

Abstract
The noninvasive assessment of kidney function and diagnosis of kidney disease have long been challenges. Traditional methods are not routinely available, because the existing protocols are cumbersome, time consuming, and invasive. In the past few years, significant progress in the area of diagnosing kidney function and disease on the basis of light‐emitting agents has been made. Herein, we briefly review light‐emitting agents, including organic fluorescent agents and inorganic renal clearable luminescent nanoparticles for the noninvasive and real‐time monitoring of kidney function and disease. Moreover, some significant requirements and strategies regarding the design of ideal glomerular filtration rate agents and renal clearable nanoparticles are discussed. Finally, we discuss future challenges in expediting clinical translation of these developed light‐emitting agents, along with considerations of the efforts that need to be made to develop new agents and diagnosing kidney disease.
Keywords: fluorescence, glomerular filtration rate, kidney disease, kidney function, nanoparticles
1. Introduction
The kidney receives an abundant blood flow of 25 % of cardiac output and eliminates xenobiotics and metabolic products from the blood into the urine.1 Three processes in kidneys are involved during the formation of urine, including glomerular filtration, tubular reabsorption, and tubular secretion (Figure 1 a).2 Glomerular filtration is essential for the kidney to remove waste products and toxins rapidly from the plasma.3 The kidney tubules are responsible for reabsorption and secretion of substances to regulate plasma pH, potassium concentrations, and other physiological environment.4 Renal damage in glomerular and tubular function affects the ability of the kidney to remove xenobiotics and metabolic products from the blood into the urine.5 Thus, evaluation of kidney function is crucial for a number of clinical situations.
Figure 1.
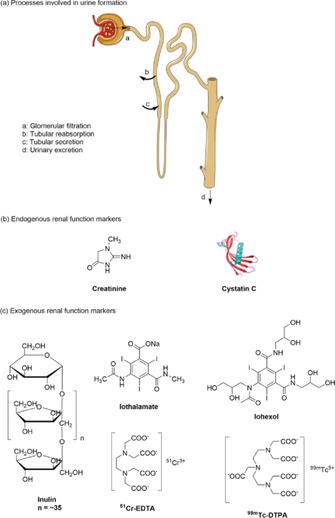
a) Three processes involved in urine formation, including glomerular filtration, tubular reabsorption, and tubular secretion. b) Structures of endogenous renal function markers. c) Structures of exogenous renal function markers.8 Reprinted with permission from Ref. 8. Copyright (2016) American Chemical Society.
Kidney disease or the loss of kidney function is often difficult to diagnose at an early stage but can cause lethal kidney failure later on. Of note, differentiation of the stages of kidney dysfunction has been a long‐term challenge. Therefore, preclinical techniques that can readily stage kidney dysfunction are essential to understand the progression of kidney disease. Furthermore, acute renal failure (ARF) has numerous causes such as hypotension, sepsis, trauma, acute tubular necrosis, drug‐induced nephrotoxicity, and urinary obstruction.6 Identifying the disease culprits in ARF is difficult and often relies on renal biopsy. However, this method is invasive and has potential risks.6 In vivo fluorescence imaging is an inexpensive, highly sensitive, and widely utilized preclinical method that is used to study various diseases. Significant developments in the area of diagnosing kidney function and disease on the basis of light‐emitting agents and fluorescence imaging has been made in the past ten years. The aim of this Review is to describe: one, traditional agents and determination methods for the assessment of kidney function; two, the utilization of organic fluorescent agents for the noninvasive real‐time assessment of kidney function; three, inorganic nanomaterials in combination with imaging technologies to identify various types of nephropathies and to differentiate the stages of kidney dysfunction. The main emphasis is placed on the design and application of light‐emitting agents, including organic fluorescent agents and inorganic nanomaterials as a means to monitor kidney function and disease.
2. Traditional Agents and Determination Methods for the Assessment of Kidney Function
The functional state of the kidneys can be assessed by using the glomerular filtration rate (GFR), renal blood flow, and tubular reabsorption or secretion of various substances.7 Among them, the GFR is often used to evaluate overall renal function and is accepted as the best indicator for kidney function.8 The GFR represents the plasma volume cleared by the nephrons per time unit during urine formation. It is generally presented in milliliter per minute.3 However, the GFR cannot be measured directly. The most common method is based on the concept of clearance.4 The concentration of endogenous creatinine in plasma is commonly used to determine the GFR (Figure 1 b), but it may result in erroneous estimates due to age, gender, muscle mass, and many other anthropometric variables.9 Determination of the plasma/urinary clearance of exogenous renal agents such as 99mTc–DTPA (diethylenetriaminepentaacetic acid, DTPA), inulin, and iothalamate (Figure 1 c) is invasive and cumbersome due to the requirement of multiple blood/urine sampling steps and tedious sample analysis by high‐performance liquid chromatography (HPLC).8 Moreover, many studies have revealed that creatinine and iothalamate are secreted by proximal tubule cells, whereas cystatin C and 99mTc–DTPA are reabsorbed by the tubular epithelial cells, which leads to a bias in the GFR.5 Urinary clearance is the traditional method to determine the GFR. However, timed urinary collections are time consuming, cumbersome, and susceptible to error. A long period of urine collection over 24 h to determine creatinine clearance is no longer routinely recommended as a means to assess kidney function.5 Alternatively, plasma clearance is used to determine the GFR to avoid inconvenience and to prevent errors from timed urine collections. Nevertheless, the main limitation of plasma clearance is the need for repeated and invasive blood sampling.4 Thus, new GFR agents and determination methods are highly desired to improve the determination of kidney function.
3. Organic Fluorescent Agents for Noninvasive Assessment of Kidney Function
Real‐time, noninvasive assessment of the GFR is essential not only to monitor ill patients at bedside in clinics but also to diagnose and stage patients with chronic kidney disease.10 Noninvasive in vivo imaging including single‐photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and computed tomography (CT) are widely used to assess kidney function by real‐time monitoring of the kidney clearance kinetics of renal‐clearable agents.11, 12, 13 However, those techniques may result in erroneous estimates, because traditional GFR agents are used and their reabsorption or secretion in proximal tubules is suspected. What is worse, the diagnosis of kidney diseases has not been greatly improved with the use of those techniques, due to their high costs, low accessibility, and the potential risk of exposure to radiation.12 Thus, novel noninvasive techniques are highly needed for the assessment of kidney function. To this end, the development of fluorescent agents and detection methods has provided new strategies. However, only a limited number of fluorescent agents have so far been developed to assess kidney function (Table 1).
Table 1.
Summary of organic fluorescent agents, their parameters, and determination methods for assessing kidney function.
| Optical window | Agent | λ ab [a] [nm] | λ em [b] [nm] | PPB[c] [%] | Plasma clearance half‐life [min] | Urinary recovery of the ID [%][d] | Filtration, reabsorption, or secretion in kidneys | Determination method |
|---|---|---|---|---|---|---|---|---|
| blue and green region | pyrazine agent 2 d | 435 | 557 | 0 | 29±1 | 90±1 (6 h) | only filtration | blood sampling and optical monitoring apparatus14, 15, 18 |
| pyrazine agent 2 h | 484 | 594 | 6 | 19±1 | 88±2 (6 h) | filtration and slight reabsorption | ||
| pyrazine agent 2 j | 486 | 597 | 0 | 20±1 | 85±2 (6 h) | filtration and slight reabsorption | ||
| PEGylated pyrazine 4 d | 439 | 559 | 5 | 20±1 | 96±1 (6 h) | only filtration | ||
| PEGylated pyrazine 5 c | 499 | 604 | 3 | 19±1 | 97±1 (6 h) | only filtration | ||
| FITC–inulin | 488 | 525 | 10.8 | NR[e] | NR[e] | only filtration | blood sampling19, 20 | |
| FITC–sinistrin | 488 | 525 | 7.4 | 22.1±1.9 | 95.4±0.8 (24 h) | only filtration | transcutaneous measurements8, 24, 25, 26, 27, 28, 35 | |
| FITC–HPαCD | 490 | 522 | 7.1 | 38.0±4.7 | 99.1±2.6 (24 h) | filtration and slight reabsorption | ||
| FITC–HPβCD | 490 | 522 | 2.3 | 24.1±3.2 | 103.4±4.1 (24 h) | only filtration | ||
| FITC–HPγCD | 490 | 522 | 2.8 | 20.2±3.2 | 100[e]±6.7 (24 h) | filtration and slight reabsorption | ||
| XITC–HPβCD | 498 | 530 | 7.7 | 31.3±7.0 | 103.2±5.4 (24 h) | only filtration | ||
| near‐infrared region | ABZWCY–HPβCD | 706 | 790 | 3.7 | 30.1±2.7 | 97±3.9 (24 h) | only filtration | |
| AAZWCY–HPβCD | 708 | 791 | 6.5 | 30.6±3.1 | 103.3±4.2 (24 h) | only filtration |
[a] λab: wavelength of maximum absorbance. [b] λem: wavelength of maximum absorbance. [c] PPB: plasma protein binding. [d] Urinary recovery of the ID: urinary recovery of the injected dose at a time period. [e] NR: not reported.
3.1. Pyrazine‐Based Fluorescent Agents for the Assessment of Kidney Function
Considerable efforts have been made by Dorshow et al., who have focused on the development of exogenous GFR agents for the real‐time assessment of renal function by using nonradioactive methods.10, 14 Two general approaches for the design of fluorescent renal agents have been considered. The first approach involves enhancing the fluorescence of known or existing renal agents that are intrinsically poor emitters such as lanthanide‐metal complexes; for example, a series of Eu3+ and In3+ complexes of DTPA–monoamide ligands bearing molecular “antennae” to enhance metal fluorescence through an intramolecular ligand–metal fluorescence resonance energy‐transfer process have been prepared and evaluated by fluorescence enhancement and biodistribution. Compound 18 a and 18 b (Figure 2 a) exhibit the highest fluorescent enhancement in those DTPA‐type metal complexes. The kidney clearance properties can be assessed by using the corresponding radioactive 111In complexes. To assess whether these complexes are reabsorbed or secreted by organic anion transporter (OAT) proteins in kidney tubules, probenecid, an inhibitor of OAT proteins, is administered (30 min prior) to block both the tubular reabsorption and secretion pathways. However, the results suggest that these complexes are excreted not only by glomerular filtration but also through secretion in the proximal tubules.10
Figure 2.
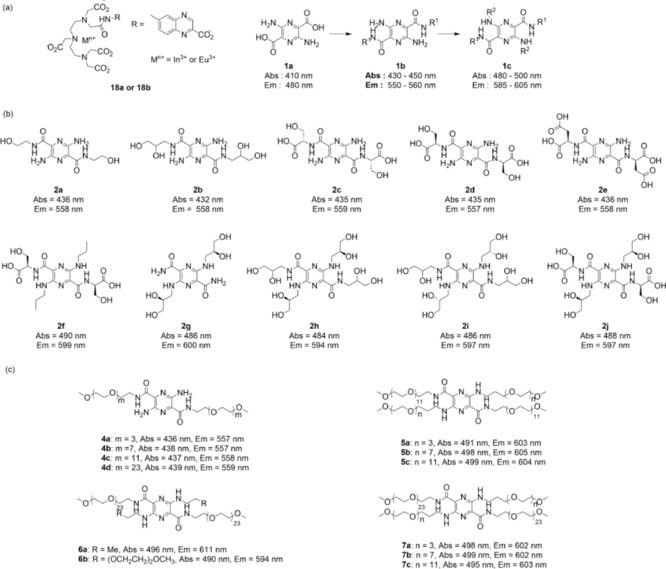
a) Structures and optical wavelengths of DTPA–monoamide complexes and pyrazine–dicarboxylic acid as well as potential derivatives.10 b) Structures and optical wavelengths of pyrazine‐based fluorescent agents.14 c) Structures and optical wavelengths of PEG–pyrazine conjugates.15
The other approach is based on transforming fluorescent lipophilic dyes into hydrophilic and anionic substances to force them to be eliminated by the kidneys. Thus, the pyrazine–dicarboxylic acid backbone (Figure 2 a, compound 1 a) is lipophilic and insoluble in water, and it can be used as a fluorophore and can be modified at the 2,5‐positions with electron‐withdrawing groups and at the 3,6‐positions with electron‐donating groups to improve its wavelength properties.14 By this concept, various neutral and anionic substituents have been designed and synthesized by condensing the pyrazine scaffold with amino ethanol, aminopropanediol, and amino acid derivatives. These pyrazine derivatives can be divided into two general categories: one, pyrazine derivatives bearing primary amino groups with the wavelength in the blue range of the spectrum (Figure 2 b, compounds 2 a–e); two, N‐alkylated pyrazines bearing electron‐donating groups with the wavelength in the green region of the spectrum (Figure 2 b, compounds 2 f–j). In vitro and in vivo studies show that pyrazine markers 2 d, 2 h, and 2 j have low plasma protein binding (PPB <7 %), plasma clearance half‐lives ranging from 20 to 30 min, high urine recovery, and are excreted by the kidneys. More importantly, renal tubular secretion is not a significant elimination pathway for these three compounds, which is testified by treating with probenecid to block the OAT pathway.14
Extensive studies to develop a new class of pyrazine derivatives based on the poly(ethylene glycol) (PEG) moiety have also been conducted.15 PEG is a hydrophilic, nontoxic, biocompatible polymer consisting of repeating ethylene oxide units. It was approved by the Food and Drug Administration (FDA) in 1990 for parenteral or topical administration as a component of various cosmetics, foods, and drug‐delivery systems.16 Particularly, PEG is eliminated through the kidneys without accumulation in the body, whereas the clearance rate is dependent on the molecular weight. It has been reported that PEGylation can significantly influence the excretion pathway and clearance rate. For example, the clearance half‐life increases from 18 min to 1 day if the molecular weight of PEG increases from 6 to 190 kDa, and PEG chains with molecular weights less than 6 kDa have been proven to be filtered by the glomerulus and not reabsorbed by kidney proximal tubules.16 These considerations have led to the investigation of a series of PEGylated pyrazine conjugates. For a systematic study of the effect of the length of PEG, PEG chains with different lengths have been conjugated to the pyrazine backbone, whereas the total molecular weight of PEG for modification has been limited to less than 5 kDa for free filtration. These conjugates can also be divided into two categories: one, pyrazine derivatives bearing primary amino groups with the wavelength in the blue region of the spectrum; two, N‐alkylated pyrazines with the wavelength in the green region of the spectrum. Similarly, both in vitro and in vivo assays including fluorescence properties, PPB, urinary recovery of the injected dose, probenecid blocking studies, and plasma clearance kinetics have been performed. PEGylated pyrazine compounds 4 d and 5 c have very low PPB values (5 and 3 %, respectively), high urinary recovery values (96 and 97 %, respectively), and short plasma clearance half‐lives (25 and 19 min, respectively) in healthy rats. These two compounds exhibit properties that are superior to those of iothalamate, which is an accepted standard for the measurement of the GFR.15 In preclinical studies, a series of in vitro and in vivo safety/toxicity studies have been performed with fluorescent agent 2 d (Figure 2 b), including blood compatibility, mutation assay, chromosomal aberration assay, and several single‐dose toxicity studies in rats and dogs to determine overall toxicity and toxicokinetics.17 Upon administering high doses of up to 200–300 times the estimated human dose, all animals survive to scheduled sacrifice. No effects are found on body weight, food consumption, and ophthalmic observations, and no abnormal anatomical pathology is observed by either macroscopic or microscopic evaluation of any organ or tissue. The results of these initial toxicology studies suggest that the safety/toxicity profiles of compound 2 d should be sufficient to begin a first‐in‐human clinical study. However, further nonclinical testing regarding biodistribution and developmental toxicity should be completed before starting the first‐in‐human studies.17
Indeed, to simplify the determination method of blood sampling, Dorshow et al. have developed an optical monitoring apparatus for noninvasive optical kidney function studies.18 The principle briefly is that one ear lobe is glued flat to a glass slide positioned approximately 2 mm beneath a fiber optic bundle that records the fluorescence signal from a test fluorescent agent passing through the ear. A λ=445 nm solid‐state laser source is directed through a chopper and into one leg of a silica‐bifurcated fiber optic bundle. After a short time period of recording the baseline, a fluorescent agent is intravenously injected into the rat, and the fluorescence signal corresponding to plasma and tissue distribution and subsequent renal clearance of the fluorescent agent is determined at the ear. Although the kidney clearance of a fluorescent agent can be detected by this noninvasive optical method, anesthesia is required during the whole measurement and can cause a decrease in the GFR.18
3.2. FITC–Inulin/Sinistrin for Noninvasive Assessment of Kidney Function
Inulin, an inert, uncharged polymer of fructose with a molecular weight of roughly 5000 Da fulfils all criteria of an exogenous agent and is regarded as the gold‐standard GFR agent. The GFR can be determined by injecting inulin intravenously. Given that inulin is neither reabsorbed nor secreted by kidney tubules after glomerular filtration, its excretion rate is directly proportional to the rate of filtration of water and solutes across the glomerular filter.4 Inulin has been successfully labeled in its fluorescein isothiocyanate‐conjugated variant (FITC–inulin, Figure 3 a).19 Its elimination kinetics are generally obtained by measuring the fluorescent values in plasma over a specific period of time after bolus injection.20 Although highly reproducible results are generally obtained from this assay and urine sampling is not required, repeated blood sampling is stressful to the animals. Furthermore, the FITC–inulin solution needs to be heated to enhance its solubility and dialyzed to remove residual unbound FITC, which make the procedure cumbersome.20
Figure 3.
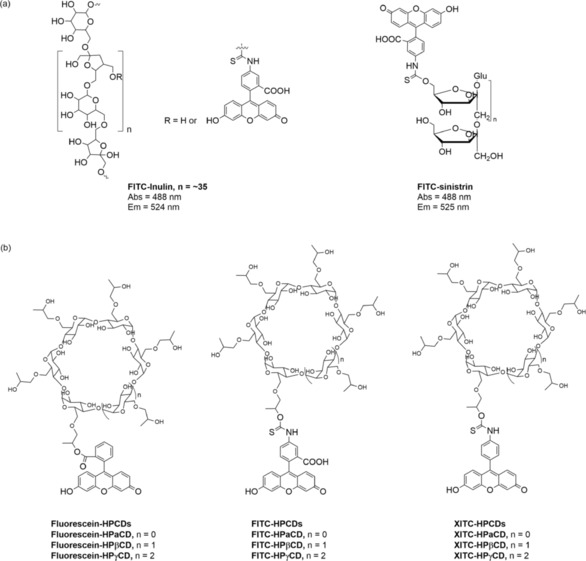
Chemical structures of FITC–inulin, FITC–sinistrin, fluorescein–HPCDs, FITC–HPCDs, and XITC–HPCDs.8
To overcome the poor water solubility of FITC–inulin, FITC–sinistrin was developed by a one‐step chemical‐labeling reaction (Figure 3 a).21 Sinistrin, an inulin analogue, was introduced in 1963 as an alternative to insulin to determine renal function due to its better water solubility.22 The better water solubility of sinistrin is attributed to its branched chemical structure rather than the linear structure of inulin. On the other hand, we have developed a noninvasive transcutaneous technique to determine the elimination kinetics of the fluorescent FITC–sinistrin agent on the basis of a miniaturized electronic device attached to the skin.23 The smart transcutaneous device comprises light‐emitting diodes that excite a fluorescent agent and a photodiode that detects the emission signal of the injected fluorescent agent.23, 24, 25 The workflow of the transcutaneous assessment of kidney function contains three main steps:
A fluorescent agent solution is injected into an animal intravenously, and the agent is diffused from the vascular space into the interstitium, where it is excreted by the kidneys.
The device is attached to the skin of an animal before the fluorescent agent is injected so that a baseline can be recorded for a short time period. The device enables the fluorescent agent to be excited repeatedly within the interstitial space by blinking each second at the appropriate wavelength. After each flash, the fluorescence emission of the fluorescent agent is detected and converted into a digital signal. These digital data are stored in an internal memory within the device.
The excretion half‐life of the fluorescent agent can be calculated after transferring and processing the data.25
Similarly to the plasma clearance kinetics, the collected data are evaluated to extrapolate the excretion kinetics of the fluorescent agent.
Extensive studies on kidney function by means of FITC–sinistrin have been conducted in preclinical animal models, including various strains of conscious mice (Balb/c, C57BL/6, SV129, and C57BL/6),26 Dahl salt‐sensitive hypertensive rats,27 and large species such as dogs and cats.28 This transcutaneous approach has been validated in different animal species and provides a more precise estimation of the GFR due to numerous data points from the transcutaneous measurements rather than a limited number of data points from blood and urine sampling. In addition, because measurements are taken transcutaneously, this method is independent of anthropometric parameters. The miniaturized transcutaneous device enables GFR measurements to be performed in conscious animals without anesthesia, and thus, it is possible to avoid an anesthesia‐related decrease in the GFR.4 Nevertheless, both inulin and sinistrin suffer from their inherent limitations, such as high costs, limited availability, and sophisticated extraction and purification from plant roots. Therefore, there is an unmet and long‐standing challenge to develop novel fluorescent GFR agents for the noninvasive real‐time assessment of kidney function.
3.3. Fluorescently Labeled Cyclodextrin Derivatives for Noninvasive Assessment of Kidney Function
An ideal exogenous fluorescent agent for transcutaneous assessment of kidney function should meet certain requirements: one, absorption and emission wavelengths in the visible region, and more preferably in the near‐infrared range; two, high hydrophilicity and very low or no PPB;29 three, neutral or weakly charged characteristics; four, no toxicity and no metabolism in vivo; five, no reabsorption and no secretion in the tubules and complete filtration by the glomerulus into the urine;30 six, easy to produce with low costs.
The reason to maintain low PPB is that binding to proteins generally influences the resulting pharmacokinetics, pharmacodynamics, and biodistribution in vivo. Moreover, tubular reabsorption and secretion may be either active or passive, depending on the particular substance and the transporter system in the tubular cells, for example, organic anion transporter (OAT) proteins and organic cation transporter (OCT) proteins. Thus, weak, charged characteristics are desirable to prevent interactions with organic transporter proteins. It is necessary to take all of these characteristics into account for the rational design of a GFR agent. Bearing this in mind, we have successfully developed 2‐hydroxypropylcyclodextrin (HPCD)‐based fluorescent agents for the transcutaneous measurement of kidney function.8 Fluorescent HPCD‐based agents are judiciously designed by combining the basic principle of renally cleared drugs and the knowledge of a cyclodextrin (CD)‐based drug‐delivery system. In this design, first, the introduction of HPCDs (including HPαCD, HPβCD, and HPγCD) increases the hydrophilicity, decreases PPB, and accelerates the elimination for fluorescent agents. The main elimination route of HPCDs is dependent on the route of administration. For both rat and dog, following oral administration, HPβCD is mainly excreted in the feces, whereas it is excreted by the kidneys after intravenous administration. HPβCD is well tolerated in animal studies (e.g. rats, mice, dogs, and rabbits) for both short‐term and long‐term studies. For humans, excretion is mainly by the kidneys. CDs are well tolerated in humans and have no adverse effects on the kidneys or other organs following either oral or intravenous administration.31 The merits of their nontoxicity resulted in FDA approval more than ten years ago.32 The reason for the utilization of HPCDs rather than CDs is that HPCDs not only have better water solubility than their native CDs, but they are also more stable to hydrolysis by α‐amylases of either porcine or human origin.33 Additionally, their narrow molecular weight distribution, low cost, and sufficient availability make them ideal backbones for GFR agents.34 Second, fluorophores belonging to the xanthene family are employed to label the HPCDs (Figure 3 b). Besides FITC and fluorescein, the panel of xanthene fluorophores has been expanded to include fluorophores based on decarboxylated FITC, referred to as XITC, for systematic study of the different conjugation bonds and sites between the fluorophore and the HPCDs. On the basis of considerable empirical knowledge that anionic and neutral substances are preferentially cleared through the renal system,14 rhodamine‐based dyes have not been selected as fluorophores to label the HPCDs due to the positive charge of the nitrogen atoms of the aniline moiety or the diethylaniline group. The three resulting categories of agents display different covalent conjugation bonds and net molecular charges and are suspected to display different pharmacokinetics and pharmacodynamics. All of the FITC–HPCDs exhibit very low PPB (<8 %), which is comparable to or even lower than that exhibited by some previous GFR agents, for example 51Cr–EDTA (12.2 %, EDTA=ethylenediaminetetraacetic acid) and 99mTc–DTPA (11 %). The extremely low PPB of the HPCD‐based agents is attributed to conjugation of the hydrophilic HPCDs on the hydrophobic fluorophores, which increases their hydrophilicity and reduces nonspecific interactions with serum proteins. These agents have no significant cytotoxic effect in representative human renal cell lines by using the 3‐(4,5‐dimethyl‐2‐thiazoly)‐2,5‐diphenyltetrazolium bromide (MTT) assay. By using the above transcutaneous technique, the plasma clearance curves and kinetic parameters of all the agents can be obtained. The clearance half‐lives of the HPCD‐based agents either in the absence or in the presence of probenecid follow the order HPαCD‐based agents > HPβCD‐based agents > HPγCD‐based agents, suggesting the rate of elimination increases with the increasing size of the HPCD. The clearance half‐lives of the tested agents also vary with the fluorophore scaffold used, for example, XITC–HPβCD > FITC–HPβCD > fluorescein–HPβCD. In the FITC–HPCD series, FITC–HPαCD and FITC–HPγCD both have slight tubular reabsorption because shorter clearance half‐lives are observed in the presence of probenecid. Fluorescein–HPCDs exhibit a clearance half‐life that is significantly higher with probenecid treatment than without probenecid treatment, which indicates that they are secreted in kidney tubules. In contrast, FITC–HPβCD and XITC–HPβCD display no tubular reabsorption or secretion and are cleared by glomerular filtration alone. A mandatory prerequisite for an ideal renal function agent is that it can obtain complete urinary recovery of the injected doses (IDs) and that it has no metabolism in vivo. Four agents, including FITC–HPαCD, FITC–HPβCD, FITC–HPγCD, and XITC–HPβCD, have high urinary recoveries of nearly 100 % of the IDs and do not undergo metabolism in vivo. However, only 40 to 60 % of the IDs of fluorescein–HPCD is recovered in urine. These results suggest that fluorescein–HPCDs are not completely excreted through the kidneys but also by other routes, including metabolism by enzymes in vivo. In sum, FITC–HPβCD is considered a promising novel exogenous fluorescent GFR agent. Of note, premature death and adverse clinical signs, as determined by measuring the body weight and food consumption of rats, are not observed during the entire set of experiments.
One of the major obstacles encountered with noninvasive real‐time transcutaneous assessment of kidney function in vivo upon using the aforementioned fluorescent GFR agents is strong intrinsic background autofluorescence from living tissue because of their short emission wavelength in the blue and green regions of the spectrum (λ<600 nm), which significantly compromises the accuracy of the measurements under physiological conditions. However, the absorption coefficient of tissue in the near‐infrared (NIR) region (λ=650–900 nm) is greatly suppressed to a minimum level, which thus drastically reduces the disturbances and noise from autofluorescence and increases tissue penetration.35 Therefore, it is highly desirable to develop GFR agents in the NIR optical window for transcutaneous assessment of kidney function. Recently, several studies have revealed that conventional organic fluorophores can persistently accumulate in the lipid membranes of the skin after intravenous injection because of their high lipophilicity.36 The large hydrophobic π‐conjugated systems of organic NIR dyes generally lead to strong binding between dyes and proteins in serum; for example, indocyanine green (ICG, Figure 4 a) displays extremely high PPB (99 %), liver uptake, and excretion. Such a high PPB hampers its kidney excretion. Many attempts have been made to overcome these obstacles, and enhanced hydrophilicity and decreased PPB for some heptamethine cyanine dyes can be achieved by increasing the number of sulfonate groups on the cyanine dyes.37 For example, the PPB of the commercial anionic dye IRDye800CW (Figure 4 a) is as high as 41 %. Although kidney clearance of IRDye800CW can be determined by multispectral optoacoustic tomography (MSOT),38 its plasma clearance curves have no decay at 90 min postinjection upon using transcutaneous measurements.35 These studies further confirm that rapid accumulation and long retention of conventional organic fluorophores in the skin is a dilemma for the noninvasive transcutaneous measurement of renal function.
Figure 4.
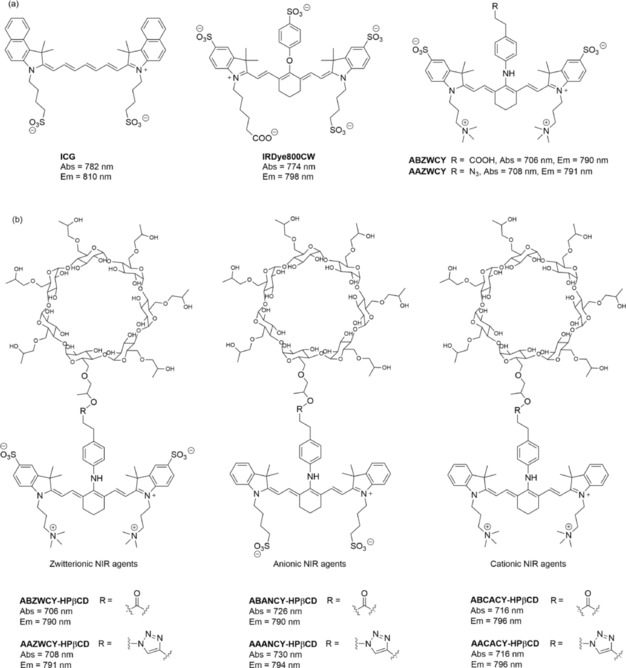
Structures of commercial cyanine dyes (ICG and IRDye800CW), zwitterionic cyanine dyes (ABZWCY and AAZWCY), and HPβCD‐based NIR agents.35 Reprinted with permission from Ref. 35. Copyright (2017) Royal Society of Chemistry.
Recently, Choi et al. reported pioneering work on zwitterionic heptamethine cyanine dyes.39 Zwitterionic dyes with a neutral overall charge exhibit reduced lipophilicity and PPB due to charge shielding. All zwitterionic analogues are excreted from the body into urine by kidney clearance with almost no significant nonspecific uptake into other organs or tissues.40 Although these zwitterionic analogues have decreased PPB and can be completely cleared by the kidneys, their PPB is still higher than that of “gold‐standard” renal function agents such as inulin and iothalamate (9.5 %)35 and cause a relatively long clearance half‐life. Inspired by that work, we have reported the first NIR GFR agents with improved hydrophilicity and much lower PPB (<7 %) through introducing zwitterionic charges and HPβCD on heptamethine cyanine dyes. To study the influence of different molecular surfaces charges on their PPB and excretion systematically, NIR fluorescent agents with three different molecular surface charge characteristics (i.e. zwitterionic, anionic, and cationic; Figure 4 b) have been designed, and they comprise two key functional components: HPβCD and NIR fluorophores. Covalent conjugation between HPβCD and the NIR fluorophores is achieved by either ester bond or click chemistry. Their spectra effectively match the configuration of the transcutaneous device, which consists of two light‐emitting diodes with an excitation wavelength at 700 nm and a photodiode for emission wavelength detection at 790 nm. In in vitro studies, the zwitterionic agents ABZWCY–HPβCD and AAZWCY–HPβCD have much lower PPB (3.7 and 6.5 %, respectively) than the anionic agents (i.e. ABANCY–HPβCD and AAANCY–HPβCD, 19.7 and 26.8 %, respectively) and the cationic agents (i.e. ABCACY–HPβCD and AACACY–HPβCD, 23.5 and 27.4 %, respectively), and even lower than some “gold‐standard” agents. In vivo, urinary recovery and fluorescent distribution investigations by small‐animal imaging experiments demonstrate that both ABZWCY–HPβCD and AAZWCY–HPβCD can be completely and rapidly excreted through the kidneys. Examination of urine samples by HPLC and matrix‐assisted laser desorption/ionization (MALDI) proves that these two zwitterionic HPβCD‐based agents have no metabolism in vivo. These two zwitterionic HPβCD‐based agents can be excreted efficiently through the kidneys into the urine without reabsorption and secretion in the tubules (Figure 5), as evidenced by studies on the blocking of both the OAT and OCT proteins in kidney tubules by treating with probenecid and cimetidine, respectively. Studies in transgenic nephropathy rat models also show that zwitterionic HPβCD‐based agents are promising agents for evaluation of kidney function. Relying on these novel zwitterionic NIR agents and a transcutaneous device, a rapid, robust, and biocompatible approach is validated for assessing kidney function in rat models that are both healthy and with kidney disease without the need for time‐consuming blood/urine sample preparation.
Figure 5.
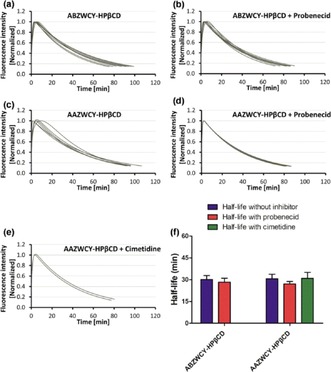
Plasma‐clearance curves of ABZWCY–HPβCD: a) in the absence and b) in the presence of probenecid treatment by transcutaneous measurements in healthy rats. Plasma clearance curves of AAZWCY–HPβCD: c) in the absence of any treatment, d) in the presence of probenecid, and e) in the presence of cimetidine by transcutaneous measurements in healthy rats. f) Clearance half‐lives of ABZWCY–HPβCD and AAZWCY–HPβCD in the absence and presence of probenecid or cimetidine treatment.35 Reprinted with permission from Ref. 35. Copyright (2017) Royal Society of Chemistry.
4. Inorganic Nanomaterials for Identifying Various Types of Nephropathies and Differentiating the Stages of Kidney Dysfunction
Numerous nanoparticle (NP)‐based agents have been utilized for biological and biomedical applications. The diverse research and applications of NPs have provided new strategies for monitoring kidney function and disease. Here, we describe both nonrenal‐clearable and renal‐clearable NPs for identifying kidney disease and monitoring kidney function, and especially, we summarize the strategies used to design renal‐clearable NPs and the growing field of renal‐clearable NPs for diagnosing various kidney diseases.
4.1. Nonrenal‐Clearable NPs for the Noninvasive Identification of Kidney Disease
Differentiation of kidney disease has long been a challenge, and currently, it often relies on renal biopsy. However, this method is invasive and has the potential risk for complications.41 Macrophage activity occurs frequently in nephritis, renal transplant rejection, and renal obstruction, but it is generally absent in normal kidneys.42, 43 Hauger et al. have used ultrasmall superparamagnetic iron oxide (USPIO) combined with MRI to determine if macrophage activity can be imaged and localized to compartments of the kidneys on the basis of disease type.44 In this study, a model of nephrotoxic nephritis induced by means of intravenous injection of sheep antirat glomerular basement membrane serum and a model of obstructive nephropathy are established. USPIO coated with dextran is injected into these two experimental rat models. In the nephrotoxic nephritis model, a significant decrease in the intensity of the MRI signal is observed only in the cortex, in which the glomerular lesions are located at 24 h postinjection of USPIO. In the obstructive nephropathy model, a decrease in the intensity of the MRI signal is found in all kidney compartments in response to diffuse interstitial lesions. The decrease in the intensity of the MRI signal is attributed to USPIO uptake by either macrophages or mesangial cells. Furthermore, the decreased signal intensity is correlated to the degree of proteinuria in the nephritis model, which suggests that USPIO‐enhanced MRI may help to identify and differentiate various types of nephropathies.44 Inspired by this study, Jo et al. have investigated if USPIO‐enhanced MRI could also detect inflammation in ischemic acute renal failure.45 The signal intensity in the outer medulla decreases after 24 and 48 h of ischemia, whereas it is not found in normal animals. USPIO is found inside the lysosomes of macrophages. Importantly, the change in the intensity of the MRI signal in the outer medulla is correlated with serum creatinine. USPIO injection does not alter renal function in both normal and ischemic animals.
Tabata et al. have designed fluorescent silica nanoparticles (SiNPs) for inflammation imaging in a mouse model with acute interstitial nephritis and unilateral ureteral obstruction (UUO). Unilateral renal obstruction has been found to cause an increase in the collagenous fibrous tissue in the renal interstitium after 6 days from time of injury.46 This change is able to be visualized with the assistance of fluorescent anti‐CD11b SiNPs (CD11b is expressed on the surface of mouse macrophages).47 After intravenous injection of fluorescent anti‐CD11b orientedly immobilized SiNPs to the mouse model with acute interstitial nephritis and UUO, the fluorescent anti‐CD11b orientedly immobilized SiNPs are accumulated to a greater extent in one kidney of the UUO model than in the normal and noninflamed kidneys. These findings are consistent with the histological results that the fluorescent anti‐CD11b orientedly immobilized SiNPs are associated with infiltration of macrophages into the inflammation site.47 Although these NPs are available to identify various types of nephropathies, their nonrenal‐clearable feature may cause long‐term retention in the organs of the reticuloendothelial system (RES) and may induce potential toxicity.
4.2. Renal‐Clearable NPs for the Noninvasive Differentiation of the Stages of Kidney Dysfunction
4.2.1. Strategies for Designing Renal‐Clearable NPs
The FDA has demanded that diagnostic agents injected into the human body be excreted completely within a reasonable amount of time.48 Although NP‐based agents show promising biomedical imaging and diagnostic features, toxicity induced by their nonspecific accumulation in vivo in the organs of the RES remains the primary roadblock to clinical translation. To avoid long‐term toxicity and nonspecific accumulation, efforts have been made to accelerate the elimination of NPs. Generally, renal excretion is a desirable pathway for the elimination of NPs, because contrast agents can be rapidly eliminated. Renal excretion relies on glomerular filtration in the kidneys.16 However, whether a nanoparticle can be cleared through the kidneys is highly dependent on its size, charge, and shape.49 As shown in Figure 6, the glomerular capillary wall mainly includes the endothelium with fenestration (70–90 nm), the glomerular basement membrane (2–8 nm), and the epithelium with a filtration slit embedded into podocyte extensions (4–11 nm). Owing to the combined effects of each layer of the glomerular capillary wall, the filtration‐size threshold of the glomeruli capillary wall is typically a hydrodynamic diameter (HD) of 6–8 nm,16 and thus, kidney excretion is exclusively possible for substances that are ultrasmall in size.
Figure 6.
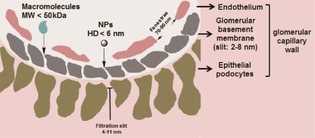
Schematic structure of the glomerular filtration barrier. Glomerular capillary wall comprises three specialized layers: endothelium with fenestration (70–90 nm), glomerular basement membrane (2–8 nm), and epithelial podocytes with filtration slit (4–11 nm).
In 2006, renal excretion of inorganic materials was first observed by Kostarelos et al. in single‐walled carbon nanotubes (SWCNTs). In this work, water‐soluble SWCNTs are functionalized with the chelating DTPA moiety and are labeled with indium (111In) for imaging.50 Although these functionalized SWCNTs have an average diameter of 1 nm and an average length of 300–1000 nm, they are not retained in any of the organs of the RES and are rapidly cleared from systemic blood circulation through the kidney excretion route.50 Choi et al. reported pioneering work on renal‐clearable quantum dots (QDs) in 2007. A series of small QDs (Figure 7 a) comprising a CdSe core/ZnS shell and coated with different charged moieties on the surface, including anionic (e.g. dihydrolipoic acid), cationic (e.g. cysteamine), zwitterionic (e.g. cysteine), and neutral small molecules (e.g. dihydrolipoic acid connected PEG), have been synthesized. This is the first study reporting that QDs with a HD less than 5.5 nm and a zwitterionic surface charge can be cleared through the kidneys.48 Since these first two landmark reports, an increasing amount of renal‐clearable NPs have been prepared (Table 2), including SiNPs,51 carbon dots,52 iron oxide NPs,53 palladium nanosheets,54 copper nanoparticles (CuNPs),55 and gold nanoparticles (AuNPs).56 Urinary recovery of these injected renal‐clearable inorganic NPs with values higher than 50 % is observed in 24 h; this value is comparable to the renal‐clearance efficiencies of some small molecular probes used in the clinic. Kidney accumulation of these renal‐clearable inorganic NPs is generally below 12 % of the ID per gram of tissue at 24 h postinjection, which is comparable to or even less than that of nonrenal‐clearable NPs in the range of 0.7 to 22 % of the ID per gram of tissue at 48 h postinjection.57 In addition, a new generation of SWCNTs has been developed (Figure 7 b), and these SWCNTs are functionalized with two fluorescent dyes (i.e. Alexa Fluor 488 and Alexa Fluor 680) and metal‐ion chelates (1,4,7,10‐tetraazacyclododecane‐1,4,7,10‐tetraacetic acid, DOTA) radiolabeled with 86Y for fluorescent and positron emission tomography imaging, respectively. These SWCNTs are rapidly renally cleared by glomerular filtration, and 65 % of the SWCNTs is observed in the urine. Importantly, competitive inhibition of the OAT, OCT, and megalin‐transport systems in the tubules does not affect clearance of the construct, which rules out tubular active secretion or reabsorption by these transporters as components of renal excretion.58 These inorganic nanomaterials with efficient renal excretion share some significant features and strategies for designing renal‐clearable NPs.
Figure 7.
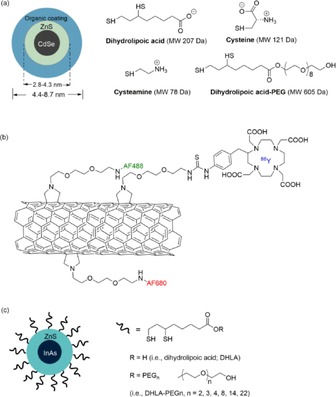
a) Chemical compositions of CdSe/ZnS QDs with DHLA (anionic), cysteamine (cationic), cysteine (zwitterionic), and DHLA–PEG (neutral) coatings.48 b) Schematic structure of the SWCNT‐[([86Y]DOTA)(AF488)(AF680)] construct. Water‐soluble SWCNT is covalently functionalized with DOTA‐86Y, AF488, and AF680.58 c) InAs(ZnS) QDs with a systematic increase in the PEG chain length attached through a DHLA linker.62
Table 2.
Renal‐clearable nanoparticles and their renal‐clearable efficiencies as well as the imaging technique.
| NP agent | Surface ligand[a] | Core size [nm]/ HD [nm] | Renal‐clearable efficiency [%] (time [h])[b] | Imaging technique | Ref. |
|---|---|---|---|---|---|
| NP agents with zwitterionic surface | |||||
| CdSe/ZnS | cysteine | 2.85/4.36 | 75 (4) | fluorescence | 48 |
| Pd nanosheets | GSH[a] | 4.4×1.8/NR[c] | 6.6 (24) | fluorescence | 54 |
| CuNPs, 64Cu‐doped | GSH | 2.0/2.2 | 62.5 (2) | fluorescence/PET | 55 |
| AuNPs | GSH | 2.5/3.3 | 50 (48) | fluorescence | 56 |
| AuNPs, 111In | DTDTP | 2.4/6.6 | 64 (24) | fluorescence/SPECT | 64 |
| NP agents with neutral surface | |||||
| SiNPs | PEG500 | NR[c]/3.3 | 73 (48) | fluorescence | 51 |
| Carbon dots, ZW800 | PEG1500 | 3/4.1 | 56 (24) | fluorescence | 52 |
| AuNPs | PEG1000 | 2.3/5.5 | 50 (24) | fluorescence | 64 |
| Au nanoclusters, 64Cu‐doped | PEG350 | 2.8/4.3 | 30 (24) | PET | 65 |
[a] GSH: glutathione. [b] Percent ID; time period postinjection is given in parentheses. [c] NR: not reported.
Size: The size of the filtration threshold of the glomerular capillary wall is typically 6–8 nm; therefore, reducing the size of the NPs is a primary strategy to enhance their renal‐clearance efficiency. With the help of inorganic synthetic chemistry, most inorganic nanoparticles with a core size below 6 nm can readily be prepared.
Shape: The efficient renal clearance of SWCNTs involves a shape effect. Although the molecular weights (300–500 kDa) and average lengths (300–1000 nm) of SWCNTs are much larger than the molecular‐weight cutoff (50 kDa) and filtration threshold (6–8 nm) for glomerular filtration, these SWNTs can still efficiently pass through the kidneys into the urine. This phenomenon can be explained by flow‐induced orientation, which makes the long axis of the SWNTs point toward the gap of the glomerular capillary pores.57 Generally, renal‐clearable NPs have a spherical shape, and spherical NPs with a diameter smaller than the kidney filtration threshold can be easily cleared into the urine.
-
Surface chemistry: NPs with ultrasmall HDs are expected to clear through the kidneys. However, many ultrasmall NPs are still nonrenal clearable and are accumulated in the organs of the RES. For example, low urinary recovery with a value of only 9 % of the ID has been determined for AuNPs coated with bis(p‐sulfonatophenyl)phenylphosphine, whereas more than 50 % of the ID of these AuNPs is found in the liver at 24 h postinjection. Besides, Choi et al. have also demonstrated that QDs coated with anionic dihydrolipoic acid or cationic cysteamine have a small HD (4 nm) and cannot be cleared through the kidneys and are mainly retained in the liver, lung, and spleen.48 The severe accumulation of ultrasmall NPs in the organs of the RES is attributed to protein adsorption, because as a result of the high surface energy and charged ligands on the NPs, nearly thousands of different kinds of plasma proteins in the blood can interact with the surfaces of the particles if the NPs are distributed in the bloodstream.59 Adsorption of those proteins can result in a remarkable increase in their HD and their uptake in the organs of the RES by macrophages.60, 61 To minimize serum protein binding, zwitterionic ligands (e.g. cysteine) and neutral ligands (e.g. PEG) have been used to modify the surfaces of NPs. More than 50 % of the ID of QDs coated with the zwitterionic cysteine ligand (HD: 4.9 nm) can be effectively cleared into the urine, and less than 5 % of the ID is observed in the liver.48 Unlike zwitterionic ligands with charged features and low molecular weights, PEG is a macromolecule with a low charge density; thus, inorganic NPs coated with PEG ligands generally have much thicker stern layers than NPs coated with zwitterionic ligands, which often leads to a HD that is larger than the kidney filtration threshold. Nevertheless, investigations have revealed that inorganic NPs coated with short PEG chains are renal clearable with a high clearance efficiency. For example, Choi et al. have found that only QDs coated with DHLA–PEG‐4 (DHLA: dihydrolipoic acid) can be cleared by the kidneys, and neither longer (DHLA–PEG‐8, ‐14, ‐22) nor shorter PEG chains (DHLA‐PEG‐2) are desirable to make QDs renal clearable (Figure 7 c).62 Furthermore, other inorganic renal‐clearable NPs coated with low‐molecular‐weight PEG (500–2000 Da) have been developed, such as PEG500‐coated SiNPs, PEG1000‐coated AuNPs (Figure 8
Figure 8.
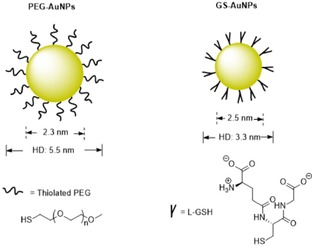
),63 and PEG1500‐coated carbon dots.52 These results indicate that fine control of the PEG chain with an optimized length is critical for developing renal‐clearable PEGylated NPs.
4.2.2. Renal‐Clearable NPs for the Noninvasive Staging of Kidney Dysfunction
Although zwitterionic‐cysteine‐coated QDs can be rapidly cleared into the urine (75 % of the ID at 4 h postinjection), the renal clearance of cysteine‐coated AuNPs is not enhanced, and (220±60) nm aggregates in phosphate‐buffered saline and accumulation of the cysteine‐coated AuNPs in the organs of the RES are observed.56 To develop renal‐clearable AuNPs, great efforts have been made by Zheng et al. by using zwitterionic glutathione (GSH, a tripeptide that is abundant in the cytoplasm and exhibits low affinity to plasma protein66) to modify the surfaces of the particles and to minimize serum protein adsorption.36, 56, 63, 67, 68, 69 The obtained GSH‐coated AuNPs (GS‐AuNPs, Figure 8) can emit near‐infrared light (core size: 2.5 nm, HD: 3.3 nm), have high resistance to PPB, and have high urinary recovery with more than 50 % of the ID at 48 h postinjection. Moreover, GSH can serve as a universal surface chemistry to minimize nonspecific accumulation of inorganic NPs in the organs of the RES, as evidenced by other GSH‐coated ultrasmall metal NPs such as palladium nanoparticles (PdNPs)54 and CuNPs55 and their renal clearance. Besides GSH‐coated AuNPs, renal‐clearable AuNPs capped by other zwitterionic ligands such as dithiolated polyaminocarboxylate (DTDTPA)57, 64 and dopamine sulfonate53 are also prepared. Among zwitterionic‐coated NPs, GS‐AuNPs have been extensively investigated for biomedical imaging and diagnosis, ranging from tumor‐targeting imaging to detection of kidney dysfunction.
On the one hand, GS‐AuNPs with a core size of 2.5 nm and a HD of 3.3 nm exhibit intrinsic NIR emission without conjugation of dyes and behave similarly to the small NIR dye IRDye800CW in terms of physiological stability and renal clearance. However, GS‐AuNPs have enhanced permeability and a retention effect because they have a much longer tumor retention time and faster normal tissue clearance than IRDye800CW. These merits enable GS‐AuNPs to detect tumors with a higher signal‐to‐noise ratio than IRDye800CW. GS‐AuNPs exhibit no severe accumulation in the organs of the RES and are desirable for cancer diagnosis and therapy.67 Additionally, NIR‐emitting radioactive GS‐[198Au]AuNPs can be synthesized by incorporating a gold radioisotope, 198Au. These GS‐[198Au]AuNPs retain the feature of renal clearance and display rapid in vivo kinetics that are comparable to those of small‐molecule contrast agents used in the clinic. These GS‐[198Au]AuNPs are NIR‐light emitters and are radioactive, and thus, they have potential applications in dual‐modality imaging.68
On the other hand, noninvasive imaging of kidney‐clearance kinetics and staging of kidney dysfunction have been validated by using GS‐AuNPs. Although the endogenous GFR marker creatinine is routinely used to assess overall kidney function and even to stage kidney dysfunction, it is considered as a late indicator of kidney impairment, because it is often insensitive to early‐stage kidney dysfunction and can vary with anthropometric factors.70 Furthermore, it is measurably abnormal only after significant GFR has been lost and cannot detect the region‐specific injury. In consequence, kidney impairment is usually detected at a late stage and a therapeutic opportunity is generally lost. Therefore, more sensitive agents to detect kidney dysfunction at an earlier stage are needed.
As mentioned above, conventional fluorophores are generally rapidly and persistently accumulated in skin tissues after intravenous injection because of their high lipophilicity and accumulation in the lipid membranes of the skin. What is more, amphiphilic fluorescent NPs including QDs,71 dye‐coated SiNPs,72 and nonluminescent plasmonic AuNPs71 also exhibit high accumulation in the skin. Such a high accumulation of agents in the skin is a major roadblock for the noninvasive imaging of kidney‐clearance kinetics. Yu et al. have found that conventional organic fluorophores such as Cy3, Cy7, and IRDye800CW fail to enhance the noninvasive kidney contrast and fluorescence imaging of kidney‐clearance kinetics.69 Luminescent inorganic NPs can exhibit NIR emissions because of quantum‐size effects. Unlike organic dyes, NIR‐emitting GS‐AuNPs can basically enhance kidney contrast and extend the noninvasive detection time period. The percentage of kidney‐contrast enhancement for GS‐AuNPs can reach 90–150 % at 12 min postinjection, and the value continuously increases to a maximum value of (240±55) % at 60 min postinjection, which is roughly 50 times higher than that obtained for IRDye800CW at 60 min postinjection [(4.7±0.8) %]. A contrast enhancement of 68 % is observed even at 10 h postinjection of GS‐AuNPs, and thus, the kidneys are still detectable after 10 h of intravenous injection. However, a similar 68 % contrast enhancement is also the maximum value that IRDye800CW can reach at 0.6 min postinjection, which indicates that the detection time of GS‐AuNPs is 1000 times longer than that of IRDye800CW. The remarkable improvement in kidney contrast and detection time is attributed to low accumulation of the hydrophilic GS‐AuNPs in the skin and rapid clearance from the skin through the kidneys to the urine. The time–fluorescence intensity curves (TFICs) of the kidneys obtained from noninvasive and invasive detection in the same mouse after GS‐AuNPs injection demonstrate that no significant differences in decay half‐life and percentage of relative renal function are observed between the two curves, and the noninvasive kidney TFICs reflect the kidney clearance of the GS‐AuNPs. These studies suggest that renal‐clearable NIR‐emitting GS‐AuNPs allow for the fluorescence imaging of kidney‐clearance kinetics and have a high potential for the noninvasive staging of kidney dysfunction.
To validate NIR‐emitting GS‐AuNPs for staging kidney dysfunction, the fundamental question of whether such fluorescence imaging techniques based on the use of GS‐AuNPs are sensitive enough for the noninvasive differentiation of the various kidney dysfunction stages should be answered. To do this, Yu et al. have used a UUO mouse model.69, 73 The UUO mouse model is a well‐established preclinical model for ureteropelvic junction obstruction and is asymptomatic at an early stage but can cause kidney impairment if not treated promptly.74, 75 In the control (sham‐operated) group, both the left and right ureters are not ligated. At 7–9 days postoperation, no significant differences in blood urea nitrogen and serum creatinine are observed between the UUO mice and the control group. However, altered kidney structures caused by the obstruction are identified by ex vivo pathological analysis. These result suggest that both blood urea nitrogen and serum creatinine are not good indicators of kidney function in a UUO model, which is consistent with previous studies.76 With the assistance of GS‐AuNPs by in vivo NIR fluorescence imaging, the UUO left kidney can be easily differentiated from the unobstructed kidneys by noninvasive imaging and analysis of the TFICs (Figure 9). The fluorescence signals of the obstructed left kidney are dramatically reduced relative to those of the right kidney in UUO mice and those of both kidneys in the control group at 1 min intravenous postinjection of GS‐AuNPs (Figure 9).73 Such a diminished accumulation of GS‐AuNPs in the UUO kidney is attributed to dramatically reduced blood perfusion after obstruction.77 However, IRDye800CW fails to distinguish so, because of its severe accumulation in the skin tissues. Aside from the detection of kidney impairment, the stages of kidney dysfunction (mild kidney damage and severe kidney damage) can also be differentiated by noninvasive imaging of the kidney‐clearance kinetics of GS‐AuNPs. For kidneys with mild damage, the imaging peak value of the UUO left kidney is slightly reduced relative to that of the left kidney in control group, and the excretion of GS‐AuNPs through the kidneys in UUO mice is slowed down. For kidneys with severe damage, the imaging peak value dramatically decreases. These observations are in agreement with the data determined by SPECT imaging of UUO and pathological analysis of kidney tissue;73 for example, the renal tubules have mild to moderate atrophy and dilatation is observed in kidneys with mild damage, whereas renal tubular damage and cortical atrophy are much more pronounced in kidneys with severe damage. These results clearly indicate that fluorescence imaging of the kidney‐clearance kinetics of GS‐AuNPs can serve as an inexpensive and highly sensitive method for the noninvasive staging of kidney dysfunction in preclinical animal models.
Figure 9.
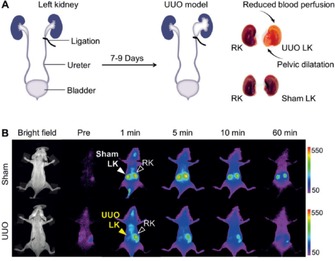
a) In the UUO mouse model, the left ureter of the mouse is completely ligated, whereas the right ureter is kept intact. In the control (sham‐operated) group, the left ureters are exposed but not ligated. b) Representative whole‐body noninvasive real‐time fluorescence images (λ ex/λ em filters: 710/830 nm) of mice before and after intravenous injection of GS‐AuNPs at preinjection: 1, 5, 10, and 60 min. Reprinted with permission from Ref. 69. Copyright (2015) Wiley‐VCH.
5. Conclusions and Perspectives
Measurement of the glomerular filtration rate (GFR) on the basis of urinary or plasma clearance of either exogenous or endogenous filtration agents is accepted as the gold‐standard approach to assess kidney function. However, it is not routinely available, because the existing protocols are cumbersome, time consuming, and/or invasive. Significant development in the area of diagnosing kidney function and disease is evident from the literature. We developed a transcutaneous detection technique that allows the rapid and convenient determination of kidney function without the need for time‐consuming blood/urine sample preparation. Impressively, a recent study revealed that this noninvasive procedure for the measurement of kidney function in unanesthetized animals did not negatively impact arterial pressure, heart rate, or locomotor activity.78 Thus, it is crucial to avoid an anesthesia‐related decrease in the GFR to acquire accurate results. Table 1 provides a collection of representative fluorescent GFR agents that have been used to determine kidney function in preclinical studies. Especially, these zwitterionic near‐infrared (NIR) agents we recently developed have a positive outlook by offering a deeper penetration depth, as the strong intrinsic background autofluorescence of living tissue is still one of the largest obstacles during transcutaneous measurements. By taking advantages of the above NIR agents and the transcutaneous detection technique, a much more rapid, robust, and convenient approach for the noninvasive real‐time assessment of kidney function is validated if compared to traditional GFR agents and determination methods. Nevertheless, further studies on the clearance and toxicity of these GFR agents in larger animals such as dogs or monkeys are needed before they can be really used in clinical practice.
Interestingly, some design strategies for fluorescent GFR agents are similar to those for inorganic renal‐clearable nanoparticles (NPs); for example, the use of either zwitterionic or neutral ligands was shown for the development of both organic GFR agents and inorganic renal‐clearable NPs. Therefore, we believe that using zwitterionic or neutral‐charge characteristics is a critical strategy for the development of renal‐clearable agents. Although a number of renally clearable NPs have been developed to date (Table 2), there are still many challenges and fundamental questions that need to be addressed. For example, glutathione‐coated gold nanoparticles (GS‐AuNPs) have been validated in differentiating the stages of kidney dysfunction; however, the exact excretion mechanisms of these GS‐AuNPs from the kidney are not clear, and the question of whether secretion or reabsorption is involved in the process of excretion needs further investigation. The low tissue penetration depth of light will remain a roadblock for further application of these renal‐clearable luminescent AuNPs in kidney functional imaging. One potential solution is to combine it with other imaging modalities, such as positron emission tomography and single‐photon emission computed tomography imaging. Of note, carbon dots are the only renal‐clearable inorganic NPs that have gained investigational new‐drug approval by the Food and Drug Administration for first‐in‐human clinical trials.79, 80 Therefore, whether other renal‐clearable NPs are biocompatible enough for future applications in humans should be addressed.
Kidney disease has numerous causes, including hypotension, trauma, acute tubular necrosis, urinary obstruction, and drug‐induced nephrotoxicity.6 Although the GFR is considered the best indicator for overall kidney function, further efforts should be made towards the development of novel light‐emitting agents for the detection of region‐specific injury in kidneys (e.g. tubular necrosis and function) so that kidney diseases can be differentiated and that kidney injury can be diagnosed at an early stage.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Jiaguo Huang was born in 1987 in China. He obtained a B.Sc. degree in Pharmacy (2009) from Nanchang University (Nanchang, China) and a Master's degree in Medicinal Chemistry (2012) from Central South University (Changsha, China) working in the field of molecular imaging. In October 2012, he joined the Medical Faculty Mannheim, University of Heidelberg (Germany), and obtained his Ph.D. degree in May of 2016 under the supervision of Prof. Norbert Gretz, focusing on the development of novel fluorescent agents and transcutaneous methods for the real‐time point‐of‐care assessment of kidney function. Currently, he is a Postdoctoral Fellow in the research group of Prof. Norbert Gretz and is focusing on developing near‐infrared fluorescent agents and tissue‐clearing protocols to visualize and count glomeruli in preclinical rodent models.

Biographical Information
Professor Norbert Gretz is the head of the Medical Research Center of the Medical Faculty Mannheim, University of Heidelberg, and is the managing director of the Institute for Medical Technology, a combined institute of the Medical Faculty Mannheim, University of Heidelberg, and the University of Applied Sciences Mannheim. After his medical studies at the Ruprecht‐Karls University Heidelberg, Norbert Gretz worked as a physician at several hospitals in Germany and abroad until he became senior physician at the University Hospital Mannheim. He was then appointed professor for experimental medicine at the University of Heidelberg. He is project head of several national and international scientific research programs and is an expert evaluator and consultant for several journals and expert associations. His key research areas are photobiomodulation, chronic wounds, kidney diseases, stem‐cell therapy, optical‐tissue clearing, gene‐expression profiling, and big data handling.

Acknowledgements
This work was supported by FP7 Marie Curie ITN project: NephroTools.
J. Huang, N. Gretz, ChemistryOpen 2017, 6, 456.
Contributor Information
Dr. Jiaguo Huang, Email: Jiaguo.Huang@medma.uni-heidelberg.de
Prof. Norbert Gretz, Email: Norbert.Gretz@medma.uni-heidelberg.de.
References
- 1. Lote C. J., Harper L., Savage C. O., Br. J. Anaesth. 1996, 77, 82–89. [DOI] [PubMed] [Google Scholar]
- 2. Tolls R. E., Dille J. M., J. Urol. 1955, 74, 197–201. [DOI] [PubMed] [Google Scholar]
- 3. Schwartz G. J., Furth S. L., Pediatr. Nephrol. 2007, 22, 1839–1848. [DOI] [PubMed] [Google Scholar]
- 4. Huang J., Gretz N., Weinfurter S., Eur. J. Pharmacol. 2016, 790, 92–98. [DOI] [PubMed] [Google Scholar]
- 5. Stevens L. A., Levey A. S., J. Am. Soc. Nephrol. 2009, 20, 2305–2313. [DOI] [PubMed] [Google Scholar]
- 6. Brede C., Labhasetwar V., Adv. Chronic Kidney Dis. 2013, 20, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brater D. C., Br. J. Clin. Pharmacol. 2002, 54, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang J., Weinfurter S., Pinto P. C., Pretze M., Kranzlin B., Pill J., Federica R., Perciaccante R., Ciana L. D., Masereeuw R., Gretz N., Bioconjugate Chem. 2016, 27, 2513–2526. [DOI] [PubMed] [Google Scholar]
- 9. Jha V., Garcia-Garcia G., Iseki K., Li Z., Naicker S., Plattner B., Saran R., Wang A. Y., Yang C. W., Lancet 2013, 382, 260–272. [DOI] [PubMed] [Google Scholar]
- 10. Chinen L. K., Galen K. P., Kuan K. T., Dyszlewski M. E., Ozaki H., Sawai H., Pandurangi R. S., Jacobs F. G., Dorshow R. B., Rajagopalan R., J. Med. Chem. 2008, 51, 957–962. [DOI] [PubMed] [Google Scholar]
- 11. Kwon S. H., Saad A., Herrmann S. M., Textor S. C., Lerman L. O., Radiology 2015, 276, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taylor A. T., J. Nucl. Med. 2014, 55, 608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krier J. D., Ritman E. L., Bajzer Z., Romero J. C., Lerman A., Lerman L. O., Am. J. Physiol. 2001, 281, F630–638. [DOI] [PubMed] [Google Scholar]
- 14. Rajagopalan R., Neumann W. L., Poreddy A. R., Fitch R. M., Freskos J. N., Asmelash B., Gaston K. R., Galen K. P., Shieh J. J., Dorshow R. B., J. Med. Chem. 2011, 54, 5048–5058. [DOI] [PubMed] [Google Scholar]
- 15. Poreddy A. R., Neumann W. L., Freskos J. N., Rajagopalan R., Asmelash B., Gaston K. R., Fitch R. M., Galen K. P., Shieh J. J., Dorshow R. B., Bioorg. Med. Chem. 2012, 20, 2490–2497. [DOI] [PubMed] [Google Scholar]
- 16. Wu H., Huang J., Curr. Protein Pept. Sci. 2016, 17, 582–595. [DOI] [PubMed] [Google Scholar]
- 17. Bugaj J. E., Dorshow R. B., Regul. Toxicol. Pharmacol. 2015, 72, 26–38. [DOI] [PubMed] [Google Scholar]
- 18.A. R. Poreddy, B. Asmelash, K. P. Galen, R. M. Fitch, J.-J. Shieh, J. M. Wilcox, T. M. Schoenstein, J. K. Wojdyla, K. R. Gaston, J. N. Freskos, W. L. Neumann, R. Rajagopalan, H.-Y. Ahn, J. G. Kostelc, M. P. Debreczeny, K. D. Belfield, R. B. Dorshow, Proc. SPIE 7190, 2009, 71900P,DOI: 10.1117/12.809287.
- 19. Lorenz J. N., Gruenstein E., Am. J. Physiol. 1999, 276, F172–177. [DOI] [PubMed] [Google Scholar]
- 20. Qi Z., Whitt I., Mehta A., Jin J., Zhao M., Harris R. C., Fogo A. B., Breyer M. D., Am. J. Physiol. 2004, 286, F590–596. [DOI] [PubMed] [Google Scholar]
- 21. Pill J., Issaeva O., Woderer S., Sadick M., Kranzlin B., Fiedler F., Klotzer H. M., Kramer U., Gretz N., Naunyn-Schmiedeberg′s Arch. Pharmacol. 2006, 373, 204–211. [DOI] [PubMed] [Google Scholar]
- 22. Chandra R., Barron J. L., Ann. Clin. Biochem. 2002, 39, 567–576. [DOI] [PubMed] [Google Scholar]
- 23. Schock-Kusch D., Xie Q., Shulhevich Y., Hesser J., Stsepankou D., Sadick M., Koenig S., Hoecklin F., Pill J., Gretz N., Kidney Int. 2011, 79, 1254–1258. [DOI] [PubMed] [Google Scholar]
- 24. Schreiber A., Shulhevich Y., Geraci S., Hesser J., Stsepankou D., Neudecker S., Koenig S., Heinrich R., Hoecklin F., Pill J., Friedemann J., Schweda F., Gretz N., Schock-Kusch D., Am. J. Physiol. 2012, 303, F783–788. [DOI] [PubMed] [Google Scholar]
- 25. Schock-Kusch D., Shulhevich Y., Xie Q., Hesser J., Stsepankou D., Neudecker S., Friedemann J., Koenig S., Heinrich R., Hoecklin F., Pill J., Gretz N., Kidney Int. 2012, 82, 314–320. [DOI] [PubMed] [Google Scholar]
- 26. Schock-Kusch D., Geraci S., Ermeling E., Shulhevich Y., Sticht C., Hesser J., Stsepankou D., Neudecker S., Pill J., Schmitt R., Melk A., PLoS One 2013, 8, e71519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. A. W. Cowley, Jr. , Ryan R. P., Kurth T., Skelton M. M., Schock-Kusch D., Gretz N., Hypertension 2013, 62, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinbach S., Krolop N., Strommer S., Herrera-Perez Z., Geraci S., Friedemann J., Gretz N., Neiger R., PLoS One 2014, 9, e111734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gleeson M. P., J. Med. Chem. 2007, 50, 101–112. [DOI] [PubMed] [Google Scholar]
- 30. Takeda M., Khamdang S., Narikawa S., Kimura H., Kobayashi Y., Yamamoto T., Cha S. H., Sekine T., Endou H., J. Pharmacol. Exp. Ther. 2002, 300, 918–924. [DOI] [PubMed] [Google Scholar]
- 31. Gould S., Scott R. C., Food Chem. Toxicol. 2005, 43, 1451–1459. [DOI] [PubMed] [Google Scholar]
- 32. Lumholdt L. R., Holm R., Jorgensen E. B., Larsen K. L., Carbohydr. Res. 2012, 362, 56–61. [DOI] [PubMed] [Google Scholar]
- 33. Sato S., Umeda Y., Fujii S., Takenaka S., Bioconjugate Chem. 2015, 26, 379–382. [DOI] [PubMed] [Google Scholar]
- 34. Takechi-Haraya Y., Tanaka K., Tsuji K., Asami Y., Izawa H., Shigenaga A., Otaka A., Saito H., Kawakami K., Bioconjugate Chem. 2015, 26, 572–581. [DOI] [PubMed] [Google Scholar]
- 35. Huang J., Weinfurter S., Daniele C., Perciaccante R., Federica R., Leopoldo D. C., Pill J., Gretz N., Chem. Sci. 2017, 8, 2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu M., Liu J., Ning X., Zheng J., Angew. Chem. Int. Ed. 2015, 54, 15434–15438; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15654–15658. [Google Scholar]
- 37. Hamann F. M., Brehm R., Pauli J., Grabolle M., Frank W., Kaiser W. A., Fischer D., Resch-Genger U., Hilger I., Mol. Imaging 2011, 10, 258–269. [DOI] [PubMed] [Google Scholar]
- 38. Scarfe L., Rak-Raszewska A., Geraci S., Darssan D., Sharkey J., Huang J., Burton N. C., Mason D., Ranjzad P., Kenny S., Gretz N., Levy R., Park B. K., Garcia-Finana M., Woolf A. S., Murray P., Wilm B., Sci. Rep. 2015, 5, 13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi H. S., Nasr K., Alyabyev S., Feith D., Lee J. H., Kim S. H., Ashitate Y., Hyun H., Patonay G., Strekowski L., Henary M., Frangioni J. V., Angew. Chem. Int. Ed. 2011, 50, 6258–6263; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6382–6387. [Google Scholar]
- 40. Njiojob C. N., Owens E. A., Narayana L., Hyun H., Choi H. S., Henary M., J. Med. Chem. 2015, 58, 2845–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cattell V., Kidney Int. 1994, 45, 945–952. [DOI] [PubMed] [Google Scholar]
- 42. Grau V., Herbst B., Steiniger B., Cell Tissue Res. 1998, 291, 117–126. [DOI] [PubMed] [Google Scholar]
- 43. Schreiner G. F., Harris K. P., Purkerson M. L., Klahr S., Kidney Int. 1988, 34, 487–493. [DOI] [PubMed] [Google Scholar]
- 44. Hauger O., Delalande C., Deminiere C., Fouqueray B., Ohayon C., Garcia S., Trillaud H., Combe C., Grenier N., Radiology 2000, 217, 819–826. [DOI] [PubMed] [Google Scholar]
- 45. Jo S. K., Hu X., Kobayashi H., Lizak M., Miyaji T., Koretsky A., Star R. A., Kidney Int. 2003, 64, 43–51. [DOI] [PubMed] [Google Scholar]
- 46. Basile D. P., Anderson M. D., Sutton T. A., Compr. Physiol. 2012, 2, 1303–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shirai T., Kohara H., Tabata Y., J. Drug Targeting 2012, 20, 535–543. [DOI] [PubMed] [Google Scholar]
- 48. Choi H. S., Liu W., Misra P., Tanaka E., Zimmer J. P., Itty Ipe B., Bawendi M. G., Frangioni J. V., Nat. Biotechnol. 2007, 25, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Longmire M., Choyke P. L., Kobayashi H., Nanomedicine 2008, 3, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh R., Pantarotto D., Lacerda L., Pastorin G., Klumpp C., Prato M., Bianco A., Kostarelos K., Proc. Natl. Acad. Sci. USA 2006, 103, 3357–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burns A. A., Vider J., Ow H., Herz E., Penate-Medina O., Baumgart M., Larson S. M., Wiesner U., Bradbury M., Nano Lett. 2009, 9, 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang X., Zhang F., Zhu L., Choi K. Y., Guo N., Guo J., Tackett K., Anilkumar P., Liu G., Quan Q., Choi H. S., Niu G., Sun Y. P., Lee S., Chen X., ACS Nano 2013, 7, 5684–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou Z., Wang L., Chi X., Bao J., Yang L., Zhao W., Chen Z., Wang X., Chen X., Gao J., ACS Nano 2013, 7, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang S., Chen M., Zheng N., Small 2014, 10, 3139–3144. [DOI] [PubMed] [Google Scholar]
- 55. Yang S., Sun S., Zhou C., Hao G., Liu J., Ramezani S., Yu M., Sun X., Zheng J., Bioconjugate Chem. 2015, 26, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou C., Long M., Qin Y., Sun X., Zheng J., Angew. Chem. Int. Ed. 2011, 50, 3168–3172; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3226–3230. [Google Scholar]
- 57. Yu M., Zheng J., ACS Nano 2015, 9, 6655–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ruggiero A., Villa C. H., Bander E., Rey D. A., Bergkvist M., Batt C. A., Manova-Todorova K., Deen W. M., Scheinberg D. A., McDevitt M. R., Proc. Natl. Acad. Sci. USA 2010, 107, 12369–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mahmoudi M., Lynch I., Ejtehadi M. R., Monopoli M. P., Bombelli F. B., Laurent S., Chem. Rev. 2011, 111, 5610–5637. [DOI] [PubMed] [Google Scholar]
- 60. Lynch I., Salvati A., Dawson K. A., Nat. Nanotechnol. 2009, 4, 546–547. [DOI] [PubMed] [Google Scholar]
- 61. Walkey C. D., Olsen J. B., Guo H., Emili A., Chan W. C., J. Am. Chem. Soc. 2012, 134, 2139–2147. [DOI] [PubMed] [Google Scholar]
- 62. Choi H. S., Ipe B. I., Misra P., Lee J. H., Bawendi M. G., Frangioni J. V., Nano Lett. 2009, 9, 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu J., Yu M., Ning X., Zhou C., Yang S., Zheng J., Angew. Chem. Int. Ed. 2013, 52, 12572–12576; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12804–12808. [Google Scholar]
- 64. Alric C., Miladi I., Kryza D., Taleb J., Lux F., Bazzi R., Billotey C., Janier M., Perriat P., Roux S., Tillement O., Nanoscale 2013, 5, 5930–5939. [DOI] [PubMed] [Google Scholar]
- 65. Zhao Y., Sultan D., Detering L., Luehmann H., Liu Y., Nanoscale 2014, 6, 13501–13509. [DOI] [PubMed] [Google Scholar]
- 66. Rosi N. L., Giljohann D. A., Thaxton C. S., Lytton-Jean A. K., Han M. S., Mirkin C. A., Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- 67. Liu J., Yu M., Zhou C., Yang S., Ning X., Zheng J., J. Am. Chem. Soc. 2013, 135, 4978–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou C., Hao G., Thomas P., Liu J., Yu M., Sun S., Oz O. K., Sun X., Zheng J., Angew. Chem. Int. Ed. 2012, 51, 10118–10122; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10265–10269. [Google Scholar]
- 69. Yu M., Zhou J., Du B., Ning X., Authement C., Gandee L., Kapur P., Hsieh J. T., Zheng J., Angew. Chem. Int. Ed. 2016, 55, 2787–2791; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2837–2841. [Google Scholar]
- 70. Lopez-Giacoman S., Madero M., World J. Nephrol. 2015, 4, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sykes E. A., Dai Q., Tsoi K. M., Hwang D. M., Chan W. C., Nat. Commun. 2014, 5, 3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kumar R., Roy I., Ohulchanskky T. Y., Vathy L. A., Bergey E. J., Sajjad M., Prasad P. N., ACS Nano 2010, 4, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Penna F. J., Chow J. S., Minnillo B. J., Passerotti C. C., Barnewolt C. E., Treves S. T., Fahey F. H., Dunning P. S., Freilich D. A., Retik A. B., Nguyen H. T., J. Urol. 2011, 185, 2405–2413. [DOI] [PubMed] [Google Scholar]
- 74. Decramer S., Wittke S., Mischak H., Zurbig P., Walden M., Bouissou F., Bascands J. L., Schanstra J. P., Nat. Med. 2006, 12, 398–400. [DOI] [PubMed] [Google Scholar]
- 75. Ulman I., Jayanthi V. R., Koff S. A., J. Urol. 2000, 164, 1101–1105. [DOI] [PubMed] [Google Scholar]
- 76. Chung S., Yoon H. E., Kim S. J., Kim S. J., Koh E. S., Hong Y. A., Park C. W., Chang Y. S., Shin S. J., Nutr. Metab. 2014, 11, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tantawy M. N., Jiang R., Wang F., Takahashi K., Peterson T. E., Zemel D., Hao C. M., Fujita H., Harris R. C., Quarles C. C., Takahashi T., BMC Neurosci. 2012, 13, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hilliard L. M., Denton K. M., Physiol. Res. 2016, 4, e12723. [Google Scholar]
- 79. Benezra M., Penate-Medina O., Zanzonico P. B., Schaer D., Ow H., Burns A., DeStanchina E., Longo V., Herz E., Iyer S., Wolchok J., Larson S. M., Wiesner U., Bradbury M. S., J. Clin. Invest. 2011, 121, 2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Phillips E., Penate-Medina O., Zanzonico P. B., Carvajal R. D., Mohan P., Ye Y., Humm J., Gönen M., Kalaigian H., Schöder H., Strauss H. W., Larson S. M., Wiesner U., Bradbury M. S., Sci. Transl. Med. 2014, 6, 260ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]