

Abstract
Omics approaches are high-throughput unbiased technologies that provide snapshots of various aspects of biological systems and include: 1) genomics, the measure of DNA variation; 2) transcriptomics, the measure of RNA expression; 3) epigenomics, the measure of DNA alterations not involving sequence variation that influence RNA expression; 4) proteomics, the measure of protein expression or its chemical modifications; and 5) metabolomics, the measure of metabolite levels. Our understanding of pulmonary diseases has increased as a result of applying these omics approaches to characterize patients, uncover mechanisms underlying drug responsiveness, and identify effects of environmental exposures and interventions. As more tissue- and cell-specific omics data is analyzed and integrated for diverse patients under various conditions, there will be increased identification of key mechanisms that underlie pulmonary biological processes, disease endotypes, and novel therapeutics that are efficacious in select individuals. We provide a synopsis of how omics approaches have advanced our understanding of asthma, chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), idiopathic pulmonary fibrosis (IPF), and pulmonary arterial hypertension (PAH), and we highlight ongoing work that will facilitate pulmonary disease precision medicine.
Keywords: Pulmonary diseases, Genomics, Transcriptomics, Epigenomics, Proteomics, Metabolomics
Background
The commoditization of high-throughput biotechnologies has enabled the collection of an unprecedentedly large number of so-called omics datasets by biomedical researchers. Starting with DNA microarrays in the 1990s and expanding to next-generation sequencing (NGS) in the 2000s, omics approaches now capture a wide variety of biological measurements, spanning DNA variation to chemical modifications of proteins [Fig. 1] [1–3]. As the repertoire of available omics approaches continues to expand with the development of methods that combine existing assays and new technologies, an unbiased characterization of biological systems at ever-increasing resolutions is possible [4, 5]. Early successes in the use of omics technologies to understand disease and enable drug development [6, 7] have resulted in optimism that many more effective diagnostic tests and treatments tailored to individuals’ genetic, environmental and lifestyle factors will be developed. The commonplace use of such tests and diagnostics is often referred to as personalized medicine, or more recently, precision medicine [8, 9].
Fig. 1.
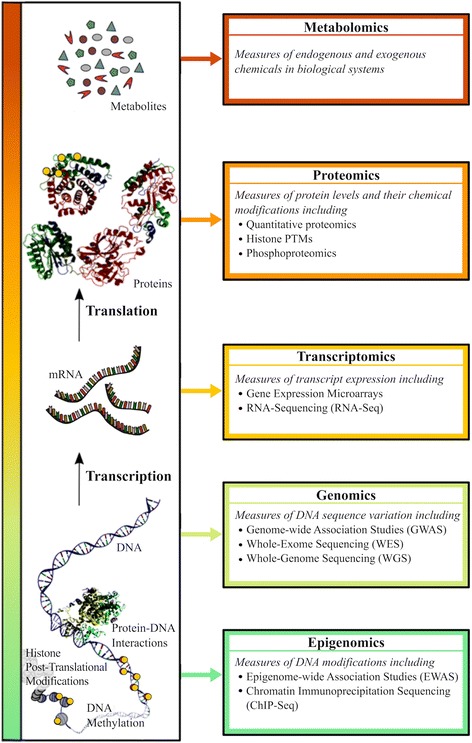
Summary of omics approaches discussed: layers of biological data (left) with corresponding omics techniques used for their characterization (right)
Decision-making in medical care for decades has often relied on a “one-size-fits-all” approach that applies mean-effect-size results from studies to individual patients [10]. The goal of precision medicine, in contrast, is to allow for more accurate treatment and prevention decisions based on matching a patient’s exposure, lifestyle and biological profile to that of similar patients with measured outcomes. The goal of many omics studies thus far has been to build a knowledgebase of omic variation using single-technology approaches that will help enable precision medicine by providing reference data to identify groups of individuals who share various attributes. In addition to collecting omics data, the application of sophisticated algorithms and use of extensive computational resources to integrate datasets are required to fully characterize diverse patients [11, 12].
Here, we provide a broad overview of how omics approaches have been used to understand five complex pulmonary diseases that stand to benefit from personalized diagnostics and treatment:
Asthma, an inflammatory disease characterized by variable airflow limitation and airway hyperresponsiveness, that affects over 25 million Americans [13].
Chronic obstructive pulmonary disease (COPD), a disease characterized by alveolar destruction, shortness of breath, cough, and sputum production, which is estimated to affect over 24 million Americans [14].
Acute respiratory distress syndrome (ARDS), a severe lung condition with high fatality rate that typically occurs in critically ill patients and is characterized by the acute development of diffuse alveolar injury leading to respiratory failure [15].
Idiopathic pulmonary fibrosis (IPF), a progressive disease that is the most common and lethal type of idiopathic interstitial pneumonias and is characterized by scarring fibrosis with an unpredictable course [16].
Pulmonary arterial hypertension (PAH), a disease that predominantly affects women and is characterized by endothelial proliferation and smooth muscle hypertrophy of small pulmonary arteries, in situ thrombosis, and plexiform lesions that lead to right ventricular failure, and ultimately, death [17].
Some of the main findings obtained by applying omics approaches to these diseases are summarized in Table 1. As our goal is to offer an extensive multi-level omics review, readers interested in learning more about specific techniques or their application to disease are encouraged to read the papers in Table 1 and other in-depth resources.
Table 1.
Summary of main findings for various omics approaches applied to the study of pulmonary diseases
| Disease | Main findings |
|---|---|
| Genomics | |
| Genome-wide/Exome-wide microarray | |
| Asthma | Prominent asthma-associated loci are 17q21 locus (including ORMDL3, GSDMB), IL33, IL1RL1, TSLP [32] Rare, potentially functional variants within GRASP, GSDMB, and MTHFR are associated differently with asthma in subjects of Latino and African ancestry [56] Severe asthma-associated loci are CDHR3, GSDMB, IL33 and IL1RL1 [43] |
| IgE levels | FCER1A and HLA-DQB1 are associated with IgE levels, the latter in asthma patients only [47] |
| Asthma drug response |
SPATS2L is associated with bronchodilator response in asthma patients [54] GLCCI1 is associated with lung function in patients treated with inhaled glucocorticoids [55] |
| COPD | Robust COPD-associated loci are FAM13A, CHRNA3/CHRNA5/IREB2, HHIP [33] Rare, potentially functional variants in MOCS3, IFIT3 and SERPINA12 are associated with COPD and airflow limitation [58] |
| COPD endotype | BICD1 is associated with emphysema [44] |
| Lung function | FAM13A, HHIP, HTR4 are associated with both lung function (i.e. FEV1 and FEV1/FVC ratio) and COPD [48] |
| IPF | TERT and MUC5B are associated with IPF [61, 62] |
| PAH | CBLN2 is associated with PAH in patients without BMPR2 mutations [65] |
| Whole exome sequencing | |
| COPD | Increased number of rare, non-silent mutations in DNAH8, ALCAM, RARS, and GBF1 are present in severe, early-onset COPD [57] |
| PAH | High penetrance missense variants in KCNK3 and TOPBP1 found in familial PAH and idiopathic PAH [67, 68] |
| Transcriptomics | |
| Gene expression microarray | |
| Asthma | Bitter taste receptors have increased expression in severe asthma [86] Distinct epithelial gene expression signature involving in interferon response found in severe childhood asthma [87] Transcriptional activation of circulating CD8+ T cells but not CD4+ T cells present in severe asthma [88] |
| Asthma endotype | Severe asthma subgroups defined based on transcriptomic and clinical characteristics [92–94] |
| Asthma drug response | KLF15 is a glucocorticoid responsive gene in ASM cells [101] |
| COPD | Distinct PBMC gene expression representing immune, inflammatory response and sphingolipid metabolism pathways, and including ASAH1, involved in COPD and emphysema [97] Sputum gene expression changes, including IL18R1, are associated with COPD severity [98] Increased gene expression of neutrophil proteases found in COPD patients with respiratory distress [99] |
| ARDS | Blood neutrophil-related genes and pre-elafin are potential biomarkers in early sepsis-induced ARDS [106] and in acute stage of ARDS [107], respectively Neutrophil gene expression changes in ARDS similar to those in sepsis and burns [108] |
| IPF |
CCNA2 and alpha-defensin genes are upregulated in lung tissue of IPF patients with acute exacerbations [109] PBMC CD28, ICOS, LCK, and ITK are predictors of poor outcomes (transplantation, death) in IPF [110] |
| PAH | Expression changes in BMP2 and BMPR2 are associated with PAH, even in tissues from patients without BMPR2 mutations [114] |
| RNA-Seq | |
| Asthma | Differential expression of SLC26A4, POSTN, and BCL2 observed in endobronchial biopsies from asthma patients [89] |
| Asthma drug response |
CRISPLD2 is a glucocorticoid responsive gene in ASM cells [103] Glucocorticoid-induced genes in ASM from asthma donors include FAM129A and SYNPO2 [104] Cytokine gene expression is modulated by vitamin D treatment in ASM [105] |
| IPF | Splicing changes in lung tissue COL6A3 and POSTN are associated with IPF [111] |
| Epigenomics | |
| Methylation microarray | |
| Asthma | Hypomethylation of IL13, RUNX3 and TIGIT observed in PBMCs of patients with persistent atopic asthma [136] SMAD3 methylation at birth is associated with asthma in children of mothers with asthma [140] |
| IgE levels |
AFPM1, ACOT7, and MND1 methylation are associated with total serum IgE levels in Hispanic children [141] Serum IgE levels are associated with low methylated loci within/near genes encoding known eosinophil products (e.g., IL5RA, IL1RL1, GATA1) [142] |
| COPD | Methylation of C10orf11, a known COPD-associated gene identified via GWAS, observed in lung of smokers who develop COPD [134] EPAS1 identified as a key regulator of COPD by combining lung methylation and gene expression data [145] |
| IPF | Methylation changes observed in CDKN2B, CAR10 and MGMT in fibroblasts from IPF patients [150] Hypermethylation of CASZ1, and subsequent gene expression changes, are observed in lung of IPF patients [153] |
| ChIP-Seq | |
| Asthma | H3K4me2-marked enhancers in T cells are enriched for asthma-associated SNPs and Th2 cell type [154] |
| Asthma drug response | Glucocorticoid receptor and p65 cooperatively regulate anti-inflammatory gene expression in airway epithelial cells [130] |
| Proteomics | |
| Asthma | Plasma protein levels of CCL5, HPGDS, NPSR are associated with childhood asthma [162] |
| COPD | CTSD, DPYSL2, TGM2, and TPP1 are potential COPD biomarkers; TGM2 in induced sputum and plasma is not associated with smoking but is associated with COPD severity [165] |
| ARDS | Pathways including inflammation and epithelial injury are associated with ARDS but ARDS-specific biomarkers have not yet been identified [167] |
| IPF | Levels of apolipoprotein A1, hemoglobin α, hemoglobin β [168], pulmonary fibrosis mediators and eosinophil- and neutrophil-derived proteins [169] differ in IPF patients vs. controls |
| PAH | TCTP is a mediator of endothelial prosurvival and growth signaling in PAH [173] |
| Metabolomics | |
| Asthma | Pathways relating to hypoxia response, oxidative stress, immunity, inflammation, lipid metabolism and the tricarboxylic acid cycle were identified as significant in at least two of 21 asthma metabolomics studies. [180] |
| COPD | Sphingolipids are highly expressed in sputum of smokers with COPD than smokers without COPD [191] |
| ARDS | Octane, acetaldehyde and 3-methylheptane in exhaled breath discriminate ARDS patients from other intensive care unit patients [194] |
| ARDS endotype | A subgroup of ARDS patients with 235 overexpressed metabolites in pulmonary edema fluid had higher mortality rate [197] |
| IPF | Distinct changes observed in IPF lung tissues vs. controls include increased lactic acid [198], and changes in adenosine triphosphate degradation, glycolysis, glutathione biosynthesis, and ornithine aminotransferase pathways [199] |
| PAH | Decreased arginine and increased nitric oxide was found in PAH lung tissues vs. healthy controls [200] |
| Integrative Omics | |
| Asthma | Asthma susceptibility loci are lung eQTLs, including a 17q21 locus associated with GSDMA mRNA expression levels. Network analyses of eQTLs and GWAS results identified SOCS3 pathway as a key driver of asthma [209] |
| COPD | eQTLs near previously reported COPD GWAS loci (FAM13A, CHRNA3/5, HHIP) help identify potential functional loci [210] COPD blood pQTLs for surfactant protein D, vitamin D binding protein, and TNFRSF10C are associated with COPD phenotypes; association between eQTLs and pQTLs was low [211] |
| Single Cell RNA-Seq | |
| IPF | Coexpression of different cell-specific markers in IPF cells demonstrating “Indeterminate” states of differentiation in IPF [224] |
Genomics
DNA sequence variation is known to cause, or confer risk for, various rare and common diseases, and genetic testing is increasingly being integrated into medical practice [18, 19]. Starting in 2005, as initial cataloging efforts of common DNA single nucleotide polymorphisms (SNPs) led to the design of commercially available microarrays [20], many investigators sought to relate SNPs to disease presence via genome-wide association studies (GWAS). Since then, over 2600 GWAS have been completed for a wide range of phenotypes [21]. With the advent of NGS in the late 2000s, attempts to relate DNA sequence variants to diseases have extended to whole-exome and whole-genome sequencing [22]. Because an individual’s DNA sequence in non-cancerous somatic cells is relatively stable over time and equal in all such cells, obtaining high quality DNA via a single peripheral blood or saliva sample at any point in a person’s life can be used to measure an individual’s sequence variation across the genome [23]. The availability of cost-effective technology, ease of sample collection, and commonality and stability of DNA sequence across a person’s cells and lifetime, have resulted in genomics studies’ sample sizes outpacing that of all other omics approaches.
Genome-wide association studies (GWAS)
Based on the hypothesis that common genetic variation underlies complex disease risk, GWAS statistically evaluate whether the frequency of SNP alleles or genotypes differs between affected and unaffected individuals [24]. Using case/control or family-based designs, current GWAS evaluate differences in more than 1 million common SNPs (i.e., those with minor allele frequency (MAF) ≥5% in a reference population) on microarrays. Because so many significance tests are performed in GWAS, multiple comparisons correction of raw scores is necessary to avoid a high false-positive rate of association findings [25]. A commonly employed threshold for genome-wide significance is a p-value <5 × 10−8, corresponding to a Bonferroni correction applied to a nominally significant p-value of 0.05 for 1,000,000 tests (i.e., =0.05/1,000,000). Although there is some merit to criticizing the inability of GWAS results to explain a large portion of complex disease risk [26], the identification of many common variants with small-to-modest effect sizes but reproducible signals that are leading to clinically useful insights has garnered strong support for GWAS among some researchers [27]. To facilitate the translation of genetic association results into functional insights, GWAS results are provided in the GWAS Catalog [21] and the National Heart, Lung, and Blood Institute (NHLBI)-curated Genome-Wide Repository of Associations between SNPs and Phenotypes (GRASP) [28]. Genotype data itself can be found in the NIH’s database of Genotypes and Phenotypes (dbGaP), which also archives individual-level phenotype, sequence data, and association results provided by investigators [29]. Additionally, the U.K. Biobank has made available to all bona fide health researchers genome-wide genotyping data for over 500,000 U.K. residents, along with in-depth health record and phenotype data [30].
Whole-exome sequencing (WES) and whole-genome sequencing (WGS)
As NGS costs decreased, WES, and subsequently WGS, have become preferred technologies to characterize the genome. Compared to genotyping microarrays, both technologies offer the advantage of being able to identify rare and novel variants (typically with MAF <1%–5%) [22]. With WES, sequencing costs are a fraction of WGS ones, as only protein-coding variants are targeted under the rationale that functional rare variants are most likely to be in regions of the genome that are translated into proteins. Ultimately, WGS will be the preferred method to characterize the genome for its ability to capture all types of sequence variation. Novel statistical approaches have been developed to analyze WES/WGS data in response to the challenges associated with detecting rare variants and measuring their association with diseases. Considerations include proper DNA sequence alignment to have confidence that detected rare variants are not sequencing errors, adequacy of sample sizes, classification of variants by their presumed function, and importantly, availability of computational resources for storing and analyzing large datasets [31]. Because WES/WGS projects for complex diseases require large sample sizes and substantial funding, some of the most notable projects have been coordinated by government agencies. The NHLBI-Exome Sequencing Project (ESP), for instance, used existing disease-specific cohorts to identify rare variants associated with complex diseases, including asthma, COPD, and acute lung injury [31]. The more recent Trans-Omics for Precision Medicine (TOPMed) Program of the NHLBI focuses on obtaining WGS and other omics data for a greater number of existing population-based studies with the goal of enabling precision medicine in heart, lung and blood-related diseases.
Genomics of pulmonary diseases
Many asthma and COPD GWAS have been published and thorough reviews have detailed their findings and limitations [32, 33]. The most prominent GWAS of both diseases are large meta-analyses that pool cohorts gathered by investigators around the world and thus have good statistical power to detect associations. Two major published asthma GWAS consortium projects are GABRIEL [34], consisting of European cohorts, and EVE [35], consisting of diverse North American cohorts. Major COPD GWAS include ECLIPSE, COPDGene, and ICGN cohorts [36, 37]. The most well-known and highly replicated asthma association signal is within the 17q21 locus that includes the ORMDL3 and GSDMB genes [34, 35]. Although this signal is specific to childhood-onset asthma, and studies have made some progress in understanding the function of these genes, the precise mechanism by which variants in this region modify asthma risk is unknown [38–40]. Other robust asthma associations have been found in and near IL33, TSLP, and IL1RL1 [34, 35], supporting the notion that epithelial cell-derived cytokines play a critical role in promoting the differentiation and activation of T helper 2 (Th2) cells in asthma pathogenesis [32]. Prominent COPD GWAS loci include FAM13A, CHRNA3/CHRNA5/IREB2, and HHIP [33]; functional work to understand the mechanisms by which they modulate COPD risk in ongoing. Notably, HHIP haploinsufficiency has been found to result in increased age-related emphysema in a mouse model [41], and variation in the CHRNA3/CHRNA5 locus is associated with nicotine dependence and lung function, suggesting its role in COPD is related to tobacco use and/or metabolism [42].
Due to the complex nature of asthma and COPD, there have been attempts to increase power to detect gene associations and clarify their functional role by measuring associations with objective secondary quantitative phenotypes and specific disease endotypes. This precise approach has been successful in some cases. A severe asthma GWAS identified a novel locus, CDHR3, that had not been observed using broader asthma definitions, in addition to the known asthma susceptibility loci GSDMB, IL33, and IL1RL1 [43]. In COPD, SNPs at the BICD1 gene were uniquely associated with emphysema [44]. GWAS has been used to study levels of IgE, an antibody that mediates allergic diseases and is elevated in some asthma cases [45, 46]. Some associations found correspond to IgE levels broadly (e.g., FCER1A), while other others (e.g., HLA-DQB1) appear to be specific to elevated IgE among asthma patients [47]. GWAS of forced expiratory volume in one second (FEV1) and FEV1-to-forced vital capacity (FVC) ratio have found that SNPs in genes modifying lung function in healthy adults overlap with some of those that confer COPD risk, including FAM13A, HHIP, and HTR4, suggesting that these genes modulate COPD risk via changes in lung function [48].
Bronchodilator and glucocorticoid medications are common drugs used in the treatment of asthma and COPD [49, 50]. Inhaled short acting bronchodilators (i.e., β2-agonists for asthma and COPD, anticholinergics for COPD) are used to provide quick symptom relief [49], while inhaled glucocorticoids are anti-inflammatory controller medications that decrease symptoms with regular use [49, 51]. Patients respond differently to these and other asthma and COPD medications, and there is evidence that genetics plays a role in determining drug response [52, 53]. While efforts to develop pharmacogenetic tests for asthma and COPD drugs have not moved beyond early stages, GWAS of pharmacogenetic traits have found novel gene associations. For example, SPATS2L has been associated with bronchodilator response [54], and GLCCI1 has been associated with lung function outcomes in asthma patients who used inhaled glucocorticoids [55].
Rare variants do not appear to confer much risk for asthma or COPD based on studies published thus far, although future efforts with larger and precisely phenotyped subjects may yield more promising results. One exome study found some evidence of population-specific low-frequency variants being associated with asthma in the following genes: GRASP and GSDMB among Latinos, and MTHFR among African Americans/African Caribbeans [56]. Findings from COPD exome studies have provided some evidence of rare variant association in novel and previously reported regions, but most results do not meet exome-wide statistical significance levels [57, 58]. Thus, rare variants are unlikely to account for a significant proportion of asthma or COPD heritability.
Compared to asthma and COPD, fewer GWAS have been published for ARDS, IPF and PAH, in large part due to their lower prevalence, which results in difficulty recruiting cohorts of sample size necessary to detect statistically significant associations. An ongoing ARDS GWAS by the iSPAAR Consortium has reported in abstract form that moderate ARDS association signals are found in FARP1 [59, 60]. IPF GWAS performed in Japanese and European individuals have identified genetic risk loci within TERT, TOLLIP/MUC5B, and SPPL2C [61, 62]; a GWAS of the broader phenotype idiopathic interstitial pneumonia identified an association within the MUC5B promoter that was also associated with IPF [63]. One form of PAH, familial PAH, is known to be caused by genetic mutations, especially in the BMPR2 gene [64], indicating that genetic loci may confer risk to other forms of PAH. Indeed, a PAH GWAS using cases without BMPR2 mutations detected an association near CBLN2 with an odds ratio of 1.97 (95% CI: 1.59–2.45) and p-value 7.5 × 10−10, suggesting that other loci might be identified via future GWAS [65]. As expected for rare diseases, in which rare loci of larger effect size may modulate disease risk in a small number of individuals, WES has been used to identify high penetrance rare variants in both familial and idiopathic PAH and in familial IPF [66–69], suggesting that there is promise in continuing to apply WGS to understand PAH and IPF.
Transcriptomics
Characterizing a transcriptome, or all transcripts expressed in a cell or tissue, entails capturing a static measure of a dynamic process that depends on many factors, including developmental stage, health status, time of day, and recent exposures [70]. As such, the goal of most transcriptomic studies is to compare cells or tissues under controlled conditions or disease states to identify major changes in gene or transcript expression that lead to specific functional hypotheses or biomarker development. In parallel with genomics, transcriptomes were first characterized using microarrays, and shortly after the advent of NGS, their measure was extended to RNA-Seq. Although microarrays have remained a cost-effective and widely used technique [71], a primary microarray supplier (Illumina, Inc., La Jolla, CA) discontinued its expression arrays in Dec. 2016, suggesting that sequencing will soon replace arrays entirely.
Because cell and tissue types have their own characteristic transcriptomes [72], selecting the proper site to collect RNA for a study is critical. For pulmonary disease transcriptomic studies, RNA is often extracted from 1) blood, either as whole blood, peripheral blood mononuclear cells (PBMCs), neutrophils, CD4+ T cells, or CD8+ T cells; 2) lung and airway tissues, either as whole lung, endobronchial biopsies, airway smooth muscle cells, or bronchial epithelial cells; 3) induced sputum; and 4) bronchoalveolar or nasal lavage fluids. Several disease-relevant genes and pathways have been identified via transcriptomic studies, and transcriptomics results have been used to identify disease sub-phenotypes – so called endotypes [73]. Although most results from transcriptomics have not yet yielded diagnostic tests or new drugs for the five diseases discussed, given the growing sample size and careful design of ongoing transcriptomic studies, translation of more results into actionable clinical insights may occur in the near future.
Gene expression microarrays
Since 1995, gene expression microarrays, which are designed based on known transcripts, have been the primary technique used for gene expression profiling. Recent human mRNA arrays measure expression of all known genes with over 40,000 probes, and additional platforms are available to measure expression of micro RNA (miRNA) and long-non-coding RNA (lnRNA). Analysis of microarrays required the development of new methods, most of which are now well established and readily accessible to investigators with various analytical backgrounds [74]. As with GWAS, multiple comparisons correction procedures are necessary to reduce false-positive findings, given the large number of tests performed. Public repositories such as Gene Expression Omnibus (GEO) that host transcriptome data of over 140,000 assays [75], have facilitated re-use of gene expression data for various purposes, including increasing transparency and ensuring reproducibility of published findings [76].
RNA-Seq
RNA-Seq allows for sequencing and quantification of transcripts in a cell or tissue at unprecedented depth [77]. Compared to microarrays, RNA-Seq is able to (1) quantify a greater portion of RNA, (2) quantify RNA at baseline, rather than only measure fold changes across conditions, and (3) cover a wider dynamic range of signal [78]. Although RNA-Seq has been shown to provide accurate and reproducible results [79], controversy about the best way to analyze data still exists and development of related methods is ongoing [80]. Along with microarray data, publicly available RNA-Seq data can be found via GEO, although RNA-Seq data is hosted in the Sequence Read Archive (SRA) along with other sequencing data [81].
Transcriptomics of pulmonary diseases
Many transcriptomic studies of asthma and COPD have been performed, with studies increasing in sample size and including a wider range of cell and tissue types over the past 10 years [82–84]. Overall, such studies have found a lot of heterogeneity in expression patterns among patients and no clear expression signature that distinguishes patients from healthy controls. Such observations have led to studies that are more restrictive in their definition of asthma or COPD, and to studies that attempt to use expression patterns to identify disease endotypes using unbiased analytic approaches [85].
One prominent asthma study compared expression profiles of white blood cells from 17 severe asthma patients, 19 well-controlled asthma patients, and 18 healthy controls and identified bitter taste transduction receptors (TAS2Rs) as highly expressed in severe asthma [86]. Due to experimental evidence showing that TAS2Rs are bronchodilators and reduce inflammation, TAS2Rs are now candidate drug targets in asthma. Other microarray studies have found that (1) genes involved in interferon response, including GSDMB, one of the genes in the 17q21 locus whose variants are strongly associated with asthma, distinguish severe asthma epithelial cells [87], and (2) CD8+, but not CD4+, T cells have gene expression changes that distinguish severe asthma vs. mild asthma [88]. An RNA-Seq study that compared transcriptome profiles of endobronchial biopsies from asthma patients vs. controls identified 46 differentially expressed genes, including SLC26A4, POSTN, and BCL2, but these results have not been further validated [89].
Transcriptomic data has been used, in combination with clinical variables, to identify asthma endotypes by utilizing unsupervised algorithms to identify expression signatures that characterize groups of patients [90–94]. The Unbiased Biomarkers in Prediction of Respiratory Disease Outcomes (U-BIOPRED) Study Group found that peripheral blood of patients with severe asthma could be divided into groups according to differential response to oral steroids [92]. Another U-BIOPRED study based on sputum transcriptomics found that patients with severe asthma could be clustered into four stable groups with distinct clinical characteristics (i.e., well-controlled moderate-to-severe asthma; late-onset severe asthma with a history of smoking and chronic airflow obstruction; late-onset severe asthma in non-smokers with chronic airflow obstruction; obese female patients with uncontrolled severe asthma and normal lung function) [93]. The genes defining such groups reflect distinct molecular mechanisms of disease and thus may lead to biomarkers and group-specific treatments. The Severe Asthma Research Program (SARP) has also sought to identify asthma endotypes, most recently finding via a weighted gene coexpression network analysis that while they could not selectively identify severe asthma patients, genes in network modules linked to epithelial growth and repair and neuronal function were markedly decreased in severe asthma [94].
While in vitro exposure to tobacco produces a strong gene expression signature [95] and some of these changes may persist in lung tissue of past smokers [96], a broad gene expression signature for COPD has not been found and may not exist, given that COPD can occur via different physiological processes. One study comparing PBMC expression changes in past/current smokers with and without COPD and emphysema found 26 genes that distinguished those with disease, representing immune and inflammatory responses as well as sphingolipid metabolism [97]. Other human transcriptomic studies of COPD have searched for differences in expression among COPD patients. One large study of sputum from 148 COPD patients found gene expression changes that characterize the extent of emphysema and airflow limitation, including in IL18R, which was also found to have increased protein expression in airway macrophages [98]. Another study found that neutrophil proteases have increased expression in COPD patients with respiratory distress [99]. Beyond COPD alone, comparison of transcriptomic changes in COPD and IPF vs. normal lungs found that both diseases were characterized by increased expression of genes in the p53/hypoxia pathway, suggesting that they share some expression changes that reflect overlapping biological processes [100].
Transcriptome profiling also been used to understand asthma and COPD drug responses. For example, papers describing glucocorticoid-induced gene expression changes have strong and consistent results, largely because the mechanism of action of glucocorticoids includes direct modulation of gene transcription within cell nuclei [101–104]. An early microarray study that investigated the effects of the glucocorticoid dexamethasone treatment on airway smooth muscle focused on functional validation of KLF15, which was identified as a novel modulator of airway hyperresponsiveness and has been the focus of various studies since its discovery [101]. RNA-Seq studies to identify transcriptomic differences in donor-derived primary airway smooth muscle cells that were untreated vs. treated with a glucocorticoid have identified other novel glucocorticoid response genes, such as CRISPLD2 [103]. The effects of a 2-week course of oral prednisolone on gene expression in patients with mild asthma, using airway smooth muscle extracted via laser caption microdissection from bronchoscopy samples, was investigated with RNA-Seq [104]. Comparing samples from 6 patients assigned to glucocorticoid treatment vs. 6 patients assigned to placebo, this study found that 15 genes were significantly differentially expressed between groups, and 2 of the 15 genes, FAM129A and SYNPO2, were also associated with airway hyperresponsiveness. Studies have also found that airway smooth muscle treated with vitamin D influences cytokine gene expression [105]. In contrast, transcriptomic studies of β2-agonist treatment response do not have as strong results, likely because their mechanism of action does not involve direct modulation of gene transcription [103, 105].
As is the case for genomics, fewer ARDS, IPF and PAH transcriptomic studies have been published compared to those of asthma and COPD. ARDS studies indicate that genes related to neutrophil response to infection are modulated in sepsis patients who develop ARDS, as well as during the acute vs. recovery stages [106, 107]. Additionally, polymorphonuclear leucocytes in ARDS patients have gene expression changes that are very similar to those in sepsis and burns [108]. IPF transcriptomic studies have attempted to find markers of acute exacerbations and poor outcome. Using RNA from lung tissue, CCNA2 and alpha-defensins were identified as upregulated genes during acute exacerbations [109], while a composite model that included levels of CD28, ICOS, LCK, and ITK in PBMCs was identified as a useful predictor of death and transplantation in IPF patients [110]. RNA-Seq analysis of lung tissue changes in IPF found that COL6A3 and POSTN have splicing changes associated with disease [111]. A variety of other IPF-associated expression changes have been identified in lung tissue [112] and peripheral blood [113], suggesting that transcriptomic changes will help identify IPF onset and outcomes. For PAH, over 25 transcriptomic studies have been published, most using RNA from lung homogenate, PBMCs and pulmonary artery smooth muscle cells [114]. Among the most significant findings from these studies are that BMP2 and its receptor, BMPR2, have expression changes associated with PAH. While mutations in BMPR2 are known to lead to familial PAH, expression changes of this gene and others related to its signaling pathway are present in tissues derived from PAH patients who do not have BMPR2 mutations [115, 116]. Consistent with what is known about PAH pathophysiology, other expression changes that have been observed include elevated expression of the estrogen receptor 1 (ESR1) gene, and genes in pathways related to vascular remodeling [114]. Future studies of PAH gene expression changes that are able to better capture changes unique to pulmonary artery smooth muscle cells may shed further light on specific biological changes that may be targeted by drugs.
Epigenomics
An epigenome refers to genome modifications that regulate gene expression activity and downstream phenotypes, but do not involve DNA sequence variation per se. Epigenomic information can be heritable, but it also varies considerably according to cell type, developmental stage, and environmental exposures [117]. Most epigenomic studies are designed to address the question of whether epigenome states are different in samples with disease or other phenotype vs. those without it. Commonly studied epigenomic phenomena are DNA methylation and histone modifications. In human methylation studies, “methylation” usually refers to the presence of covalently attached methyl groups to 5′ cytosine DNA positions in CpG dinucleotides, a change that typically represses gene expression [118]. Global profiling of DNA methylation can be achieved using quantitative molecular assays such as bisulfite treatment [119] and methylated DNA immunoprecipitation (MeDIP), followed by genotyping or sequencing of DNA [120]. Although whole-genome bisulfite sequencing (WGBS) is the most accurate way to measure DNA methylation, it is an expensive technique. Hence, the considerably more affordable DNA methylation arrays have been widely used. Histone post-translational modifications (PTMs), including methylation, phosphorylation, acetylation and ubiquitynation that occur on N-terminal tails of histones, modulate gene expression by affecting chromatin condensation and the ability of transcriptional proteins to access DNA [121]. To create global maps of genomic locations where histones with modifications of a specific type are present, chromatin immunoprecipitation (ChIP) to select DNA bound to histones with specific modifications is used followed by microarray analysis [120] or sequencing (ChIP-Seq) [122]. More broadly, techniques such as ChIP-Seq can be used to characterize the cistrome, that is, the genome-wide catalog of all short DNA sequences where a transcription factor binds [123]. Because epigenomes are cell-specific, pulmonary epigenomic studies use DNA extracted from disease-related tissues and cells, including blood, PBMCs, lung, and airway. While epigenomic findings related to most pulmonary diseases have not yet yielded diagnostic tests or therapies that are used in clinical practice, some of the modifications identified have provided insights into disease mechanisms that with further study may yield actionable insights.
Epigenome-wide association studies (EWAS)
Akin to the concept of GWAS, epigenome-wide association studies (EWAS) are unbiased studies that measure associations between epigenetic modifications across the genome with diseases or phenotypes [124]. To date, EWAS have typically focused on DNA methylation due to the availability of affordable methylation microarrays, including Illumina’s HumanMethylation450 BeadChip, which contains >450,000 probes and, more recently, the MethylationEPIC BeadChip, which contains >850,000 probes [125]. Important considerations related to EWAS design and interpretation include having appropriate sample sizes, accounting for cellular heterogeneity, and noting that causality cannot be inferred from association results [126]. The GEO and SRA resources mentioned earlier host hundreds of methylation datasets obtained via microarray and high throughput sequencing, respectively [127].
Chromatin Immunoprecipitation sequencing (ChIP-Seq)
Experimental and analytical protocols for ChIP-Seq have matured as this technique was used for hundreds of Encyclopedia of DNA Elements (ENCODE) experiments, resulting in evidence-based published standards and guidelines [128, 129]. Although ENCODE assayed 118 cell types, neither this consortium nor others have published many ChIP-Seq results specific to cells that are key to pulmonary phenotypes. ChIP-Seq has been used, however, to study gene regulation mechanisms of important pulmonary disease-related drugs that affect gene transcription. The global characterization of NF-κB and glucocorticoid receptor cistromes using ChIP-Seq, for example, has provided insights into how glucocorticoids alter immune response [130]. Recent studies that measure transcription factor binding sites employ more direct techniques, such as Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-Seq). Unlike ChIP-Seq, which indirectly measures chromatin structure via overlapping histone tail modifications, ATAC-Seq probes open chromatin and provides increased resolution of binding sites [131]. ChIP-Seq data for various studies can be found in GEO and SRA, while results for some, most notably those from ENCODE, are available in the UCSC genome browser [127, 132].
Epigenomics of pulmonary diseases
Epigenomic studies of asthma and COPD mostly consist of case/control designs that used arrays to measure genome-wide methylation changes [133], with recent studies simultaneously measuring gene expression profiles to more directly link methylation status with gene expression levels [134–136]. One study comparing PBMC methylation changes of 97 inner-city children with persistent atopic asthma to 97 healthy controls found 81 differentially methylated regions, including hypomethylated immune genes in asthma (i.E. il13, RUNX3, TIGIT) [136]. Analyses of methylation status of 2484 genes that were also correlated with expression levels led to the identification of asthma-associated CpG markers in RUNX3, IL4, and CAT [136].
Exposures to environmental factors, such as air pollution and maternal tobacco smoking during pregnancy, have been associated with asthma development and exacerbation in late childhood via alterations of DNA methylation [137–139]. A recent EWAS comparing methylation status in cord blood mononuclear cells from 36 children born to mothers with asthma, of whom 18 did and 18 did not develop asthma by age 9 years, identified 589 differentially methylated regions, including one near the asthma-associated gene SMAD3, implying that epigenetic changes may contribute to asthma pathogenesis long before disease develops [140].
Total serum IgE EWAS conducted in asthma cohorts found that methylation status of some genes was associated with total IgE levels [136]. In Hispanic children, AFPM1, ACOT7, and MND1 were associated with total IgE levels in PBMCs [141], while a family-based EWAS of blood leukocytes obtained from European nuclear pedigrees found associations between IgE and low methylation at 36 loci annotated to genes (e.g., IL5RA, IL1RL1, GATA1) encoding known eosinophil products and phospholipid inflammatory mediators. Loci within genes encoding eosinophil products were also found to be hypomethylated in eosinophils of asthmatics with high IgE levels vs. asthmatics with low IgE levels and controls, suggesting potential targets for asthma patient stratification [142].
Epigenomic studies of COPD have identified methylation changes associated with COPD severity, decreased lung function, and systemic glucocorticoid use [143, 144]. Combined analysis of genome-wide DNA methylation and gene expression data in lung tissues obtained from patients with COPD vs. healthy controls identified EPAS1 as a key regulator of COPD pathogenesis that has been confirmed via functional studies [145]. Because tobacco smoking alters DNA methylation in cells/tissues [146], with changes that persist after cessation [147], epigenomic studies of COPD match cases and controls by smoking status. DNA methylation changes associated with the development of COPD among smokers were observed in airway epithelial cells and lung tissues [134, 135]. The loci involved were enriched for transcription factors and overlapped with known COPD GWAS hits, such as C10orf11 [134]. Changes in methylation among COPD patients have also been linked to the development of lung cancer, as tumors from non-small-cell patients with COPD had more methylated CCDC37 and MAP1B promoters than those of patients without COPD [148].
An ARDS genome-wide methylation profiling study of whole blood from 114 intensive care unit patients, 39 of whom developed ARDS, used a candidate-gene approach to determine that methylation changes in MYLK were associated with genetic variation and modified by ethnicity between ARDS cases and intensive care unit controls [149]. IPF EWAS conducted using lung tissues and fibroblasts have provided preliminary evidence that widespread DNA methylation changes were present in IPF, including in CDKN2B, CARD10, and MGMT, genes that were also differentially expressed at the mRNA and protein levels [150–152]. Combined genome-wide DNA methylation and gene expression data of lung tissues from 94 IPF patients vs. 67 controls identified 738 differentially methylated regions with significant changes in gene expression, also suggesting that DNA methylation changes affecting gene expression contribute to the pathogenesis of IPF, although precise mechanisms of this change are unknown [153]. Genome-wide methylation studies of PAH have not yet been published.
Beyond the ENCODE datasets described above, two salient pulmonary-disease related ChIP-Seq studies have been published. A ChIP-Seq study of H3K4me2 in naïve and memory CD4+ T cells obtained from 12 asthma patients and 12 healthy controls found that H3K4me2-marked enhancers were associated with both asthma susceptibility and Th2 cell type, and that asthma GWAS SNPs were enriched in the Th2 enhancers [154]. Secondly, ChIP-Seq data for glucocorticoid receptor (GR) and NF-κB (p65) in Beas-2B bronchial epithelial cells treated with the glucocorticoid dexamethasone found that GR and p65 cooperatively regulate the expression of anti-inflammatory genes [130].
Proteomics
Protein expression levels reflect the metabolic state of, and physical processes experienced by, cells. In addition to measures of protein levels themselves, assays to measure critical aspects of protein function, including localization, protein-protein interactions, and post-translational modifications (PTMs) have also been developed. Characterizing the full proteome is still challenging, but technological innovations are improving our ability to obtain cross-sectional time and space snapshots of protein levels. These snapshots reflect observed phenotypes more closely than those of genomic, transcriptomic, or epigenomic techniques [3]. Protein microarrays designed based on known proteins or peptides were the first to increase high-throughput capacity to discover protein biomarkers, analogous to the microarray technologies that were first used to measure global gene expression and variation [155]. Beside array-based assays, high-throughput technologies for proteomics include mass spectrometry (MS)-based techniques (e.g., tandem-MS (MS/MS)) and gel-based techniques (e.g., differential in-gel electrophoresis (DIGE)) [3].
Pulmonary disease proteomics studies have been carried out using induced sputum, pulmonary epithelial lining fluid, bronchoalveolar and nasal lavage fluids, exhaled breath condensate, and blood plasma and serum [156]. Most proteomic studies of pulmonary diseases have case-control designs, with sample sizes ranging from tens to hundreds of samples [156]. While some of the statistical issues relevant to other omics techniques apply to proteomics, the technologies used for proteomics are quite different than those used for next-generation sequencing and arrays, necessitating tailored analytical approaches and introducing new limitations. Additionally, the greater diversity of technologies used to obtain proteomics data has resulted in a slower adoption of standards to identify and report findings. While the creation and use of a public repository analogous to GEO or SRA to store and catalog proteomics data contributed by researchers has lagged relative to other omics data types, the European Bioinformatics Institute’s PRoteomics IDEntifications (PRIDE) database, a standards-compliant repository that now contains data from over 70,000 assays, is a comprehensive and widely utilized resource [157].
Quantitative proteomics
Although MS-based approaches have been widely used since the 1980s, recent advances referred to as “next-generation proteomics,” allow for the quantitative characterization of nearly complete proteomes [3]. These newer approaches involve pre-fractionating protein samples or enriching specific subpopulations of peptides (e.g., by selecting for PTMs), and then using liquid chromatography (LC)-MS, which consists of measuring peptides via MS/MS within fractionated portions separated by LC. After MS/MS spectra are converted into corresponding peptide sequences via comparison to a known database, peptides are assembled into proteins [3]. The ProteomicsDB resource contains a draft of the human proteome, consisting of 18,097 human protein-coding genes, identified via the collection of MS-based assays [158].
Histone post-translational modifications (PTMs)
In addition to characterizing DNA binding sites for specific histone PTMs via ChIP-Seq, recent high-throughput quantitative proteomics approaches can measure modified forms of histone amino terminal tails using online LC-MS. Via these state-of-the-art techniques, both single and combinatorial histone codes can be interrogated to quantify global changes in PTMs under different conditions [159]. Because analyses of histone PTMs have revealed that a large number of modified residues in histones act as “histone codes” that are associated with specific physiological processes, identifying combinations of PTMs provides a more complete view of how histone states influence gene transcription and lead to specific physiological processes or disease states [160]. Following the identification of histone PTMs that are associated with an outcome, ChIP-Seq can be used to identify specific genes whose expression is altered by the PTMs.
Phosphoproteomics
Changing the catalytic activity of proteins via phosphorylation is a ubiquitous mechanism used to control many biological pathways. Over 30,000 phosphorylation sites have been quantitatively identified via MS-based technology, and it is estimated that an additional 500,000 phosphorylation sites exist in the proteome [3]. Phosphoproteomics is a technique that attempts to quantify levels of all phosphorylation sites simultaneously by eluting phosphorylated peptides from solution prior to performing MS. Although phosphoproteomic studies of pulmonary diseases have not been reported so far, this technique has shown promise in other areas. Global phosphoproetomic profiles of thrombin response in human endothelial cells, for instance, were used to identify known and novel phosphorylation sites that may play a role in platelet aggregation [161].
Proteomics in pulmonary diseases
A proteomic study of plasma from 106 children with asthma and 68 controls identified three proteins (CCL5, HPGDS, NPSR) that had different plasma levels in asthmatic children compared to controls, suggesting they could be biomarkers [162]. Studies of the induced sputum proteome from asthma and COPD patients vs. healthy controls have found many potential biomarkers, including calgranulin A and B [163, 164]. Comparison of the lung tissue proteome of nonsmokers, smokers, smokers with mild to moderate COPD, and those with severe to very severe COPD found and validated potential COPD biomarkers, including CTSD, DPYSL2, TGM2, and TPP1 [165]. Additionally, increased sputum and plasma levels of TGM2, which were not associated with smoking, were correlated with COPD severity. Although histone PTM studies related to pulmonary diseases have not been published, such studies would provide helpful insights. For example, reduced responsiveness to glucocorticoids in patients with severe asthma and COPD has been attributed to GR modifications mediated via histone deacetylase 2 (HDAC2) [166]. Because HDAC2 modifies histone PTMs widely, understanding its role on a global level would provide a more comprehensive view of how it alters glucocorticoid response.
Proteomics studies have been conducted to find biomarkers for ARDS, and poor outcomes among those who develop it, as the need for drug development and understanding disease pathobiology are particularly high for this disease [167]. Although some pathways have been identified via these studies (e.g., inflammation and epithelial injury), ARDS-specific markers have not yet been identified [167]. Proteomic changes in lung tissue, nasal lavage fluid and bronchoalveolar lavage fluid from patients with IPF compared to healthy controls include differences in apolipoprotein A1, hemoglobin α, and hemoglobin β [168], pulmonary fibrosis mediators (osteopontin, MMP7, CXCL7, CCL18) and eosinophil- and neutrophil-derived proteins [169]. Proteomic changes identified in PAH thus far also suggest proteins (e.g., TCTP) and pathways (e.g., cell contraction, oxidative stress) that may be important, but there are no validated markers to identify patients or subclassify those with PAH [170–174].
Metabolomics
Metabolites are small molecules (<1 kDa) that participate in chemical reactions within living organisms, and they include endogenous (amino acids, nucleic acids, vitamins) and exogeneous (drugs, toxins) chemicals. Metabolomics refers to the measure of all metabolites in a biological system [175]. Similar to proteomics, metabolomics provides a snapshot of the active physiological status of a cell or tissue. The most common techniques used to characterize the metabolome are nuclear magnetic resonance (NMR) and high-resolution MS [176, 177]. While metabolomics studies are unbiased, the identification of specific metabolites relies on having references to link measured spectra to them. The public repository Human Metabolome Database (HMDB) provides curated metabolomic data, currently listings 42,000 metabolites [178]. This resource allows users to search metabolites by categories (e.g., metabolite name, disease, and biofluid), and it expedites the process of mining metabolomic data. A wide range of biological samples have been used for pulmonary disease metabolomics studies, including blood serum and plasma, induced sputum, exhaled breath condensate, bronchoalveolar lavage fluid, and lung tissue [179]. Due to the non-invasive and convenient nature of acquiring urine in a clinical setting, urine is also sought as a target to identify metabolomics biomarkers.
Metabolomics in pulmonary diseases
Metabolomics studies related to asthma have focused on differences between patients and healthy controls, and between those who are responsive to glucocorticoid treatment vs. those who have low responsiveness [180–182]. Despite small sample sizes, varied biospecimens, profliling technologies, and populations, there were consistencies across 21 asthma biomarker studies: several identified the metabolites acetate, adenosine, hippurate, succinate, alanine, and threonine as related to asthma, and the pathways hypoxia response, oxidative stress, immunity, inflammation, lipid metabolism, and TCA cycle [180]. Although high accuracy tests based on metabolic markers in exhaled breath condensate, serum, and urine have been proposed for noninvasive asthma diagnostics and therapeutic monitoring, most have not been validated in independent samples [183–186]. For example, a metabolomic study of volatile organic compounds in exhaled breath condensate samples from 63 children with asthma vs. 57 healthy controls found that eight volatile organic compounds could classify children with asthma with a sensitivity of 89% and a specificity of 95% [187].
Metabolomic studies of COPD have identified potential serum/plasma metabolic markers used for early recognition of COPD development and exacerbation, independently of smoking status, with markers consistently representing chronic inflammation and oxidative stress pathways [188–190]. A study that focused on lipidome metabolites, found that sphingolipids were highly expressed in sputum of smokers with COPD compared to smokers without COPD [191]. A subsequent targeted study of plasma sphingolipids, found that two specific subgroups, sphingomyelins and glycosphingolipids, were associated with emphysema and COPD exacerbations, respectively [192]. A study of urine metabolomic data from patients with asthma and COPD created a diagnostic model to discriminate asthma from COPD that had 90% accuracy on the dataset used for model creation [193]. Validation of this and other previously reported metabolomics-based biomarker tests in independent cohorts is necessary to verify their potential clinical utility.
Metabolomics has also been applied to the search for ARDS biomarkers, with reports based on exhaled breath, plasma, and bronchoalveolar lavage fluid that showed promise to discriminate between 1) ARDS patients and healthy controls, 2) ventilated intensive care unit patients who did or did not develop ARDS, and 3) sepsis patients who did or did not develop ARDS [194–196]. A recent study comparing undiluted pulmonary edema fluid in ARDS patients and control patients with hydrostatic pulmonary edema, identified an endotype of ARDS patients based on a signature of 235 overrepresented metabolites that was associated with a higher mortality rate [197]. IPF metabolomics studies comparing lung tissue of IPF patients vs. healthy controls have found increased levels of lactic acid in lung tissue of patients suggesting a pH-dependent TGF-β activation mechanism that drives myofibroblast differentiation in IPF [198], and metabolite signatures involving the pathways adenosine triphosphate degradation, glycolysis, glutathione biosynthesis, and ornithine aminotransferase [199]. A PAH metabolomics study comparing lung tissues of 8 PAH patients vs. 8 healthy controls found disruption of arginine and oxidative pathways in PAH lung tissues, including decreased arginine and increased nitric oxide [200].
Integrative omics
The integration of diverse omics datasets provides an opportunity to gain insights beyond those that are possible from individual datasets. Thus far, most integrative omics analyses involve the pairing of two data types, but increasingly complex analyses have been conducted as novel methods have been developed [201]. Genome-wide expression quantitative trait loci (eQTL) studies, the most common paired approach, involve the measurement of associations between genetic variants and gene expression levels. An assumption of eQTL studies is that differing levels of mRNA result in different observable phenotypic outcomes. Thus, if a GWAS hit is also an eQTL, one can hypothesize that the SNP modulates an associated phenotype via changes to level of expression of a specific gene. Due to the cell- and exposure-specific nature of gene expression, eQTLs are most relevant to a GWAS if their results were obtained in a disease-relevant context. The most comprehensive eQTL database currently available is the Genotype-Tissue Expression (GTEx) resource, which includes data for 1641 samples across 43 tissues from 175 individuals [202]. Although this resource is helpful, it does not ideal for pulmonary disease studies, as results for lung are derived from lung tissue, rather than cell-specific lung components. The design of eQTL studies has been extended to relate genetic variants to protein and methylation levels, resulting in so-called protein quantitative trait loci (pQTL) [203] and methylation quantitative trait loci (meQTL) [204] studies. Approaches that consider more than two data types often involve multi-staged analyses, where relationships among datasets are sought in a hierarchical fashion [205, 206]. Prior to integration, individual datasets must be analyzed carefully to reduce propagation of errors, as these compound when performing integrated analyses [11]. Additionally, overfitting errors are more prominent because the number of observations in integrative analyses are much greater than the number of individual samples [11].
Most integrative omics studies in pulmonary diseases are eQTL studies [207, 208]. A large-scale asthma eQTL study of 1111 human lung tissues identified an eQTL within GSDMA that was a risk allele for asthma in the GABRIEL GWAS study, and created a network of relationships with eQTL and GWAS data that identified SOCS3 as a key asthma pathway [209]. A large COPD eQTL study that used whole blood and sputum gene expression data from 121 ECLIPSE subjects found eQTLs near previously reported GWAS loci (FAM13A, CHRNA3/5, HHIP), suggesting hypotheses for how association signals are functionally related to COPD [210]. A COPD pQTL study based on expression levels of 88 blood proteins and a COPD GWAS, found 527 pQTLs, including surfactant protein D, vitamin D binding protein, and TNFRSF10C [211]. Despite not using unbiased proteomic measures, this study showed that pQTLs provide helpful functional links that were absent at the level of eQTLs for most of the genes in question. A broader integrative study of longitudinal FEV1 in children with asthma combined GWAS, RNA-Seq, and ChIP-Seq data to identify seven nominally significant variants that could be related to this phenotype [212]. Although this study did not fully integrate datasets and it suffered from having a small sample size, it demonstrated that leveraging multiple datasets can lead to helpful disease insights.
Another approach taken in integrative omics studies is to use networks to model higher-order interactions among biological, environmental, and clinical characteristics of patient groups to identify hypotheses regarding disease mechanisms. A common network approach is to represent molecules (e.g. genes, proteins) and diseases as nodes, and assign edges between nodes according to known or measured relationships. From such networks, various structural measures are made, including the identification of the most highly connected nodes (i.e., the key nodes) and of modules consisting of highly connected groups of nodes that are thought to participate in specific biological or pathogenic processes or share other commonalities [213, 214]. Such measures are used to prioritize relationships for further validation studies. Network-based analyses focused on transcriptomic data are most common, and have been applied to identify clinically distinct asthma and COPD subgroups [94, 215, 216].
Single cell approaches
In contrast to the omics techniques already discussed that use cells or tissue in bulk as starting materials, newer assays are enabling the characterization of single cells [217]. Such approaches offer a more thorough understanding of physiological processes, as single-cell resolution omics data enables the characterization of intra-cellular populations, cell states, and cell transitions that are not observable with population-averaged cell data [218, 219]. The workflow for single-cell techniques is similar to that for their bulk counterparts, but with additional key steps to isolate single cells and amplify the genome component of interest [220]. Novel computational and statistical methods have arisen to deal with challenges related to these techniques. For example, analysis of single-cell RNA-Seq (scRNA-Seq) data, the most widely used single cell approach, involves dealing with a large number of undetected transcripts (i.e., an abundance of zeros), increased variability, and multi-modal expression distributions [221].
Although the number of scRNA-Seq studies is quickly expanding, few related to pulmonary diseases have been published. Mouse model scRNA-Seq studies have been used to 1) reconstruct cell lineage hierarchies of lung epithelial cells, unveiling progenitors of two alveolar type cells and novel cell-type specific markers [222], and 2) identify innate lymphoid cell precursor subsets using mouse bone marrow progenitors [223]. An IPF scRNA-Seq study found that IPF cells often coexpressed markers of alveolar type 1, alveolar type 2, and conducting airway cells, and were thus in “indeterminate” states of differentiation, in contrast to normal lung epithelial cells that expressed alveolar type 2 markers [224]. Future single-cell omics studies will be especially helpful to understand cell populations that are drivers of disease and characterize developmental processes.
Conclusions
A growing number of pulmonary disease omics studies have been reported over the last decade, covering an ever-increasing number of tissues and using newer technologies. Omics studies thus far have led to insights into disease mechanisms and laid a foundation for biomarker and therapeutic discovery, but our ability to generate hypotheses from omics-based studies has quickly outpaced our ability to translate hypotheses into actionable biologic insights. Genomic and transcriptomic studies of asthma, COPD, ARDS, IPF, and PAH, have identified replicable findings that are the subject of ongoing functional validation studies. Prominent genomics results include the 17q21 locus, HHIP and MUC5B genetic variants that confer risk for asthma, COPD and IPF, respectively, while prominent transcriptomic results include the differential expression of bitter taste receptor and KLF5 genes observed in asthma that may lead to novel therapeutic targets. Epigenomic studies have identified methylation patterns specific to COPD (e.g., C10orf11 in lung), asthma (e.g., IL13, RUNX3, TIGIT in PBMCs) and IPF (e.g., CASZ1 in lung), although much work remains to characterize cell-specific changes and include more ARDS and PAH samples. Relatively few proteomic and metabolomic studies have been published for pulmonary diseases, but sphingolipids are candidate biomarkers for COPD and a combination of exhaled breath condensates, including octane and acetaldehyde, show promise to become ARDS biomarkers. Studies that have identified asthma, COPD and IPF endotypes thus far show promise in our ability to reliably classify diseases using omics data, and such approaches will extend to ARDS and PAH.
As the knowledgebase derived from omics profiles of healthy, diseased and treated cells from diverse donors grows, defining subgroups of patients with distinct molecular and clinical characteristics will facilitate the development of biomarkers to accurately predict whether a patient has or will develop a specific disease type, or whether a patient is responding or will respond to a particular drug or form of therapy. Integrative analyses of omics data that combine a wide variety of data types will help prioritize mechanistic studies to understand the function of various observed relationships, as well as identify the most promising candidates for prospective biomarker trials. Ongoing and future omics studies covering a broader and diverse set of patients and data types and leveraging integrative analytic strategies will accelerate the advance of pulmonary disease precision medicine.
Acknowledgements
We thank Samantha Zauscher for help with figure design.
Funding
This work was supported by NIH R00 HL105663 and R01 HL133433. Funders played no role in the design, collection, analysis, and interpretation of data presented in this review.
Availability of data and materials
Not applicable.
Abbreviations
- ARDS
Acute respiratory distress syndrome
- ChIP-Seq
Chromatin Immunoprecipitation Sequencing
- COPD
Chronic obstructive pulmonary disease
- eQTL
Expression quantitative trait loci
- EWAS
Epigenome-wide association studies
- GEO
Gene Expression Omnibus
- GWAS
Genome-wide association study
- IPF
Idiopathic pulmonary fibrosis
- LC
Liquid chromatography
- MS
Mass spectrometry
- NGS
Next-generation sequencing
- NHLBI
National Heart, Lung, and Blood Institute
- PAH
Pulmonary arterial hypertension
- PBMC
Peripheral blood mononuclear cell
- pQTL
Protein quantitative trait loci
- PTM
Post-translational modification
- RNA-Seq
RNA sequencing
- scRNA-Seq
Single-cell RNA-Seq
- SNP
single nucleotide polymorphisms
- SRA
Sequence Read Archive
- WES
Whole-exome sequencing
- WGS
Whole-genome sequencing
Authors’ contributions
MK, MS, and BEH wrote and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mengyuan Kan, Email: mengykan@mail.med.upenn.edu.
Maya Shumyatcher, Email: mayashum@sas.upenn.edu.
Blanca E. Himes, Phone: (215) 573-3282, Email: bhimes@upenn.edu
References
- 1.Duggan DJ, Bittner M, Chen Y, Meltzer P, Trent JM. Expression profiling using cDNA microarrays. Nat Genet. 1999;21:10–14. doi: 10.1038/4434. [DOI] [PubMed] [Google Scholar]
- 2.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 3.Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14:35–48. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 4.Shendure J, Lieberman Aiden E. The expanding scope of DNA sequencing. Nat Biotechnol. 2012;30:1084–1094. doi: 10.1038/nbt.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramani V, Shendure J, Duan Z. Understanding spatial genome organization: methods and insights. Genomics Proteomics Bioinformatics. 2016;14:7–20. doi: 10.1016/j.gpb.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–594. doi: 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- 7.Collins DC, Sundar R, Lim JS, Yap TA. Towards precision medicine in the clinic: from biomarker discovery to novel therapeutics. Trends Pharmacol Sci. 2017;38:25–40. doi: 10.1016/j.tips.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Nimmesgern E, Benediktsson I, Norstedt I. Personalized medicine in Europe. Clin Transl Sci. 2017;10:61–63. doi: 10.1111/cts.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.https://ghr.nlm.nih.gov/primer/precisionmedicine/definition. Accessed 20 July 2017.
- 11.Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. 2015;16:85–97. doi: 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- 12.Brookes AJ, Robinson PN. Human genotype-phenotype databases: aims, challenges and opportunities. Nat Rev Genet. 2015;16:702–715. doi: 10.1038/nrg3932. [DOI] [PubMed] [Google Scholar]
- 13.Akinbami L, Moorman J, Bailey C, Zahran H, King M, Johnson C, Liu X. NCHS data brief, no 94. Hyattsville, MD: National Center for Health Statistics; 2012. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. [PubMed] [Google Scholar]
- 14.Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance--United States, 1971-2000. MMWR Surveill Summ. 2002;51:1–16. [PubMed] [Google Scholar]
- 15.Stapleton RD, Wang BM, Hudson LD, Rubenfeld GD, Caldwell ES, Steinberg KP. Causes and timing of death in patients with ARDS. Chest. 2005;128:525–532. doi: 10.1378/chest.128.2.525. [DOI] [PubMed] [Google Scholar]
- 16.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Respir J. 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 18.Berg JS, Amendola LM, Eng C, Van Allen E, Gray SW, Wagle N, Rehm HL, DeChene ET, Dulik MC, Hisama FM, et al. Processes and preliminary outputs for identification of actionable genes as incidental findings in genomic sequence data in the clinical sequencing exploratory research consortium. Genet Med. 2013;15:860–867. doi: 10.1038/gim.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The International HapMap Consortium The international HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 21.Hindorff LA, MacArthur J, Morales J, Junkins HA, Hall PN, Klemm AK, Manolio TA. A Catalog of Published Genome-Wide Association Studies. Available at: http://www.genome.gov/gwastudies. Accessed 20 July 2017.
- 22.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinds DA, McMahon G, Kiefer AK, Do CB, Eriksson N, Evans DM, St Pourcain B, Ring SM, Mountain JL, Francke U, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. 2013;45:907–911. doi: 10.1038/ng.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 25.Spencer CC, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477. doi: 10.1371/journal.pgen.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 27.Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549–558. doi: 10.1038/nrg3523. [DOI] [PubMed] [Google Scholar]
- 28.Eicher JD, Landowski C, Stackhouse B, Sloan A, Chen W, Jensen N, Lien JP, Leslie R, Johnson AD. GRASP v2.0: an update on the genome-wide repository of associations between SNPs and phenotypes. Nucleic Acids Res. 2015;43:D799–D804. doi: 10.1093/nar/gku1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auer PL, Reiner AP, Wang G, Kang HM, Abecasis GR, Altshuler D, Bamshad MJ, Nickerson DA, Tracy RP, Rich SS, et al. Guidelines for large-scale sequence-based complex trait association studies: lessons learned from the NHLBI exome sequencing project. Am J Hum Genet. 2016;99:791–801. doi: 10.1016/j.ajhg.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardin M, Silverman E. Chronic obstructive pulmonary disease genetics: a review of the past and a look into the future. Chronic Obstruct Pulm Dis: Journal of the COPD Foundation. 2014;1:33–46. doi: 10.15326/jcopdf.1.1.2014.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson WO. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, Himes BE, Levin AM, Mathias RA, Hancock DB, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse north American populations. Nat Genet. 2011;43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua AA, Sparrow D, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, Hersh CP, Demeo DL, Sylvia JS, Ziniti J, Laird NM, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2:214–225. doi: 10.1016/S2213-2600(14)70002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ono JG, Worgall TS, Worgall S. 17q21 locus and ORMDL3: an increased risk for childhood asthma. Pediatr Res. 2014;75:165–170. doi: 10.1038/pr.2013.186. [DOI] [PubMed] [Google Scholar]
- 39.Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, Doherty TA, MD MG, Pena CA, Suzukawa M, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. 2014;192:3475–3487. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, Rosenthal P, Mueller JL, Hoffman HM, Suzukawa M, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci. 2012;109:16648–16653. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lao T, Jiang Z, Yun J, Qiu W, Guo F, Huang C, Mancini JD, Gupta K, Laucho-Contreras ME, Naing ZZ, et al. Hhip haploinsufficiency sensitizes mice to age-related emphysema. Proc Natl Acad Sci U S A. 2016;113:E4681–E4687. doi: 10.1073/pnas.1602342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaur-Knudsen D, Nordestgaard BG, Bojesen SE. CHRNA3 genotype, nicotine dependence, lung function and disease in the general population. Eur Respir J. 2012;40:1538–1544. doi: 10.1183/09031936.00176811. [DOI] [PubMed] [Google Scholar]
- 43.Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, den Dekker HT, Husby A, Sevelsted A, Faura-Tellez G, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46:51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 44.Kong X, Cho MH, Anderson W, Coxson HO, Muller N, Washko G, Hoffman EA, Bakke P, Gulsvik A, Lomas DA, et al. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med. 2011;183:43–49. doi: 10.1164/rccm.201004-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levin AM, Mathias RA, Huang L, Roth LA, Daley D, Myers RA, Himes BE, Romieu I, Yang M, Eng C, et al. A meta-analysis of genome-wide association studies for serum total IgE in diverse study populations. J Allergy Clin Immunol. 2013;131:1176–1184. doi: 10.1016/j.jaci.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Granada M, Wilk JB, Tuzova M, Strachan DP, Weidinger S, Albrecht E, Gieger C, Heinrich J, Himes BE, Hunninghake GM, et al. A genome-wide association study of plasma total IgE concentrations in the Framingham heart study. J Allergy Clin Immunol. 2012;129:840–845. doi: 10.1016/j.jaci.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potaczek DP, Kabesch M. Current concepts of IgE regulation and impact of genetic determinants. Clin Exp Allergy. 2012;42:852–871. doi: 10.1111/j.1365-2222.2011.03953.x. [DOI] [PubMed] [Google Scholar]
- 48.Kheirallah AK, Miller S, Hall IP, Sayers I. Translating lung function genome-wide association study (GWAS) findings: new insights for lung biology. Adv Genet. 2016;93:57–145. doi: 10.1016/bs.adgen.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Fanta CH. Asthma. N Engl J Med. 2009;360:1002–1014. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 50.Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 51.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, Committee GS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 52.Quax RA, Manenschijn L, Koper JW, Hazes JM, Lamberts SW, van Rossum EF, Feelders RA. Glucocorticoid sensitivity in health and disease. Nat Rev Endocrinol. 2013;9:670–686. doi: 10.1038/nrendo.2013.183. [DOI] [PubMed] [Google Scholar]
- 53.McGeachie MJ, Stahl EA, Himes BE, Pendergrass SA, Lima JJ, Irvin CG, Peters SP, Ritchie MD, Plenge RM, Tantisira KG. Polygenic heritability estimates in pharmacogenetics: focus on asthma and related phenotypes. Pharmacogenet Genomics. 2013;23:324–328. doi: 10.1097/FPC.0b013e3283607acf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Himes BE, Jiang X, Hu R, Wu AC, Lasky-Su JA, Klanderman BJ, Ziniti J, Senter-Sylvia J, Lima JJ, Irvin CG, et al. Genome-wide association analysis in asthma subjects identifies SPATS2L as a novel bronchodilator response gene. PLoS Genet. 2012;8:e1002824. doi: 10.1371/journal.pgen.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, Lange C, Lazarus R, Sylvia J, Klanderman B, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365:1173–1183. doi: 10.1056/NEJMoa0911353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Igartua C, Myers RA, Mathias RA, Pino-Yanes M, Eng C, Graves PE, Levin AM, Del-Rio-Navarro BE, Jackson DJ, Livne OE, et al. Ethnic-specific associations of rare and low-frequency DNA sequence variants with asthma. Nat Commun. 2015;6:5965. doi: 10.1038/ncomms6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiao D, Lange C, Beaty TH, Crapo JD, Barnes KC, Bamshad M, Hersh CP, Morrow J, Pinto-Plata VM, Marchetti N, et al. Exome sequencing analysis in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;193:1353–1363. doi: 10.1164/rccm.201506-1223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson VE, Ntalla I, Sayers I, Morris R, Whincup P, Casas JP, Amuzu A, Choi M, Dale C, Kumari M, et al. Exome-wide analysis of rare coding variation identifies novel associations with COPD and airflow limitation in MOCS3, IFIT3 and SERPINA12. Thorax. 2016;71:501–509. doi: 10.1136/thoraxjnl-2015-207876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wurfel MM, Christie JD, Holden TD, O'Keefe GE, Calfee CS, Matthay MA, Hakonarson H, Jarvik GP, Crosslin DR, Lin X, et al. Genome-Wide Association Study Of Acute Lung Injury Risk Loci In The Ispaar Consortium [abstract] Am J Respir Crit Care Med. 2011;183:A5536. [Google Scholar]
- 60.Wurfel MM, Christie JD, Calfee CS, Matthay MA, Barneet I, Meyer NJ, O'Mahony DD, O'Keefe GE, Hakonarson H, Jarvik GP, et al. Validation Of Genome-Wide Association Study Of Acute Respiratory Distress Syndrome Risk Loci In The Ispaar Consortium [abstract] Am J Respir Crit Care Med. 2014;189:A6716. [Google Scholar]
- 61.Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, Taniguchi H, Kubo M, Kamatani N, Nakamura Y. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–656. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 62.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 2015;36:1113–1127. doi: 10.1002/humu.22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Germain M, Eyries M, Montani D, Poirier O, Girerd B, Dorfmuller P, Coulet F, Nadaud S, Maugenre S, Guignabert C, et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013;45:518–521. doi: 10.1038/ng.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, Garnett ET, Montgomery KH, Mason WR, McKean DF, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191:646–655. doi: 10.1164/rccm.201408-1510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, Shuffle EM, Mathur M, Yancy L, Jr, Rojas V, Li CG, et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:1260–1272. doi: 10.1164/rccm.201310-1749OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Tregouet DA, Borczuk A, Rosenzweig EB, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–361. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47:512–517. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 71.Uziela K, Honkela A. Probe region expression estimation for RNA-Seq data for improved microarray comparability. PLoS One. 2015;10:e0126545. doi: 10.1371/journal.pone.0126545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.GTEx Consortium. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. [DOI] [PMC free article] [PubMed]
- 73.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 74.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 75.Barrett T, Suzek TO. Troup DB, Wilhite SE, Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W, Edgar R. NCBI GEO: mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ioannidis JP, Khoury MJ. Improving validation practices in “omics” research. Science. 2011;334:1230–1232. doi: 10.1126/science.1211811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Consortium SM-I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat Biotechnol. 2014;32:903–914. doi: 10.1038/nbt.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teng M, Love MI, Davis CA, Djebali S, Dobin A, Graveley BR, Li S, Mason CE, Olson S, Pervouchine D, et al. A benchmark for RNA-seq quantification pipelines. Genome Biol. 2016;17:74. doi: 10.1186/s13059-016-0940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leinonen R, Sugawara H, Shumway M. International nucleotide sequence database C: the sequence read archive. Nucleic Acids Res. 2011;39:D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berube JC, Bosse Y. Future clinical implications emerging from recent genome-wide expression studies in asthma. Expert Rev Clin Immunol. 2014;10:985–1004. doi: 10.1586/1744666X.2014.932249. [DOI] [PubMed] [Google Scholar]
- 83.Bunyavanich S, Schadt EE. Systems biology of asthma and allergic diseases: a multiscale approach. J Allergy Clin Immunol. 2015;135:31–42. doi: 10.1016/j.jaci.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ghosh N, Dutta M, Singh B, Banerjee R, Bhattacharyya P, Chaudhury K. Transcriptomics, proteomics and metabolomics driven biomarker discovery in COPD: an update. Expert Rev Mol Diagn. 2016;16:897–913. doi: 10.1080/14737159.2016.1198258. [DOI] [PubMed] [Google Scholar]
- 85.Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, Bansal AT, Lutter R, Sousa AR, Corfield J, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum -omics. J Allergy Clin Immunol. 2016;139(6):1797–1807. doi: 10.1016/j.jaci.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 86.Orsmark-Pietras C, James A, Konradsen JR, Nordlund B, Soderhall C, Pulkkinen V, Pedroletti C, Daham K, Kupczyk M, Dahlen B, et al. Transcriptome analysis reveals upregulation of bitter taste receptors in severe asthmatics. Eur Respir J. 2013;42:65–78. doi: 10.1183/09031936.00077712. [DOI] [PubMed] [Google Scholar]
- 87.Giovannini-Chami L, Marcet B, Moreilhon C, Chevalier B, Illie MI, Lebrigand K, Robbe-Sermesant K, Bourrier T, Michiels JF, Mari B, et al. Distinct epithelial gene expression phenotypes in childhood respiratory allergy. Eur Respir J. 2012;39:1197–1205. doi: 10.1183/09031936.00070511. [DOI] [PubMed] [Google Scholar]
- 88.Tsitsiou E, Williams AE, Moschos SA, Patel K, Rossios C, Jiang X, Adams OD, Macedo P, Booton R, Gibeon D, et al. Transcriptome analysis shows activation of circulating CD8+ T cells in patients with severe asthma. J Allergy Clin Immunol. 2012;129:95–103. doi: 10.1016/j.jaci.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 89.Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Dijkhuis A, Bel EH, Baas F, Lutter R, Sterk PJ. Transcriptome sequencing (RNA-Seq) of human endobronchial biopsies: asthma versus controls. Eur Respir J. 2013;42:662–670. doi: 10.1183/09031936.00115412. [DOI] [PubMed] [Google Scholar]
- 90.Modena BD, Tedrow JR, Milosevic J, Bleecker ER, Meyers DA, Wu W, Bar-Joseph Z, Erzurum SC, Gaston BM, Busse WW, et al. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am J Respir Crit Care Med. 2014;190:1363–1372. doi: 10.1164/rccm.201406-1099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rossios C, Pavlidis S, Hoda U, Kuo CH, Wiegman C, Russell K, Sun K, Loza MJ, Baribaud F, Durham AL, et al. Sputum transcriptomics reveal upregulation of IL-1 receptor family members in patients with severe asthma. J Allergy Clin Immunol. 2017 May 18. pii: S0091-6749(17)30764-9. [DOI] [PubMed]
- 92.Bigler J, Boedigheimer M, Schofield JPR, Skipp PJ, Corfield J, Rowe A, Sousa AR, Timour M, Twehues L, Hu X, et al. A severe asthma disease signature from gene expression profiling of peripheral blood from U-BIOPRED cohorts. Am J Respir Crit Care Med. 2017;195:1311–1320. doi: 10.1164/rccm.201604-0866OC. [DOI] [PubMed] [Google Scholar]
- 93.Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, Bansal AT, Lutter R, Sousa AR, Corfield J, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol. 2017;139:1797–1807. doi: 10.1016/j.jaci.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 94.Modena BD, Bleecker ER, Busse WW, Erzurum SC, Gaston BM, Jarjour NN, Meyers DA, Milosevic J, Tedrow JR, Wu W, et al. Gene expression correlated with severe asthma characteristics reveals heterogeneous mechanisms of severe disease. Am J Respir Crit Care Med. 2017;195:1449–1463. doi: 10.1164/rccm.201607-1407OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hackett NR, Butler MW, Shaykhiev R, Salit J, Omberg L, Rodriguez-Flores JL, Mezey JG, Strulovici-Barel Y, Wang G, Didon L, Crystal RG. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beane J, Sebastiani P, Liu G, Brody JS, Lenburg ME, Spira A. Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol. 2007;8:R201. doi: 10.1186/gb-2007-8-9-r201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bahr TM, Hughes GJ, Armstrong M, Reisdorph R, Coldren CD, Edwards MG, Schnell C, Kedl R, LaFlamme DJ, Reisdorph N, et al. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;49:316–323. doi: 10.1165/rcmb.2012-0230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh D, Fox SM, Tal-Singer R, Plumb J, Bates S, Broad P, Riley JH, Celli B, Investigators E. Induced sputum genes associated with spirometric and radiological disease severity in COPD ex-smokers. Thorax. 2011;66:489–495. doi: 10.1136/thx.2010.153767. [DOI] [PubMed] [Google Scholar]
- 99.Almansa R, Socias L, Sanchez-Garcia M, Martin-Loeches I, del Olmo M, Andaluz-Ojeda D, Bobillo F, Rico L, Herrero A, Roig V, et al. Critical COPD respiratory illness is linked to increased transcriptomic activity of neutrophil proteases genes. BMC Res Notes. 2012;5:401. doi: 10.1186/1756-0500-5-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kusko RL, Brothers JF, 2nd, Tedrow J, Pandit K, Huleihel L, Perdomo C, Liu G, Juan-Guardela B, Kass D, Zhang S, et al. Integrated genomics reveals convergent Transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:948–960. doi: 10.1164/rccm.201510-2026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Masuno K, Haldar SM, Jeyaraj D, Mailloux CM, Huang X, Panettieri RA, Jr, Jain MK, Gerber AN. Expression profiling identifies Klf15 as a glucocorticoid target that regulates airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2011;45:642–649. doi: 10.1165/rcmb.2010-0369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Misior AM, Deshpande DA, Loza MJ, Pascual RM, Hipp JD, Penn RB. Glucocorticoid- and protein kinase A-dependent transcriptome regulation in airway smooth muscle. Am J Respir Cell Mol Biol. 2009;41:24–39. doi: 10.1165/rcmb.2008-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, Whitaker RM, Duan Q, Lasky-Su J, Nikolos C, et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;9:e99625. doi: 10.1371/journal.pone.0099625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Fluiter K, Bel EH, Lutter R, Baas F, Sterk PJ. Glucocorticoid-induced changes in gene expression of airway smooth muscle in patients with asthma. Am J Respir Crit Care Med. 2013;187:1076–1084. doi: 10.1164/rccm.201210-1886OC. [DOI] [PubMed] [Google Scholar]
- 105.Himes BE, Koziol-White C, Johnson M, Nikolos C, Jester W, Klanderman B, Litonjua AA, Tantisira KG, Truskowski K, MacDonald K, et al. Vitamin D modulates expression of the airway smooth muscle Transcriptome in fatal asthma. PLoS One. 2015;10:e0134057. doi: 10.1371/journal.pone.0134057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kangelaris KN, Prakash A, Liu KD, Aouizerat B, Woodruff PG, Erle DJ, Rogers A, Seeley EJ, Chu J, Liu T, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1102–L1113. doi: 10.1152/ajplung.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Z, Beach D, Su L, Zhai R, Christiani DC. A genome-wide expression analysis in blood identifies pre-elafin as a biomarker in ARDS. Am J Respir Cell Mol Biol. 2008;38:724–732. doi: 10.1165/rcmb.2007-0354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Juss J, Herre J, Begg M, Bradley G, Lennon M, Amour A, House D. Hessel EM, Summers C, Condliffe AM, Chilvers ER. Genome-wide transcription profiling in neutrophils in acute respiratory distress syndrome. Lancet. 2015;385(Suppl 1):S55. doi: 10.1016/S0140-6736(15)60370-1. [DOI] [PubMed] [Google Scholar]
- 109.Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, Bisceglia M, Gilbert S, Yousem SA, Song JW, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;180:167–175. doi: 10.1164/rccm.200810-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, Feingold E, Juan-Guardela BM, Richards TJ, Lussier Y, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5:205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G, Montgomery SB. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One. 2014;9:e92111. doi: 10.1371/journal.pone.0092111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, Rosen R, Neidermyer AJ, McKean DF, Groshong SD, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–1121. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang IV, Luna LG, Cotter J, Talbert J, Leach SM, Kidd R, Turner J, Kummer N, Kervitsky D, Brown KK, et al. The peripheral blood transcriptome identifies the presence and extent of disease in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e37708. doi: 10.1371/journal.pone.0037708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoffmann J, Wilhelm J, Olschewski A, Kwapiszewska G. Microarray analysis in pulmonary hypertension. Eur Respir J. 2016;48:229–241. doi: 10.1183/13993003.02030-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1235–H1248. doi: 10.1152/ajpheart.00254.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Gao B, Loyd JE, Tuder RM, Voelkel NF. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. doi: 10.1161/01.RES.88.6.555. [DOI] [PubMed] [Google Scholar]
- 117.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 119.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 121.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 122.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C, Clark SJ. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Birney E, Smith GD, Greally JM. Epigenome-wide association studies and the interpretation of disease -Omics. PLoS Genet. 2016;12:e1006105. doi: 10.1371/journal.pgen.1006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, et al. NCBI GEO: archive for functional genomics data sets--10 years on. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kadiyala V, Sasse SK, Altonsy MO, Berman R, Chu HW, Phang TL, Gerber AN. Cistrome-based cooperation between airway epithelial Glucocorticoid receptor and NF-kappaB orchestrates anti-inflammatory effects. J Biol Chem. 2016;291:12673–12687. doi: 10.1074/jbc.M116.721217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. The UCSC genome browser database: 2016 update. Nucleic Acids Res. 2016;44:D717–D725. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim YJ, Park SW, Kim TH, Park JS, Cheong HS, Shin HD, Park CS. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: relationship to atopy. BMC Med Genet. 2013;14:39. doi: 10.1186/1471-2350-14-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morrow JD, Cho MH, Hersh CP, Pinto-Plata V, Celli B, Marchetti N, Criner G, Bueno R, Washko G, Glass K, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics. 2016:1–10. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 135.Vucic EA, Chari R, Thu KL, Wilson IM, Cotton AM, Kennett JY, Zhang M, Lonergan KM, Steiling K, Brown CJ, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50:912–922. doi: 10.1165/rcmb.2013-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang IV, Pedersen BS, Liu A, O'Connor GT, Teach SJ, Kattan M, Misiak RT, Gruchalla R, Steinbach SF, Szefler SJ, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015;136:69–80. doi: 10.1016/j.jaci.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ji H, Biagini Myers JM, Brandt EB, Brokamp C, Ryan PH, Khurana Hershey GK. Air pollution, epigenetics, and asthma. Allergy Asthma Clin Immunol. 2016;12:51. doi: 10.1186/s13223-016-0159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gref A, Kebede Merid S, Gruzieva O, Ballereau S, Becker A, Bellander T, Bergstrom A, Bosse Y, Bottai M, Chan-Yeung M, et al. Genome-wide interaction analysis of air pollution exposure and childhood asthma with functional follow-up. Am J Respir Crit Care Med. 2017;195(10):1373–83. doi: 10.1164/rccm.201605-1026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu CJ, et al. DNA Methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98:680–696. doi: 10.1016/j.ajhg.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.DeVries A, Wlasiuk G, Miller SJ, Bosco A, Stern DA, Lohman IC, Rothers J, Jones AC, Nicodemus-Johnson J, Vasquez MM, et al. Epigenome-wide analysis links SMAD3 methylation at birth to asthma in children of asthmatic mothers. J Allergy Clin Immunol. 2016. pii:S0091-6749(16)32459-9. [DOI] [PMC free article] [PubMed]
- 141.Chen W, Wang T, Pino-Yanes M, Forno E, Liang L, Yan Q, Hu D, Weeks DE, Baccarelli A, Acosta-Perez E, et al. An epigenome-wide association study of total serum IgE in Hispanic children. J Allergy Clin Immunol. 2017. pii: S0091-6749(16)32546-5. [DOI] [PMC free article] [PubMed]
- 142.Liang L, Willis-Owen SA, Laprise C, Wong KC, Davies GA, Hudson TJ, Binia A, Hopkin JM, Yang IV, Grundberg E, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature. 2015;520:670–674. doi: 10.1038/nature14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, Rennard S, Agusti A, Anderson W, Lomas DA, DeMeo DL. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185:373–381. doi: 10.1164/rccm.201108-1382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson WH, Lomas DA, DeMeo DL. Systemic steroid exposure is associated with differential methylation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:1248–1255. doi: 10.1164/rccm.201207-1280OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yoo S, Takikawa S, Geraghty P, Argmann C, Campbell J, Lin L, Huang T, Tu Z, Foronjy RF, Spira A, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11:e1004898. doi: 10.1371/journal.pgen.1004898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wan ES, Qiu W, Carey VJ, Morrow J, Bacherman H, Foreman MG, Hokanson JE, Bowler RP, Crapo JD, DeMeo DL. Smoking-associated site-specific differential Methylation in Buccal mucosa in the COPDGene study. Am J Respir Cell Mol Biol. 2015;53:246–254. doi: 10.1165/rcmb.2014-0103OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tessema M, Yingling CM, Picchi MA, Wu G, Liu Y, Weissfeld JL, Siegfried JM, Tesfaigzi Y, Belinsky SA. Epigenetic repression of CCDC37 and MAP1B links chronic obstructive pulmonary disease to lung cancer. J Thorac Oncol. 2015;10:1181–1188. doi: 10.1097/JTO.0000000000000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Szilagyi KL, Liu C, Zhang X, Wang T, Fortman JD, Zhang W, Garcia JG. Epigenetic contribution of the myosin light chain kinase gene to the risk for acute respiratory distress syndrome. Transl Res. 2017;180:12–21. doi: 10.1016/j.trsl.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang SK, Scruggs AM, McEachin RC, White ES, Peters-Golden M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS One. 2014;9:e107055. doi: 10.1371/journal.pone.0107055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, Yakhini Z, Kaminski N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, Bray M, Zhang K, Thannickal VJ, Hagood JS. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–535. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yang IV, Pedersen BS, Rabinovich E, Hennessy CE, Davidson EJ, Murphy E, Guardela BJ, Tedrow JR, Zhang Y, Singh MK, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:1263–1272. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Seumois G, Chavez L, Gerasimova A, Lienhard M, Omran N, Kalinke L, Vedanayagam M, Ganesan AP, Chawla A, Djukanovic R, et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat Immunol. 2014;15:777–788. doi: 10.1038/ni.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yu X, Schneiderhan-Marra N, Joos TO Protein microarrays for personalized medicine. Clin Chem. 2010;56:376–387. doi: 10.1373/clinchem.2009.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Terracciano R, Pelaia G, Preiano M, Savino R. Asthma and COPD proteomics: current approaches and future directions. Proteomics Clin Appl. 2015;9:203–220. doi: 10.1002/prca.201400099. [DOI] [PubMed] [Google Scholar]
- 157.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 159.Young NL, PA DM, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8:2266–2284. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Molden RC, Garcia BA. Middle-down and top-down mass spectrometric analysis of co-occurring Histone modifications. Curr Protoc Protein Sci. 2014;77:23.7.1–23.728. doi: 10.1002/0471140864.ps2307s77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.van den Biggelaar M, Hernandez-Fernaud JR, van den Eshof BL, Neilson LJ, Meijer AB, Mertens K, Zanivan S. Quantitative phosphoproteomics unveils temporal dynamics of thrombin signaling in human endothelial cells. Blood. 2014;123:e22–e36. doi: 10.1182/blood-2013-12-546036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hamsten C, Haggmark A, Grundstrom J, Mikus M, Lindskog C, Konradsen JR, Eklund A, Pershagen G, Wickman M, Grunewald J, et al. Protein profiles of CCL5, HPGDS, and NPSR1 in plasma reveal association with childhood asthma. Allergy. 2016;71:1357–1361. doi: 10.1111/all.12927. [DOI] [PubMed] [Google Scholar]
- 163.Gharib SA, Nguyen EV, Lai Y, Plampin JD, Goodlett DR, Hallstrand TS. Induced sputum proteome in healthy subjects and asthmatic patients. J Allergy Clin Immunol. 2011;128:1176–1184. doi: 10.1016/j.jaci.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gray RD, MacGregor G, Noble D, Imrie M, Dewar M, Boyd AC, Innes JA, Porteous DJ, Greening AP. Sputum proteomics in inflammatory and suppurative respiratory diseases. Am J Respir Crit Care Med. 2008;178:444–452. doi: 10.1164/rccm.200703-409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ohlmeier S, Nieminen P, Gao J, Kanerva T, Ronty M, Toljamo T, Bergmann U, Mazur W, Pulkkinen V. Lung tissue proteomics identifies elevated transglutaminase 2 levels in stable chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1155–L1165. doi: 10.1152/ajplung.00021.2016. [DOI] [PubMed] [Google Scholar]
- 166.Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–645. doi: 10.1016/j.jaci.2012.12.1564. [DOI] [PubMed] [Google Scholar]
- 167.Levitt JE, Rogers AJ. Proteomic study of acute respiratory distress syndrome: current knowledge and implications for drug development. Expert Rev Proteomics. 2016;13:457–469. doi: 10.1586/14789450.2016.1172481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Priyadharshini VS, Teran LM. Personalized medicine in respiratory disease: role of proteomics. Adv Protein Chem Struct Biol. 2016;102:115–146. doi: 10.1016/bs.apcsb.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 169.Foster MW, Morrison LD, Todd JL, Snyder LD, Thompson JW, Soderblom EJ, Plonk K, Weinhold KJ, Townsend R, Minnich A, Moseley MA. Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res. 2015;14:1238–1249. doi: 10.1021/pr501149m. [DOI] [PubMed] [Google Scholar]
- 170.Abdul-Salam VB, Wharton J, Cupitt J, Berryman M, Edwards RJ, Wilkins MR. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation. 2010;122:2058–2067. doi: 10.1161/CIRCULATIONAHA.110.972745. [DOI] [PubMed] [Google Scholar]
- 171.Terrier B, Tamby MC, Camoin L, Guilpain P, Broussard C, Bussone G, Yaici A, Hotellier F, Simonneau G, Guillevin L, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:1128–1134. doi: 10.1164/rccm.200707-1015OC. [DOI] [PubMed] [Google Scholar]
- 172.Abdul-Salam VB, Paul GA, Ali JO, Gibbs SR, Rahman D, Taylor GW, Wilkins MR, Edwards RJ. Identification of plasma protein biomarkers associated with idiopathic pulmonary arterial hypertension. Proteomics. 2006;6:2286–2294. doi: 10.1002/pmic.200500510. [DOI] [PubMed] [Google Scholar]
- 173.Lavoie JR, Ormiston ML, Perez-Iratxeta C, Courtman DW, Jiang B, Ferrer E, Caruso P, Southwood M, Foster WS, Morrell NW, Stewart DJ. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation. 2014;129:2125–2135. doi: 10.1161/CIRCULATIONAHA.114.008777. [DOI] [PubMed] [Google Scholar]
- 174.Meyrick BO, Friedman DB, Billheimer DD, Cogan JD, Prince MA, Phillips JA, 3rd, Loyd JE. Proteomics of transformed lymphocytes from a family with familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:99–107. doi: 10.1164/rccm.200703-499OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17:451–459. doi: 10.1038/nrm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Markley JL, Bruschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi: 10.1016/j.copbio.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Junot C, Fenaille F, Colsch B, Becher F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom Rev. 2014;33:471–500. doi: 10.1002/mas.21401. [DOI] [PubMed] [Google Scholar]
- 178.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. HMDB 3.0--the human Metabolome database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nobakht MGBF, Aliannejad R, Rezaei-Tavirani M, Taheri S, Oskouie AA. The metabolomics of airway diseases, including COPD, asthma and cystic fibrosis. Biomarkers. 2015;20:5–16. doi: 10.3109/1354750X.2014.983167. [DOI] [PubMed] [Google Scholar]
- 180.Kelly RS, Dahlin A, McGeachie MJ, Qiu W, Sordillo J, Wan ES, Wu AC, Lasky-Su J. Asthma Metabolomics and the potential for integrative Omics in research and the clinic. Chest. 2017;151:262–277. doi: 10.1016/j.chest.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Carraro S, Giordano G, Reniero F, Carpi D, Stocchero M, Sterk PJ, Baraldi E. Asthma severity in childhood and metabolomic profiling of breath condensate. Allergy. 2013;68:110–117. doi: 10.1111/all.12063. [DOI] [PubMed] [Google Scholar]
- 182.Fitzpatrick AM, Park Y, Brown LA, Jones DP. Children with severe asthma have unique oxidative stress-associated metabolomic profiles. J Allergy Clin Immunol, 2014. 133:258–61. e251-258 [DOI] [PMC free article] [PubMed]
- 183.Saude EJ, Skappak CD, Regush S, Cook K, Ben-Zvi A, Becker A, Moqbel R, Sykes BD, Rowe BH, Adamko DJ. Metabolomic profiling of asthma: diagnostic utility of urine nuclear magnetic resonance spectroscopy. J Allergy Clin Immunol. 2011;127:757–764. doi: 10.1016/j.jaci.2010.12.1077. [DOI] [PubMed] [Google Scholar]
- 184.Comhair SA, McDunn J, Bennett C, Fettig J, Erzurum SC, Kalhan SC. Metabolomic Endotype of asthma. J Immunol. 2015;195:643–650. doi: 10.4049/jimmunol.1500736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ibrahim B, Marsden P, Smith JA, Custovic A, Nilsson M, Fowler SJ. Breath metabolomic profiling by nuclear magnetic resonance spectroscopy in asthma. Allergy. 2013;68:1050–1056. doi: 10.1111/all.12211. [DOI] [PubMed] [Google Scholar]
- 186.Carraro S, Rezzi S, Reniero F, Heberger K, Giordano G, Zanconato S, Guillou C, Baraldi E. Metabolomics applied to exhaled breath condensate in childhood asthma. Am J Respir Crit Care Med. 2007;175:986–990. doi: 10.1164/rccm.200606-769OC. [DOI] [PubMed] [Google Scholar]
- 187.Dallinga JW, Robroeks CM, van Berkel JJ, Moonen EJ, Godschalk RW, Jobsis Q, Dompeling E, Wouters EF, van Schooten FJ. Volatile organic compounds in exhaled breath as a diagnostic tool for asthma in children. Clin Exp Allergy. 2010;40:68–76. doi: 10.1111/j.1365-2222.2009.03343.x. [DOI] [PubMed] [Google Scholar]
- 188.Chen Q, Deeb RS, Ma Y, Staudt MR, Crystal RG, Gross SS. Serum metabolite biomarkers discriminate healthy smokers from COPD smokers. PLoS One. 2015;10:e0143937. doi: 10.1371/journal.pone.0143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Paige M, Burdick MD, Kim S, Xu J, Lee JK, Shim YM. Pilot analysis of the plasma metabolite profiles associated with emphysematous chronic obstructive pulmonary disease phenotype. Biochem Biophys Res Commun. 2011;413:588–593. doi: 10.1016/j.bbrc.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ubhi BK, Riley JH, Shaw PA, Lomas DA, Tal-Singer R, MacNee W, Griffin JL, Connor SC. Metabolic profiling detects biomarkers of protein degradation in COPD patients. Eur Respir J. 2012;40:345–355. doi: 10.1183/09031936.00112411. [DOI] [PubMed] [Google Scholar]
- 191.Telenga ED, Hoffmann RF, Ruben TK, Hoonhorst SJ, Willemse BW, van Oosterhout AJ, Heijink IH, van den Berge M, Jorge L, Sandra P, et al. Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. Am J Respir Crit Care Med. 2014;190:155–164. doi: 10.1164/rccm.201312-2210OC. [DOI] [PubMed] [Google Scholar]
- 192.Bowler RP, Jacobson S, Cruickshank C, Hughes GJ, Siska C, Ory DS, Petrache I, Schaffer JE, Reisdorph N, Kechris K. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am J Respir Crit Care Med. 2015;191:275–284. doi: 10.1164/rccm.201410-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Adamko DJ, Nair P, Mayers I, Tsuyuki RT, Regush S, Rowe BH. Metabolomic profiling of asthma and chronic obstructive pulmonary disease: a pilot study differentiating diseases. J Allergy Clin Immunol. 2015;136:571–580. doi: 10.1016/j.jaci.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 194.Bos LD, Weda H, Wang Y, Knobel HH, Nijsen TM, Vink TJ, Zwinderman AH, Sterk PJ, Schultz MJ. Exhaled breath metabolomics as a noninvasive diagnostic tool for acute respiratory distress syndrome. Eur Respir J. 2014;44:188–197. doi: 10.1183/09031936.00005614. [DOI] [PubMed] [Google Scholar]
- 195.Evans CR, Karnovsky A, Kovach MA, Standiford TJ, Burant CF, Stringer KA. Untargeted LC-MS metabolomics of bronchoalveolar lavage fluid differentiates acute respiratory distress syndrome from health. J Proteome Res. 2014;13:640–649. doi: 10.1021/pr4007624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Stringer KA, Serkova NJ, Karnovsky A, Guire K, Paine R, 3rd, Standiford TJ. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma (1)H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L4–L11. doi: 10.1152/ajplung.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rogers AJ, Contrepois K, Wu M, Zheng M, Peltz G, Ware LB, Matthay MA: Profiling of ARDS pulmonary edema fluid identifies a metabolically distinct subset. Am J Physiol Lung Cell Mol Physiol 2017:ajplung 00438 02016. [DOI] [PMC free article] [PubMed]
- 198.Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med. 2012;186:740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kang YP, Lee SB, Lee JM, Kim HM, Hong JY, Lee WJ, Choi CW, Shin HK, Kim DJ, Koh ES, et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res. 2016;15:1717–1724. doi: 10.1021/acs.jproteome.6b00156. [DOI] [PubMed] [Google Scholar]
- 200.Zhao YD, Chu L, Lin K, Granton E, Yin L, Peng J, Hsin M, Wu L, Yu A, Waddell T, et al. A biochemical approach to understand the pathogenesis of advanced pulmonary arterial hypertension: Metabolomic profiles of Arginine, Sphingosine-1-phosphate, and Heme of human lung. PLoS One. 2015;10:e0134958. doi: 10.1371/journal.pone.0134958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5. [DOI] [PMC free article] [PubMed]
- 203.Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, Tylavsky FA, Conneely KN. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics. 2014;15:145. doi: 10.1186/1471-2164-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Holzinger ER, Ritchie MD. Integrating heterogeneous high-throughput data for meta-dimensional pharmacogenomics and disease-related studies. Pharmacogenomics. 2012;13:213–222. doi: 10.2217/pgs.11.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bosse Y. Genome-wide expression quantitative trait loci analysis in asthma. Curr Opin Allergy Clin Immunol. 2013;13:487–494. doi: 10.1097/ACI.0b013e328364e951. [DOI] [PubMed] [Google Scholar]
- 208.Hobbs BD, Hersh CP. Integrative genomics of chronic obstructive pulmonary disease. Biochem Biophys Res Commun. 2014;452:276–286. doi: 10.1016/j.bbrc.2014.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hao K, Bosse Y, Nickle DC, Pare PD, Postma DS, Laviolette M, Sandford A, Hackett TL, Daley D, Hogg JC, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 2012;8:e1003029. doi: 10.1371/journal.pgen.1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Castaldi PJ, Cho MH, Zhou X, Qiu W, McGeachie M, Celli B, Bakke P, Gulsvik A, Lomas DA, Crapo JD, et al. Genetic control of gene expression at novel and established chronic obstructive pulmonary disease loci. Hum Mol Genet. 2015;24:1200–1210. doi: 10.1093/hmg/ddu525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sun W, Kechris K, Jacobson S, Drummond MB, Hawkins GA, Yang J, Chen TH, Quibrera PM, Anderson W, Barr RG, et al. Common genetic polymorphisms influence blood biomarker measurements in COPD. PLoS Genet. 2016;12:e1006011. doi: 10.1371/journal.pgen.1006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wu K, Gamazon ER, Im HK, Geeleher P, White SR, Solway J, Clemmer GL, Weiss ST, Tantisira KG, Cox NJ, et al. Genome-wide interrogation of longitudinal FEV1 in children with asthma. Am J Respir Crit Care Med. 2014;190:619–627. doi: 10.1164/rccm.201403-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gustafsson M, Nestor CE, Zhang H, Barabasi AL, Baranzini S, Brunak S, Chung KF, Federoff HJ, Gavin AC, Meehan RR, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014;6:82. doi: 10.1186/s13073-014-0082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Diez D, Agusti A, Wheelock CE. Network analysis in the investigation of chronic respiratory diseases. From basics to application. Am J Respir Crit Care Med. 2014;190:981–988. doi: 10.1164/rccm.201403-0421PP. [DOI] [PubMed] [Google Scholar]
- 215.Chang Y, Glass K, Liu YY, Silverman EK, Crapo JD, Tal-Singer R, Bowler R, Dy J, Cho M, Castaldi P. COPD subtypes identified by network-based clustering of blood gene expression. Genomics. 2016;107:51–58. doi: 10.1016/j.ygeno.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Obeidat M, Nie Y, Chen V, Shannon CP, Andiappan AK, Lee B, Rotzschke O, Castaldi PJ, Hersh CP, Fishbane N, et al. Network-based analysis reveals novel gene signatures in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Res. 2017;18:72. doi: 10.1186/s12931-017-0558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Macaulay IC, Ponting CP, Voet T. Single-cell Multiomics: multiple measurements from single cells. Trends Genet. 2017;33:155–168. doi: 10.1016/j.tig.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, Holmes C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol. 2013;31:748–752. doi: 10.1038/nbt.2642. [DOI] [PubMed] [Google Scholar]
- 220.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 221.Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016;17:63. doi: 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, Brandt C, Kane L, Campos LS, Lu L, et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature. 2016;539:102–106. doi: 10.1038/nature20105. [DOI] [PubMed] [Google Scholar]
- 224.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.