

Abstract
Rationale
Kappa-opioid receptor (KOPr) agonists have pre-clinical anti-cocaine and analgesic effects. However, side-effects including sedation, dysphoria, aversion, anxiety and depression limit their therapeutic development. The unique structure of Salvinorin A has been used to develop longer-acting KOPr agonists.
Objectives
We evaluate two novel C-2 analogues of Salvinorin A, ethoxymethyl ether Sal B (EOM Sal B) and β-tetrahydropyran Sal B (β-THP Sal B) alongside U50,488 for their ability to modulate cocaine-induced behaviours and side-effects, pre-clinically.
Methods
Anti-cocaine properties of EOM Sal B were evaluated using the reinstatement model of drug-seeking in self-administering rats. EOM Sal B and β-THP Sal B were evaluated for effects on cocaine-induced hyperactivity, spontaneous locomotor activity and sucrose self-administration. EOM Sal B and β-THP Sal B were evaluated for aversive, anxiogenic and depressive-like effects using conditioned place aversion (CPA), elevated plus maze (EPM) and forced swim tests (FST) respectively.
Results
EOM Sal B (0.1, 0.3 mg/kg, i.p.) dose-dependently attenuated drug-seeking and EOM Sal B (0.1 mg/kg, i.p.) and β-THP Sal B (1 mg/kg, i.p.) attenuated cocaine-induced hyperactivity. No effects on locomotor activity, open arm times (EPM) or swimming behaviours (FST), were seen with EOM (0.1 or 0.3 mg/kg, i.p.) or β-THP Sal B (1 or 2 mg/kg, i.p.). However, β-THP Sal B decreased time spent in the drug-paired chamber.
Conclusion
EOM Sal B is more potent than Sal A and β-THP Sal B in reducing drug-seeking behaviour with fewer side-effects. EOM Sal B showed no effects on sucrose self-administration (0.1 mg/kg), locomotor, depressive-like, aversive-like or anxiolytic effects.
Keywords: Salvinorin A, Behavioural Pharmacology, Self-Administration, Cocaine, Drug-Seeking, Anxiolytic, Conditioned Place Aversion, Rat, Elevated Plus Maze, Forced Swim Test, Sucrose Self-Administration
Introduction
Kappa opioid receptor (KOPr) agonists have well-established anti-reward and cocaine antagonistic actions in pre-clinical models of drug use (Glick et al. 1998; Morani et al. 2009; Morani et al. 2012; Schenk et al. 1999). These effects have been attributed to their ability to decrease dopamine (DA) release (Ebner et al. 2010) and increase DA reuptake via regulation of the dopamine transporter (DAT) (Kivell et al. 2014). Unfortunately, activation of KOPr's by traditional arylacetamide agonists such as U50,488 and U69,593 also cause side effects that include sedation (Gallantine and Meert 2008), aversion (Land et al. 2009), anxiety (Gillett et al. 2013), and depression (Mague et al. 2003), effects that have limited their use clinically (Walsh et al. 2001a; Walsh et al. 2001b).
Salvinorin A (Sal A), is a structurally unique KOPr agonist with a non-nitrogenous structure (Prisinzano 2005; Roth et al. 2002), isolated from the plant Salvia divinorum and is known to produce hallucinogenic effects in humans (MacLean et al. 2013). Sal A, like traditional KOPr agonists, has been shown to have anti-cocaine actions in preclinical models of drug abuse (Kivell et al. 2013; Morani et al. 2009). Sal A is a full agonist at the KOPr, with similar binding affinity and efficacy as U69,593 and U50,488 (Chavkin et al. 2004). Sal A does not display bias (White et al. 2015) and acts via both G-protein and β-arrestin pathways, similar to U69,593 and U50,488. Interestingly, Sal A has been found to produce fewer depressive-like and sedative side effects compared to classic agonists (Braida et al. 2009; Braida et al. 2008; Harden et al. 2012; Morani et al. 2009; Morani et al. 2012). However, the short half-life of Sal A in vivo (approximately 50 min) (Butelman et al. 2009; Hooker et al. 2008; Schmidt et al. 2005; Teksin et al. 2009), likely due to clearance from the CNS by P-glycoprotein (Butelman et al. 2012) and rapid metabolism at the C-2 position to an inactive metabolite, Salvinorin B (Beguin et al. 2005; Chavkin et al. 2004), has limited its clinical development. This has led to the development of Sal A derivatives with increased metabolic stability and the hypothesis that analogues of Sal A with improved pharmacokinetic properties and fewer side effects will lead to the development of KOPr therapeutics for drug-abuse, pain, pruritus and other neurological disorders.
Numerous analogues of Sal A have been synthesised in recent years. Derivatives synthesised by modifying the C-2 group include β-tetrahydropyran Sal B (β-THP Sal B) (Prevatt-Smith et al. 2011), Mesyl Sal B (Harding et al. 2005b), 2-methoxymethyl ether Salvinorin B (MOM Sal B) (Baker et al. 2009; Morani et al. 2013; Wang et al. 2008) and 2-ethoxymethyl ether Sal B (EOM Sal B) (Peet and Baker 2011; Prevatt-Smith et al. 2011) (See (Cunningham et al. 2011) for a review of structural modifications). The improved in vivo stability of these analogues, in comparison to Sal A has been attributed to the lack of a hydrolysable ester at the C-2 position (Wang et al. 2008). MOM Sal B was found to be seven-fold more potent than Sal A (Lee et al. 2005) and had a longer half-life in vivo (Baker et al. 2009; Wang et al. 2008). Studies have shown that MOM Sal B (0.3 mg/kg) was effective in reducing cocaine-seeking behaviours in rats However, MOM Sal B, at the same doses that were effective in attenuation of cocaine-seeking behaviours, exhibited side effects that included attenuation of natural reward in sucrose intake tests and pro-depressive effects in the forced swim test (FST) (Morani et al. 2013). These KOPr mediated side effects currently limit therapeutic development.
In the present study we chose to compare the C-2 analogues, EOM Sal B and β-THP Sal B. EOM Sal B was selected as it has been found to be 60 times more potent at KOPr than Sal A (ED50 in GTP-γS assays EOM Sal B =0.65 nM; Sal A =40 nM) (Munro et al. 2008) and displays improved metabolic stability in vivo, likely due to a combination of improved metabolic stability and decreased plasma protein affinity, although pharmacokinetics in the brain was found to be similar to Sal A (Hooker et al. 2009). β-THP Sal B (Prevatt-Smith et al. 2011) is a full agonist at KOPr with similar KOPr binding affinity and efficacy compared to Sal A. β-THP Sal B (1 mg/kg) has also been shown to attenuate cocaine- and cue- induced reinstatement of drug-seeking in a similar manner to Sal A (0.3 mg/kg) in rats, although at a higher dose (Prevatt-Smith et al. 2011). The side effects of β-THP Sal B and EOM Sal B compounds have not been fully evaluated.
Therefore, the purpose of the present study was to investigate the anti-cocaine effects and side effect profile of both EOM Sal B and β-THP Sal B to fully evaluate their therapeutic utility. We utilised the analgesic properties of KOPr agonists to evaluate the potency and efficacy dose-response in vivo. We chose to evaluate the anti-cocaine effects of EOM Sal B using the cocaine and cue-prime-induced reinstatement test in rats to model human drug relapse (Bossert et al. 2013) and compare the effects of EOM Sal B and β-THP Sal B in cocaine hyperactivity tests. Because KOPr agonists are known to have sedative, aversive, anxiogenic and pro-depressive side-effects we also examined the effects of EOM Sal B and β-THP Sal B on sucrose responding, aversion, anxiety and depression. Pre-clinical evaluation of both the desirable anti-cocaine effects and side effects is a crucial step in the development of clinically effective anti-addiction therapeutics in addition to the development of non-addictive analgesics and effective treatments for pruritus.
Materials and Methods
Subjects
Male Sprague-Dawley rats (Rattus norvegicus) weighing 250-400 g were housed individually (self-administration studies), two per cage (sucrose reinforcement), or three-four per cage (depending on size for all other behavioural experiments) (N=382). Adult male C57bl/6J mice (N=24) weighing 25-35 g were used to evaluate analgesic dose-response effects in the warm-water tail-withdrawal assay. Animals were housed within the animal facility of the School of Biological Sciences, Victoria University of Wellington, within temperature (19–21°C) and humidity (55%) controlled rooms with lights set to a 12 h light: 12 h dark cycle with lights on at 07:00 h. Rats tested for sucrose reinforcement were maintained at 85% of their initial feeding weight by food restriction. Food and water was available ad libitum to all other animals except during testing. Motor activity experiments were carried out in the dark between 09:00 and 17:00 h in the presence of white noise. Separate groups of rats were used for each experimental test and drug dose, except in cocaine and sucrose self-administration tests where within-subject Latin square design was utilised, as indicated in relevant methods section. All experimental procedures were reviewed and approved by the Animal Ethics Committee of Victoria University of Wellington, New Zealand.
Analgesic dose-response effects
Analgesic effects in the 50°C warm-water tail-withdrawal assay were evaluated in male C57bl/6J mice using a 10 sec cutoff time to prevent tissue injury. Mice were habituated to a plexiglass restrainer in 4 daily 5 min trials. A within animal design was used to reduce animal numbers, as previously described (Bohn et al. 2000). Briefly, following baseline latency calculations, mice were given s.c. injections every 30 min at increasing concentrations to create the following cumulative doses: 0.1, 0.3, 0.6, 1.0, 2.5, 5.0, 7.5, 10 and 12.5 mg/kg for EOM Sal B and Sal A and 0.5, 1.0, 2.5, 5, 7.5, 10, 20, 30, 40 mg/kg for U50,488 and tail-withdrawal latencies recorded 30 min following each dose (N=6-9 per treatment group). The following formula was used to calculate the Maximum Possible Effect (MPE): MPE (%) × 100 × (test latency – control latency / (10 – mean control latency). U50,488 is a selective, non-peptide, full agonist at KOPr, with similar binding affinity and potency to Sal A and used as a control, alongside Sal A to evaluate typical KOPr mediated responses.
Metabolic stability in liver microsomes
Incubations were carried out in 96-well plates with a total volume of 250 μL per well. The reaction mixtures were in a 50 mM potassium phosphate buffer solution with 2 mM MgCl2 and buffered to pH 7.4. Rat liver microsomes solution, 220 μL, (protein suspended in buffered system for final concentration of 0.25 mg/mL) was added to all wells (N=4 per group, performed in duplicate). The assay plate was pre-warmed to 37°C in an incubator (no CO2 injection or humidity) for 20 min, except for the 0 time points which was performed on ice. To begin the assay, 20 μL of test compound (5 mM stock solution in MeCN (HPLC grade) diluted in assay buffer to 125 μM, for final compound concentration of 10 μM and less than 1% MeCN in well) and 10 μL of NADPH in buffer (final concentration of 1 mM) were added to the wells. At the end of each time point (0, 10, 30, 60, 90, 120, and 150 min), 150 μL of reaction mix was removed and mixed in a separate 96-well plate containing 150 μL of quench solution per well (1 μM tolbutamide in MeCN with 1% formic acid). The quench plate was stored on ice until the end of the assay. For the test samples without NADPH, the same procedure as above was conducted with the addition of 10 μL buffer instead of the NADPH solution, and time points were collected at 0, 30, 90, and 150 minutes. At the end of all time points, the plate was centrifuged at 1500 rpm for 10 min. to precipitate any protein remaining in the solution. The amount of compound remaining was quantified using LCMS. The plate wells were analysed using liquid chromatography-high resolution mass spectrometry on a Waters Acquity UPLC coupled to a Waters LCT Premier TOF mass spectrometer. The chromatography utilized a Waters Acquity HSS T3 C18 column (2.1 × 50 mm, 1.8 μm) with a guard column of the same stationary phase (2.1 × 5 mm, 1.8 μm) and was run with gradient elution, using water and acetonitrile with 0.1% formic acid, from 40% to 80% acetonitrile over 3.4 minutes and held at 80% acetonitrile for 0.4 min. The flow rate was 0.25 ml/min and the injection volume was 15 μL. Each well was sampled twice.
Verapamil and dextromethorphan were included as controls in the stability assays. Both compounds are known substrates for CYP-450s, and both were significantly metabolised in the presence of NADPH (8.5 ± 0.9 % and 26 ± 5 % remaining at 150 minutes, respectively) compared to in the absence of NADPH (101 ± 7 % and 110 ± 5 % remaining at 150 minutes, respectively), p <0.0001 for both sets of data.
Surgery and self-administration training
Rats that underwent cocaine self-administration were anaesthetised via intraperitoneal (i.p.) injection with ketamine/xylazine (90/9 mg/kg), and a silastic catheter inserted into the right jugular vein. The distal end of the catheter was passed subcutaneously (s.c.) to an exposed portion of the skull, attached to a 22-gauge stainless steel tubing, and fastened to embedded jeweler's screws using dental acrylic. Rats received carprofen (5 mg/kg, s.c.) and 6 ml of warm sodium lactate on each flank (s.c.) post-surgery. Carprofen was also given at 24 and 48 h post-surgery for analgesia. All catheters were infused daily with 0.2 ml of sterile saline containing heparin (30 U/ml) and penicillin G potassium (250,000 U/ml) to prevent clot formation and infection. Self-administration training commenced five days post-surgery.
Rats were trained to self-administer cocaine (0.1 ml of 0.5 mg/kg cocaine, per infusion, with a 12 s infusion duration) in standard operant chambers equipped with two levers (Med Associates ENV-001, VT, USA) during daily 2 h sessions, 6 days per week as previously described (N=5) (Prevatt-Smith et al. 2011; Simonson et al. 2015). Briefly, following acquisition and training to Fixed Ratio-5 (FR5), rats were tested for cocaine- and cue-produced reinstatement. Criterion for stable responding on FR5 was set at a minimum of 20 infusions in a 2 hr self-administration session over 3 consecutive days with less the 20% variation. Animals that did not have a ratio of active:inactive lever presses of at least 2:1 were excluded. Phase 1 consisted of cocaine self-administration on FR5. Rats progressed to phase 2 when responses were within 20% of baseline responses for 3 consecutive days. In phase 2 (extinction), the light cue was removed and the cocaine replaced with heparinised saline (3 U/ml). Rats remained on this schedule until lever responses dropped below 20 active lever responses in a single session (3–4 days). Upon reaching extinction criteria, rats were subjected to reinstatement (phase 3), where they were injected with either vehicle or EOM Sal B (0.1, 0.3 mg/kg, i.p.) prior to a priming injection of cocaine (20 mg/kg, i.p.). Rats were returned to the operant chamber and the light cue restored. Infusions were delivered using mechanical pumps (Razel Scientific, Model A with 1.0 rpm motor equipped with 20 ml syringe, VT, USA; 0.1 ml infusion delivered over 12 s) and active lever responses throughout the session were recorded. Some rats were pre-treated with the KOPr antagonist, nor-BNI (10 mg/kg, s.c.) 24 h prior to reinstatement testing described above with EOM Sal B (0.3 mg/kg, i.p.) followed by cocaine prime to confirm that effects were KOPr mediated (N=3). All treatments were administered using a within subject, Latin square design except for nor-BNI, which was administered last due to its long-lasting effects (Endoh et al. 1992; Horan et al. 1992). Some animals did not receive nor-BNI due to loss of catheter patency.
Locomotor activity
For all motor activity tests, rats were habituated in activity chambers for 30 min prior to testing. For spontaneous locomotor activity tests, rats were then injected with either EOM Sal B (0.1 or 0.3 mg/kg i.p.), β-THP Sal B (1.0 or 2.0 mg/kg, i.p.) or vehicle and returned to the activity chambers for 60 min (N=6-9). Motor activity was assessed by measuring horizontal locomotion for 60 min using open field chambers equipped with 16 infrared beams and sensors placed evenly on each wall (Med Associates ENV-520). Breaks in infrared beams were recorded by the Med Associates Activity Monitor software (Med Associates, SOF-811) as ambulatory counts.
In the cocaine-induced hyperactivity test, groups of rats were habituated for 30 min, injected with either EOM Sal B (0.1 mg/kg, i.p.), β-THP Sal B (1.0 mg/kg, i.p.) or vehicle prior to an injection of cocaine (20 mg/kg, i.p.) (N=5-9). They were returned immediately into the activity chambers for 60 min.
Sucrose self-administration
Rats were trained to self-administer 45 mg sucrose pellets (Dustless Precision Pellet, Able Scientific, Perth, Australia) in daily 45 min sessions. Rats were initially trained on a FR1 schedule of reinforcement, where active lever presses produced a sucrose pellet and responses on the inactive lever produced no programmed consequences. Following acquisition of the self-administration behaviour (at least 20 pellets in a session with an active:inactive lever ratio of >2:1), rats progressed to FR5. After stable responding was achieved (<20% variation in responding for three consecutive days) rats were injected with either vehicle, or KOPr agonists, (U50,488, Sal A, EOM Sal B or β-THP Sal B (i.p.)) prior to self-administration and responses recorded for 45 min. All treatments were administered in a counterbalanced manner (N=7).
Conditioned place aversion
Conditioned place aversion (CPA) was evaluated using a 3-chamber apparatus (PanLab, Harvard Apparatus, USA). Two large chambers (30 × 30 × 34 cm) were connected by a small corridor (8 × 10 × 34 cm) with removable sliding doors. One of the large chambers had a textured black floor with black dotted patterns on its walls (black chamber) and the other had a smooth white floor with black striped patterns on its walls (white chamber). The corridor was a neutral zone with grey walls and floor and illuminated at an intensity of 70 lux. The average light intensity in both conditioning chambers was 20 lux. Experiments were conducted in the presence of white noise.
The CPA procedure took place over 9 days, following previously described methods (Tejeda et al. 2013). On day 0, rats were habituated to the CPA apparatus for 15 min. On day 1 (pre-conditioning day), rats were again allowed free access to all chambers for 15 min. All activity was tracked using SMART 3.0 software (PanLab). Animals that showed >80% preference for a particular chamber or >40% preference for the corridor were excluded from testing. Conditioning was conducted on days 2-7 using a biased procedure. Injection of EOM Sal B (0.1 mg/kg), β-THP Sal B (1.0 mg/kg) or vehicle was followed by confinement in the preferred chamber with vehicle injections in the less preferred chamber on alternate days in a counterbalanced order for 45 min (N=7-11). Post-conditioning tests were carried out on day 8. Rats were placed in the corridor and allowed free access to both chambers for 15 min. Time spent in each chamber on pre- and post-conditioning days were compared to determine changes in preference.
Elevated plus maze
The elevated plus maze (EPM) apparatus consisted of four arms (50 cm long × 10 cm wide) elevated 55 cm above the ground. Two of the arms included a small parapet measuring 2.5 cm in height surrounding them (open arms) and the other two arms were enclosed by high 40 cm black walls (closed arms). Rats were habituated to conditions in the testing room 60 min prior to the study, administered Sal A (0.3 mg/kg), EOM Sal B (0.1 or 0.3 mg/kg), β-THP Sal B (1.0 or 2.0 mg/kg) or vehicle 10 min prior to placement in the centre of the apparatus facing an open arm (N=12-29). Open arm time was calculated when rats had all four paws on the open arm (Walf and Frye 2007). Activity was recorded for 5 min using a Sony HDR-SR5E digital camera recorder. Time spent on each arm was calculated by an observer blinded to experimental treatment.
Forced swim tests
Forced swim tests (FST) were conducted according to methods described in Slattery and Cryan (2012), using a cylindrical swim chamber measuring 44 cm in height and 20 cm in diameter filled with water (maintained at a temperature of 25 ± 1°C) to a depth of 35 cm. Rats were habituated to conditions in the experimental room 60 min prior to testing (N=6-8). On day 1, drug-naïve rats were habituated to forced swimming conditions for 15 min. The following day, rats were injected with EOM Sal B (0.1 or 0.3 mg/kg) or β-THP Sal B (1.0 or 2.0 mg/kg) or vehicle before a 5 min testing session. All test sessions were recorded using a Sony HDR-SR5E digital camera recorder. Test videos were analysed in 5 sec intervals by an observer blinded to experimental treatments.
Drugs
Sal A was isolated and purified from dried Salvia divinorum leaves (>98% pure by HPLC) (Butelman et al. 2007) and EOM Sal B and β-THP Sal B were synthesised from Sal A as previously described (Prevatt-Smith et al. 2011). For reinstatement and FST tests, EOM Sal B and β-THP Sal B were dissolved in 75% dimethyl sulfoxide (DMSO). For all other behavioural tests, EOM Sal B and β-THP Sal B and the prototypical KOPr agonist U50,488 (Sigma Aldrich, Auckland, New Zealand) were dissolved in DMSO:Tween-80:water (MilliQ) in a ratio of 2:1:7. U50,488, Sal A, EOM Sal B and β-THP Sal B were administered via intraperitoneal (i.p.) injection 5-10 min prior to testing as indicated for each experiment. Cocaine HCl (BDH Ltd, Wellington, NZ) was dissolved in sterile heparinised saline (3.0 U/ml) for intravenous (i.v.) infusions and in physiological saline for i.p. injections. Nor-binaltorphimine (nor-BNI) was dissolved in physiological saline and injected s.c. at a volume of 1 ml/kg 24 h prior to testing. The prototypical KOPr agonist U50,488 (Sigma Aldrich, Auckland, New Zealand), is a full agonist at KOPr used as a comparison alongside Sal A to evaluate typical KOPr mediated responses. Micrososomal stability assays utilised pooled IGS Sprague-Dawley rat liver microsomes (male, 20 mg/mL) (Sekisui XenoTech, LLC, Kansas City, KS), nicotinamide adenine dinucleotide phosphate tetrasodium salt hydrate (reduced form, NADPH, Fisher Scientific), tolbutamide, dextromethorphan hydrobomide, and (±)-verapamil hydrochloride (Sigma Aldrich, St. Louis, MO), and MeCN, (HPLC grade, Fisher Scientific). Unless otherwise stated all drug weights refer to the salt. Table 1 shows the vehicle and injection administration route of drugs and time interval utilised for each behavioural experiment.
Table 1. Summary of drug administration routes used in behavioural tests.
Test interval is the time between injection and start of test, with 0 denoting immediate testing. Tests of short duration of action requires an interval prior to testing. DMSO, dimethyl sulfoxide; i.p., intraperitoneal, s.c., sub cutaneous.
| Test type | Drug | Vehicle | Injection route | Injection to test interval (min) | Test duration (min) |
|---|---|---|---|---|---|
|
| |||||
| Tail Withdrawal (mice) | EOM Sal B | DMSO:Tween-80:water 2:1:7 | s.c. | 30 | 0.167 (10 s) |
| Sal A | ″ | ″ | ″ | ″ | |
| U50,488 | ″ | ″ | ″ | ″ | |
|
| |||||
| Cocaine Prime Reinstatement | EOM Sal B | 75% DMSO | i.p. | 0 | 120 |
| Sal A | ″ | ″ | ″ | ″ | |
| nor-BNI | s.c. | s.c. | 24 | - | |
|
| |||||
| Spontaneous locomotor activity | EOM Sal B | 75% DMSO | i.p. | 0 | 60 |
| βTHP Sal B | ″ | i.p. | 0 | 60 | |
|
| |||||
| Cocaine-induced hyperactivity | EOM Sal B | 75% DMSO | i.p. | 0 | 60 |
| βTHP Sal B | ″ | i.p. | 0 | 60 | |
| nor-BNI | Saline (0.9%) | s.c. | 24 | - | |
|
| |||||
| Sucrose Self-Administration | EOM Sal B | DMSO:Tween-80:water 2:1:7 | i.p. | 0 | 45 |
| Sal A | ″ | ″ | ″ | ″ | |
| U50,488 | ″ | ″ | ″ | ″ | |
|
| |||||
| Conditioned Place Aversion | EOM Sal B | DMSO:Tween-80:water 2:1:7 | i.p. | 0 | 45 |
| βTHP Sal B | ″ | ″ | ″ | ″ | |
|
| |||||
| Elevated plus maze | EOM Sal B | DMSO:Tween- | i.p. | 5-10 | 5 |
| βTHP Sal B | 80:water 2:1:7 | ″ | ″ | ″ | |
| Sal A | ″ | ″ | ″ | ″ | |
|
| |||||
| Forced swim test | EOM Sal B | DMSO:Tween-80:water 2:1:7 | i.p. | 5-10 | 5 |
| βTHP Sal B | ″ | ″ | ″ | ″ | |
Statistical analyses
All statistical analysis was performed using GraphPad Prism software version 7 unless otherwise indicated (GraphPad, La Jolla, CA). For the dose-response experiment, non-linear regression analysis was performed on each individual dose-response curve to calculate the potency (ED50) and the efficacy (Emax), with data normalized to U50,488 (Emax =100%), a full KOR agonist. A p value of ≤0.05 was used to define statistical significance. For micrososomal stability assays, the ratio between the target area peak to internal standard peak was used throughout the data to normalize the amount of sample in each well. This ratio at time 0 was set to 100%, and the amount of sample present at the rest of the time points were analysed as percent remaining. The ln of the % remaining was taken, and data were analysed using linear regression analysis in GraphPad Prism 5.0 software (GraphPad, La Jolla, CA). The linear equation generated in GraphPad was solved for y = 150 and the inverse ln of this value was taken to give the % remaining at 150 minutes. Statistical analysis of the % remaining at 150 minutes was performed using a two-tailed, unpaired t-test. All compounds were run in parallel assays in duplicate in > 2 individual experiments. % remaining values are reported as the means ± S.E.M. and represent the average of each individual experiment.
Data for cocaine-primed reinstatement and sucrose reinforcement tests were analysed using one-way ANOVA with repeated measures. Paired comparisons using Student's t-tests with an applied Bonferroni correction was then used to compare the effects of the treatment to controls. For motor activity tests, repeated measures one-way ANOVA was used to evaluate time-course effects and one-way ANOVA was used to compare total ambulatory counts, with a post-hoc of planned comparisons with the Bonferroni correction used where applicable. EPM data was analysed using one-way ANOVA followed by Bonferroni, planned comparisons between treatment and vehicle controls. Student's t-test was performed on FST data, whereas a paired Student's t-test was used to analyse the pre- and post- conditioning time in CPA experiments. Unless otherwise stated, all values are presented as mean ± SEM. Data were considered statistically significant when p≤0.05.
Results
Pharmacology
EOM Sal B and β-THP Sal B are neoclerodane diterpenes derived from Sal A. EOM Sal B has an ethoxymethyl ether and β-THP Sal B a tetrahydropyran modification at the C-2 position (Fig. 1). Previous in vitro studies have shown both are full KOPr agonists Table 1, with EOM Sal B having increased binding at KOPr and increased efficacy in [35S]GTP-ϒ-S assays in CHO cells stably expressing KOPr (Ki =3.1 ± 0.4 nM; EC50 =0.65 ± 0.17) compared to β-THP Sal B (Ki =6.2 ± 0.4 nm; EC50 =60 ± 6) and Sal A (Ki =7.4 ± 0.7; EC50 =40 ± 10) (Prevatt-Smith et al. 2011). Preliminary experiments using the hot water tail-withdrawal assay were performed to determine optimal pretreatment times for each compound with an onset of action for both compounds of around 5 min (data not shown). To explore the potency of EOM Sal B in vivo, we assessed tail-withdrawal latencies following cumulative dosing and show that EOM Sal B has the highest potency (ED50=0.8336, 95% CI [0.6606 to 1.168] . Both EOM Sal B and Sal A (ED50=1.433, 95% CI [0.9581 to 3.055]) have increased potency compared to U50,488 (ED50=5.008 95% CI [4.103, 6.408]. EOM Sal B (Emax=87% ± 10%) and Sal A (Emax=87% ± 3 %) show similar efficacy to U50,488 (Emax=100% ± 12%) (Fig. 2). β-THP Sal B (EC50, 1.4 mg/kg) has been evaluated previously in this assay with increased potency compared to Sal A (EC50=2.1 mg/kg; Emax 107% ± 7%) in male B6-SJL mice (Paton et al. 2017).
Fig. 1. Chemical structures of Sal A, EOM Sal B and β-THP Sal B.
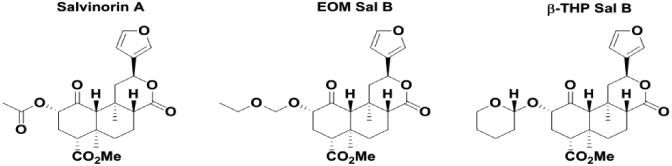
Fig. 2.
Dose response effects in the warm-water tail-withdrawal assay in mice. Nonlinear regression analysis of tail-withdrawal latencies following cumulative dosing of EOM Sal B, Sal A or U50,488 Values presented as mean ± SEM (N=6-9)
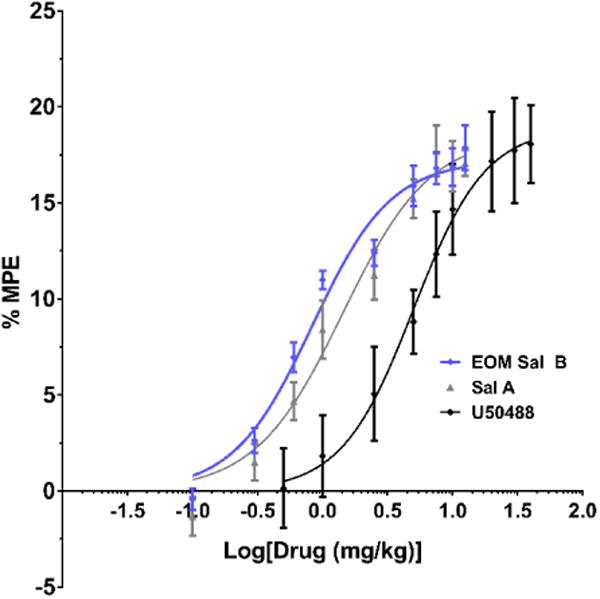
The metabolic stability of EOM Sal B and β-THP Sal B in rat liver microsome assays was compared to Sal A. EOM Sal B showed a significant increase in metabolism compared to salvinorin A in the presence of NADPH indicating that the CYP450-mediated oxidative potentials are increased (t(8)=3.365, p=0.0099). There was also a non-significant trend towards and increase with β-THP-Sal B (t(8)=2.116, p=0.067). However, other metabolic liabilities have been improved. Sal A is highly metabolised without the CYP450 cofactor NADPH (50 ± 10% remaining at 150 min), in contrast to both EOM Sal B and β-THP Sal B analogues which are not readily metabolised in the absence of NADPH (105 ± 6% and 104 ± 8% remaining at 150 min respectively) (Fig. 3).
Fig. 3.
Metabolic stability of EOM Sal B and β-THP Sal B in liver microsomes in vitro. Compounds (10 μM) were incubated in rat liver microsomes in either the presence (stripes) or absence (no shading) of NADPH and quantified via LCMS over time. Data is presented as percent remaining at 150 min. **p<0.005, drug compared to SalA in the absence of NADPH. two-tailed, unpaired t-test (N=4)
Attenuation of cocaine-prime induced reinstatement
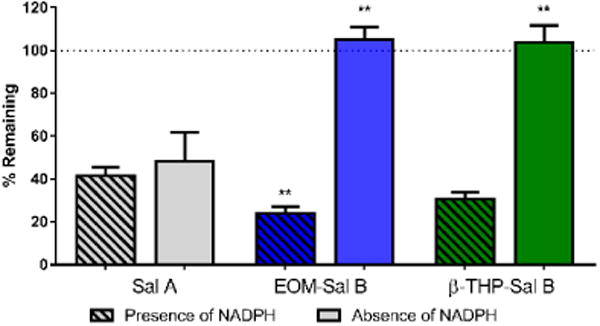
EOM Sal B significantly attenuated cocaine-primed drug-seeking responses on the previously active lever in a dose-dependent manner (Fig. 4) with a significant effect of treatment [F (3, 12) =7.382, p=0.0046]. Priming injections of cocaine produced robust reinstatement (350 ± 77 active lever presses), which was attenuated by pre-treatment with EOM Sal B (150 ± 53 and 43 ± 11 for EOM Sal B 0.1 and 0.3 mg/kg, respectively (values expressed as mean ± SEM)). Sal A (0.3 mg/kg) also attenuated reinstatement of drug-seeking behavior (151 ± 104 active lever presses), consistent with our previously published work (p<0.05) (Morani et al. 2009). Pre-treatment of rats with the selective KOPr antagonist nor-BNI (10 mg/kg, s.c.) prevented attenuation in drug seeking behaviour seen with EOM Sal B (0.3 mg/kg, p<0.01).
Fig. 4.
Effects of EOM Sal B on cocaine-primed reinstatement in rats. Rats received vehicle (75% DMSO) or EOM Sal B (0.1, light blue or 0.3 mg/kg, dark blue, i.p.) prior to cocaine (20 mg/kg, i.p.), and reinstatement was tested in a 2 h session. EOM Sal B significantly attenuated cocaine- and cue-induced reinstatement of drug-seeking compared to vehicle controls (N=5). Pre-treatment of rats with nor-BNI 24 h prior (10 mg/kg, s.c.) abolished the effects of EOM Sal B (0.3 mg/kg) (N=3). *p<0.05, **p<0.01 compared to vehicle, ##p<0.01 compared to 0.1 mg/kg EOM Sal B
Effects of EOM Sal B and β-THP Sal B on locomotor activity
Time course analysis of spontaneous locomotor activity revealed no significant interaction between time and EOM Sal B (0.1 mg/kg, i.p.) [F (17, 170) =0.60, p=0.8896] (Fig. 5a) or EOM Sal B (0.3 mg/kg, i.p.); [F (12, 180) =0.49, p=0.8162] (Fig. 5b). Similarly, β-THP Sal B at both 1 mg/kg [F (17, 238) =0.98, p=0.4788] (Fig. 5e) and 2 mg/kg [F (11, 154) =1.129, p=0.3421] (Fig 5f) showed no significant changes in locomotor behaviours. In addition, neither dose of EOM Sal B (Fig. 5c and d) nor β-THP Sal B (Fig. 5g and h), significantly altered total ambulatory counts in the 60 min locomotor activity test.
Fig. 5.
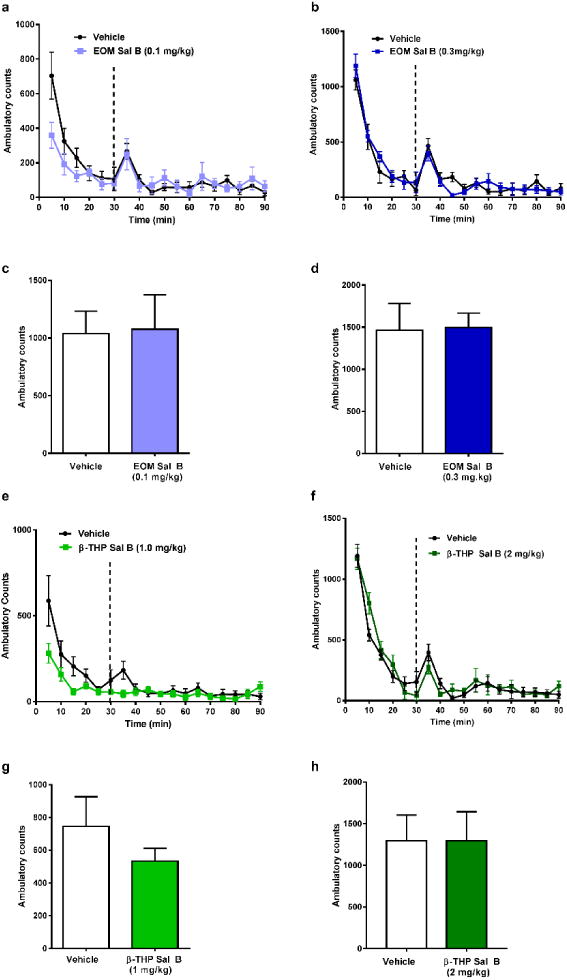
Effects of EOM Sal B and β-THP Sal B on spontaneous locomotor activity in rats. Rats were habituated in activity chambers for 30 min prior to vehicle (75% DMSO) and time-course and total locomotor activity counts evaluated following EOM Sal B at a dose of 0.1 mg/kg, i.p. (light blue) (a, c), and 0.3 mg/kg, i.p. (dark blue) (b. d) and β-THP Sal B (1.0 mg/kg, i.p.) (light green) (e, g), 2 mg/kg (dark green) (f, h). Values represent mean ± SEM (N=6-9)
The effects of EOM Sal B (0.1 mg/kg) and β-THP Sal B (1.0 mg/kg) on cocaine-induced hyperactivity are presented in Fig. 6 (N=5-9). Analysis of the time course data showed that a significant interaction was found when rats were pre-treated with EOM Sal B [F (34, 255) =1.62, p=0.0205] (Fig. 6a) or β-THP Sal B [F (34, 153) =2.49, p<0.0001] (Fig. 6c). Total ambulatory counts also revealed a significant effect of treatment for EOM Sal B [F (2, 15) =9.872, p=0.0018] (Fig. 6b) and β-THP Sal B [F (2, 9) =9.875, p=0.0054] (Fig. 6d). Bonferroni post-hoc analyses showed that EOM Sal B (Fig. 6b) and β-THP Sal B (Fig. 6d) significantly attenuated cocaine-induced hyperactivity. Pre-treatment of rats with the KOPr antagonist, nor-BNI, abolished the decrease in cocaine-induced hyperactivity (Fig. 6b and 6d).
Fig. 6.
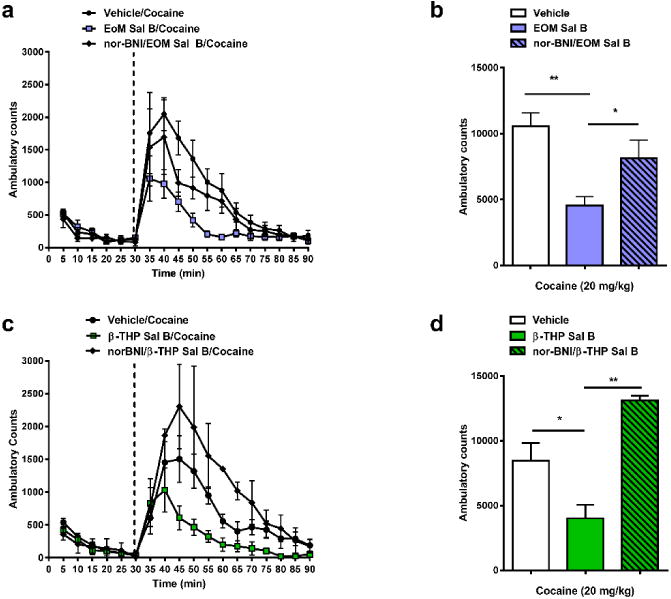
Effects of EOM Sal B and β-THP Sal B on cocaine-induced hyperactivity. EOM Sal B (0.1 mg/kg, i.p.) (a, b) and β-THP Sal B (1.0 mg/kg, i.p.) (c, d) produced significant attenuation of cocaine-induced hyperactivity compared to vehicle (N=5-9), an effect inhibited by nor-BNI pre-treatment (10 mg/kg s.c.) (N=5) *p<0.05, **p<0.01
Modulation of sucrose reinforcement
The effect of U50,488, Sal A, EOM Sal B and β-THP Sal B on responding maintained by sucrose is shown in Fig. 7a. There was a significant effect of treatment on the number of active lever presses for sucrose [F (4, 24) =59.44, p<0.0001]. Bonferroni post-hoc comparisons showed that U50,488 significantly attenuated responding (21 ± 11 lever press responses) compared to vehicle treated controls (497 ± 54). Neither Sal A (600 ± 46) nor EOM Sal B (616 ± 33) or β-THP Sal B (626 ± 53) showed significant changes compared to vehicle-treated controls (values represent mean ± SEM).
Fig. 7.
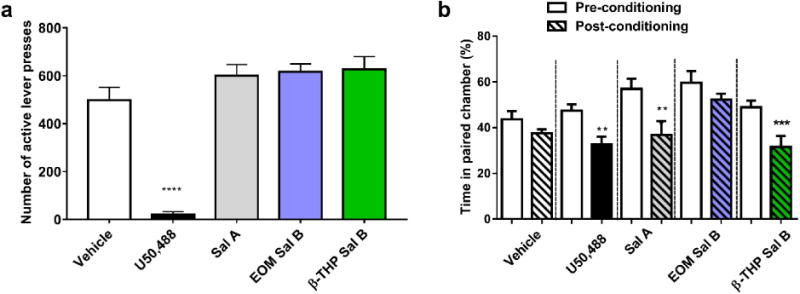
Effects of U50,488, Sal A, EOM Sal B and β-THP Sal B on sucrose responding and CPA in rats. One way ANOVA revealed a significant effect of treatment on sucrose responding (a). Bonferroni post-tests revealed U50,488 significantly attenuated sucrose responding (p<0.001) but no significant effects were seen with either Sal A (0.3 mg/kg, i.p.) or EOM Sal B (0.1 mg/kg, i.p.) or β-THP Sal B (1.0 mg/kg, i.p.) compared to vehicle controls (N=7). Sal A (0.3 mg/kg), and β-THP Sal B (1.0 mg/kg, i.p.), but not EOM Sal B (0.1 mg/kg, i.p.), produced a significant decrease in time spent in the paired chamber in the post-conditioning compared to pre-conditioning in CPA tests (b) (N=7-11) *p<0.05, ***p<0.001, ****p<0.0001
Effects of EOM Sal B and β-THP Sal B on tests of aversion-like, anxiety-like and depressive-like effects
The effect of EOM Sal B and β-THP Sal B on CPA is shown in Fig. 7b. Rats treated with EOM Sal B (0.1 mg/kg, i.p.) produced no significant changes in percentage of time spent in the paired chamber (t(7)=1.496, p=0.1783). However, comparison of pre- and post-test times of rats conditioned with β-THP Sal B (1 mg/kg. i.p.) showed that rats spent significantly less time in the β-THP Sal B-paired chamber (p<0.001) during the post-test (t(6)=7.655, p=0.0003), similar to the place aversion effects observed with Sal A (0.3 mg/kg, i.p.) (t(8)=3.742, p=0.0057) and U50,488 (10 mg/kg, i.p.) t(10)=3.835, p=0.0033).
Fig. 8 shows the effect of EOM Sal B (0.1 and 0.3 mg/kg) and β-THP Sal B (1 and 2 mg/kg) on anxiogenic behaviour in rats in the EPM test. There was a significant effect of treatment on the amount of time spent on the open arm [F (3, 66) =3.919, p=0.0123]. Bonferroni post-hoc analyses showed that only Sal A (0.3 mg/kg, i.p.) (p<0.05) produced a significant anxiogenic effect in rats (Fig. 8).
Fig. 8.
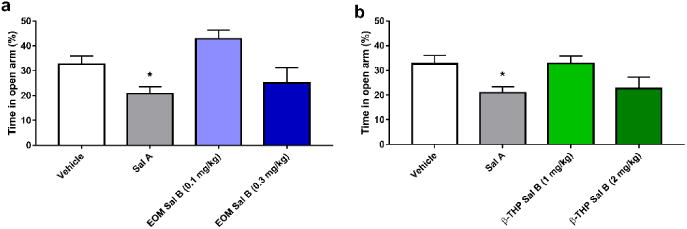
Dose-response effects of EOM Sal B and β-THP Sal B anxiogenic behaviours. Sal A (0.3 mg/kg, i.p.) significantly decreased time spent in the open arm in the EPM but neither EOM Sal B (0.1, or 0.3 mg/kg i.p.) (a) or β-THP Sal B (1.0 or 2 mg/kg, i.p.) changed open arm times compared to vehicle (b). (N=12-29) *p<0.05
The effects of EOM Sal B (Fig. 9a and b) and β-THP Sal B (Fig. 9c and d) on depressive-like behaviour was evaluated in rats in the FST. Neither EOM Sal B (0.1 and 0.3 mg/kg, i.p) (t(15)=0.09377, p=0.9265 and t(14)=0.14, p=0.8907, respectively), nor β-THP Sal B (1 and 2 mg/kg, i.p.) (t(12)=0.1426, p=0.8890 and t(14)=0.8477, p=0.4169, respectively) treated rats displayed significant changes in swimming behaviour (time spent mobile or immobile) when compared to vehicle treated controls.
Fig. 9.
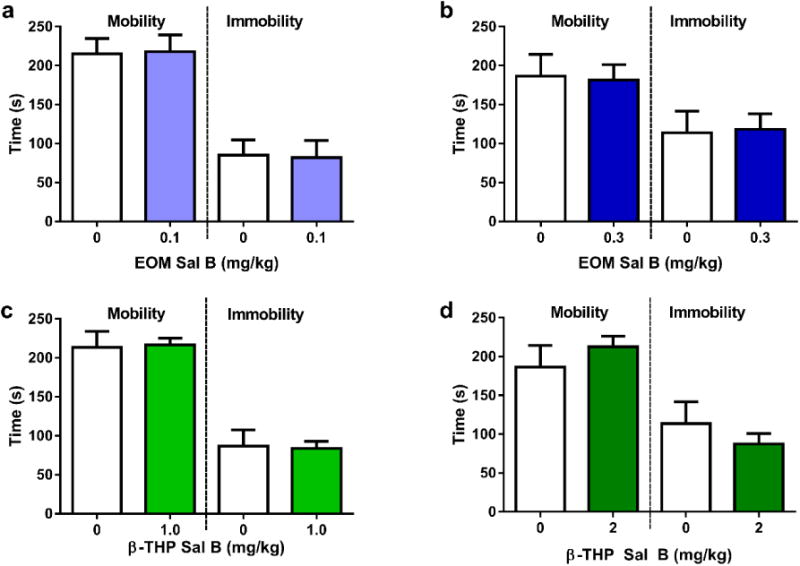
Effects of EOM SalB and β-THP Sal B on depressive-like behaviours in the FST. Neither EOM Sal B treated rats at 0.1 mg/kg (a) or 0.3 mg/kg (b) or β-THP Sal B at 1.0 mg/kg, (i.p.) (c) or 2.0 mg/kg (d) displayed any significant changes in time spent mobile or immobile compared to vehicle treated rats. (N=6-8)
Discussion
Results from the present study show that the C-2 analogue of Sal A, EOM Sal B is a potent full agonist in behavioural assays of tail-withdrawal, a reflex modulated by spinal reflexes in vivo. Both EOM Sal B and Sal A have significantly higher potency (lower EC50 values) compared to U50,488, although the differences between EOM Sal B and Sal A are not statistically significant. This is consistent with recently reported values for β-THP Sal B which showed no change in Emax values but increased potency (EC50 values of 1.4 mg/kg compared to Sal A 2.1 mg/kg in B6-SJL mice) (Paton et al. 2017). Evaluation of metabolic stability in rat liver microsomes, showed a small but significant increase in CYP450-mediated metabolism. However, other metabolic liabilities have been improved. Sal A is highly metabolised without the CYP450 cofactor NADPH but EOM Sal B and β-THP-Sal B analogues are not. Hydrolysis of the C2 acetate of Sal A to salvinorin B, results in significantly less potent metabolite (Hooker et al. 2009). The rapid metabolism of Sal A in plasma has been previously attributed to carboxylesterase activity (Tsujikawa et al. 2009). This improved metabolic stability ex vivo is likely to contribute to the longer duration of action observed in vivo compared to Sal A.
The reinstatement model was utilised in this study due to concordance between rat studies and human laboratory findings. Studies using rat models of stress, cue and drug-primed reinstatement models have been increasingly utilised to better understand drug use in humans, for recent review see (Bossert et al. 2013). The cue- and drug-prime relapse methods employed in this study are identical to previous models evaluating the KOPr agonists U50,488, U69,593 and Sal A (Schenk et al. 1999; Morani et al. 2009), allowing for direct comparisons to the present study. Both EOM Sal B and β-THP Sal B display anti-cocaine effects with reduced side effects. EOM Sal B dose-dependently reduced reinstatement of drug-seeking following extinction at doses as low as 0.1 mg/kg, which is equally potent to the Sal A analogue 16-Ethynyl Sal A under the same experimental conditions (Riley et al. 2014). Pre-administration of the KOPr antagonist, nor-BNI, prevented the attenuation of drug-seeking, confirming that the effects are KOPr mediated, supporting known selectivity and activity at KOPr obtained in previous in vitro experiments. Sal A has consistently been shown to be effective at attenuating cue plus cocaine-primed drug-seeking at a dose of 0.3 mg/kg (Morani et al. 2009; Prevatt-Smith et al. 2011; Riley et al. 2014). The same dose of U69,593 was also effective (0.3 mg/kg), however, U50,488 was only effective at 30 mg/kg (Morani et al. 2009) despite all having similar binding efficacy and potency at KOPr. It is important to note that previous studies have shown that a cue-stimulus, paired with a cocaine-prime is required for U69,593 attenuation of cocaine self-administration, an effect that was suggested to be due to modulation of stimulus and reward associations (Schenk et al. 2001). The increased effectiveness of EOM Sal B in its ability to modulate drug-seeking behaviour correlates with its increased potency in activating KOPr in vitro (EC50=0.65 nM) compared to Sal A (EC50=40 nM) (Table 1)(Prevatt-Smith et al. 2011), and also in vivo in the warm water tail-withdrawal assay. The increased effectiveness is also supported by discrimination studies where EOM Sal B was shown to fully substitute for Sal A at a greater potency than, and at longer post-injection intervals, than Sal A in Long Evans rats (Peet and Baker 2011). In contrast, β-THP Sal B was only effective in attenuating cocaine-primed reinstatement at 1 mg/kg using identical experimental protocols despite similar potency to Sal A in vitro (Prevatt-Smith et al. 2011). Other C-2-derived KOPr agonists, including Mesyl Sal B, have similar efficacy at KOPr to Sal A in vitro (Mesyl Sal B: EC50=30 nM; SalA: EC50=40 nM; respectively) (Harding et al. 2005b), with a matching ability to attenuate drug-seeking behaviour, with an effective dose of 0.3 mg/kg (Simonson et al. 2015).
Attenuation of the rewarding effects of cocaine by KOPr agonists have been attributed to their ability to induce aversive and/or dysphoric effects (Koob et al. 2014; Shippenberg 2009). While KOPr agonists reduce drug-induced reinstatement (Schenk et al. 1999) they may potentiate stress-induced reinstatement (Valdez et al. 2007), whereas, KOPr antagonists block stress-induced reinstatement, but not drug-induced reinstatement (Beardsley et al. 2005). This highlights different stages of addiction where both kappa agonists and antagonists may have beneficial effects (Mantsch et al. (2016). There is evidence to suggest that Sal A and its analogues may also have differing side effects to traditional KOPr agonists (Braida et al. 2009; Carr et al. 2009; Harden et al. 2012; Kivell et al. 2014; Simonson et al. 2015). Therefore, in this study we also chose to evaluate EOM Sal B and β-THP Sal B for side effects associated with KOPr activation. We used a dose range that was effective in attenuating drug-seeking behavior to screen these side effects. We evaluated EOM Sal B (0.1 mg/kg) and β-THP Sal B (1 mg/kg) for behavioural responding in tests evaluating natural reward (responding for sucrose) and aversion (CPA) and evaluated dose-response effects (EOM Sal B, 0.1 mg/kg and 0.3 mg/kg; β-THP Sal B, 1 mg/kg and 2 mg/kg) in behavioural tests of sedation (spontaneous locomotor tests), anxiety-like (EPM), and depression-like (FST) effects.
Neither EOM Sal B (0.1 mg/kg and 0.3 mg/kg) nor β-THP Sal B (1 mg/kg and 2 mg/kg) showed sedative effects in spontaneous locomotor activity tests. A previous study conducted by (Wang et al. 2008), found that MOM Sal B (1–5 mg/kg, s.c.) increased ambulation and exploration on the Y-maze in rats. Also the Sal A analogues Mesyl Sal B (0.3 mg/kg, i.p.) (Simonson et al. 2015), 16-ethynyl Sal A (0.1 mg/kg) and 16-bromo Sal B showed no change in spontaneous locomotor activity (Riley et al. 2014), and 22-thiocyanatosalvinorin (RB-64, 3 mg/kg), a potent G-protein biased KOPr agonist, did not decrease locomotor activity in accelerating rotarod tests or alter novelty-induced locomotion in mice (White et al. 2015). We therefore conclude that the attenuation of drug-seeking behaviour observed in Prevatt-Smith et al. (2011), utilising identical experimental conditions and here, cannot be attributed to sedation and/or inability to perform an operant task, as lever responding for sucrose was also maintained at these doses. This data confirms that the anti-cocaine effects of EOM Sal B and β-THP Sal B in the cocaine-induced locomotor activity test are not due to sedation, but specific effects on attenuation of cocaine-induced behaviors.
In contrast to the traditional KOPr agonist U50,488, which almost abolished responding to sucrose pellets, neither Sal A, EOM Sal B nor β-THP Sal B showed significant changes in sucrose self-administration. U50,488 (5, 10 mg/kg) has previously been shown to disrupt responses for a natural sucrose reward in Long Evans rats (Henderson-Redmond and Czachowski 2014) consistent with our findings. These results suggest that there are clear differences between U50,488, Sal A and its analogues in modulation of natural reward, despite similar in vivo binding and KOPr activity data. Previously, Sal A (0.3 mg/kg, i.p.) was shown to have no significant effects on the intake of a 10% sucrose solution (Morani et al. 2009). However, MOM Sal B (0.3 mg/kg) attenuated self-administration of a 10% sucrose solution in rats (Morani et al. 2013), indicating that despite their similar anti-cocaine properties, Sal A and its analogues can also evoke different responses in the modulation of natural reward. The specific KOPr mediated signalling pathways responsible for these effects remain to be determined. The KOPr system plays a varied role in modulation of appetitive natural rewards such as sucrose. Although it is agreed, based on a large body of evidence, that KOPr agonists produce effects opposite to that of reward (Ebner et al. 2010; Knoll and Carlezon Jr 2010; McLaughlin et al. 2006; Shippenberg et al. 2007; Wee and Koob 2010) by decreasing DA neurotransmission (Di Chiara and Imperato 1988; Ebner et al. 2010). KOPr agonists have also been shown to stimulate appetite, food consumption, and sucrose intake due to KOPr and orexin co-localisation within synaptic vesicles in the hypothalamus (Muschamp et al. 2014). The heterodimerisation (Chen et al. 2015) and co-transmission of both peptides may be a mechanism by which KOPr regulates feeding (Muschamp et al. 2014), a relationship that warrants further investigation.
To further determine the adverse effect profile of EOM Sal B and β-THP Sal B their aversive properties were examined using CPA. In this study, β-THP Sal B but not EOM Sal B-treated rats decreased the amount of time spent in the paired chamber, indicating that it has aversive properties, even though EOM Sal B is more potent in modulating KOPr activity in vivo and in vitro. We also show Sal A at a dose of 0.3 mg/kg produces significant CPA in rats. Previously, Sal A has shown CPA in Sprague-Dawley rats (0.3–1.0 mg/kg, i.p.) (Sufka et al. 2014) and mice (1.0–3.2 mg/kg, i.p.) (Zhang et al. 2005). However, work in our laboratory has also shown that this same dose of Sal A (0.3 mg/kg) did not produce aversion in the conditioned taste aversion paradigm (CTA) in rats (Morani et al. 2012). It is important to note that traditional KOPr agonists such as U50,488 (0.90, 1.60 mg/kg, s.c.) have shown CTA in rats (Davis et al. 2009), indicating that this model can be used to evaluate aversive properties. However, a recent study comparing CTA and CPA models found that they are not directly comparable due to the contrast in temporal presentation of conditioned cues and drug injection (taste cues presented prior to drug injection in the CTA vs. place cues presented after drug injection in the CPA) (Gore-Langton et al. 2015).
In EPM tests evaluating anxiety, a single administration of EOM Sal B (0.1 or 0.3 mg/kg) or β-THP Sal B (1 or 2 mg/kg) did not produce anxiety in the EPM, although a non-significant decrease in open arm time was seen with the highest dose of EOM Sal B and β-THP Sal B, and a non-significant increase in open arm time with the low dose of EOM Sal B. Previous work conducted using low doses of the parent compound Sal A (0.1–160 μg/kg, s.c.) (Braida et al. 2009) have shown that Sal A produced increased exploratory behaviour by increasing open arm time in the EPM, an indication of anxiolytic effects (Braida et al. 2009). However, we found that when the minimum dose that produces cocaine antagonistic effects was used, Sal A significantly reduced the amount of time rats spent on the open arm of the EPM. Taken together, these studies indicate that the dose of KOPr agonist administered determines the presence or absence of anxiety. We therefore conclude that both EOM Sal B and β-THP Sal B have an improved side-effect profile compared to Sal A, with no anxiogenic effects seen at doses that were effective in attenuating cocaine seeking.
KOPr agonists including Sal A and its analogues (e.g. MOM Sal B) have been shown previously to display depressive-like effects (Carlezon et al. 2006; Morani et al. 2013; Morani et al. 2012). Here we show that both EOM Sal B (0.1 and 0.3 mg/kg) and β-THP Sal B (1 mg/kg and 2 mg/kg) do not produce depressive-like effects in the FST with no changes in mobility or immobility observed.
The cellular mechanisms responsible for the differences observed in the behavioural effects between traditional KOPr agonists, Sal A and novel analogues EOM Sal B and β-THP Sal B are still not understood. However, the theory of biased agonism is one mechanism that may explain such effects (Kenakin 2007). It has been hypothesised that the desirable anti-addiction and analgesic effects associated with KOPr activation are due to G-protein mediated signalling, whereas negative side effects of KOPr agonists results from β-arrestin mediated signalling (Bruchas and Chavkin 2010; Kivell et al. 2013). Sal A, U50,488 and U69,593 do not show KOPr ligand bias. Neither EOM Sal B nor β-THP Sal B have been tested for KOPr ligand bias in the CNS and it remains to be determined if ligand bias explains the reduction in side effects observed. A recent study has shown that Sal A and EOM Sal B modulate kinase signalling pathways differently (extracellular signal-related kinase and c-Jun N-terminal kinase) in cultured peripheral neurons. This effect was shown to differentially modulate the peripheral analgesic effects of KOPr in vivo in the rat (Jamshidi et al. 2015), however, β-arrestin bias was not evaluated. A recent study by White et al. 2015 showed that the C-22 analogue of Sal A, RB-64 (Yan et al. 2009) showed anti-nociceptive effects in the hot plate assay in mice with fewer side effects including reduced effects on motor incoordination, compared to traditional KOPr agonists (White et al. 2015). This data suggests that the sedative effects are mediated through β-arrestin. However, RB-64 (and Sal A at 3 mg/kg) produced aversion in CPA tests in both wild type and β-arrestin KO mice suggesting that aversion is at least partly regulated via G-protein activation (White et al. 2015). There is additional evidence to suggest that aversion is regulated via p38 MAP kinase signalling pathways (Ehrich et al. 2015). It is possible that differences in p38 signalling between EOM Sal B and β-THP Sal B may explain the aversive effects seen with β-THP Sal B but not EOM Sal B. These studies are ongoing in our laboratory. A recent study by Schattauer et al. (2017) showed that the KOPr agonist nalfurafine is a G-protein biased agonist. This new finding in combination with known safety and lack of side effect data for nalfurafine in humans (Kumagai et al. 2012) , gives additional to support for the development of G-protein biased KOPr agonists as therapeutics with fewer side-effects.
In conclusion, this is the first study to characterise both the anti-cocaine and side-effect profiles of the Sal A-derived analogues, EOM Sal B and β-THP Sal B. Both compounds are longer acting Sal A analogues that attenuate drug-seeking behaviour and cocaine induced locomotor activities with reduced adverse effect profiles, an improvement over both traditional KOPr agonists and their parent compound, Sal A. The behavioural data from this study will aid in further Sal A-derived analogue development. The combination of ‘functionally selective’ or ‘biased agonists’ with the novel structure of Sal A-derived agonists may be the key to identifying new therapeutics with specific anti-addiction effects and fewer side effects.
Table 2. Pharmacology of Sal A and its C-2 analogues at KOPr.
EC50 and Emax values obtained using the [35S]GTP-γ-S functional assay in Chinese hamster ovary (CHO) cells stably expressing human KOPr; Ki values were obtained using [3H]U69,593 as a radioligand (*or using [125I]IOXY as radioligand); # Binding affinity using [3H]U69,593 as a radioligand in HEK cells stably expressing KOPr; ^Neuro 2A cells stably expressing human KOPr evaluated in [35S]GTP-γ-S assay. EC50= effective dose that produces 50% of the maximal response, Emax= % at which compound stimulates [35S]GTP-γ-S binding compared to U50,488 (500 nM). Values presented as mean ± SD..
| Sal A | U50,488 | Sal A | EOM Sal B | β-THP Sal B |
|---|---|---|---|---|
| Potency at KOPr, EC50 (nM) | ^30(4) | 40 ± 10(1,
2,
3) ^23(4) |
0.65 ± 0.17(2) | 60 ± 6(2) |
| Binding affinity at KOPr, Ki (nM) | #0.42 ± 0.22(4) | 7.4 ± 0.7(1,
2) *1.9 ± 0.2(3) #0.28 ± 0.22(4) |
3.13 ± 0.40(2) | 6.21 ± 0.40(2) |
| Binding efficiency at KOPr, Emax (%) | ^101 4.2(4) | 120 ± 2(1,
2,
3) ^101 ± 2.9(4) |
127 ± 5(2) | 109 ± 3(2) |
Acknowledgments
Funding was provided by the Neurological Foundation of New Zealand and the Health Research Council of New Zealand (to BMK), DA018151 (to TEP). GM008545 (to RSC), and an AFPE Pre-doctoral Fellowship in Pharmaceutical Sciences (to RSC). A Postgraduate Research Scholarship was provided by Victoria University of Wellington (to AE). We would like to acknowledge Andrew Biggerstaff, Kelly Paton and Stephen Mathew for technical assistance.
Footnotes
Compliance with ethical standards: All animal use and procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Victoria University of Wellington Animal Ethics Committee, Wellington, New Zealand.
Conflict of interest: The authors have no conflict of interest to declare
References
- Baker LE, Panos JJ, Killinger BA, Peet MM, Bell LM, Haliw LA, Walker SL. Comparison of the discriminative stimulus effects of salvinorin A and its derivatives to U69,593 and U50,488 in rats. Psychopharmacology. 2009;203:203–211. doi: 10.1007/s00213-008-1458-3. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183(1):118–26. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Beguin C, Richards MR, Wang YL, Chen Y, Liu-Chen LY, Ma ZZ, Lee DYW, Carlezon WA, Cohen BM. Synthesis and in vitro pharmacological evaluation of salvinorin A analogues modified at C(2) Bioorganic and Medicinal Chemistry Letters. 2005;15:2761–2765. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- Bohn L, Gainetdinov R, Lin F, Lefkowitz R, Caron M. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408(6813):720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, Shaham Y. The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl) 2013;229(3):453–76. doi: 10.1007/s00213-013-3120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braida D, Capurro V, Zani A, Rubino T, Vigano D, Parolaro D, Sala M. Potential anxiolytic- and antidepressant-like effects of salvinorin A, the main active ingredient of Salvia divinorum, in rodents. British Journal of Pharmacology. 2009;157:844–853. doi: 10.1111/j.1476-5381.2009.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braida D, Limonta V, Capurro V, Fadda P, Rubino T, Mascia P, Zani A, Gori E, Fratta W, Parolaro D, Sala M. Involvement of kappa-opioid and endocannabinoid system on salvinorin A-induced reward. Biological Psychiatry. 2008;63:286–292. doi: 10.1016/j.biopsych.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology. 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelman ER, Caspers M, Lovell KM, Kreek MJ, Prisinzano TE. Behavioral effects and central nervous system levels of the broadly available kappa-agonist hallucinogen salvinorin A are affected by p-glycoprotein modulation in vivo. Journal of Pharmacology and Experimental Therapeutics. 2012;341:802–808. doi: 10.1124/jpet.112.193227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelman ER, Mandau M, Tidgewell K, Prisinzano TE, Yuferov V, Kreek MJ. Effects of salvinorin A, a kappa-opioid hallucinogen, on a neuroendocrine biomarker assay in nonhuman primates with high kappa-receptor homology to humans. Journal of Pharmacology and Experimental Therapeutics. 2007;320:300–306. doi: 10.1124/jpet.106.112417. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Prisinzano TE, Deng H, Rus S, Kreek MJ. Unconditioned behavioral effects of the powerful kappa-opioid hallucinogen salvinorin A in nonhuman primates: Fast onset and entry into cerebrospinal fluid. Journal of Pharmacology and Experimental Therapeutics. 2009;328:588–597. doi: 10.1124/jpet.108.145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma ZZ, Lee DYW, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. Journal of Pharmacology and Experimental Therapeutics. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, Lucki I. Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology. 2009;35:752–763. doi: 10.1038/npp.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Sud S, Jin WZ, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. Salvinorin A, an active component of the hallucinogenic sage Salvia divinorum is a highly efficacious kappa-opioid receptor agonist: Structural and functional considerations. Journal of Pharmacology and Experimental Therapeutics. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang R, Chen X, Wang C, Cai X, Liu H, Jiang Y, Liu C, Bai B. Heterodimerization of human orexin receptor 1 and kappa opioid receptor promotes protein kinase A/cAMP-response element binding protein signaling via a Galphas-mediated mechanism. Cell Signalling. 2015;27:1426–1438. doi: 10.1016/j.cellsig.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Cunningham CW, Rothman RB, Prisinzano TE. Neuropharmacology of the naturally occurring κ-opioid hallucinogen salvinorin A. Pharmacological Reviews. 2011;63:316–347. doi: 10.1124/pr.110.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CM, Rice KC, Riley AL. Opiate-agonist induced taste aversion learning in the Fischer 344 and Lewis inbred rat strains: Evidence for differential mu opioid receptor activation. Pharmacology, Biochemistry and Behavior. 2009;93:397–405. doi: 10.1016/j.pbb.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Opposite effects of mu-opiate and kappa-opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. Journal of Pharmacology and Experimental Therapeutics. 1988;244:1067–1080. [PubMed] [Google Scholar]
- DiMattio KM, Ehlert FJ, Liu-Chen LY. Intrinsic relative activities of k opioid agonists in activating Gαproteins and internalising receptor: differences betweenhuman and mouse receptors. European Journal of Pharmacology. 2015;761:235–44. doi: 10.1016/j.ejphar.2015.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology. 2010;210:241–252. doi: 10.1007/s00213-010-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:12917–31. doi: 10.1523/JNEUROSCI.2444-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: A potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Archives Internationales de Pharmacodynamie et de Thérapie. 1992;316:30–42. [PubMed] [Google Scholar]
- Gallantine EL, Meert TF. Antinociceptive and adverse effects of μ- and κ-opioid receptor agonists: A comparison of morphine and U50488-H. Basic and Clinical Pharmacology and Toxicology. 2008;103:419–427. doi: 10.1111/j.1742-7843.2008.00306.x. [DOI] [PubMed] [Google Scholar]
- Gillett K, Harshberger E, Valdez GR. Protracted withdrawal from ethanol and enhanced responsiveness stress: Regulation via the dynorphin/kappa opioid receptor system. Alcohol. 2013;47:359–65. doi: 10.1016/j.alcohol.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick SD, Visker KE, Maisonneuve IM. Effects of cyclazocine on cocaine self-administration in rats. European Journal of Pharmacology. 1998;357:9–14. doi: 10.1016/s0014-2999(98)00548-2. [DOI] [PubMed] [Google Scholar]
- Gore-Langton JK, Flax SM, Pomfrey RL, Wetzell BB, Riley AL. Measures of the aversive effects of drugs: A comparison of conditioned taste and place aversions. Pharmacology Biochemistry and Behavior. 2015;134:99–105. doi: 10.1016/j.pbb.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Harden MT, Smith SE, Niehoff JA, McCurdy CR, Taylor GT. Antidepressive effects of the kappa-opioid receptor agonist salvinorin A in a rat model of anhedonia. Behavioural Pharmacology. 2012;23:710–5. doi: 10.1097/FBP.0b013e3283586189. [DOI] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. Journal of Medicinal Chemistry. 2005a;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Salvinicins A and B, new neoclerodane diterpenes from Salvia divinorum. Organic Letters. 2005b;7:3017–3020. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson-Redmond A, Czachowski C. Effects of systemic opioid receptor ligands on ethanol-and sucrose seeking and drinking in alcohol-preferring (P) and Long Evans rats. Psychopharmacology. 2014;231:4309–4321. doi: 10.1007/s00213-014-3571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker JM, Munro TA, Béguin C, Alexoff D, Shea C, Xu Y, Cohen BM. Salvinorin A and derivatives: Protection from metabolism does not prolong short-term, whole-brain residence. Neuropharmacology. 2009;57:386–391. doi: 10.1016/j.neuropharm.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker JM, Xu Y, Schiffer W, Shea C, Carter P, Fowler JS. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. NeuroImage. 2008;41:1044–1050. doi: 10.1016/j.neuroimage.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. Journal of Pharmacology and Experimental Therapeutics. 1992;260:1237–43. [PubMed] [Google Scholar]
- Jamshidi RJ, Jacobs BA, Sullivan LC, Chavera TA, Saylor RM, Prisinzano TE, Clarke WP, Berg KA. Functional selectivity of kappa opioid receptor agonists in peripheral sensory neurons. The Journal of pharmacology and experimental therapeutics. 2015;355:174–82. doi: 10.1124/jpet.115.225896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Collateral efficacy in drug discovery: Taking advantage of the good (allosteric) nature of 7TM receptors. Trends in Pharmacological Sciences. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Kivell B, Ewald A, Prisinzano T. Salvinorin A analogs and other kappa-opioid receptor compounds as treatments for cocaine abuse. Advances in Pharmacology. 2013;69:481–511. doi: 10.1016/B978-0-12-420118-7.00012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell B, Uzelac Z, Sundaramurthy S, Rajamanickam J, Ewald A, Chefer V, Jaligam V, Bolan E, Simonson B, Annamalai B. Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism. Neuropharmacology. 2014;86:228–240. doi: 10.1016/j.neuropharm.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Research. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, Jr, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76 Pt B:370–82. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, Kurihara M, Yanagita T, Suzuki H. Efficacy and safety of a novel K-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol. 2012;36(2):175–83. doi: 10.1159/000341268. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19168–73. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Karnati VV, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA. Synthesis and in vitro pharmacological studies of new C (2) modified salvinorin A analogues. Bioorganic and Medicinal Chemistry Letters. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- Lozama A, Cunningham CW, Caspers MJ, Douglas JT, Dersch CM, Rothman RB, Prisinzano TE. Opioid receptor probes derived from cycloaddition of the hallucinogen natural product salvinorin A. Journal of Natural Products. 2011;74:718–26. doi: 10.1021/np1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jones RM, Portoghese PS, Carlezon WA. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. Journal of Pharmacology and Experimental Therapeutics. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacology. 2006;31:787–94. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean KA, Johnson MW, Reissig CJ, Prisinzano TE, Griffiths RR. Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology (Berl) 2013;226(2):381–92. doi: 10.1007/s00213-012-2912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Funk D, Lê AD, Shaham Y. Stress-Induced Reinstatement of Drug Seeking: 20 Years of Progress. Neuropsychopharmacology Reviews. 2016;41:335–356. doi: 10.1038/npp.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani A, Ewald A, Prevatt-Smith K, Prisinzano TE, Kivell B. The 2-methoxy methyl analogue of salvinorin A attenuates cocaine-induced drug seeking and sucrose reinforcements in rats. European Journal of Pharmacology. 2013 doi: 10.1016/j.ejphar.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani AS, Kivell B, Prisinzano TE, Schenk S. Effect of kappa-opioid receptor agonists U69593, U50488H, spiradoline and salvinorin A on cocaine-induced drug-seeking in rats. Pharmacology Biochemistry and Behavior. 2009;94:244–249. doi: 10.1016/j.pbb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morani AS, Schenk S, Prisinzano TE, Kivell BM. A single injection of a novel kappa opioid receptor agonist salvinorin A attenuates the expression of cocaine-induced behavioral sensitization in rats. Behavioural Pharmacology. 2012;23:162–170. doi: 10.1097/FBP.0b013e3283512c1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen LY, Carlezon WA, Jr, Cohen BM, Beguin C. Standard protecting groups create potent and selective kappa opioids: Salvinorin B alkoxymethyl ethers. Bioorganic & Medicinal Chemistry. 2008;16:1279–1286. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, Kamenecka TM, Borgland SL, Kenny PJ, Carlezon WA. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E1648–E1655. doi: 10.1073/pnas.1315542111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton KF, Kumar N, Crowley RS, Harper JL, Prisinzano TE, Kivell BM. The analgesic and anti-inflammatory effects of Salvinorin A analogue β-tetrahydropyran Salvinorin B in mice. European Journal of Pain. 2017 doi: 10.1002/ejp.1002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet MM, Baker LE. Salvinorin B derivatives, EOM-Sal B and MOM-Sal B, produce stimulus generalization in male Sprague-Dawley rats trained to discriminate salvinorin A. Behavioural Pharmacology. 2011;22:450–457. doi: 10.1097/FBP.0b013e328349fc1b. [DOI] [PubMed] [Google Scholar]
- Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, Dersch CM, Rothman RB, Kivell B, Prisinzano TE. Potential drug abuse therapeutics derived from the hallucinogenic natural product salvinorin A. Medicinal Chemistry Communications. 2011;2:1217–1222. doi: 10.1039/C1MD00192B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sciences. 2005;78:527–531. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Riley AP, Groer CE, Young D, Ewald AW, Kivell BM, Prisinzano TE. Synthesis and κ-opioid receptor activity of furan-substituted salvinorin A analogues. Journal of Medicinal Chemistry. 2014;57:10464–10475. doi: 10.1021/jm501521d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer SS, Kuhar JR, Song A, Chavkin C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal. 2017;32:59–65. doi: 10.1016/j.cellsig.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S, Partridge B, Shippenberg TS. Effects of the kappa-opioid receptor agonist, U69593, on the development of sensitization and on the maintenance of cocaine self-administration. Neuropsychopharmacology. 2001;24(4):441–50. doi: 10.1016/S0893-133X(00)00190-1. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B, Shippenberg TS. U69593, a kappa-opioid agonist, decreases cocaine self-administration and decreases cocaine-produced drug-seeking. Psychopharmacology. 1999;144:339–346. doi: 10.1007/s002130051016. [DOI] [PubMed] [Google Scholar]
- Schmidt MD, Schmidt MS, Butelman ER, Harding WW, Tidgewell K, Murry DJ, Kreek MJ, Prisinzano TE. Pharmacokinetics of the plant-derived kappa-opioid hallucinogen salvinorin A in nonhuman primates. Synapse. 2005;58:208–210. doi: 10.1002/syn.20191. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS. The dynorphin/kappa opioid receptor system: A new target for the treatment of addiction and affective disorders? Neuropsychopharmacology. 2009;34:247–247. doi: 10.1038/npp.2008.165. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacology and Therapeutics. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson B, Morani AS, Ewald AWM, Walker L, Kumar N, Simpson D, Miller JH, Prisinzano TE, Kivell BM. Pharmacology and anti-addiction effects of the novel κ opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin A. British Journal of Pharmacology. 2015;172:515–531. doi: 10.1111/bph.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery DA, Cryan JF. Using the rat forced swim test to assess antidepressant-like activity in rodents. Nature Protocols. 2012;7:1009–1014. doi: 10.1038/nprot.2012.044. [DOI] [PubMed] [Google Scholar]
- Sufka KJ, Loria MJ, Lewellyn K, Zjawiony JK, Ali Z, Abe N, Khan IA. The effect of Salvia divinorum and Mitragyna speciosa extracts, fraction and major constituents on place aversion and place preference in rats. Journal of Ethnopharmacology. 2014;151:361–364. doi: 10.1016/j.jep.2013.10.059. [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Bäckman CM, Chefer V, O'Donnell P, Shippenberg TS. Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology. 2013;38:1770–1779. doi: 10.1038/npp.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teksin ZS, Lee IJ, Nemieboka NN, Othman AA, Upreti VV, Hassan HE, Syed SS, Prisinzano TE, Eddington ND. Evaluation of the transport, in vitro metabolism and pharmacokinetics of Salvinorin A, a potent hallucinogen. European Journal of Pharmaceutics and Biopharmaceutics. 2009;72:471–7. doi: 10.1016/j.ejpb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa K, Kuwayama K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H. In vitro stability and metabolism of salvinorin A in rat plasma. Xenobiotica. 2009;39(5):391–8. doi: 10.1080/00498250902769967. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Platt DM, Rowlett JK, Rüedi-Bettschen D, Spealman RD. Kappa agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J Pharmacol Exp Ther. 2007;323(2):525–33. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nature Protocols. 2007;2:322–8. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SL, Geter-Douglas B, Strain EC, Bigelow GE. Enadoline and butorphanol: Evaluation of kappa-agonists on cocaine pharmacodynamics and cocaine self-administration in humans. Journal of Pharmacology and Experimental Therapeutics. 2001a;299:147–158. [PubMed] [Google Scholar]
- Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: Comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001b;157:151–162. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen Y, Xu W, Lee DYW, Ma Z, Rawls SM, Cowan A, Liu-Chen LY. 2-methoxymethyl-salvinorin B is a potent kappa opioid receptor agonist with longer lasting action in vivo than salvinorin A. Journal of Pharmacology and Experimental Therapeutics. 2008;324:1073–1083. doi: 10.1124/jpet.107.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. Journal of Pharmacology and Experimental Therapeutics. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Bikbulatov RV, Mocanu V, Dicheva N, Parker CE, Wetsel WC, Mosier PD, Westkaemper RB, Allen JA, Zjawiony JK, Roth BL. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry. 2009;48:6898–6908. doi: 10.1021/bi900605n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology. 2005;179:551–558. doi: 10.1007/s00213-004-2087-0. [DOI] [PubMed] [Google Scholar]