

Abstract
Obtaining DNA nanostructures with potential applications in drug discovery, diagnostics, and electronics in a simple and affordable way represents one of the hottest topics in nanotechnological and medical sciences. Herein, we report a novel strategy to obtain structurally homogeneous DNA G‐wire nanostructures of known length, starting from the short unmodified G‐rich oligonucleotide d(5′‐CGGT‐3′–3′‐GGC‐5′) (1) incorporating a 3’–3′ inversion of polarity site. The reported approach allowed us to obtain long G‐wire assemblies through 5′–5′ π–π stacking interactions in between the tetramolecular G‐quadruplex building blocks that form when 1 is annealed in the presence of potassium ions. Our results expand the repertoire of synthetic methodologies to obtain new tailored DNA G‐wire nanostructures.
Keywords: DNA nanostructures, G-quadruplexes, G-wires, inversion of polarity sites, self-assembly
1. Introduction
In the development of new nanotechnologies and biomaterials, the possibility of assembling ordered supramolecular structures starting from small building blocks and the exploitation of self‐assembly driving forces are very important and much studied topics.1, 2, 3 In the bottom‐up approach, DNA strands have been demonstrated to be very useful building blocks for the assembly of supramolecular structures of various dimensions and shapes. For example, the DNA nanotechnology known as DNA origami allows the design and the construction of tailored three‐dimensional superstructures by exploiting the Watson–Crick base‐pairing scheme.4, 5, 6, 7 Besides the Watson–Crick base‐pairing scheme, also the Hoogsteen hydrogen bonding one, which allows the formation of G‐quadruplex DNA, can be exploited to obtain DNA supramolecular structures. G‐Quadruplexes are unusual secondary structures of DNA, which form when guanine‐rich DNA strands are annealed in the presence of suitable monovalent or divalent cations.
The building block of G‐quadruplexes is the G‐quartet (Figure 1 A), a planar arrangement of four guanines held together by a cyclic array of eight Hoogsteen hydrogen bonds.8, 9, 10 The onset of π–π interactions among the stacked G‐quartets greatly stabilizes the G‐quadruplex assembly, which is generally more stable than a DNA duplex of the same length. A G‐quadruplex can be formed by one, two, or four G‐rich DNA strands and can be classified as either parallel, antiparallel, or hybrid type depending on the mutual orientation of the strands involved in the G‐quartets formation.11, 12 Further factors that contribute to the wide polymorphism of G‐quadruplexes are the length and the base composition of the loops (when present), as well as the nature of the cations used to stabilize the quadruple helix structure.13, 14, 15 The biological implications of G‐quadruplexes in cellular processes16, 17, 18 and their use as promising drugs19, 20, 21, 22 or in drugs delivery23 and diagnostics23, 24, 25 are well known. In addition, G‐quadruplexes possess greater conductivity than DNA duplexes, thus suggesting their use for the obtainment of electronic nano‐biomaterials and nanodevices.26, 27, 28 These properties indicate the G‐quadruplex scaffold as a useful structural motif to obtain supramolecular self‐assemblies, including the so‐called G‐wires.29, 30, 31, 32 G‐wire superstructures can reach the length of thousands of nanometers along the axis perpendicular to the G‐tetrad planes. Depending on the topological arrangement of the G‐rich strands participating in their formation, G‐wires can be classified into two categories: i) interlocked G‐wires, characterized by the cooperative assembly of interlocked slipped strands (Figure 1 B) and ii) stacked G‐wires, characterized by the multimerization of G‐quadruplex building blocks held together by end‐to‐end π–π stacking (Figure 1 C). Besides the well‐known interlocked G‐wires29, 30, 31, 32, 33, 34, 35 and G‐wires containing both topological motifs,36, 37, 38 only a few examples of exclusively stacked G‐wires have been reported so far.39, 40, 41
Figure 1.
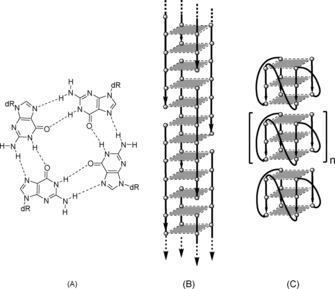
Schematic representations of a G‐tetrad (A) and of interlocked (B) and stacked (C) G‐wire polymers.
In a previous study, we reported that in the presence of K+ cations the 7‐mer d(CGGTGGT) can assemble into the octameric higher‐ordered G‐quadruplex complex d(CGGTGGT)8 (Figure S1 in the Supporting Information) through π–π stacking of two unusual G(:C):G(:C):G(:C):G(:C) planar octads belonging to two identical tetramolecular parallel G‐quadruplexes.42 Later, we demonstrated that the same dimerization pathway is also possible for all the other d(CGGXGGT) DNA strands, with X=A, G, or C.43 Herein, we report on the achievement of a new type of stacked G‐wire, here indicated as Q n (Figure 2) obtained by exploiting the 5’–5′ π–π stacking interactions between the G(:C):G(:C):G(:C):G(:C) octads formed at both ends of the tetramolecular G‐quadruplex building block Q1 (Figure 2). The last forms when the d(5′‐CGGT‐3’–3′‐GGC‐5′) DNA strand (1, Figure 2), incorporating a 3’–3′ inversion of polarity site, is annealed in the presence of K+ ions. The resulting G‐wires were obtained as a distribution of quadruplex multimers of different length.
Figure 2.
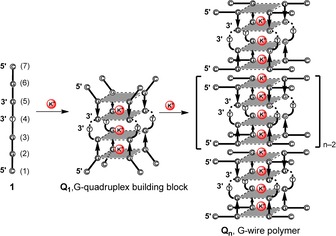
Formation of the G‐quadruplex building block Q1 and its multimerization into Q n G‐wire polymers starting from the ODN 1. The expected stabilizing K+ ions are shown as red spheres.
The n subscript in Q n indicates the number of tetramolecular G‐quadruplex building blocks participating in the G‐wires elongation. The actual formation of the target G‐wires was confirmed by polyacrylamide gel electrophoresis (PAGE), HPLC size exclusion chromatography (HPLC‐SEC), circular dichroism (CD), 1H nuclear magnetic resonance (NMR) spectroscopy, and atomic force microscopy (AFM) studies. The analytical results allowed us also to gather information on the G‐wires assembly and stability, and to isolate and characterize the three shortest Q n species (n=2–4).
2. Results and Discussion
2.1. Synthesis of d(5′‐CGGT‐3’–3′‐GGC‐5′) and G‐Wires
The d(5′‐CGGT‐3’–3′‐GGC‐5′) (1) incorporating a 3’–3′ inversion of polarity site was synthesized by using a solid‐phase automated DNA synthesizer as described in the Experimental Section. The inversion of polarity site was achieved by performing the first four coupling cycles with 5′‐phosphoramidites and the remaining three with standard 3′‐phosphoramidites. The Q n species were obtained by heating 1—dissolved in 1.0 m K+ containing buffer at the single strand concentration of 0.1 or 1.6 mm—at 90 °C for 10 min and then rapidly cooling it to 4 °C (fast annealing procedure). All samples were stored at 4 °C for 24 h before further investigation.
2.2. PAGE Studies
The annealed 1.6 mm 1 was analyzed by PAGE to obtain information on its propensity to form the tetramolecular G‐quadruplex building block Q1 and/or the target Q n multimers. For this purpose, we compared the electrophoretic mobility of 1 (lane 3 in Figure 3) with those of the G‐quadruplexes d(TGGGGT)4 (lane 1) and d(CGGTGGT)8 (lane 2), used as size markers for the tetramolecular G‐quadruplex building block Q1 and for its eight‐stranded dimer Q2, respectively. At the studied conditions, the annealed 1 migrated as a ladder of bands, thus confirming its propensity to form G‐quadruplex multimers. The electrophoretic mobility of the two fastest bands matched almost perfectly with that of the size markers in lanes 1 and 2, thus confirming the formation of a tetra‐stranded Q1 and octa‐stranded Q2; whereas the remaining slower bands in lane 3 were likely a result of longer Q n species (n≥3). The electrophoretic mobility of the isolated Q1 –4 species, obtained by HPLC‐SEC fractionation (see the following section), was also assessed (lanes 4–7). Taken together, the PAGE data suggested that the oligodeoxynucleotide (ODN) 1 in the presence of K+ ions is capable of self‐assembling in a distribution of G‐wire species of different lengths (Q n) obtained by the sequential stacking of the tetramolecular G‐quadruplex building block Q1 presenting two “sticky” G(:C):G(:C):G(:C):G(:C) planar octads at both 5′‐ends.
Figure 3.
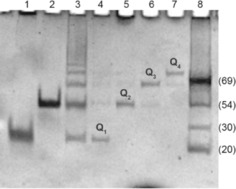
Electrophoretic mobility of the ODNs under study annealed in 1.0 m K+ buffer. Lane 1: d(TGGGGT)4; lane 2: d(CGGTGGT)8; lane 3: ODN 1; lanes 4–7: isolated peaks from the HPLC‐SEC fractionation of annealed 1; lane 8: single‐stranded DNA reference ladder.
2.3. SEC Studies
The Q n G‐wire population was analyzed by HPLC‐SEC at room temperature on a ReproSil 200 SEC column 24 h after annealing and storage at 4 °C. The HPLC profile of the annealed 0.1 mm 1 showed a distribution of peaks in which low molecular weight species had greater retention times (Figure S2 A in the Supporting Information). To obtain information on the M W of the observed product peaks, we used the same HPLC‐SEC conditions (see the Experimental Section) to determine the retention times (R t) of i) the single‐stranded dT7 (panel B), ii) the tetramolecular quadruplex d(TGGGGT)4 (panel C), and iii) the dimeric quadruplex d(CGGTGGT)8 (panel D). Based on the HPLC‐SEC data, the peak at the highest retention time (R t=21.45 min) was attributed to the single‐stranded 1, whereas the two preceding peaks (at R t=19.89 and 19.02 min) were assigned to the G‐quadruplex building block Q1 and to its dimer Q2, respectively. From these data, it was reasonable to hypothesize that each peak in the HPLC‐SEC profile belongs to a species differing from those of the adjacent peaks for a M W corresponding to the tetramolecular G‐quadruplex building block Q1 (i.e. 28 nucleotides). Exploiting the good peak resolution on the HPLC‐SEC column, the species likely corresponding to Q1 –4 were recovered and their purity and electrophoretic mobility assessed by PAGE (Figure 3, lanes 4–7). PAGE results confirmed the purity and allowed us to match the species responsible for the four fast‐moving bands in the PAGE (i.e. Q4, Q3, Q2, and Q1 from the slowest to the fastest band in Figure 3) with the four corresponding peaks in the HPLC‐SEC distribution (i.e. Q4, Q3, Q2, and Q1 from the least to the most retained in Figure S2 A in the Supporting Information). Furthermore, HPLC‐SEC re‐injection of the isolated Q1 –4 species, performed 24 h after their isolation and storage at 4 °C (Figure S3 in the Supporting Information), disclosed that the G‐quadruplex building block Q1 and the first three Q n species (Q2 –4) do not interconvert each other and can be stored as single G‐wire segments of known length for at least 24 h. However, considering the contribution of entropy to the formation of Q n G‐wires, different behaviors could be anticipated for samples of 1 annealed at different ODN concentrations. Thus, we exploited the good chromatographic separation of Q n species to assess the effect of the ODN annealing concentration on the formation and size distribution of Q n species. As expected, the HPLC‐SEC profile of Q n obtained 24 h after the annealing of 1 at 1.6 mm concentration (Figure 4) confirmed that the formation of longer G‐wires is strongly dependent on the ODN concentration during the annealing procedure. Indeed, in Figure 4 all Q n species with n≤8 are clearly distinguishable and almost equally populated, and an intense envelope peak attributable to longer G‐wires is present at R t=13.6 min. Conversely, in the HPLC‐SEC profile obtained from the sample annealed at 0.1 mm ODN concentration (Figure S2 A in the Supporting Information), the envelope peak is absent and only the Q n species with n≤3 are significantly populated.
Figure 4.
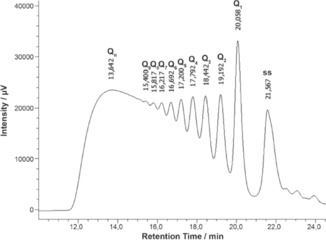
HPLC‐SEC profile of Q n distribution obtained by annealing 1.6 mm 1 in 1.0 m K+ containing buffer. ss=single‐stranded 1.
The effect of the temperature on the distribution of Q n species was investigated by HPLC‐SEC 24 h after the annealing of 0.1 mm 1 in 1.0 m K+ buffer. HPLC‐SEC analyses were performed by injecting the sample 30 min after heating and equilibration at 25, 45, 65, and 85 °C (Figure S4 in the Supporting Information). The profile obtained from the sample injected at 25 °C showed almost the same Q n distribution as the sample injected at 4 °C, but showed that at room temperature the dimeric Q2 species not only is more abundant than the building block Q1 (whereas the opposite was observed in the sample stored and injected at 4 °C), but represents the most abundant species. The progressive heating of the sample determined the reduction of the amount of Q n species and the contextual increase of the single‐stranded 1, which resulted in the only observable peak at 85 °C. However, two significant outcomes emerged from the profiles obtained at 45 and 65 °C: 1) at 45 °C, although the most abundant species was represented by the single‐stranded 1, we observed that the distribution of Q n species shifted towards longer G‐wires, with the appearance of a new peak at R t=13.3 min and with the Q3 species being even more abundant than Q2; 2) the analysis of the profile obtained at 65 °C suggested that the Q n species melt through a cooperative process, which drives directly to the random coil 1 (the peak of the G‐quadruplex building block Q1 was even less abundant than that of Q5 species). The higher thermal stability of Q n species relative to Q1 was also confirmed by CD melting data (see the following section).
2.4. CD Studies
CD is a well‐established diagnostic technique to provide preliminary information about the topology of G‐quadruplexes in solution. Generally, the CD profile of parallel G‐quadruplexes, in which all guanosines in the G‐tetrads are in the anti glycosidic bond conformation (Type 1 stem), is characterized by a positive signal at around 264 nm and a negative signal at around 240 nm; whereas that of antiparallel G‐quadruplexes with alternating syn–anti glycosidic bonds along the stem (Type 3 stem) shows a positive signal at 245 nm, a trough at around 260 nm, and a second positive signal at 290 nm.41, 44, 45, 46 Considering the formation of stacking interactions between the terminal G‐tetrads of Q1 building blocks (required to obtain the Q n G‐wires) and the presence of the 3’–3′ inversion of polarity site within the stem of Q1 and its multimers, the CD profile obtained for the annealed 1 (containing a mixture of Q n of different lengths), showing positive signals at 246 nm and 300 nm and a negative signal at 270 nm (Figure 5, dashed line), was somewhat unexpected. In fact, although the latter structural features anticipated for a Type 2 CD profile,44 the experimental result resembled that of a Type 3 stem. A Type 3 stem could be obtainable from 1 only if the thymines of two ODN 1 strands would participate in the formation of a propeller loop, thus allowing the formation of a bimolecular antiparallel quadruplex stem. However, this structural hypothesis has been ruled out by PAGE and HPLC‐SEC evidence, which indicated the exclusive formation of the tetra‐stranded quadruplex building block and its multimers.
Figure 5.
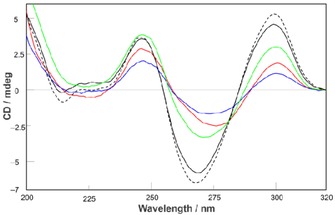
CD spectra of Q n distribution (dashed curve) and isolated Q1 (black curve), Q2 (green curve), Q3 (red curve), Q4 (blue curve) G‐quadruplexes.
Indeed, the CD profile of Q n resembles those reported for the G‐quadruplexes formed by d(5′TG3’–3′GGT)4 47 and d(TGG3’–3′GGT)4 48 incorporating a 3’–3′ inversion of polarity site and characterized by the presence of all anti G‐tetrads. The occurrence of the same system of uninterrupted stacked G‐tetrads, including both clockwise and counter‐clockwise directionality, in Q1 and Q n could explain the CD profiles shown in Figure 5. The resulting structural hypothesis requires that the thymines are projected outside the quadruplex stem to form bulge loops,49 as depicted in Figure 2. Unfortunately, the strong positive signal at 300 nm induced by the presence of the 3’–3′ inversion of polarity site precluded the observation of the negative band at 290 nm, which, in our previous studies, proved diagnostic for the formation of higher‐ordered G‐quadruplex assemblies obtained by non‐covalent π–π stacking of planar G(:C):G(:C):G(:C):G(:C) octads.42, 43 Similar CD profiles were obtained for the isolated Q1 –4 (Figure 5, colored lines), thus confirming that the CD profile of Q n was the result of the presence of quadruplex assemblies of different lengths incorporating the 3’–3′ inversion of polarity site. The overall thermal stability of the species populating the Q n distribution and that of the isolated Q1 –4 complexes was assessed by means of CD denaturation experiments (Figure S5 in the Supporting Information). The resulting apparent melting curves clearly provided evidence that the thermal stability of the quadruplex multimers was higher than that of the parent quadruplex building block Q1.
2.5. NMR Spectroscopy
The formation of G‐quadruplex assemblies from suitable G‐rich ODN sequences is usually confirmed by water‐suppressed 1H NMR spectroscopy. When the N‐1 imino protons of guanine bases are engaged in Hoogsteen‐type hydrogen bonding with the O‐6 carbonyl oxygen atoms of flanking guanines in each G‐tetrad, they are protected from exchange with the hydrogen atoms of the solvent and are thus visible in the 1H NMR spectrum as quadruplex‐diagnostic slightly broad signals resonating between 10 and 12 ppm.50, 51 Depending on the orientation of the strands and on the glycosidic torsion angle of guanosines participating in G‐tetrads, the four imino protons in each G‐tetrad resonate as one, two, or four NMR signals.52 The downfield region of the water‐suppressed 1H NMR spectra of the Q n distribution—obtained by annealing 1 in 1 m K+ buffer at 1.6 mm single strand concentration—recorded at 25, 45, 65, and 85 °C are shown in Figure S6 in the Supporting Information. In agreement with the CD evidence, the observation of the G‐quadruplex diagnostic imino protons signals in the 11–12 ppm region confirmed the presence in solution of G‐quadruplex species at temperatures lower than 65 °C. However, the intensity and the shape of imino and anomeric protons signals differed significantly from those we observed in the 1H NMR spectra of the dimeric higher‐ordered G‐quadruplexes formed by CGGTGGT42 and CGGAGGT.43 In the two latter, four well‐resolved imino protons (11.0–12.0 ppm) and intense anomeric protons signals (5.5–6.5 ppm) were visible at temperatures up to 65 °C. Conversely, in the NMR spectra of 1 recorded at 25 and 45 °C, all imino and anomeric signals appeared severely broadened, thus suggesting the involvement of imino and anomeric protons in chemical or conformational exchange phenomena. The line broadening of the imino protons was so intense as to require 10× software amplification for signal observation (see insets in Figure S6 in the Supporting Information). As far as the low intensity of the imino proton signals is concerned, we hypothesize that the presence of the bulge loops connecting the stacked G3 and G5 tetrads destabilizes the G‐quadruplex stem, thus speeding up the imino proton exchange with water. The anomeric proton signal broadening, instead, could be the result of side‐by‐side aggregation of G‐quadruplex units, as seen by Hu et al. in G‐wire assemblies formed by dGMP annealed in the presence of Sr2+.53 As we have shown in our previous papers,42, 43 the anomeric protons are located at the outer shell of 2Q‐like quadruplexes, thus it is conceivable that lateral contacts between flanking G‐quadruplex structures participating in the Q n G‐wire distribution could be responsible for the observed anomeric signal broadening. As we will see in the following section, AFM evidence supports the formation of extended quadruplex layers by lateral aggregation of quadruplex units. Although AFM gives us a picture of quadruplex aggregates deposited on mica surfaces, we believe that such aggregates could also form in solution considering the relatively high ODN concentration used for the preparation of the NMR samples (6.0 mm). In the light of this hypothesis, the comparison of the NMR spectra recorded in the 25–85 °C temperature range indicates that the breakdown of quadruplex aggregates and the melting of quadruplex units to the random coil 1 are cooperative processes, which start at temperatures between 45 and 65 °C and complete at temperatures lower than 85 °C.
2.6. AFM Studies
Q n quadruplexes interact and self‐assemble on mica surfaces in different ways depending on their concentration in the starting solution. The morphology of the obtained films has been investigated by AFM by using 1.6 mm and 16 μm solutions of 1 in 0.1 m phosphate buffer, at pH 7.0. Muscovite mica was used as the AFM support both for the super‐hydrophilic property of its surface, which guarantees a lower interaction between suspended molecules in aqueous solution during the evaporation of the solvent, and for its flatness, with less than 0.5 nm of root mean‐square (r.m.s.) roughness for a 1000×1000 nm2 surface area. The AFM images of the G‐quadruplexes spontaneously adsorbed from 1.6 mm ODN freshly prepared solutions showed a densely populated surface (Figure S7 in the Supporting Information), characterized by aggregates of different lengths and widths (x,y coordinates on panels A–C), whereas their heights (z coordinate) are always around 2 nm. These structures are due to the formation, after adsorption on the mica surface, of rod‐like shaped quadruplex aggregates grown during the evaporation of a 2 μL drop of 1.6 mm solution (panel A, scale bar=1 μm). The preferential direction of alignment of the G‐rodlets is well evident in panel B—the scale bar is 200 nm—where aggregates with lengths from 21 up to 166 nm (average value of 60±40 nm) are shown; widths are in the tenths of nanometers range and are always less than 100 nm; heights are almost all about 2 nm (see the statistics panels reported in Figure S7 in the Supporting Information). The AFM results are compatible with reported SEC results considering that, differently from height measurements, the length and width measurements are affected by the curvature radius of the AFM tip. The preferred orientation can be ascribed to the interaction between G‐quadruplexes and the mica surface. The homogeneous values of heights recorded suggested the formation of self‐assembled monolayers in the x,y plane owing to the evaporation of the buffer solution, which promoted lateral and longitudinal aggregation of G‐quadruplexes, the first one being favored by side‐packing.
A 100‐times diluted solution gave different results, as can be noted in Figure S8 in the Supporting Information. Even if a preferred orientation could still be envisaged, the structures seemed sparser and less ordered (see panel A); lengths are in the same interval as before, whereas the widths are smaller, never exceeding 30 nm, as evident from the measurements in panel B. Also in this case, a tip radius of curvature of about 10 nm must be taken in account. This result confirmed a side‐packing mechanism in the assembly of G‐structures during evaporation of the solution. The heights are always about 2 nm as before (see measurements in panel C), again endorsing the hypothesis of spontaneous formation of a monolayer. In the case of diluted starting solution, the longitudinal aggregation is competitive compared with the lateral one, owing to steric conditions during the evaporation.
3. Conclusions
In this paper, we reported the successful results of our study aimed at the obtainment of a new kind of G‐wire DNA by exploitation of π–π stacking interactions between tetramolecular G‐quadruplex building blocks incorporating a 3’–3′ inversion of polarity site and exposing G(:C):G(:C):G(:C):G(:C) planar “sticky” octads at both 5′‐ends. The main findings of the study can be summarized as follows: 1) the obtained G‐wires are unprecedented and are achievable by using the short 5′CGGT3’–3′GGC5′ G‐rich ODN as the starting material in a facile and affordable way; 2) the use of a tetramolecular G‐quadruplex building block to obtain DNA G‐wire polymers represents a major improvement over the use of monomolecular G‐quadruplexes, which, possessing different end‐faces and lateral nucleotide loops, could induce the formation of different kinds of aggregates thus reducing the amount of target G‐wires; 3) the reported G‐wires can be easily monitored in their formation and length‐growth by PAGE and HPLC‐size exclusion chromatography. We also demonstrated that the HPLC‐SEC technique can be used to determine the number of quadruplex building blocks in each Q n G‐wire, as well as to isolate and recover the three shortest Q n species (n=2–4).
Experimental Section
DNA Synthesis and Purification
DNA sequences d(TGGGGT) and d(CGGTGGT) were chemically synthesized with an Expedite 8909 DNA synthesizer (PerSeptive Biosystems, USA) using a universal CPG support purchased from Glen Research. The syntheses were performed by adopting the standard β‐cyanoethyl phosphoramidite chemistry at 10–15 μm scale and the products were purified as previously described.54 The synthesis of d(5′‐CGGT‐3’–3′‐GGC‐5′) (1) was performed with the same DNA synthesizer. The inversion of polarity site within the sequence was achieved by initially assembling the 5′‐CGGT‐3′ tract by using 5′‐phorphoramidites and then the 3′‐GGC‐5′ tract with standard 3′‐phosphoramidites. After completion of the ODN sequence, the support was treated with concentrated aqueous ammonia at 55 °C for 15 h. The combined filtrates and washings were concentrated under reduced pressure and purified through HPLC (JASCO PU2089 pumps equipped with the JASCO 2075 UV detector) with an anion exchange column (Macherey–Nagel, 1000‐8/46, 4.4×50 mm, 5 μm) using a linear gradient from 0 to 100 % B in 30 min, flow rate=1 mL min−1 and detection at 260 nm (buffer A: 20 mm NaH2PO4 aq. solution pH 7.0, containing 20 % (v/v) CH3CN; buffer B: 20 mm NaH2PO4 aq. solution pH 7.0, containing 1 m NaCl and 20 % (v/v) CH3CN).
Annealing Procedure
The ODN concentrations were determined in water by measuring the absorbance at 260 nm at 90 °C by using the nearest‐neighbor calculated molar extinction coefficient of 5′‐CGGTGGC‐3′ (ϵ=63 100 m −1 cm−1). The 0.1 and 1.6 mm solutions of 1 were obtained by dissolving the lyophilized sample in 900 mm KCl and 100 mm KH2PO4. The samples were annealed by heating at 90 °C for 10 min and then quickly cooling to 4 °C. After the annealing procedure, the samples were stored at 4 °C before measurements.
PAGE
Native gel electrophoresis experiments were performed on 20 % polyacrylamide gels containing TBE (8.9 mm Tris, 8.9 mm borate, 0.2 mm EDTA, from BIORAD) and 30 mm KCl, at room temperature, 120 V for 2 h. The ODN samples, annealed at 1.6 mm single strand concentration in 1.0 m K+ buffer, were diluted at 0.6 mm loading concentration just before the PAGE runs. Glycerol was added (10 % final) to facilitate sample loading in the wells. The bands were finally visualized by ethidium bromide staining in a Bio‐Rad Laboratories Gel DocTM XR+ image system.
HPLC‐SEC Analyses and Isolation of Qn Species
HPLC‐SEC analyses and purifications were performed with a ReproSil 200 SEC column operating in the M W range of 2000–70 000 Dalton (Dr. Maisch GmbH, 300×8 mm, 5 μm) eluted with 90 mm KCl and 10 mm KH2PO4/CH3CN (80:20, v/v), flow rate 0.5 mL min−1, detector at 260 nm. The analyses were performed at room temperature.
CD
CD spectra and CD melting profiles were recorded with a Jasco 715 CD spectrophotometer (Jasco, Tokyo, Japan) equipped with a Jasco JPT423S Peltier temperature controller in 1 mm optical path quartz cuvettes (100 nm min−1 scanning speed, 1 s response time). The spectra were recorded in triplicate at 4 °C from 220 to 320 nm. CD samples were prepared in potassium buffer (90 mm KH2PO4 and 10 mm KCl) at 20 μm final single strand concentration. The buffer baseline was subtracted from each spectrum and the spectra were normalized to have zero at 320 nm. CD melting curves were registered at 268 nm, 1 °C min−1 heating rate, temperature range 5–90 °C.
NMR Spectroscopy
NMR data were recorded with a Varian UNITYINOVA 500 MHz spectrometer equipped with a broadband inverse probe with z‐field gradient. The data were processed by using the iNMR software package (http://www.inmr.net). One‐dimensional NMR spectra were acquired as 16384 data points with a recycle delay of 1.0 s at 25, 45, 65, and 85 °C and the spectra were apodized with a shifted sine bell squared window function. Water suppression was achieved by including a double pulsed‐field gradient spin‐echo (DPFGSE) module55, 56 in the pulse sequence prior to acquisition. NMR samples were prepared at the concentration of 1.6 mm single strand in 200 μL of H2O/D2O 9:1 containing 900 mm KCl and 100 mm KH2PO4.
AFM
A XE‐100 Park Systems instrument was used for the AFM imaging of Q n G‐wires. Surface imaging was obtained in non‐contact mode by using 125 μm long silicon/aluminium‐coated cantilevers (PPP‐NCHR 10 m; Park Systems; tip radius lower than 10 nm), with a resonance frequency of 200 to 400 kHz and nominal force constant of 42 N m−1. The scan frequency was typically 0.5 Hz per line. When necessary, the AFM images were processed by flattening to remove the background slope, and the contrast and brightness were adjusted. Muscovite mica of about 1 cm2 surface was used as the substrate in the AFM study. Muscovite mica surfaces are typically used as AFM substrates owing to their perfect cleavage along a <001> plane, yielding large atomically flat areas. Mica consists of layers of an aluminium phyllosilicate lattice ionically bonded through interstitial K+ ions. Upon cleavage, the K+ ions are highly mobile and are readily exchanged with divalent cation species at the solid–liquid interface. This exchange results in a positive overcharging of the mica surface, which enables the deposition of molecules that hold a net negative charge, such as DNA.57 Moreover, the positive charge distribution after cleavage enables a super‐hydrophilic surface that guarantees a lower interaction between suspended biomolecules during the evaporation of aqueous solvent. Mica was freshly cleaved by using adhesive tape prior to each deposition to establish its cleanliness. Aliquots (2 μL) of the DNA/imaging buffer were directly deposited by casting onto freshly cleaved muscovite mica. After 2 min, every sample was gently washed with deionized water and then dried by evaporation at room temperature under a ventilated fume hood.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to Dr Maria Marzano for her technical assistance in the processing of HPLC‐SEC data.
G. Oliviero, S. D'Errico, B. Pinto, F. Nici, P. Dardano, I. Rea, L. De Stefano, L. Mayol, G. Piccialli, N. Borbone, ChemistryOpen 2017, 6, 599.
References
- 1. Ozin G. A., Hou K., Lotsch B. V., Cademartiri L., Puzzo D. P., Scotognella F., Ghadimi A., Thomson J., Mater. Today 2009, 12, 12–23. [Google Scholar]
- 2. Busseron E., Ruff Y., Moulin E., Giuseppone N., Nanoscale 2013, 5, 7098–7140. [DOI] [PubMed] [Google Scholar]
- 3. Kim J. K., Yang S. Y., Lee Y., Kim Y., Prog. Polym. Sci. 2010, 35, 1325–1349. [Google Scholar]
- 4. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 5. Marras A. E., Zhou L., Su H.-J., Castro C. E., Proc. Natl. Acad. Sci. USA 2015, 112, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Linko V., Dietz H., Curr. Opin. Biotech. 2013, 24, 555–561. [DOI] [PubMed] [Google Scholar]
- 7. Yang D., Campolongo M. J., Nhi Tran T. N., Ruiz R. C. H., Kahn J. S., Luo D., Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 648–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phan A. T., Kuryavyi V., Luu K. N., Patel D. J., in Quadruplex Nucleic Acids (Eds.: S. Neidle, S. Balasubramanian), Royal Society of Chemistry, Cambridge, 2006, pp. 81–99. [Google Scholar]
- 9. Simonsson T., Biol. Chem. 2001, 382, 621–628. [DOI] [PubMed] [Google Scholar]
- 10. Parkinson G. N., in Quadruplex Nucleic Acids (Eds.: S. Neidle, S. Balasubramanian), Royal Society of Chemistry, Cambridge, 2006, pp. 1–30. [Google Scholar]
- 11. Searle M. S., Williams H. E. L., Gallagher C. T., Grant R. J., Stevens M. F. G., Org. Biomol. Chem. 2004, 2, 810–812. [DOI] [PubMed] [Google Scholar]
- 12. Guédin A., De Cian A., Gros J., Lacroix L., Mergny J.-L., Biochimie 2008, 90, 686–696. [DOI] [PubMed] [Google Scholar]
- 13. Risitano A., Fox K. R., Nucleic Acids Res. 2004, 32, 2598–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rachwal P. A., Findlow I. S., Werner J. M., Brown T., Fox K. R., Nucleic Acids Res. 2007, 35, 4214–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hazel P., Huppert J., Balasubramanian S., Neidle S., J. Am. Chem. Soc. 2004, 126, 16405–16415. [DOI] [PubMed] [Google Scholar]
- 16. Maizels N., Nat. Struct. Mol. Biol. 2006, 13, 1055–1059. [DOI] [PubMed] [Google Scholar]
- 17. Lipps H. J., Rhodes D., Trends Cell Biol. 2009, 19, 414–422. [DOI] [PubMed] [Google Scholar]
- 18. Biffi G., Tannahill D., McCafferty J., Balasubramanian S., Nat. Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borbone N., Bucci M., Oliviero G., Morelli E., Amato J., D′Atri V., D′Errico S., Vellecco V., Cirino G., Piccialli G., Fattorusso C., Varra M., Mayol L., Persico M., Scuotto M., J. Med. Chem. 2012, 55, 10716–10728. [DOI] [PubMed] [Google Scholar]
- 20. Choi E. W., Nayak L. V., Bates P. J., Nucleic Acids Res. 2010, 38, 1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. D′Atri V., Oliviero G., Amato J., Borbone N., D′Errico S., Mayol L., Piccialli V., Haider S., Hoorelbeke B., Balzarini J., Piccialli G., Chem. Commun. 2012, 48, 9516–9518. [DOI] [PubMed] [Google Scholar]
- 22. Reyes-Reyes E. M., Teng Y., Bates P. J., Cancer Res. 2010, 70, 8617–8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Musumeci D., Piccialli G., Oliviero G., Roviello G. N., Bucci E. M., Bioconjugate Chem. 2012, 23, 382–391. [DOI] [PubMed] [Google Scholar]
- 24. Lv L., Guo Z., Wang J., Wang E., Curr. Pharm. Des. 2012, 18, 2076–2095. [DOI] [PubMed] [Google Scholar]
- 25. Lin X., Chen Q., Liu W., Li H., Lin J. M., Biosens. Bioelectron. 2014, 56, 71–76. [DOI] [PubMed] [Google Scholar]
- 26. Livshits G. I., Stern A., Rotem D., Borovok N., Eidelshtein G., Migliore A., Penzo E., Wind S. J., Di Felice R., Skourtis S. S., Cuevas J. C., Gurevich L., Kotlyar A. B., Porath D., Nat. Nanotechnol. 2014, 9, 1040–1046. [DOI] [PubMed] [Google Scholar]
- 27. Liu S.-P., Weisbrod S. H., Tang Z., Marx A., Scheer E., Erbe A., Angew. Chem. Int. Ed. 2010, 49, 3313–3316; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3385–3388. [Google Scholar]
- 28. Cohen H., Sapir T., Borovok N., Molotsky T., Di Felice R., Kotlyar A. B., Porath D., Nano Lett. 2007, 7, 981–986. [DOI] [PubMed] [Google Scholar]
- 29. Marsh T. C., Vesenka J., Henderson E., Nucleic Acids Res. 1995, 23, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vesenka J., Henderson E., Marsh T., in DNA-Based Molecular Construction, International Workshop on DNA-Based Molecular Construction (Ed.: W. Fritzsche), Jena, 2002, pp. 109–122. [Google Scholar]
- 31. Marsh T. C., Henderson E., Biochemistry 1994, 33, 10718–10724. [DOI] [PubMed] [Google Scholar]
- 32. Kotlyar A. B., Borovok N., Molotsky T., Cohen H., Shapir E., Porath D., Adv. Mater. 2005, 17, 1901–1905. [Google Scholar]
- 33. Dai T.-Y., Marotta S. P., Sheardy R. D., Biochemistry 1995, 34, 3655–3662. [DOI] [PubMed] [Google Scholar]
- 34. Batalia M. A., Protozanova E., Macgregor R. B., Erie D. A., Nano Lett. 2002, 2, 269–274. [Google Scholar]
- 35. Krishnan-Ghosh Y., Liu D., Balasubramanian S., J. Am. Chem. Soc. 2004, 126, 11009–11016. [DOI] [PubMed] [Google Scholar]
- 36. Ilc T., Šket P., Plavec J., Webba da Silva M., Drevenšek-Olenik I., Spindler L., J. Phys. Chem. C 2013, 117, 23208–23215. [Google Scholar]
- 37. Hessari N. M., Spindler L., Troha T., Lam W.-C., Drevenšek-Olenik I., Webba da Silva M., Chem. Eur. J. 2014, 20, 3626–3630. [DOI] [PubMed] [Google Scholar]
- 38. Tóthová P., Krafčíková P., Víglaský V., Biochemistry 2014, 53, 7013–7027. [DOI] [PubMed] [Google Scholar]
- 39. Shi Y., Luo H. Q., Li N. B., Chem. Commun. 2013, 49, 6209–6211. [DOI] [PubMed] [Google Scholar]
- 40. Saintomé C., Amrane S., Mergny J.-L., Alberti P., Nucleic Acids Res. 2016, 44, 2926–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smargiasso N., Rosu F., Hsia W., Colson P., Baker E. S., Bowers M. T., De Pauw E., Gabelica V., J. Am. Chem. Soc. 2008, 130, 10208–10216. [DOI] [PubMed] [Google Scholar]
- 42. Borbone N., Amato J., Oliviero G., D′Atri V., Gabelica V., De Pauw E., Piccialli G., Mayol L., Nucleic Acids Res. 2011, 39, 7848–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D′Atri V., Borbone N., Amato J., Gabelica V., D′Errico S., Piccialli G., Mayol L., Oliviero G., Biochimie 2014, 99, 119–128. [DOI] [PubMed] [Google Scholar]
- 44. Karsisiotis A. I., Hessari N. M., Novellino E., Spada G. P., Randazzo A., Webba da Silva M., Angew. Chem. Int. Ed. 2011, 50, 10645–10648; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10833–10836. [Google Scholar]
- 45. Vorlíčková M., Kejnovská I., Sagi J., Renčiuk D., Bednárová K., Motlová J., Kypr J., Methods 2012, 57, 64–75. [DOI] [PubMed] [Google Scholar]
- 46. Marchand A., Gabelica V., Nucleic Acids Res. 2016, 44, 10999–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Esposito V., Virgilio A., Pepe A., Oliviero G., Mayol L., Galeone A., Bioorg. Med. Chem. 2009, 17, 1997–2001. [DOI] [PubMed] [Google Scholar]
- 48. Esposito V., Virgilio A., Randazzo A., Galeone A., Mayol L., Chem. Commun. 2005, 3953–3955. [DOI] [PubMed] [Google Scholar]
- 49. Mukundan V. T., Phan A. T., J. Am. Chem. Soc. 2013, 135, 5017–5028. [DOI] [PubMed] [Google Scholar]
- 50. Jin R., Gaffney B. L., Wang C., Jones R. A., Breslauer K. J., Proc. Natl. Acad. Sci. USA 1992, 89, 8832–8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feigon J., Koshlap K. M., Smith F. W., Methods Enzymol. 1995, 261, 225–255. [DOI] [PubMed] [Google Scholar]
- 52. Adrian M., Heddi B., Phan A. T., Methods 2012, 57, 11–24. [DOI] [PubMed] [Google Scholar]
- 53. Hu D., Ren J., Qu X., Chem. Sci. 2011, 2, 1356–1361. [Google Scholar]
- 54. Oliviero G., Borbone N., Amato J., D′Errico S., Galeone A., Piccialli G., Varra M., Mayol L., Biopolymers 2009, 91, 466–477. [DOI] [PubMed] [Google Scholar]
- 55. Hwang T. L., Shaka A. J., J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar]
- 56. Dalvit C., J. Biomol. NMR 1998, 11, 437–444. [Google Scholar]
- 57. Lee A. J., Szymonik M., Hobbs J. K., Wälti C., Nano Res. 2015, 8, 1811–1821. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary