-Fawcett M, Kasabov N, Toro T, Merrie AE, van Rij A, Yoon HS, McCall JL, Siewert JR, Holzmann B, Reeve AE. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(2 Pt 1):498–507. doi: 10.1158/1078-0432.CCR-05-2734. [DOI] [PubMed] [Google Scholar]
- 51.Schwarze S, Rangnekar VM. Targeting plasma membrane GRP78 for cancer growth inhibition. Cancer biology & therapy. 2010;9(2):153–155. doi: 10.4161/cbt.9.2.10760. [DOI] [PubMed] [Google Scholar]
- 52.Matsuo K, Gray MJ, Yang DY, Srivastava SA, Tripathi PB, Sonoda LA, Yoo EJ, Dubeau L, Lee AS, Lin YG. The endoplasmic reticulum stress marker, glucose-regulated protein-78 (GRP78) in visceral adipocytes predicts endometrial cancer progression and patient survival. Gynecologic oncology. 2013;128(3):552–559. doi: 10.1016/j.ygyno.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu R, Yang P, Wu HL, Li ZW, Li ZY. GRP78 secreted by colon cancer cells facilitates cell proliferation via PI3K/Akt signaling. Asian Pacific journal of cancer prevention : APJCP. 2014;15(17):7245–7249. doi: 10.7314/apjcp.2014.15.17.7245. [DOI] [PubMed] [Google Scholar]
- 54.Kaira K, Toyoda M, Shimizu A, Imai H, Sakakura K, Nikkuni O, Suzuki M, Iijima M, Asao T, Chikamatsu K. Decreasing expression of glucose-regulated protein GRP78/BiP as a significant prognostic predictor in patients with advanced laryngeal squamous cell carcinoma. Head & neck. 2016;38(10):1539–1544. doi: 10.1002/hed.24471. [DOI] [PubMed] [Google Scholar]
- 55.Shao Q, Ren P, Li Y, Peng B, Dai L, Lei N, Yao W, Zhao G, Li L, Zhang J. Autoantibodies against glucose-regulated protein 78 as serological diagnostic biomarkers in hepatocellular carcinoma. International journal of oncology. 2012;41(3):1061–1067. doi: 10.3892/ijo.2012.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma X, Guo W, Yang S, Zhu X, Xiang J, Li H. Serum GRP78 as a Tumor Marker and Its Prognostic Significance in Non-Small Cell Lung Cancers: A Retrospective Study. Disease markers. 2015;2015:814670. doi: 10.1155/2015/814670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Current opinion in cell biology. 2011;23(2):150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langer R, Feith M, Siewert JR, Wester HJ, Hoefler H. Expression and clinical significance of glucose regulated proteins GRP78 (BiP) and GRP94 (GP96) in human adenocarcinomas of the esophagus. BMC cancer. 2008;8:70. doi: 10.1186/1471-2407-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chroma K, Mistrik M, Moudry P, Gursky J, Liptay M, Strauss R, Skrott Z, Vrtel R, Bartkova J, Kramara J, Bartek J. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene. 2016 doi: 10.1038/onc.2016.392. [DOI] [PubMed] [Google Scholar]
- 60.Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician's perspective. Journal of hematology & oncology. 2011;4:8. doi: 10.1186/1756-8722-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 62.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 63.Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer letters. 2013;332(2):249–264. doi: 10.1016/j.canlet.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 64.Hill DS, Lovat PE, Haass NK. Induction of endoplasmic reticulum stress as a strategy for melanoma therapy: is there a future? Melanoma Management. 2014;1(2):127–137. doi: 10.2217/mmt.14.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H, Passeron T, Hofman V, Mograbi B, Dabert-Gay AS, Debayle D, Alcor D, Rabhi N, Annicotte JS, Heliot L, Gonzalez-Pisfil M, Robert C, Morera S, Virougoux A, Gual P, Ali MM, Bertolotto C, Hofman P, Ballotti R, Benhida R, Rocchi S. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer cell. 2016;29(6):805–819. doi: 10.1016/j.ccell.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 66.Cerezo M, Rocchi S. New anti-cancer molecules targeting HSPA5/BIP to induce endoplasmic reticulum stress, autophagy and apoptosis. Autophagy. 2017;13(1):216–217. doi: 10.1080/15548627.2016.1246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. Discovery of 7-methyl-5-(1- {[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4- amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) Journal of medicinal chemistry. 2012;55(16):7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 68.Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E, Mencken T, Gaul N, Goetz A, Stanley T, Hassell AM, Gampe RT, Atkins C, Kumar R. Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. ACS medicinal chemistry letters. 2013;4(10):964–968. doi: 10.1021/ml400228e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, Diehl JA. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Molecular and cellular biology. 2012;32(24):5129–5139. doi: 10.1128/MCB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Volkmann K, Lucas JL, Vuga D, Wang X, Brumm D, Stiles C, Kriebel D, Der-Sarkissian A, Krishnan K, Schweitzer C, Liu Z, Malyankar UM, Chiovitti D, Canny M, Durocher D, Sicheri F, Patterson JB. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286(14):12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(15):E869–878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, Kiziltepe T, Ikeda H, Kawano Y, French M, Blumenthal M, Tam V, Kertesz NL, Malyankar UM, Hokenson M, Pham T, Zeng Q, Patterson JB, Richardson PG, Munshi NC, Anderson KC. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119(24):5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ri M, Tashiro E, Oikawa D, Shinjo S, Tokuda M, Yokouchi Y, Narita T, Masaki A, Ito A, Ding J, Kusumoto S, Ishida T, Komatsu H, Shiotsu Y, Ueda R, Iwawaki T, Imoto M, Iida S. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress- induced XBP1 mRNA splicing. Blood cancer journal. 2012;2(7):e79. doi: 10.1038/bcj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang D, Tam AB, Alagappan M, Hay MP, Gupta A, Kozak MM, Solow-Cordero DE, Lum PY, Denko NC, Giaccia AJ, Le QT, Niwa M, Koong AC. Acridine Derivatives as Inhibitors of the IRE1alpha-XBP1 Pathway Are Cytotoxic to Human Multiple Myeloma. Molecular cancer therapeutics. 2016;15(9):2055–2065. doi: 10.1158/1535-7163.MCT-15-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanches M, Duffy NM, Talukdar M, Thevakumaran N, Chiovitti D, Canny MD, Lee K, Kurinov I, Uehling D, Al-awar R, Poda G, Prakesch M, Wilson B, Tam V, Schweitzer C, Toro A, Lucas JL, Vuga D, Lehmann L, Durocher D, Zeng Q, Patterson JB, Sicheri F. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nature communications. 2014;5:4202. doi: 10.1038/ncomms5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR, Hu CC. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. The Journal of clinical investigation. 2014;124(6):2585–2598. doi: 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, Wiestner A, Trenkle WC, Ye Y. The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PloS one. 2010;5(11):e15479. doi: 10.1371/journal.pone.0015479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Q, Li L, Ye Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem. 2008;283(12):7445–7454. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, Ye Y. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2200–2205. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, Bode AM, Dong Z. (-)- Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer research. 2006;66(18):9260–9269. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 83.Cook KL, Clarke R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med Chem. 2015;7(12):1529–1534. doi: 10.4155/fmc.15.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schonthal AH, Chen TC, Hofman FM, Louie SG, Petasis NA. Preclinical development of novel anti- glioma drugs targeting the endoplasmic reticulum stress response. Curr Pharm Des. 2011;17(23):2428–2438. doi: 10.2174/138161211797249242. [DOI] [PubMed] [Google Scholar]
- 85.Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, Lehman NL, Rempel SA, Mikkelsen T, Brodie C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-oncology. 2008;10(3):236–243. doi: 10.1215/15228517-2008-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin S, Hill DS, Paton JC, Paton AW, Birch-Machin MA, Lovat PE, Redfern CP. Targeting GRP78 to enhance melanoma cell death. Pigment Cell Melanoma Res. 2010;23(5):675–682. doi: 10.1111/j.1755-148X.2010.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gupta P, Walter MR, Su ZZ, Lebedeva IV, Emdad L, Randolph A, Valerie K, Sarkar D, Fisher PB. BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer research. 2006;66(16):8182–8191. doi: 10.1158/0008-5472.CAN-06-0577. [DOI] [PubMed] [Google Scholar]
- 88.Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, Krasnoperov V, Dong D, Liu S, Li D, Zhu G, Louie S, Conti PS, Li Z, Lee AS, Gill PS. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(24):6802–6811. doi: 10.1158/1078-0432.CCR-13-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hensel F, Eckstein M, Rosenwald A, Brandlein S. Early development of PAT-SM6 for the treatment of melanoma. Melanoma Res. 2013;23(4):264–275. doi: 10.1097/CMR.0b013e328362cbc8. [DOI] [PubMed] [Google Scholar]
- 90.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene. 2013;32(12):1518–1529. doi: 10.1038/onc.2012.174. [DOI] [PubMed] [Google Scholar]
- 92.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, Lazova R, Klump V, Pawelek JM, Xu X, Xu W, Schuchter LM, Davies MA, Herlyn M, Winkler J, Koumenis C, Amaravadi RK. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. The Journal of clinical investigation. 2014;124(3):1406–1417. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meng XX, Yao M, Zhang XD, Xu HX, Dong Q. ER stress-induced autophagy in melanoma. Clinical and experimental pharmacology & physiology. 2015;42(8):811–816. doi: 10.1111/1440-1681.12436. [DOI] [PubMed] [Google Scholar]
- 94.Wang J, Kang R, Huang H, Xi X, Wang B, Wang J, Zhao Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy. 2014;10(5):766–784. doi: 10.4161/auto.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29(31):4424–4435. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 96.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and cellular biology. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford NDP, Shaw RJ. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Molecular cell. 2015;59(2):285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rothe K, Lin H, Lin KBL, Leung A, Wang HM, Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM, Jiang X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123(23):3622–3634. doi: 10.1182/blood-2013-07-516807. [DOI] [PubMed] [Google Scholar]
- 99.Ohwada J, Ebiike H, Kawada H, Tsukazaki M, Nakamura M, Miyazaki T, Morikami K, Yoshinari K, Yoshida M, Kondoh O, Kuramoto S, Ogawa K, Aoki Y, Shimma N. Discovery and biological activity of a novel class I PI3K inhibitor, CH5132799. Bioorganic & medicinal chemistry letters. 2011;21(6):1767–1772. doi: 10.1016/j.bmcl.2011.01.065. [DOI] [PubMed] [Google Scholar]
- 100.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, Piao S, Heitjan DF, Tan KS, Pontiggia L, O'Dwyer PJ, Davis LE, Amaravadi RK. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359–1368. doi: 10.4161/auto.28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, Paul TM, Nichols CW, Porter DL, Kaplan J, Mallon G, Bradner JE, Amaravadi RK. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10(8):1380–1390. doi: 10.4161/auto.29264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, Giles FJ, Carew JS. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10(8):1403–1414. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, Piao S, Troxel AB, Evans TL, DeMichele AM, Nathanson KL, O'Dwyer PJ, Kaiser J, Pontiggia L, Davis LE, Amaravadi RK. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1369–1379. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, Tan KS, Heitjan DF, DeMichele AM, Vaughn DJ, Redlinger M, Alavi A, Kaiser J, Pontiggia L, Davis LE, O'Dwyer PJ, Amaravadi RK. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1391–1402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10(8):1415–1425. doi: 10.4161/auto.29165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17(2):69–82. doi: 10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Developmental cell. 2012;23(6):1129–1140. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 108.Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harbor perspectives in biology. 2011;3(9):a004424. doi: 10.1101/cshperspect.a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19(11):1880–1891. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Molecular biology of the cell. 2011;22(22):4390–4405. doi: 10.1091/mbc.E11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Molecular biology of the cell. 2005;16(12):5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Science signaling. 2016;9(415):ra18. doi: 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, Allen JE, Wang Z, Ma W, Lee HC, Orlowski R, Sarbassov dos D, Lorenzi PL, Huang X, Neelapu SS, McDonnell T, Miranda RN, Wang M, Kantarjian H, Konopleva M, Davis RE, Andreeff M. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Science signaling. 2016;9(415):ra17. doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Allen JE, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, Stogniew M, Schalop L, Benes C, Kaufman HL, Pottorf RS, Nallaganchu BR, Olson GL, Al- Mulla F, Duvic M, Wu GS, Dicker DT, Talekar MK, Lim B, Elemento O, Oster W, Bertino J, Flaherty K, Wang ML, Borthakur G, Andreeff M, Stein M, El-Deiry WS. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016 doi: 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hanson BE, Vesole DH. Retaspimycin hydrochloride (IPI-504): a novel heat shock protein inhibitor as an anticancer agent. Expert opinion on investigational drugs. 2009;18(9):1375–1383. doi: 10.1517/13543780903158934. [DOI] [PubMed] [Google Scholar]