

Abstract
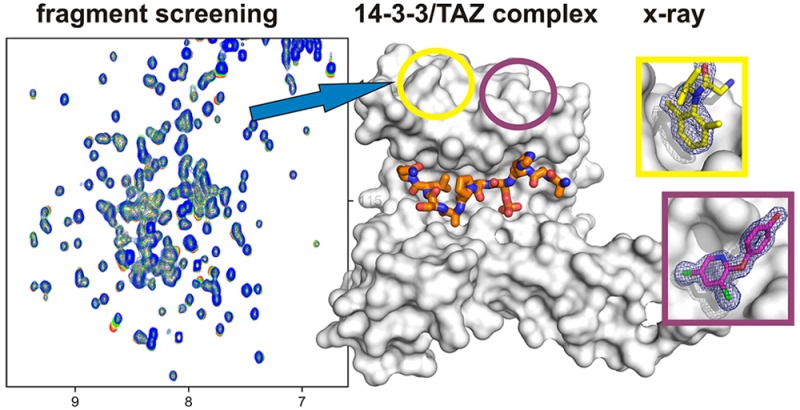
Proteins typically interact with multiple binding partners, and often different parts of their surfaces are employed to establish these protein–protein interactions (PPIs). Members of the class of 14-3-3 adapter proteins bind to several hundred other proteins in the cell. Multiple small molecules for the modulation of 14-3-3 PPIs have been disclosed; however, they all target the conserved phosphopeptide binding channel, so that selectivity is difficult to achieve. Here we report on the discovery of two individual secondary binding sites that have been identified by combining nuclear magnetic resonance-based fragment screening and X-ray crystallography. The two pockets that these fragments occupy are part of at least three physiologically relevant and structurally characterized 14-3-3 PPI interfaces, including those with serotonin N-acetyltransferase and plant transcription factor FT. In addition, the high degree of conservation of the two sites implies their relevance for 14-3-3 PPIs. This first identification of secondary sites on 14-3-3 proteins bound by small molecule ligands might facilitate the development of new chemical tool compounds for more selective PPI modulation.
14-3-3 proteins play an important role in many physiological processes and are implicated in a number of human diseases ranging from cancer and neurodegeneration to metabolic diseases and infection.1−3 These widespread functions are explained by the fact that 14-3-3 proteins interact with several hundred proteins in human cells.4 Hence, small molecule modulation of the extensive protein–protein interaction network of 14-3-3 proteins is of significant biomedical interest, but at the same time, obtaining specificity is highly challenging. In recent years, it has been shown that 14-3-3 proteins can be targeted by natural products and their derivatives,5−10 modified peptides,11,12 “classic” small molecules,13,14 and supramolecular ligands.15,16 These compounds act as inhibitors or stabilizers of 14-3-3 PPIs, to obtain the desired effect on the activity, dimerization, or cellular localization of the binding partner and depending on its normal effector function in cellular pathways. For example, it was shown that stabilization of the inhibitory 14-3-3/C-Raf interaction by the natural product Cotylenin A leads to tumor shrinkage in a mouse model.6 Likewise, a related natural product, Fusicoccin A, can stabilize the interaction of 14-3-3 with CFTR, a chloride channel functionally impaired in cystic fibrosis. Stabilization of this interaction increases the level of plasma membrane localization of this channel, which could be a new therapeutic strategy in the treatment of cystic fibrosis.10 One example in which inhibition of binding to 14-3-3 would be desirable is Pseudomonas aeruginosa pathogenicity factor Exoenzyme S.17 Here, modification of the original Exoenzyme S-derived 14-3-3 binding motifs shows significantly increased affinity for 14-3-3 and could be a valuable starting point for the development of 14-3-3/Exoenzyme S PPI inhibitors.11,18
So far, all reported 14-3-3 binding molecules are accommodated in the central groove of the 14-3-3 proteins and thereby directly interfere with binding of the mostly phosphorylated, primary interaction motifs of their target proteins, resulting in the lack of specificity. Therefore, we believe it would be of great value to identify small molecules binding outside of the central binding groove, as they may provide anchor points for the development of modulators affecting smaller subsets of the 14-3-3 interactome. In PPI drug discovery, most efforts have resulted in inhibitors, whereas in many interactions of 14-3-3 with binding partners, it would be highly relevant to stabilize, as described above for C-Raf and CFTR. Interestingly, stabilizing a protein–protein interaction has the added benefit of being more selective, because a small molecule stabilizer targets a select interface formed by multiple proteins.
One of the proteins for which 14-3-3 PPI stabilization would be desirable but has not yet been achieved is TAZ (transcriptional coactivator with a PDZ binding motif), which was first reported in 2000 as a 14-3-3 binding protein.19 TAZ function has been linked to mesenchymal stem cell differentiation, to the development of limb, heart, bone, muscle, fat, and lung tissues,20 and to mechanotransduction.21 Like the related transcriptional coactivator YAP (Yes-associated protein), TAZ is a major effector of the Hippo pathway that is named after a Drosophila mutant showing significant aberrations in organ size control.22,23 Active TAZ and YAP migrate from the cytoplasm to the nucleus where they can form hybrid transcription factors with TEA domain (TEAD) family proteins. In this way, TAZ drives the expression of genes that lead to cell proliferation, survival, migration, and invasion, and thus, an increased level of activation of TAZ is observed in many cancers.24 Regulation of TAZ/YAP activity is complex and involves a number of upstream kinases,22,24,25 of which large tumor suppressor 1/2 (LATS1/2) directly phosphorylates TAZ at Ser89 and YAP at Ser127, thus facilitating binding of 14-3-3 proteins.26 When in complex with 14-3-3, both TAZ and YAP are sequestered in the cytoplasm and are thus functionally inactivated.19,27 Although in recent years a flurry of studies of the complexity of influences on the Hippo pathway and YAP/TAZ activity have been published, including the involvement of mechanotransduction21 and metabolic and nutrient inputs,28 nuclear availability and transcriptional responses of YAP/TAZ remain the ultimate outcome of all these impacts. Thus, compounds that can interfere with these nuclear activities may represent a “universal” anti-YAP/TAZ approach.29 For this reason, we aim for a small molecule stabilizer of the TAZ/14-3-3 interaction, which is expected to prevent translocation of the complex into the nucleus. We crystallized 14-3-3 bound to a TAZ-derived phosphopeptide, which was found to fully occupy the binding groove, analogous to the binding mode of a YAP phosphopeptide.30 Subsequently, we aimed to screen for chemical matter that could potentially modulate the interaction with TAZ, which required a different approach as the prominent binding site on 14-3-3 where PPI modulators bind is unavailable in this binary structure.
A recent study by Astex Pharmaceuticals, in which more than 5000 in-house crystal structures determined in the context of fragment-based ligand discovery projects were analyzed, revealed the existence of previously unrecognized secondary binding sites in each of the 24 analyzed protein targets.31 In addition, for 16 of these targets, multiple sites (up to six) could be discovered, suggesting that the occurrence of secondary sites that can accommodate small molecules is a general feature of most, if not all, proteins. This finding caught our attention and made us wonder if this feature also holds for and could be exploited in the case of 14-3-3 proteins and the search for new, more selective 14-3-3 PPI modulators. Therefore, we set out to screen the Novartis in-house fragment library against 14-3-3 in its apo form and against 14-3-3 in complex with a TAZ phosphopeptide. These complementary screens serve two purposes. The first is to identify novel fragments binding outside of the known 14-3-3 central binding groove, as it is blocked by the peptide. For the second, we hoped to detect fragments only interacting simultaneously with both 14-3-3 and the TAZ peptide as these could provide starting points for the development of 14-3-3/TAZ interaction stabilizers.
Here, we report on the results of this pioneering study and highlight the discovery of two individual secondary sites on 14-3-3 proteins. By employing nuclear magnetic resonance (NMR)-based fragment screening, we identified hit molecules binding to the binary complex of 14-3-3 and the TAZ phosphopeptide. X-ray crystallography then revealed the binding of these hit molecules at two surface-exposed sites, ∼20 Å from the central phospho-accepting groove of 14-3-3. The biological relevance of these sites is supported by their high degree of conservation among the 14-3-3 isoforms, as well as the previously elucidated crystal structures of 14-3-3 with serotonin N-acetyltransferase,32 H+-ATPase PMA2,33 and plant transcription factor FT,34 which clearly show that these sites are part of an interaction surface. This study opens up new possibilities for more specific 14-3-3 PPI modulation without targeting the central phosphoserine/phosphothreonine binding pocket of this adapter protein.
Materials and Methods
Peptide Synthesis
Peptides were synthesized via Fmoc solid phase peptide synthesis making use of an Intavis MultiPep RSi peptide synthesizer. The phosphorylated peptide was synthesized using Rink amide resin (Novabiochem), acetylated before deprotection and cleavage of the resin, and purified using preparative high-performance liquid chromatography with mass spectrometry detection.
Protein Expression and Purification
His6-tagged 14-3-3 proteins (full-length and ΔC) were expressed in NiCo21(DE3) competent cells with a pPROEX HTb plasmid and purified using Ni2+-affinity chromatography. The ΔC variant meant for crystallization was treated with TEV protease to cleave off the His6 tag, followed by a second Ni2+-affinity column and size exclusion chromatography.
Fragment Library
The Novartis fragment library screened for this study was described previously by Kutchukian et al.35 and comprises a core set of 1408 compounds. It was assembled by querying the Novartis archive for fragment-sized compounds (molecular weight of ≤300) that satisfied multiple property cutoffs (ClogP < 3, between 1 and 3 rings, ≤3 rotatable bonds, ≤3 H-bond donors, and ≤5 H-bond acceptors, more than 30 mg of solid and 20 or more fragment-sized analogues in the collection) and lacked undesirable substructures based on in-house and external knowledge (such as epoxides, Michael acceptors, S–S single bond, acyclic acetals, and phosphonamides). The fragments were further filtered on the basis of the number of chemical handles, diversity, and chemical attractiveness (based on in-house Bayesian models trained on medicinal chemists assessing HTS hit compounds). In addition, the fragments were required to have analogues in the archive. Quality control (QC) was then performed on the fragments: the identity, purity, and solubility of the compounds were determined by NMR, and additional profiling included binding to a SA BiaCore chip. After compounds had been filtered for acceptable QC and solubility (200 μM or better), ∼3700 compounds were submitted for review by chemists. Chemists were surveyed to assess whether they would be willing to carry forward fragments if they were identified as hits in a campaign, and the results of the survey were used to impact the final design of the library as follows. The 3700 fragments were clustered, and representative fragments from clusters were selected on the basis of either high solubility or high desirability by chemists, yielding a core set of 1408 fragments, termed the Novartis fourth-generation library.
NMR Spectroscopy
The Novartis fragment core library was screened in mixtures of eight against 10 μM 14-3-3ζ isoform, with a 20-fold molar excess of each ligand over protein. One-dimensional ligand-observed waterLOGSY36 and T1ρ(37) experiments were recorded to obtain binding information for the apo protein. The phosphorylated TAZ peptide was then added to a final concentration of 15 μM, and the experiments were repeated. Primary hits were chosen on the basis of comparison of the two data sets (e.g., in the presence and absence of the peptide) obtained for mixtures, and binding of each of the ligands was subsequently confirmed by testing them as singles. All ligand-observed experiments were performed at 296 K on a 600 MHz Bruker AVANCE III spectrometer, equipped with a triple-resonance cryogenic probe head.
Protein-observed experiments were performed at 310 K on an 800 MHz Bruker AVANCE III spectrometer, equipped with a triple-resonance cryo-probe. 1H–15N heteronuclear single-quantum coherence (HSQC) spectra were collected with 160 scans per increment and an acquisition time of 40 ms in the indirect dimension, resulting in total experimental time of 6.5 h per compound. For the analysis of chemical shift perturbations, the samples contained 50 μM uniformly 15N-labeled 14-3-3, 75 μM TAZ peptide, and 1 mM ligand. Titrations to determine binding affinity were performed on samples containing 100 μM 14-3-3 and 150 μM TAZpS89, with the ligand concentration increasing in five steps to 4 mM (protein:ligand molecular ratios of 1:2.5, 1:5, 1:10, 1:20, and 1:40). Data were processed with NMRPipe38 and analyzed in Sparky.39
Crystallography
The 14-3-3σ protein was C-terminally truncated (ΔC) after T231 to enhance crystallization. The 14-3-3 protein and TAZpS89 peptide were dissolved in complexation buffer [25 mM HEPES, 2 mM MgCl2, and 2 mM BME (pH 7.5)] and mixed in a stoichiometry of 1:1.5 (protein:peptide molar ratio) at a final protein concentration of 12.5 mg/mL. The complex was set up for hanging-drop crystallization after overnight incubation at 4 °C, in a homemade crystallization liquor [0.095 M HEPES, 0.19 M CaCl2, 28% (v/v) PEG 400, and 5% (v/v) glycerol (pH 7.1)]. Crystals grew within 10–14 days at 4 °C. Soaking of the fragment hits was performed by adding 0.6 μL of a 100 mM stock solution in dimethyl sulfoxide to 4 μL drops containing multiple crystals. Crystals were removed after overnight incubation at 4 °C and flash-cooled in liquid nitrogen. Diffraction data were collected either in-house with a Rigaku Compact HomeLab (TAZpS89, NV1 and NV2) or PETRA III, beamline P11, DESY, Hamburg, Germany (NV3).
Data sets were indexed and integrated using XDS40 and scaled using SCALA.41 The structures were phased by molecular replacement, using Protein Data Bank (PDB) entry 3MHR,30 in Phaser.42 Coot43 and phenix.refine44 were used in alternating cycles of model building and refinement. See Table S1 for data collection, structure determination, and refinement. The crystal structures were submitted to the PDB as entries 5N75, 5N5R, 5N5T, and 5N5W for the complexes of 14-3-3σ with TAZpS89, TAZpS89/NV1, TAZpS89/NV2, and TAZpS89/NV3, respectively.
Results and Discussion
The 14-3-3σ/TAZpS89 Complex as a Model System for Studying the “Ligandability” of 14-3-3 PPIs
In the context of the biological activities and biomedical importance of the transcriptional coactivator TAZ, we initially set out to determine the structure of the 14-3-3/TAZ complex and evaluate its “ligandability” using a fragment-based approach. TAZ is highly homologous with Yes-associated protein (YAP). However, whereas a crystal structure has been reported for the YAP-derived phosphopeptide (PDB entry 3MHR),30 no structure has been described for TAZ. To obtain a first picture of the interface of TAZ binding to 14-3-3, we co-crystallized a synthetic peptide derived from the TAZ sequence surrounding the phosphorylated Ser89 (TAZpS89, residues 86–98) with 14-3-3. To this end, 14-3-3 and TAZpS89 were mixed in a molar ratio of 1:1.5 and set up for crystallization at a protein concentration of 12.5 mg/mL. We tested several isoforms of 14-3-3 for co-crystallization and obtained robust and highly diffracting crystals with the σ isoform of 14-3-3 (14-3-3σ). A data set was collected to 1.80 Å resolution, and the structure was determined using the nearly identical 14-3-3σ/YAPpS127 structure (PDB entry 3MHR)30 as a search model. The electron density allowed us to build almost all 231 residues of the 14-3-3σ construct that has been used for crystallization. Of the TAZ peptide, 10 of 13 residues could be built showing how TAZpS89 binds in an elongated conformation to 14-3-3σ and revealing an interface that spans the entire binding channel (Figure 1a). As in other crystal structures with phosphorylated 14-3-3 binding motifs, key polar contacts between the peptide and 14-3-3 are 14-3-3’s K49, R56, and R128 that together with Y130 form the phospho-accepting pocket in 14-3-3, and N42, S45, N175, and N226 establishing contacts with the peptide’s main chain. In addition, side-chain to side-chain contacts are observed between D215 from 14-3-3σ and Q95 from TAZpS89. A hydrophobic contact surface for S90, P91, and L94 of the TAZpS89 peptide is formed by I168, G171, N175, I219, and L222 of 14-3-3σ (Figure 1b), reflecting a PPI interface that is dominated by polar interactions but also shows important hydrophobic elements. As mentioned above, this structure is nearly identical with that of the 14-3-3 binding motif of YAP surrounding pS12730 (Figure S1). The only sequence deviation in the YAP and TAZ peptides used in co-crystallography with 14-3-3 can be found at position −2 with respect to the phosphorylated serine. In YAP, this residue is an alanine (Ala125), and in TAZ, it is a serine (Ser87). This serine allows an additional polar contact to W230 of 14-3-3σ (Figure 1b). From the structure, it can be additionally observed that the glutamine at position 6 is clearly visible in the structure of 14-3-3σ with TAZpS89 but not in that with YAPpS127. An additional polar contact can be observed with the aspartate at position 215 of 14-3-3σ (Figure 1b).
Figure 1.

Crystal structure of the 14-3-3σ/TAZpS89 complex. (a) Final 2Fo – Fc electron density (blue mesh, contoured at 1.0σ) of the TAZpS89 peptide (orange sticks) bound to 14-3-3σ (white surface). (b) Details of the 14-3-3σ/TAZpS89 interaction. Residues of 14-3-3σ that are important for binding of TAZpS89 are shown as white sticks; polar interactions are depicted as black dotted lines, and hydrophobic contacts are displayed as a semitransparent surface. (c) Surface representation of the 14-3-3σ/TAZpS89 interface in overlay with ERα (green sticks) and Fusicoccin A (green lines and semitransparent spheres). The small molecule binding site is unavailable in the 14-3-3σ/TAZpS89 structure as the central binding channel of 14-3-3 is almost entirely filled by the TAZ peptide.
Given the great biomedical potential of a 14-3-3/TAZ stabilizing compound, we undertook a combined NMR- and crystallography-based fragment screening. We have shown previously how small molecules of the fusicoccin family stabilize the interaction of 14-3-3 with targets like C-Raf, ERα, Gab2, and CFTR.6,7,9,10 In all these cases, a prominent binding pocket is established between the C-terminus and the phosphorylated serine or threonine at the 14-3-3/peptide interface. As this pocket or parts thereof are entirely missing in the 14-3-3σ/TAZpS89 interface (Figure 1c), it suggests that targeting of this interface with previously developed approaches might be challenging but at the same time creates an opportunity to find novel binding pockets for small molecules by fragment-based ligand discovery.
Identification of 14-3-3 Binding Fragments via NMR
To first assess the overall ability of 14-3-3 to bind fragments, we screened the Novartis core fragment library (∼1400 molecules) using the apo form of the protein. Ligand-observed NMR experiments revealed a relatively high hit rate of nearly 8%. The screen was then repeated in the presence of the TAZ phosphopeptide described above (TAZpS89). Strikingly, only a handful of fragment mixtures displayed significant differences in ligand-observed NMR spectra between the apo form of the protein and the 14-3-3/TAZpS89 complex, and these differences were only small, suggesting a modulation of fragment binding affinity by the peptide rather than full displacement (Figure S2). Together, this suggests that binding of the vast majority of the hits is in fact independent of the peptide. Six fragments were confirmed to bind to the 14-3-3/TAZpS89 complex as singles, and these were further investigated by protein-observed NMR experiments. Two fragments induced considerable chemical shift perturbations in two-dimensional (2D) 1H–15N HSQC spectra (Figure 2). Interestingly, for both fragments, the observed chemical shift perturbations were of comparable magnitude when measured in the presence and absence of the phosphorylated TAZ peptide, again suggesting a lack of full displacement by the peptide. Despite the results from protein-observed NMR experiments, all six initial fragment hits were used to start crystal soaking experiments.
Figure 2.

Chemical shift perturbations induced by binding of fragments NV1 (left) and NV2 (right) to 14-3-3 in the apo form (top) and complexed with the TAZpS89 peptide (bottom). The reference 1H–15N HSQC spectrum of the 14-3-3/TAZpS89 complex is colored gray, that of the 14-3-3/TAZpS89/NV1 complex yellow, and that of the 14-3-3/TAZpS89/NV2 complex magenta. In addition to 100 μM 14-3-3, the samples contained (A) 1 mM NV1, (B) 150 μM TAZ and 1 mM NV1, (C) 1 mM NV2, and (D) 150 μM TAZ and 1 mM NV2. 2D 1H–15N HSQC spectra were recorded as described in Materials and Methods.
Identification of Two Secondary Binding Sites in 14-3-3 by X-ray Crystallography
The six fragment hits from the NMR-based screen were soaked into crystals of 14-3-3σ and the synthetic TAZ-derived peptide described above (TAZpS89). Additional electron density could be observed for fragments NV1 and NV2 (Figure 3a,b), which correspond to the two fragments for which chemical shift changes were observed in HSQC experiments. Both fragments were found not to bind in the proximity of the central binding groove of 14-3-3 or even close to the TAZ phosphopeptide, but at two neighboring but distinct pockets located on the “upper” rim of the 14-3-3 monomer ∼20 Å from the basic cluster that accepts the phosphorylated serine or threonine residue (Figure 3c,d). NV1 binds near the loop connecting helices 8 and 9 of 14-3-3 with its dimethylbenzyl ring pointing into a hydrophobic surface pocket lined by two methionine side chains (Met202 and Met229) with further contact surface contributed by Phe198, Thr217, Gln221, and Tyr213 (Figure 3e). Fragment NV2 occupies a shallow pocket also formed between helices 8 and 9 and comprised of Ile191, Phe198, Leu223, Leu227, and Thr231 as well as the hydrocarbon parts of Lys195 and Arg224 (Figure 3f).
Figure 3.

Crystal structures of fragments NV1 and NV2 binding to 14-3-3. (a) NV1 (yellow sticks) and (b) NV2 (magenta sticks) binding to 14-3-3σ (gray surface). The final 2Fo – Fc electron density map (contoured at 1.0σ) is shown as blue mesh. 14-3-3σ monomer (gray surface) in complex with (c) TAZ (residues 87–95, orange sticks) and NV1 (yellow spheres) or (d) NV2 (magenta spheres). Detailed view of the 14-3-3 residues in the binding site of (e) NV1 and (f) NV2.
Binding of both fragments to such remote pockets is somewhat unexpected considering they were selected on the basis of differential behavior in wLOGSY experiments (Figure S2); however, it corresponds well with the observation that ligand-observed NMR experiments suggested no full displacement by the peptide, and chemical shift perturbations induced by binding of these fragments to 14-3-3 occurred in the presence and absence of the TAZ peptide (Figure 2).
Binding Affinity of Fragments for the 14-3-3/TAZpS89 Complex
Considering the novelty of the binding sites and the fact that crystallography can detect even weak interactions, binding affinities were determined by NMR. Titration experiments with apo 14-3-3 and the 14-3-3/TAZpS89 complex were performed for fragment NV2, as the chemical shift perturbations induced by fragment NV2 were larger in magnitude than those for fragment NV1. In line with the observations of binding to a distant pocket illustrated above (Figure 3) and similar chemical shift perturbations induced by fragment hits in the presence and absence of the TAZ peptide (Figure 2), the NMR binding affinities for apo 14-3-3 and the binary 14-3-3/TAZpS89 complex were very similar and in the range of 1 mM (Figure S3), which corresponds to a moderate ligand efficiency of 0.27.
Fragment Hit Expansion
To probe the chemical space around the secondary binding sites and potential evolution of the fragment hits, further analogues of NV1 and NV2 were tested by NMR. Ligand-observed experiments confirmed binding of 12 additional fragments, which were then soaked into crystals of the 14-3-3/TAZpS89 complex, yielding an additional crystal structure for fragment NV3 (Figure 4). Like NV2, the fragment bound to a shallow pocket comprised of Ile191, Lys195, Asp199, Phe198, Leu223, Arg224, Leu227, and Thr231 (Figure 4b). In comparison with the apo structure, binding of NV2 and NV3 induces a 90° shift of the Arg224 side chain away from the fragment and a less prominent rotamer shift of Lys195 toward the fragment. Besides NV3, which is a close analogue of NV2, no additional structures could be obtained for the NV2 series. We believe that this is due to the crystal packing of 14-3-3 molecules that does not leave much space to accommodate larger fragments.
Figure 4.
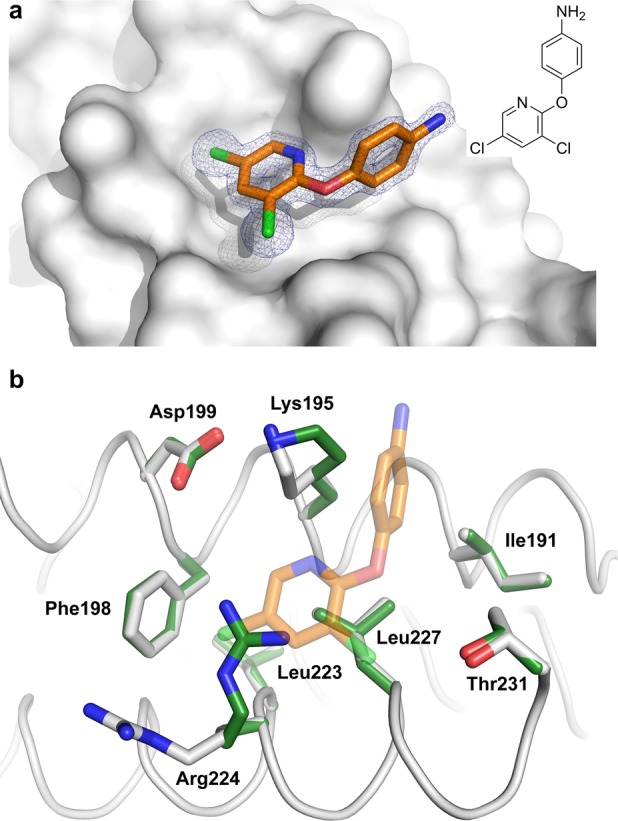
Crystal structure of fragment NV3 binding to 14-3-3. (a) NV3 (orange sticks) binding to 14-3-3σ (gray surface). The final 2Fo – Fc electron density map (contoured at 1.0σ) is shown as blue mesh. (b) Structural comparison of the 14-3-3 residues in the NV3 binding pocket in the presence (white sticks) and absence (green sticks) of NV3.
Conservation of the Secondary Binding Sites among Human 14-3-3 Isoforms
14-3-3 proteins are highly conserved among eukaryotes with sequence similarities between different species and intraspecies isoforms of around 50%.45 In humans, seven 14-3-3 isoforms can be found, all of which have been structurally elucidated by protein crystallography.46 Structure-based sequence comparisons reveal that the high degree of total sequence similarity is distributed unevenly among the protein surface of the 14-3-3 isoforms (Figure 5). In the central binding groove, which mediates the interaction with their mostly phosphorylated partner proteins, the degree of conservation is almost 100%. However, regions that are not directly involved in recognition of these primary interaction sites are significantly more variable, namely the “outer” surface of 14-3-3, opposite of the peptide binding grooves (Figure 5a).
Figure 5.

Conservation of surface-exposed residues among human 14-3-3 isoforms. (a) Residue conservation of the seven human isoforms of 14-3-3 determined with ConSurf47 after amino acid sequence alignment of the seven human isoforms of 14-3-3 performed with ClustalOmega.48 Corresponding protein entries (UniProt) P62258 for 14-3-3ε, P31947 for 14-3-3σ, Q04917 for 14-3-3η, P61981 for 14-3-3γ, P27348 for 14-3-3τ, P31946 for 14-3-3β, and P63104 for 14-3-3ζ. The TAZpS89 peptide is shown as green sticks. (b) The positions of the binding pockets of the fragments are indicated by yellow circles.
Interestingly, the binding sites of the fragments are highly conserved, although they are not directly part of the peptide binding groove (Figure 5b). The only differences for the identified secondary sites among the seven human 14-3-3 isoforms are Met202, in the binding pocket of NV1, and Ile191, in the binding pocket of NV2 and NV3. All six other 14-3-3 isoforms display an isoleucine at the M202 position of 14-3-3σ (Figure S4) and a cysteine at position Ile191 of 14-3-3σ (Figure S5). Whereas the methionine-to-isoleucine change can be expected to be less relevant, the isoleucine-to-cysteine difference is more substantial and could, for example, also be exploited for covalent fragment or compound binding. Such a high degree of conservation in protein surfaces usually suggests a functional relevance in, for example, ligand binding or protein–protein interactions. Indeed, three crystal structures of 14-3-3 in complex with a partner protein extending the primary, peptidic binding motif have been reported: serotonin N-acetyltransferase (AANAT),32 plant proton pump PMA2,33 and plant transcription factor FT.34
Involvement of Secondary Sites in 14-3-3 PPIs
Most of the crystal structures published in the PDB until now for 14-3-3 have been binary complexes of the 14-3-3 protein bound to a phosphorylated peptide derived from the interaction partner. As stated before, these phosphorylated motifs bind in the central recognition groove of 14-3-3, and this has become the state of the art in crystallographic studies of 14-3-3 PPIs. However, as it is becoming more apparent that additional interaction surfaces are involved in the affinity and recognition of PPIs and may contain hot spots for more specific PPI modulation, the examples of AANAT,32 PMA2,33 and plant transcription factor FT,34 describing larger protein domains bound to 14-3-3, are highly relevant with respect to 14-3-3 drug discovery efforts. We were interested in seeing if the fragment-bound secondary sites we identified are part of the interaction interfaces in these binary structures. Indeed, AANAT and PMA2 bind to 14-3-3 in the proximity of the NV1 pocket (Figures 6 and 7), and the Hd3a protein docks onto 14-3-3 at the surface in which both secondary site pockets are located (Figure 8). In the next section, we will go into some detail describing these structures.
Figure 6.

Position of the secondary binding sites of NV1 and NV2 in relation to the 14-3-3ζ/AANAT complex. (a) Overview of the complex of a 14-3-3 dimer (gray surface) bound to two AANAT monomers (orange surface) (PDB entry 1IB1). (b) Superimposition of the 14-3-3 structure with NV1 (yellow spheres and sticks) and NV2 (magenta spheres and sticks), with one monomer of AANAT (orange cartoon and semitransparent surface) bound to one monomer of 14-3-3ζ. (c) Detailed view of Glu43 of AANAT binding in the vicinity of the NV1 pocket in 14-3-3.
Figure 7.

Position of the secondary binding sites of NV1 and NV2 in relation to the tobacco 14-3-3/PMA2-CT52 complex. (a) Overview of the complex of the tobacco T14-3c dimer (gray surface) bound to two PMA-CT52 molecules (blue surface) (PDB entry 2O98). (b) Superimposition of the 14-3-3 structure with NV1 (yellow spheres and sticks) and NV2 (magenta spheres and sticks), with PMA2-CT52 (blue cartoon and semitransparent surface) bound to one monomer of T14-3c (gray surface). (c) Detailed view of the main helix of CT52 binding in the vicinity of the NV1 pocket in 14-3-3.
Figure 8.

Position of the secondary binding sites of NV1 and NV2 in relation to the rice 14-3-3/FT complex. (a) Overview of the complex of the rice 14-3-3 GF14c dimer (gray surface) bound to two FT molecules (green surface) (PDB entry 3AXY). (b) Superimposition of the 14-3-3 structure with NV1 (yellow spheres and sticks) and NV2 (magenta spheres and sticks) bound to one monomer of 14-3-3σ (gray surface) with FT (green cartoon and semitransparent surface) bound to GF14c (gray surface). (c) Detailed view of FT interactions at the surface containing both the NV1 and NV2 pockets in 14-3-3.
The 14-3-3ζ/AANAT Complex
Serotonin N-acetyltransferase (AANAT) catalyzes the transfer of an acetyl group from acetyl-coenzyme A to serotonin, thereby producing the precursor of melatonin. Melatonin levels are strictly coupled to the daily rhythm, with high levels occurring at night and thus providing a hormonal analogue signal of environmental lighting, which can be used to optimize circadian physiology.49,50 In 2001, the group of Dyda published the crystal structure of 14-3-3ζ in complex with AANAT showing that binding to 14-3-3 significantly increases its affinity for its substrate serotonin, thus activating the enzyme.32 In the crystal structure, two AANAT molecules (residues 18–196) bind to the central channel of a 14-3-3 dimer (Figure 6a). In addition to the phosphorylation-dependent interaction of the N-terminus of AANAT in the central groove, the well-structured C-terminal part of the enzyme makes extensive contacts with the inner wall of the 14-3-3 dimer (Figure 6b). Here, the side chain of Glu43 in AANAT helix 1 points into the NV1 binding site for interaction with 14-3-3 (Figure 6c).
The T14-3c/PMA2-CT52 Complex
Almost all transport mechanisms at the plasma membrane of plant cells are energetically dependent on the electrochemical proton gradient, which is generated by H+-ATPase PMA.51−54 One of the most important regulatory events for PMA is phosphorylation of the penultimate C-terminal threonine followed by binding of 14-3-3 proteins, which activates PMA.55,56 This activation is strongly enhanced by the natural product fusicoccin,44,45 which fills a gap in the interface of 14-3-3 with the regulatory C-terminus (CT) of PMA.33,59 The structure of the last 52 amino acids of the C-terminus of PMA isoform 2 (PMA2-CT52) from tobacco (Nicotiana plumbaginifolia) in complex with 14-3-3 revealed two PMA C-termini bound to one 14-3-3 dimer (Figure 7a). The 30 C-terminal amino acids bind as an elongated peptide and a short helix in the amphipathic groove of each 14-3-3 monomer, whereas the 22 N-terminal amino acids of PMA2-CT52 form a helix that perpendicularly leaves the 14-3-3 binding channel (Figure 7b). This helix comes into the proximity of the NV1 binding pocket (Figure 7c), which suggests that a slightly extended molecule based on NV1 could make direct physical contact with PMA-CT52 to modulate its interaction with 14-3-3.57,58
The 14-3-3/FT Complex
“Florigen” is a flowering-inducing molecule that is encoded by the highly conserved plant gene FLOWERING LOCUS T (FT).60,61 In 2011, it was shown that the FT protein from rice (Hd3a) binds to 14-3-3 proteins in the apical cells of shoots to form a complex that migrates into the nucleus where it interacts with basic leucine zipper (bZIP) transcription factor FD.34 In the crystal structure, the entire construct of Hd3a (residues 6–170) is visible, making it the second-largest 14-3-3 partner protein after AANAT (residues 18–196) that has been co-crystallized with 14-3-3. Two Hd3a molecules bind to one 14-3-3 (GF14c) dimer and occupy an interesting position, which is not part of the central binding channel as seen with all other 14-3-3 ligands, including AANAT. Rather, Hd3a binds to the “upper” edges of the horseshoe-like 14-3-3 dimer (Figure 8a). This site buries the pockets of NV1 and NV2 (Figure 8b). In particular, the hydrophobic interface in 14-3-3 that is used by the ring system of NV1 is occupied by Met63 and Thr98 of Hd3a in the GF14c/Hd3a complex (Figure 8c). Likewise, the pocket of NV2 is used by Ser102 and Phe103 of Hd3a to bind to GF14c (Figure 8c). Given the importance of the regulation of transcription factor FT by 14-3-3 proteins in plant physiology, small molecules that modulate this PPI might be useful for studying the molecular details of plant flowering.
Very recently, the crystal structure of 14-3-3σ in complex with small heat-shock protein HSPB6 has been published.62 Here, the 14-3-3 dimer forms an asymmetric complex with the dimer of HSPB6. The pockets of fragments NV1–NV3 are also part of the interface between the compact HSPB6 dimer binding to 14-3-3, further strengthening the significance of these secondary binding sites.
Conclusion
Small molecule modulation of protein–protein interactions is currently one of the most promising and active fields in drug discovery and chemical biology. Hub protein 14-3-3 interacts with several hundred partner proteins and is thus involved in nearly every physiological process and human disease. Hence, small molecules that can modulate these interactions are of great interest. We applied a fragment-based screen combining NMR and X-ray crystallography to identify chemical starting points for the modulation of the interaction between 14-3-3 and TAZ. Aside from a genuine interest in finding stabilizers of the 14-3-3/TAZ interaction, another reason to select this system was the binding mode of the peptide, in which it occupies the entire phosphopeptide binding groove on 14-3-3. The natural product pocket, which is targeted by fusicoccin in the 14-3-3/ERα complex7 or cotylenin A in the 14-3-3/CRAF complex,6 was therefore found to be inaccessible for 14-3-3 bound to TAZpS89, permitting the discovery of novel sites.
In this study, we present the first identification of two individual secondary binding sites on 14-3-3 proteins. We crystallized fragments bound to these novel pockets at a surface ∼20 Å from the central binding groove of 14-3-3. This discovery holds promise for the development of more selective 14-3-3 modulators, because on the basis of our current understanding, these sites are used by only a few partner proteins. We believe that our results provide solid chemical starting points and convincingly warrant future ligand optimization campaigns for the development of 14-3-3 modulators with an unprecedented mode of action.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00153.
Crystallographic data (Table S1), overlay of the TAZ/YAP crystal structure binding to 14-3-3, one-dimensional ligand-observed waterLOGSY NMR spectra and 2D 1H–15N HSQC NMR spectra of validated hits and titration experiments, and structural alignments of NV1 and NV2 binding pockets (PDF)
Author Contributions
E.S. and L.S. contributed equally to this work.
We acknowledge the financial support of the Collaborative Research Centre 1093, funded by the Deutsche Forschungsgemeinschaft (DFG), Marie Curie Action PIAPP-GA-2011-286418 14-3-3Stabs, and The Netherlands Organization for Scientific Research (NWO) via Gravity Program 024.001.035 and VICI Grant 016.150.366.
The authors declare no competing financial interest.
Supplementary Material
References
- Milroy L.-G.; Brunsveld L.; Ottmann C. (2013) Stabilization and inhibition of protein-protein interactions: the 14–3-3 case study. ACS Chem. Biol. 8, 27–35. 10.1021/cb300599t. [DOI] [PubMed] [Google Scholar]
- Tinti M.; Madeira F.; Murugesan G.; Hoxhaj G.; Toth R.; Mackintosh C. (2014) ANIA: ANnotation and Integrated Analysis of the 14–3-3 interactome. Database 2014, bat085. 10.1093/database/bat085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghazadeh Y.; Papadopoulos V. (2016) The role of the 14–3-3 protein family in health, disease, and drug development. Drug Discovery Today 21, 278–287. 10.1016/j.drudis.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Johnson C.; Crowther S.; Stafford M. J.; Campbell D. G.; Toth R.; MacKintosh C. (2010) Bioinformatic and experimental survey of 14–3-3-binding sites. Biochem. J. 427, 69–78. 10.1042/BJ20091834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C.; Higuchi Y.; Koschinsky K.; Bartel M.; Schumacher B.; Thiel P.; Nitta H.; Preisig-Müller R.; Schlichthörl G.; Renigunta V.; Ohkanda J.; Daut J.; Kato N.; Ottmann C. (2013) A semisynthetic fusicoccane stabilizes a protein-protein interaction and enhances the expression of K+ channels at the cell surface. Chem. Biol. 20, 583–593. 10.1016/j.chembiol.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Molzan M.; Kasper S.; Röglin L.; Skwarczynska M.; Sassa T.; Inoue T.; Breitenbuecher F.; Ohkanda J.; Kato N.; Schuler M.; Ottmann C. (2013) Stabilization of physical RAF/14–3-3 interaction by cotylenin A as treatment strategy for RAS mutant cancers. ACS Chem. Biol. 8, 1869–1875. 10.1021/cb4003464. [DOI] [PubMed] [Google Scholar]
- De Vries-van Leeuwen I. J.; da Costa Pereira D.; Flach K. D.; Piersma S. R.; Haase C.; Bier D.; Yalcin Z.; Michalides R.; Feenstra K. A.; Jiménez C. R.; de Greef T. F. A.; Brunsveld L.; Ottmann C.; Zwart W.; de Boer A. H. (2013) Interaction of 14–3-3 proteins with the estrogen receptor alpha F domain provides a drug target interface. Proc. Natl. Acad. Sci. U. S. A. 110, 8894–8899. 10.1073/pnas.1220809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skwarczynska M.; Molzan M.; Ottmann C. (2013) Activation of NF-κB signalling by fusicoccin-induced dimerization. Proc. Natl. Acad. Sci. U. S. A. 110, E377–386. 10.1073/pnas.1212990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier D.; Bartel M.; Sies K.; Halbach S.; Higuchi Y.; Haranosono Y.; Brummer T.; Kato N.; Ottmann C. (2016) Small-Molecule Stabilization of the 14–3-3/Gab2 Protein-Protein Interaction (PPI) Interface. ChemMedChem 11, 911–918. 10.1002/cmdc.201500484. [DOI] [PubMed] [Google Scholar]
- Stevers L. M.; Lam C. V.; Leysen S. F. R.; Meijer F. A.; van Scheppingen D. S.; de Vries R. M. J. M.; Carlile G. W.; Milroy L.-G.; Thomas D. Y.; Brunsveld L.; Ottmann C. (2016) Characterization and small-molecule stabilization of the multisite tandem binding between 14–3-3 and the R domain of CFTR. Proc. Natl. Acad. Sci. U. S. A. 113, E1152–1161. 10.1073/pnas.1516631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas A.; Bier D.; Hahne G.; Rademacher C.; Ottmann C.; Grossmann T. N. (2014) Constrained peptides with target-adapted cross-links as inhibitors of a pathogenic protein-protein interaction. Angew. Chem., Int. Ed. 53, 2489–2493. 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]
- Milroy L.-G.; Bartel M.; Henen M. A.; Leysen S.; Adriaans J. M. C.; Brunsveld L.; Landrieu I.; Ottmann C. (2015) Stabilizer-Guided Inhibition of Protein-Protein Interactions. Angew. Chem., Int. Ed. 54, 15720–15724. 10.1002/anie.201507976. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Du Y.; Horton J. R.; Upadhyay A. K.; Lou B.; Bai Y.; Zhang X.; Du L.; Li M.; Wang B.; Zhang L.; Barbieri J. T.; Khuri F. R.; Cheng X.; Fu H. (2011) Discovery and structural characterization of a small molecule 14–3-3 protein-protein interaction inhibitor. Proc. Natl. Acad. Sci. U. S. A. 108, 16212–16216. 10.1073/pnas.1100012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel P.; Röglin L.; Meissner N.; Hennig S.; Kohlbacher O.; Ottmann C. (2013) Virtual screening and experimental validation reveal novel small-molecule inhibitors of 14–3-3 protein-protein interactions. Chem. Commun. 49, 8468–8470. 10.1039/c3cc44612c. [DOI] [PubMed] [Google Scholar]
- Bier D.; Rose R.; Bravo-Rodriguez K.; Bartel M.; Ramirez-Anguita J. M.; Dutt S.; Wilch C.; Klärner F.-G.; Sanchez-Garcia E.; Schrader T.; Ottmann C. (2013) Molecular tweezers modulate 14–3-3 protein-protein interactions. Nat. Chem. 5, 234–239. 10.1038/nchem.1570. [DOI] [PubMed] [Google Scholar]
- de Vink P. J.; Briels J. M.; Schrader T.; Milroy L.-G.; Brunsveld L.; Ottmann C. (2017) A binary bivalent supramolecular assembly platform based on cucurbit[8]uril and dimeric adaptor protein 14-3-3. Angew. Chem., Int. Ed. 56, n/a. 10.1002/anie.201701807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottmann C.; Yasmin L.; Weyand M.; Veesenmeyer J. L.; Diaz M. H.; Palmer R. H.; Francis M. S.; Hauser A. R.; Wittinghofer A.; Hallberg B. (2007) Phosphorylation-independent interaction between 14–3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26, 902–913. 10.1038/sj.emboj.7601530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromm P. M.; Wallraven K.; Glas A.; Bier D.; Fürstner A.; Ottmann C.; Grossmann T. N. (2016) Constraining an Irregular Peptide Secondary Structure through Ring-Closing Alkyne Metathesis. ChemBioChem 17, 1915–1919. 10.1002/cbic.201600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F.; Marignani P. A.; Sarbassova D.; Yagi R.; Hall R. A.; Donowitz M.; Hisaminato A.; Fujiwara T.; Ito Y.; Cantley L. C.; Yaffe M. B. (2000) TAZ: a novel transcriptional co-activator regulated by interactions with 14–3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791. 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C. G.; Moroishi T.; Guan K.-L. (2015) YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 25, 499–513. 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S. (2016) Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 343, 42–53. 10.1016/j.yexcr.2015.10.034. [DOI] [PubMed] [Google Scholar]
- Yu F.-X.; Zhao B.; Guan K.-L. (2015) Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey K. F.; Pfleger C. M.; Hariharan I. K. (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467. 10.1016/S0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- Moroishi T.; Hansen C. G.; Guan K.-L. (2015) The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 15, 73–79. 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F.; Battilana G.; Cordenonsi M.; Piccolo S. (2016) YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 29, 26–33. 10.1016/j.coph.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z.; Moroishi T.; Guan K.-L. (2016) Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Wei X.; Li W.; Udan R. S.; Yang Q.; Kim J.; Xie J.; Ikenoue T.; Yu J.; Li L.; Zheng P.; Ye K.; Chinnaiyan A.; Halder G.; Lai Z.-C.; Guan K.-L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santinon G.; Pocaterra A.; Dupont S. (2016) Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 26, 289–299. 10.1016/j.tcb.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Zanconato F.; Battilana G.; Cordenonsi M.; Piccolo S. (2016) YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 29, 26–33. 10.1016/j.coph.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B.; Skwarczynska M.; Rose R.; Ottmann C. (2010) Structure of a 14–3-3σ-YAP phosphopeptide complex at 1.15 A resolution. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 66, 978–984. 10.1107/S1744309110025479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow R. F.; Verdonk M. L.; Saini H. K.; Tickle I. J.; Jhoti H. (2015) Detection of secondary binding sites in proteins using fragment screening. Proc. Natl. Acad. Sci. U. S. A. 112, 15910–15915. 10.1073/pnas.1518946112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T.; Ghirlando R.; Klein D. C.; Ganguly S.; Dyda F. (2001) Crystal structure of the 14–3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell 105, 257–267. 10.1016/S0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
- Ottmann C.; Marco S.; Jaspert N.; Marcon C.; Schauer N.; Weyand M.; Vandermeeren C.; Duby G.; Boutry M.; Wittinghofer A.; Rigaud J.-L.; Oecking C. (2007) Structure of a 14–3-3 coordinated hexamer of the plant plasma membrane H+ -ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell 25, 427–440. 10.1016/j.molcel.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Taoka K.; Ohki I.; Tsuji H.; Furuita K.; Hayashi K.; Yanase T.; Yamaguchi M.; Nakashima C.; Purwestri Y. A.; Tamaki S.; Ogaki Y.; Shimada C.; Nakagawa A.; Kojima C.; Shimamoto K. (2011) 14–3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature 476, 332–335. 10.1038/nature10272. [DOI] [PubMed] [Google Scholar]
- Kutchukian P. S.; Wassermann A. M.; Lindvall M. K.; Wright S. K.; Ottl J.; Jacob J.; Scheufler C.; Marzinzik A.; Brooijmans N.; Glick M. (2015) Large scale meta-analysis of fragment-based screening campaigns: privileged fragments and complementary technologies. J. Biomol. Screening 20, 588–596. 10.1177/1087057114565080. [DOI] [PubMed] [Google Scholar]
- Gossert A. D.; Henry C.; Blommers M. J. J.; Jahnke W.; Fernández C. (2009) Time efficient detection of protein-ligand interactions with the polarization optimized PO-WaterLOGSY NMR experiment. J. Biomol. NMR 43, 211–217. 10.1007/s10858-009-9303-5. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Meadows R. P.; Fesik S. W. (1997) Discovering high-affinity ligands for proteins. Science 278, 497–499. 10.1126/science.278.5337.497. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Lee W.; Tonelli M.; Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327. 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjarland E.; Keen T. J.; Kleppe R. (2006) Does isoform diversity explain functional differences in the 14–3-3 protein family?. Curr. Pharm. Biotechnol. 7, 217–223. 10.2174/138920106777549777. [DOI] [PubMed] [Google Scholar]
- Yang X.; Lee W. H.; Sobott F.; Papagrigoriou E.; Robinson C. V.; Grossmann J. G.; Sundström M.; Doyle D. A.; Elkins J. M. (2006) Structural basis for protein-protein interactions in the 14–3-3 protein family. Proc. Natl. Acad. Sci. U. S. A. 103, 17237–17242. 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser F.; Morris R. J.; Najmanovich R. J.; Laskowski R. A.; Thornton J. M. (2006) A method for localizing ligand binding pockets in protein structures. Proteins: Struct., Funct., Genet. 62, 479–488. 10.1002/prot.20769. [DOI] [PubMed] [Google Scholar]
- Bodenhofer U.; Bonatesta E.; Horejš-Kainrath C.; Hochreiter S. (2015) msa: an R package for multiple sequence alignment. Bioinformatics 31, 3997–3999. 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- Arendt J. (2005) Melatonin in humans: it’s about time. J. Neuroendocrinol. 17, 537–538. 10.1111/j.1365-2826.2005.01333.x. [DOI] [PubMed] [Google Scholar]
- Ooms S.; Ju Y.-E. (2016) Treatment of Sleep Disorders in Dementia. Current Treatment Options in Neurology 18, 40. 10.1007/s11940-016-0424-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren M. G. (2001) PLANT PLASMA MEMBRANE H+-ATPases: Powerhouses for Nutrient Uptake. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 817–845. 10.1146/annurev.arplant.52.1.817. [DOI] [PubMed] [Google Scholar]
- Arango M.; Gévaudant F.; Oufattole M.; Boutry M. (2003) The plasma membrane proton pump ATPase: the significance of gene subfamilies. Planta 216, 355–365. 10.1007/s00425-002-0856-8. [DOI] [PubMed] [Google Scholar]
- Sondergaard T. E.; Schulz A.; Palmgren M. G. (2004) Energization of transport processes in plants. roles of the plasma membrane H+-ATPase. Plant Physiol. 136, 2475–2482. 10.1104/pp.104.048231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duby G.; Boutry M. (2009) The plant plasma membrane proton pump ATPase: a highly regulated P-type ATPase with multiple physiological roles. Pfluegers Arch. 457, 645–655. 10.1007/s00424-008-0457-x. [DOI] [PubMed] [Google Scholar]
- Jahn T.; Fuglsang A. T.; Olsson A.; Brüntrup I. M.; Collinge D. B.; Volkmann D.; Sommarin M.; Palmgren M. G.; Larsson C. (1997) The 14–3-3 protein interacts directly with the C-terminal region of the plant plasma membrane H(+)-ATPase. Plant Cell 9, 1805–1814. 10.2307/3870526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski M.; Morsomme P.; Boutry M.; Oecking C. (1998) Complementation of the Saccharomyces cerevisiae plasma membrane H+-ATPase by a plant H+-ATPase generates a highly abundant fusicoccin binding site. J. Biol. Chem. 273, 30018–30023. 10.1074/jbc.273.45.30018. [DOI] [PubMed] [Google Scholar]
- Olsson A.; Svennelid F.; Ek B.; Sommarin M.; Larsson C. (1998) A phosphothreonine residue at the C-terminal end of the plasma membrane H+-ATPase is protected by fusicoccin-induced 14–3-3 binding. Plant Physiol. 118, 551–555. 10.1104/pp.118.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svennelid F.; Olsson A.; Piotrowski M.; Rosenquist M.; Ottman C.; Larsson C.; Oecking C.; Sommarin M. (1999) Phosphorylation of Thr-948 at the C terminus of the plasma membrane H(+)-ATPase creates a binding site for the regulatory 14–3-3 protein. Plant Cell 11, 2379–2391. 10.2307/3870962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würtele M.; Jelich-Ottmann C.; Wittinghofer A.; Oecking C. (2003) Structural view of a fungal toxin acting on a 14–3-3 regulatory complex. EMBO J. 22, 987–994. 10.1093/emboj/cdg104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y.; Weigel D. (2007) Move on up, it’s time for change--mobile signals controlling photoperiod-dependent flowering. Genes Dev. 21, 2371–2384. 10.1101/gad.1589007. [DOI] [PubMed] [Google Scholar]
- Tamaki S.; Matsuo S.; Wong H. L.; Yokoi S.; Shimamoto K. (2007) Hd3a protein is a mobile flowering signal in rice. Science 316, 1033–1036. 10.1126/science.1141753. [DOI] [PubMed] [Google Scholar]
- Sluchanko N. N.; Beelen S.; Kulikova A. A.; Weeks S. D.; Antson A. A.; Gusev N. B.; Strelkov S. V. (2017) Structural Basis for the Interaction of a Human Small Heat Shock Protein with the 14–3-3 Universal Signaling Regulator. Structure 25, 305–316. 10.1016/j.str.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.