

Summary
Cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) ‐mediated regulation of already tolerized autoreactive T cells is critical for understanding autoimmune responses. Although defects in CTLA‐4 contribute to abnormal FOXP3+ regulatory T (Treg) cell function in rheumatoid arthritis, its role in autoreactive T cells remains elusive. We studied immunity towards the dominant collagen type II (CII) T‐cell epitope in collagen‐induced arthritis both in the heterologous setting and in the autologous setting where CII is mutated at position E266D in mouse cartilage. CTLA‐4 regulated all stages of arthritis, including the chronic phase, and affected the priming of autologous but not heterologous CII‐reactive T cells. CTLA‐4 expression by both conventional T (Tconv) cells and Treg cells was required but while Tconv cell expression was needed to control the priming of naive autoreactive T cells, CTLA‐4 on Treg cells prevented the inflammatory tissue attack. This identifies a cell‐type‐specific time window when CTLA‐4‐mediated tolerance is most powerful, which has important implications for clinical therapy with immune modulatory drugs.
Keywords: arthritis, autoimmunity, cytotoxic T‐lymphocyte antigen 4, regulatory T cell, tolerance
Abbreviations
- APC
antigen‐presenting cell
- CIA
collagen‐induced arthritis
- CII
collagen type II
- CTLA‐4
cytotoxic T‐lymphocyte antigen 4
- GalHy264
galactosylated form of immunodominant T‐cell epitope on CII
- iCKO
inducible conditional CTLA‐4 knockout mice
- ICOS
inducible T‐cell costimulator
- IFN‐γ
interferon‐γ
- iKO
inducible CTLA‐4 knockout mice
- IL‐17
interleukin‐17
- K264
native form of immunodominant T‐cell epitope on CII
- MMC
mouse‐mutated collagen
- PD‐1
programmed death I
- RA
rheumatoid arthritis
- Tconv
conventional T cells
- Treg
FOXP3+ regulatory T cell
- WT
wild‐type
Introduction
Despite major advances, we still only have an incomplete understanding of the origin, cause and regulation of rheumatoid arthritis (RA), a chronic multifactorial disease that leads to severe inflammation and destruction of articular joints and affects up to 0·5% of the human population. The disease process leading to RA is believed to be initiated many years before clinical onset by the loss of tolerance to self antigens.1 Collagen type‐II (CII), the predominant component of articular joint cartilage, has been identified as an interesting target for the inflammatory attack on joints as both T and B cells reactive to glycosylated CII can be found in patients with RA.2, 3, 4, 5
Since their discovery, FOXP3+ regulatory T (Treg) cells have been attractive targets to restore tolerance in autoimmune diseases. In collagen‐induced arthritis (CIA), a commonly used model for RA that relies on immunization of mice with heterologous CII,6 Treg cells have been shown to modulate disease.7, 8, 9 In patients with RA, however, the role of Treg cells has not been as clear. Although there is no consensus on frequencies of peripheral blood Treg cells in patients with RA, most studies report increased frequencies in synovial fluid.10 Yet it remains controversial whether Treg cells at the site of inflammation are fully functional11, 12, 13, 14, 15 or if it is the synovial effector T cells that have become resistant.16
A crucial mechanism of suppression by Treg cells involves cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4). CTLA‐4, a co‐inhibitory member of the CD28 family, is up‐regulated on activated conventional T (Tconv) cells but constitutively expressed on Treg cells.17 CTLA‐4 has a vital function in immune regulation and is critical for Treg cells, as shown by the development of lethal autoimmunity in full CTLA‐4 knockout (KO) mice,18, 19 as well as in Treg‐specific CTLA‐4 KO mice.20 CTLA‐4 has been suggested to function either cell autonomously, directly suppressing T‐cell effector functions, or non‐autonomously by for example down‐regulation of co‐stimulatory molecules CD80/86 on antigen‐presenting cells (APC).21 Defects in CTLA‐4 regulation have been reported to contribute to the abnormal Treg cell function observed in the synovial fluid of patients with RA22, 23 and certain polymorphisms in Ctla‐4 are associated with an increased risk of developing RA.24 Its importance in RA is further underlined by successful therapeutic treatment with CTLA‐4–immunoglobulin fusion proteins (abatacept, belatacept).25, 26 Despite the widespread use of CTLA‐4–immunoglobulin in the treatment of autoimmunity and the use of anti‐CTLA‐4 antibodies (ipilimumab) in the treatment of cancer,27, 28 the underlying mechanisms have not been fully clarified. For example, it remains unclear whether Tconv or Treg cell‐specific CTLA‐4 is involved and at which time‐point during disease CTLA‐4‐mediated regulation is most important.
To address these questions we have used inducible CTLA‐4 KO (iKO) mice in which CTLA‐4 can be depleted at will to investigate the role of CTLA‐4 at various stages during CIA induced with heterologous rat CII.29 To also study the role of CTLA‐4 in the response towards an autologous antigen, we use mouse mutated collagen (MMC) mice that express CII with a mutation replacing aspartic acid with glutamic acid at position 266 in joints, thereby mimicking the heterologous rat CII used for immunization.30 The mutation leads to a stronger binding of the immunodominant CII256‐270 epitope to the MHC class II molecule.31 This leads to a strong T‐cell tolerance, similar to the situation in human RA, and MMC mice are therefore more resistant to CIA induced with heterologous CII.30 A previous study has shown this tolerance to be partly dependent on CTLA‐4 but not on CD4+ CD25+ T cells.32 To further investigate the origin of protective CTLA‐4 expression in the tolerization towards an auto‐antigen we generated MMC mice that either could be depleted of CTLA‐4 on all cells (iKO mice) or only on Treg cells through tamoxifen‐inducible Foxp3‐driven Cre expression in CTLA‐4fl/fl mice. Collectively, our data identify a time window during autoimmune disease in which CTLA‐4‐mediated tolerance is most powerful and this could have important implications for the treatment of patients with CTLA‐4‐modifying drugs.
Materials and methods
Mice
CTLA‐4fl/fl 20 and Rosa26Cre/ERT2 mice (Jackson Laboratory, Bar Harbor, ME) were backcrossed to C57BL/10Q for at least six generations. Offspring of these colonies were intercrossed to obtain Crewt/wt CTLA‐4fl/fl (WT) and Cre+/wt CTLA‐4fl/fl (iKO) genotypes. To generate inducible Treg‐specific loss of CTLA‐4, CTLA‐4fl/fl mice were intercrossed with Foxp3tm9(EGFP/cre/ERT2)Ayr/J mice (Jackson Laboratory) to obtain Foxp3wt/wt CTLA‐4fl/fl or fl/wt (WT) and Foxp3Cre/Cre CTLA‐4fl/fl (iCKO) genotypes. MMC mice have been described previously.30, 33 All mice were maintained under specific pathogen‐free conditions. All experimental animal procedures were approved by the local ethics committee.
CTLA‐4 depletion
To induce CTLA‐4 depletion, mice were intraperitoneally injected with 4 mg Tamoxifen in corn oil (+ 5% ethanol) on days 0, 1 and 5. Complete depletion was achieved by day 6.
Antibodies
The following reagents were purchased from Biolegend (San Diego, CA) or BD Biosciences (San Jose, CA): anti‐CD3 (145‐2C11), anti‐CD4 (H129.19), anti‐CTLA‐4 (UC10‐4B9), anti CD62L (MEL‐14), anti‐CD44 (Ly24), anti‐inducible T‐cell co‐stimulator (anti‐ICOS; C398.4A), anti‐programmed death 1 (anti‐PD‐1; RMPI‐30), anti‐Ki67 (B56), anti‐CD8 (53‐6.7), anti‐B220 (RA3‐6B2), anti‐interferon‐γ (anti‐IFN‐γ; R46A2), anti‐interleukin‐17 (anti‐IL‐17; TC1118H10.1). FOXP3 (FJK‐16s), anti‐CD73 (eBioTY/11.8) and anti‐FR4 (eBIo12A5) were purchased from eBioscience (San Diego, CA).
Intracellular cytokine staining
Spleen and lymph node cell suspensions were stimulated with 50 ng/ml PMA, 250 ng/ml ionomycin and 10 μg/ml brefeldin A for 4 hr in vitro. Then, surface antigens were stained, and cells were fixed and permeabilized according to the manufacturer's instructions (eBioscience) and stained for FOXP3, Ki67 and intracellular cytokines.
Serology
Serum antibody titres against rat CII were determined by coating ELISA plates (NUNC, Roskilde, Denmark) with 10 μg rat CII and serum diluted 1 : 20 000. Bound antibodies were detected with horseradish‐peroxidase‐conjugated rat anti‐mouse immunoglobulin κ antibody (Southern Biotech, Birmingham, AL) and ABTS substrate (Roche, Basel, Switzerland). The absorbance was read at 405 nm (Synergy‐2). Pooled serum from immunized mice with known concentration was used as a standard.
ELISPOT
Either 5 × 105 draining lymph node cells/well or 1 × 106 mixed draining lymph node and spleen cells/well were plated in ELISPOT plates (#MSIPS4W10; Millipore, Billerica, MA) coated with anti‐IL‐17 (TC11‐18H10, 5 μg/ml) or anti‐IFN‐γ (AN18, 10 μg/ml) and stimulated with 20 μg/ml K264 or GalHyK264 peptide or 3 μg/ml concanavalin A for 24 hr. Bound cytokines were detected with either biotinylated anti‐IL‐17 (TC11‐8H4.1, 1 μg/ml) or biotinylated anti‐IFN‐γ (R46A2, 2 μg/ml) and alkaline phosphatase‐conjugated streptavidin. Plates were developed with BCIP/NBT (Sigma‐Aldrich, St Louis, MO) and wells were scanned and analysed with immunospot software (Cellular Technology Ltd, Shaker Heights, OH). Spots appearing in non‐stimulated wells were taken as background and substracted from all other wells within the same sample.
Histology
Fixed (4% PFA) organs were dehydrated, mounted in paraffin, sectioned (5 μm) and stained with haematoxylin & eosin. Joints were first decalcified in either EDTA or 50% formic acid for 2–5 days.
CIA induction and evaluation
Collagen‐induced arthritis was induced as previously described. In short, animals (both female and male) were intradermally injected with an emulsion of 100 μg rat CII (purified from the Swarm rat chondrosarcoma, as described previously,34 in 50 μl complete Freund's adjuvant. After 35 days, mice received a booster injection of 50 μg CII in 25 μl incomplete Freund's adjuvant. Arthritis was monitored macroscopically, i.e. each red and swollen toe or knuckle equals 1 point and each red and swollen ankle or wrist equals 5 points, resulting in a maximum score of 60 points/mouse.34 All experiments were scored blind.
Statistical analysis
graphpad prism software was used for statistical analysis. For pairwise comparisons either Student's unpaired two‐tailed t‐test or Mann–Whitney U‐test was used depending on whether the data followed a Gaussian distribution or not. For more than two groups, one‐way analysis of variance or Kruskal–Wallis test was used. Fisher's exact test was used to compare CIA incidences. Error bars represent mean ± SEM. Differences were considered statistically significant with a P‐value of <0·05.
Results
CTLA‐4 regulates CIA severity
To study the impact of CTLA‐4 on CIA, we crossed mice with a floxed Ctla‐4 gene (i.e. CTLA‐4fl/fl)20 to mice expressing the tamoxifen‐inducible Cre recombinase gene at the Rosa26 locus (i.e. Rosa26Cre/ERT2+) on the CIA‐susceptible C57BL/10.Q background. These Rosa26CreERT2+/− CTLA‐4fl/fl mice, hereafter called inducible CTLA‐4 knockout (iKO) mice, express CTLA‐4 levels comparable with WT (Cre−/− CTLA‐4fl/fl) littermates but injection of tamoxifen will lead to complete and permanent depletion within 6 days. These iKO mice were previously shown to have increased CIA susceptibility and severity compared with WT littermates if CTLA‐4 depletion occurred before the immunization with heterologous rat CII.29 To investigate if loss of CTLA‐4 enhances the priming or effector mechanisms of CII‐reactive T cells or both, a new set of experiments was conducted where mice were depleted of CTLA‐4 either before immunization or at the time of the secondary booster immunization, which correlates with arthritis onset (Fig. 1a). As expected, iKO mice depleted before immunization (denoted iKO Prime), developed more severe disease compared with WT controls (Fig. 1b), which was also evident in histological analysis of articular joints (Fig. 1c). Interestingly, iKO mice depleted of CTLA‐4 day 35 after immunization, around the onset of arthritis (denoted iKO Onset), showed the same profound effect on arthritis development (Fig. 1a–c). Hence, loss of CTLA‐4 around the onset of arthritis was enough, indicating that the main effect of CTLA‐4 is to dampen autoreactive T cells and to prevent the inflammatory attack on the joints.
Figure 1.
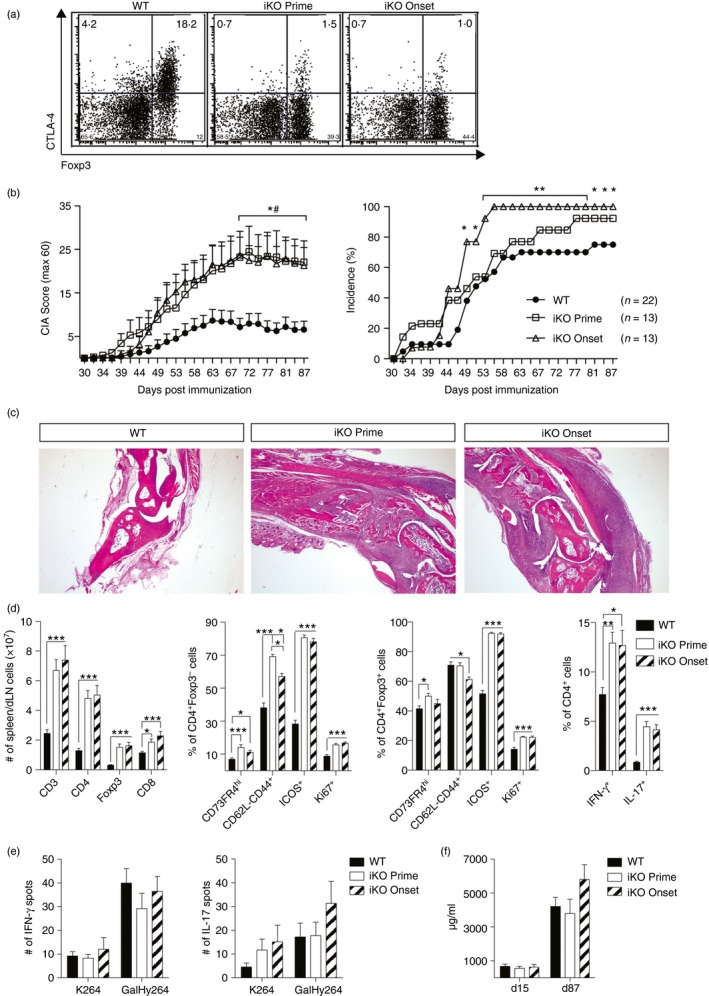
Cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) regulates collagen‐induced arthritis (CIA) severity. (a) Representative examples of CTLA‐4 levels in draining lymph nodes (dLN) of wild‐type (WT) and inducible knockout (iKO) mice 87 days after CIA. Mice were treated with tamoxifen either before immunization (iKO Prime; treated day –6, –5, –1) or at the onset (iKO Onset, treated day 35, 36, 40). (b) Mean arthritis score and incidence of WT and iKO mice that were CTLA‐4 depleted either before CIA immunization or at disease onset. # Mann–Whitney U‐test WT versus iKO Prime; *Mann–Whitney (Score) or Fisher's exact test (Incidence) WT versus iKO Onset. (c) Representative images of WT, iKO Prime and iKO Onset joint sections stained with haematoxylin & eosin, 25× magnification. (d) Absolut number of T‐cell subsets (far left), activation markers of CD4+ FOXP3− conventional T (Tconv) cells (middle left), and CD4+ FOXP3+ regulatory T (Treg) cells (middle right) and frequencies of cytokine‐producing CD4+ T cells stimulated with PMA and ionomycin (far right) in mixed spleen/dLN cells 87 days after CIA. Kruskal–Wallis test with Dunn's multiple comparison. (e) Interferon‐γ (IFN‐γ) (left) and interleukin‐17 (IL‐17) (right) T‐cell responses against unmodified (K264) or galactosylated (GalHy264) collagen type‐II (CII) peptides, after deduction of the response to RPMI medium, determined by ELISPOT from pooled dLN and spleen cells 87 days after immunization. (f) Total anti‐CII antibody titres in serum of WT and iKO mice depleted before immunization or at onset. Error bars represent mean ± SEM. Differences were considered statistically significant with *P < 0·05, **P < 0·01, or ***P < 0·001. [Colour figure can be viewed at wileyonlinelibrary.com]
Deletion of CTLA‐4 induces a lymphoproliferative disorder29 and as expected both groups of iKO mice presented with marked lymphadenopathy and splenomegaly, caused by an increase in CD4+ T cells including FOXP3+ Treg cells (Fig. 1d). Upon termination, day 87 after immunization, CD4+ FOXP3− Tconv cells of both depletion groups were more proliferative and showed an increase in effector memory T cells (CD62llow CD44+) compared with WT controls. A small but significant increase in Tconv cells with an anergic phenotype (CD73hi FR4hi) was also seen. In addition, CTLA‐4 deletion induced increased ICOS but not PD‐1 expression (Fig. 1d and data not shown). Similarly, FOXP3+ Treg cells in both CTLA‐4 depletion groups had increased frequencies of ICOS+ cells, CD73hi FR4hi anergic cells and were also actively proliferating (Ki67+) compared with immunized WT controls (Fig. 1d). The increased general activation of Tconv cells in iKO mice of both groups was also apparent in their increased production of pro‐inflammatory cytokines IFN‐γ and IL‐17 (Fig. 1d). Notably, no difference in either activation or cytokine production could be detected between mice that lacked CTLA‐4 for the duration of the entire disease course and those that only lacked it from the arthritis onset and onwards. Since total T cells and not only the antigen‐specific population were analysed it is likely that the major impact is due to loss of CTLA‐4 and not the immunization.
Therefore, we next investigated the specific T‐ and B‐cell responses to CII. The T‐cell response after CII immunization in normal WT mice is restricted to the heterologous CII256‐270 epitope.31 This epitope is either galactosylated (GalHyK264) or present in its native form (K264) and tolerance is typically more easily broken to the post‐translationally modified GalHyK264 peptide than to K264.35 However, despite an exacerbated disease and an overall activated T‐cell compartment, neither of the iKO groups showed increased T‐cell recall responses to the dominant T‐cell epitope GalHyK264, or its native form as determined by IFN‐γ and IL‐17 ELISPOT analysis (Fig. 1e). Moreover, the antibody response against CII was also not significantly different between iKO and WT mice (Fig. 1f). In summary, loss of CTLA‐4 led to an overall immune activation resulting in increased susceptibility to and severity of CIA, independent of whether CTLA‐4 was absent during the primary T‐cell activation (i.e. depletion before immunization) or during the secondary antigen response (i.e. depletion at arthritis onset). This disease exacerbation was not apparent in the measurable antigen‐specific heteroreactive T‐cell response to CII nor the anti‐CII antibody response, again indicating that CTLA‐4 did not influence antigen priming but rather the immune activation preceding the arthritis onset.
CTLA‐4 regulates the spreading of joint inflammation in the chronic phase of CIA
As these results indicate that CTLA‐4 in CIA may predominantly have a regulatory role on lymphocytes that prevent the onset of arthritis rather than affecting T‐cell priming, we went further to elucidate whether CTLA‐4 was also involved in the chronic phase of disease. Hence, mice were depleted of CTLA‐4 starting from day 65 after immunization when the disease has passed its peak and a healing response rather than active inflammation predominates. Interestingly, the incidence of newly affected limbs increased in iKO mice (Fig. 2a). So, although no impact could be seen on clinical disease score or incidence (data not shown), CTLA‐4 was shown to have an apparent role when it came to the spreading of joint inflammation to previously unaffected limbs, i.e. the role of CTLA‐4 expression on T cells is to prohibit inflammatory flares. Consistent with the earlier depletion time‐points, loss of CTLA‐4 did not impact on serum anti‐CII antibody titres (Fig. 2b), which was expected as the level of B‐cell response is largely determined from the immune priming. In summary, loss of CTLA‐4 appears to play its critical role in T cells that regulate the inflammatory attack on the tissue, both the initial attack and new relapses.
Figure 2.
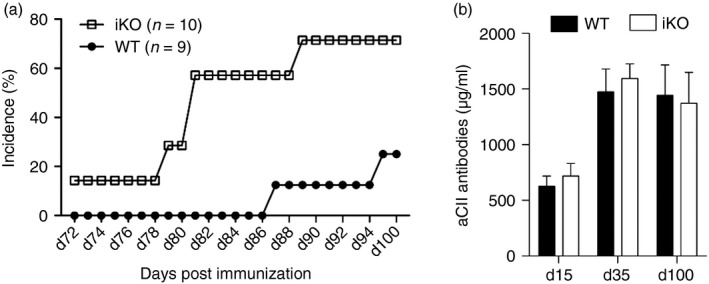
Cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) regulates the spreading of joint inflammation in the chronic phase of collagen‐induced arthritis (CIA). (a) Incidence of newly affected limbs in inducible knockout (iKO) mice depleted of CTLA‐4 from day 65 after immunization, and wild‐type (WT) littermate controls. (b) Total anti‐CII antibody titres in serum of iKO mice depleted at day 65 after immunization, and WT littermates. Error bars represent mean ± SEM.
Loss of CTLA‐4 before immune priming breaks established tolerance of autoreactive T cells
As CIA is induced with heterologous CII it will not reflect all aspects of tolerance to a true self‐antigen, which could impact on the role of CTLA‐4. To circumvent this limitation, we chose to study the effect of CTLA‐4 depletion in the MMC model. MMC mice transgenically express CII with a single nucleotide mutation leading to reversal of the aspartic acid at amino acid position 266 to glutamic acid. This minimal change of one methyl group leads to a stronger binding to the MHC class II molecule and mimics heterologous (i.e. rat, bovine, chicken or human) CII.31 Hence, the rat CII256‐270 T‐cell epitope is now expressed as a self antigen in a cartilage‐specific fashion mimicking self CII30 and T cells are naturally tolerized towards this specific epitope.36 Depending on the genetic background this leads to more or less complete protection of MMC mice from CIA induced with rat CII.30, 35, 37
In a first set of experiments, CTLA‐4 was depleted before immunization in iKO MMC+ or MMC− mice. Loss of CTLA‐4 broke tolerance and led to arthritis in iKO MMC+ mice, which confirms previous work using blocking anti‐CTLA‐4 antibodies.32 The iKO MMC− mice developed more severe disease than iKO MMC+ littermates but as the latter had indistinguishable disease from WT MMC− mice (Fig. 3a,b), this aggravated disease in iKO MMC− mice did not appear to be the result of a direct CTLA‐4‐mediated T‐cell response.
Figure 3.
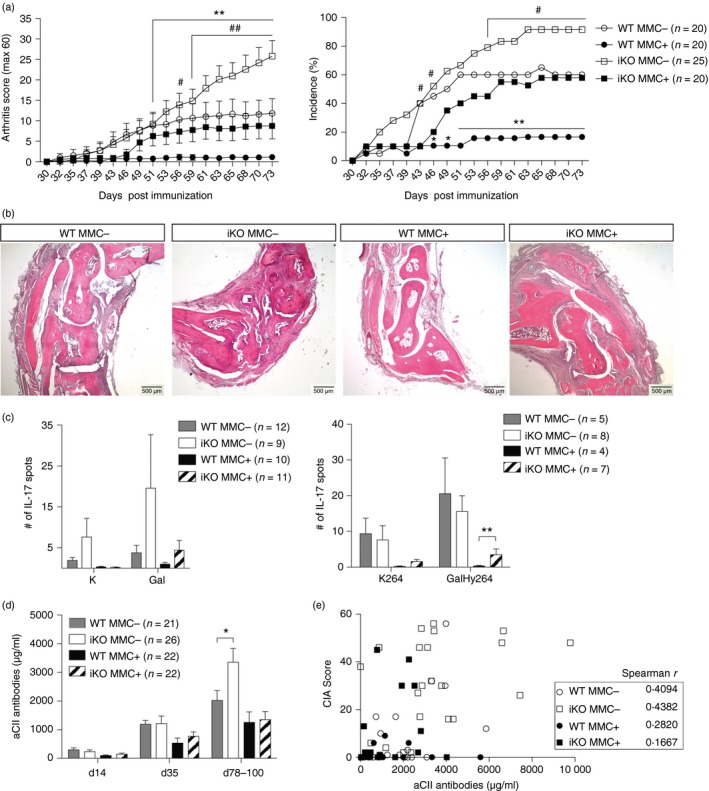
Loss of cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) before immune priming breaks the established tolerance of autoreactive T cells. (a) Mean arthritis score (left) and incidence (right) of wild‐type (WT) MMC + or MMC − and inducible knockout (iKO) MMC + or MMC − mice that were depleted of CTLA‐4 before immunization. The graphs depict pooled data from two independent experiments. *Mann–Whitney or Fisher's exact test WT MMC − versus MMC +; #Mann–Whitney or Fisher's exact test iKO MMC − versus iKO MMC +. (b) Representative images of WT MMC −, iKO MMC −, WT MMC + and iKO MMC + joint sections stained with haematoxylin & eosin, 25× magnification. (c) Interleukin‐17 (IL‐17) T‐cell recall responses against unmodified (K264) or galactosylated (GalHy264) collagen type‐II (CII) peptides after deduction of the response to RPMI medium, determined by ELISPOT from pooled draining lymph node (dLN) and spleen cells 10 days (left) and 78 days (right) after immunization. (d) Total anti‐CII antibody titres in serum of WT and iKO mice depleted before immunization. (e) Spearman correlation of collagen‐induced arthritis (CIA) Score and anti‐CII antibody titres in serum 78 days after CIA. Statistical analysis: Mann–Whitney WT MMC − versus iKO MMC − and WT MMC + versus iKO MMC + if not depicted otherwise. Error bars represent mean ± SEM. Differences were considered statistically significant with *P < 0·05, **P < 0·01. [Colour figure can be viewed at wileyonlinelibrary.com]
On the T‐cell level, MMC‐mediated tolerance in WT animals resulted in decreased T‐cell recall responses against the immunodominant GalHy264 epitope as well as its native form K264 (Fig. 3c). CTLA‐4‐dependent loss of tolerance in iKO MMC+ mice was established shortly after priming and persisted as shown by an increased IL‐17 T‐cell recall response against the GalHy264 epitope 10 days (Fig. 3c left) after priming and at day 78 (Fig. 3c right). Interestingly, the loss of tolerance seemed to be specific for the galactosylated rather than the native form of the epitope (Fig. 3c).
Disease protection in WT MMC+ mice could also be seen on the B‐cell level, as serum anti‐CII antibody titres were decreased compared with WT MMC− littermates. Loss of CTLA‐4 led to significantly increased anti‐CII titres late in disease, in iKO MMC− compared with WT MMC− mice (Fig. 3d). However, since anti‐CII titres in individual mice did not correlate with disease scores (Fig. 3e) it is doubtful whether higher anti‐CII titres in iKO MMC− mice could fully explain their aggravated disease compared with MMC+ littermates. In fact very similar antibody titres were detected in protected WT MMC+ mice and non‐protected iKO MMC+ mice (Fig. 3d).
Taken together, loss of CTLA‐4 before immunization broke self‐antigen‐specific T‐cell tolerance and enhanced autoreactive T‐cell responses against CII.
Loss of CTLA‐4 at arthritis onset also breaks tolerance in autoreactive T cells
Next, we addressed whether tolerance could also be broken after the priming of autoreactive T cells in a tolerogenic environment. Hence, CTLA‐4 was depleted starting from day 35, which is during disease onset in MMC− mice or from day 49, which is during established arthritis. This means that depletion was complete on day 41 or 55 after immunization, respectively. From the time of complete loss of CTLA‐4, both groups of iKO MMC+ mice developed arthritis (Fig. 4a). As already seen with depletion at priming, loss of CTLA‐4 in MMC+ mice resulted in a dramatic increase of disease similar to that observed in WT MMC− animals (Figs 3a and 4a). Loss of tolerance in CTLA‐4‐depleted mice could also be seen at the T‐cell level by increased IFN‐γ as well as IL‐17 recall responses to CII (Fig. 4b). As this was observed in MMC+ but not MMC− mice this indicates that CTLA‐4 has an impact on the priming of autoreactive but not heteroreactive T cells (Figs 3c and 4b). However, in both the heterologous and the autologous settings, CTLA‐4 seems to have important regulatory functions during the secondary encounter with antigen at the onset of arthritis.
Figure 4.
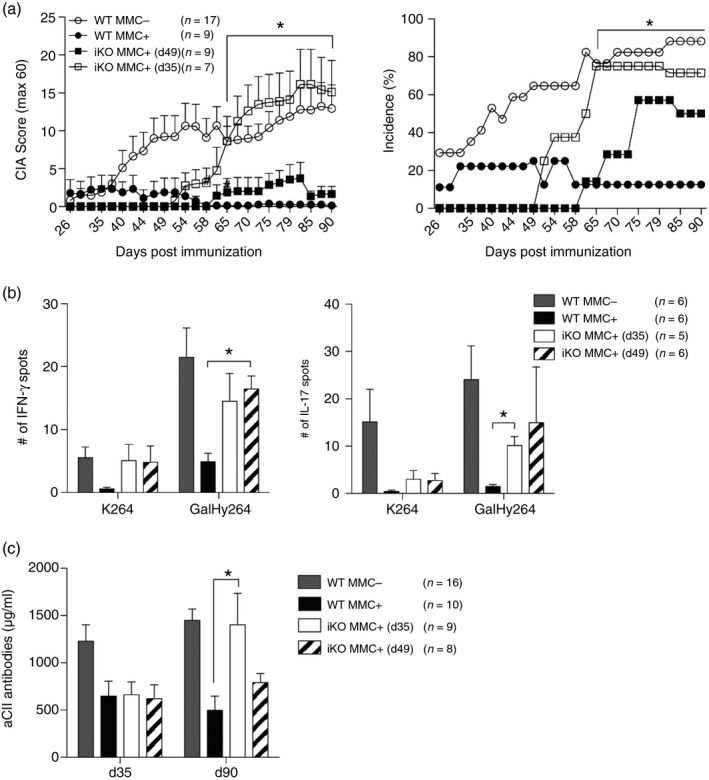
Loss of cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) at arthritis onset also breaks tolerance in autoreactive T cells. (a) Arthritis score (left) and incidence (right) of wild‐type (WT) MMC + or MMC − and inducible knockout (iKO) MMC + mice that were CTLA‐4 depleted either on days 35, 36, 40 (iKO MMC + d35) or on days 49, 50, 54 (iKO MMC + d49). Kruskal–Wallis with Dunn's multiple comparison test for score; * depicts WT + versus iKO MMC + (d35) and Fisher's exact test for incidence; * depicts WT + versus iKO MMC+ (d35). (b) Interferon‐ γ (IFN‐γ) (left) and IL‐17 (right) T‐cell responses against unmodified (K264) or galactosylated (GalHy264) collagen type‐II (CII) peptides after deduction of the RPMI medium response, determined by ELISPOT from draining lymph node (dLN) cells on day 90 after immunization. Kruskal–Wallis with Dunn's multiple comparison. WT + versus both iKO + (c) Total anti‐CII antibody titres in serum of WT MMC + or MMC − and iKO MMC + or MMC − mice depleted day 35 or 49 post immunization. Kruskal–Wallis with Dunn's multiple comparison test. Error bars represent mean ± SEM. Differences were considered statistically significant with *P < 0·05.
Contrary to depletion before immunization, the B‐cell response was also affected by loss of CTLA‐4 at disease onset in MMC+ mice. At day 35 post immunization both iKO MMC+ groups had virtually identical anti‐CII antibody titres (Fig. 4c), which was expected as both iKO groups were still CTLA‐4 sufficient at this time‐point. At day 90, however, antibody titres in iKO MMC+ mice depleted at day 35 were significantly increased compared with WT MMC+ mice, whereas there was only a trend towards increased titres in mice depleted at day 49 (Fig. 4c). In summary, loss of CTLA‐4 after T‐cell priming was sufficient to break tolerance in self‐antigen‐specific T cells, resulting in increased adaptive responses to CII.
CTLA‐4 expression on both Tconv and Treg cells regulates tolerance to CII but at different stages
To analyse the specific role of CTLA‐4 expressed on Tconv cells or on Treg cells in CIA, we introduced tamoxifen inducible Foxp3‐Cre with floxed CTLA‐4 on the MMC background, denoted iCKO mice. Following the same tamoxifen treatment protocol as before, CTLA‐4 was specifically depleted in FOXP3+ Treg cells before immunization with CII (Fig. 5a). Similar to iKO mice, the loss of CTLA‐4 on Treg cells only, led to a general immune cell activation and proliferation (Fig. 5b). Both iCKO MMC− and MMC+ mice suffered from comparable lymphoproliferation mainly caused by an expansion of CD4+ T cells, of which 40% were FOXP3+ Treg cells. Frequencies but not absolute numbers of CD8+ T cells were decreased in iCKO mice whereas CD19+ B‐cell numbers but not frequencies were increased (Fig. 5b). With regard to the general T‐cell activation, immunized iCKO MMC− and MMC+ mice were also similar. Both Tconv and Treg cells were more proliferative and activated than WT controls, as seen in the up‐regulation of Ki67, ICOS and PD‐1 and here was also a significant increase in CD73+ FR4+ anergic T cells (Fig. 5c). Interestingly, iCKO MMC+ mice immunized with CII remained protected from CIA, as were WT (Foxp3Cre+/− or Foxp3Cre −/−) MMC+ controls (Fig. 5d). This confirms the lack of a regulatory effect mediated by CTLA‐4 expressed on Treg cells on the priming of autoreactive T cells.32 MMC− mice, on the other hand developed a mild yet histologically evident arthritis that was not significantly influenced by loss of CTLA‐4 expression (Fig. 5d,e). Evaluation of the T‐cell recall response 84 days after immunization revealed that both WT and iCKO T cells responded predominantly to the GalHy264 peptide with no influence by the removal of CTLA‐4 (Fig 5f). Sustained tolerance to CII was also seen at the B‐cell level in iCKO MMC+ mice compared with WT MMC+ littermates (Fig. 5g).
Figure 5.
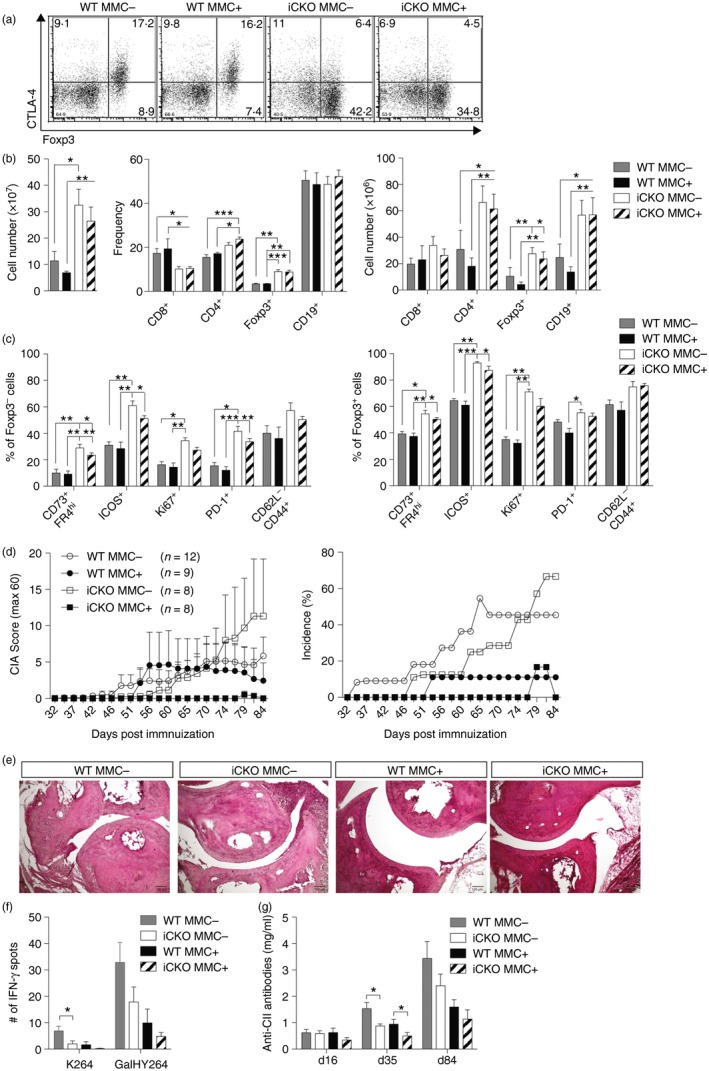
Cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) expression on both conventional T (Tconv) and regulatory T (Treg) cells regulates tolerance to collagen type‐II (CII) but at different stages. (a) Dot plots of CTLA‐4 expression levels in wild‐type (WT) and inducible conditional knockout (iCKO) mice depleted before immunization, day 84 after collagen‐induced arthritis (CIA). (b) Absolute number (left) of mixed spleen and draining lymph node (dLN) cells and frequencies (middle) as well as total numbers (right) of lymphocyte subsets 84 days after CIA and CTLA‐4 depleted before immunization. (c) Activation markers of CD4+ FOXP3− Tconv cells (left) and CD4+ FOXP3+ Treg cells (right) 84 days after CIA. (d) Arthritis score (left) and incidence (right) of WT MMC + or MMC − and iCKO MMC + or MMC − mice that were CTLA‐4 depleted before immunization. (e) Representative images of WT MMC + or MMC − and iCKO MMC + or MMC − joint sections stained with haematoxylin & eosin, 25× magnification. (f) Interferon‐γ (IFN‐γ) T‐cell response against unmodified (K264) or galactosylated (GalHy264) CII peptides, minus response to RPMI medium, determined by ELISPOT from pooled draining lymph node and spleen cells 84 days after immunization. (g) Total anti‐CII antibody titres in serum of WT MMC + or MMC − and iCKO MMC + or MMC − mice depleted before immunization. Statistical analysis: (b,c) Kruskal–Wallis with Dunn's multiple comparison test, (f,g) Mann–Whitney comparing WT MMC − with iCKO MMC − and WT MMC + with iCKO MMC +. Error bars represent mean ± SEM. Differences were considered statistically significant with *P < 0·05, **P < 0·01, or ***P < 0·001. [Colour figure can be viewed at wileyonlinelibrary.com]
Next CTLA‐4 was depleted only from the boost onwards to specifically study the role of CTLA‐4 expressed by Treg cells around disease onset. Interestingly, Treg‐specific loss of CTLA‐4 at disease onset completely broke tolerance in iCKO MMC+ mice, rendering them as susceptible as iCKO MMC− littermates (Fig. 6a). This loss of tolerance was, however, not visible at the antigen‐specific T‐ or B‐cell level ex vivo (Fig. 6b,c). In summary, expression of CTLA‐4 on Treg cells in a truly autoreactive setting critically controls T‐cell‐mediated autoimmune attacks on joint tissue leading to the development of arthritis.
Figure 6.
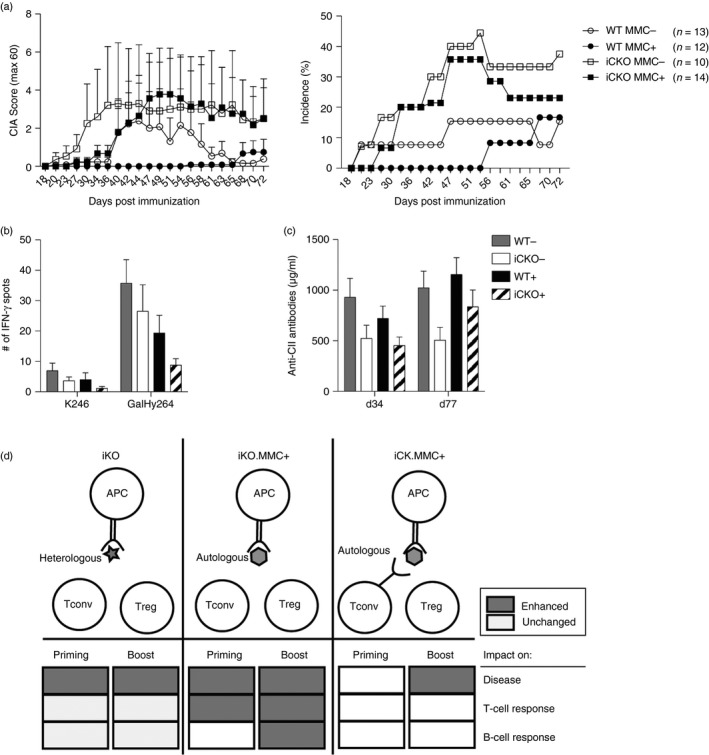
Cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) expression on regulatory T (Treg) cells prevents autoimmune attack on tissue. (a) Arthritis score (left) and incidence (right) of wild‐type (WT) MMC + or MMC − and inducible conditional knockout (iCKO) MMC + or MMC − mice that were CTLA‐4 depleted at the boost, days 35, 36, 40. (b) interferon‐γ (IFN‐γ) T‐cell response against unmodified (K264) or galactosylated (GalHy264) collagen type‐II (CII) peptides, after deduction of RPMI medium response, determined by ELISPOT from pooled draining lymph node and spleen cells on day 77 after immunization. (c) Total anti‐CII antibody titres in serum of WT MMC + or MMC − and iCKO MMC + or MMC − mice depleted at the boost. (d) Visual summary of the role of CTLA‐4 expression on conventional T (Tconv) and Treg cells towards heterologous and autologous CII‐peptides in collagen‐induced arthritis. Error bars represent mean ± SEM.
Discussion
In this study, we found that the major impact of CTLA‐4 was not to control the immune priming but rather to regulate the autoimmune inflammatory response as induced CTLA‐4 deficiency enhanced disease independent of the expression at priming. This is based on a model where CII, a tissue‐specific autoantigen of relevance for arthritis, was available in two variants. One that mimics heterologous CII used for immunization, which allows high‐affinity peptide binding to MHC class II and the induction of tolerance in autoreactive T cells (i.e. MMC mice). And its natural variant with lower affinity for MHC class II, which is subsequently ignored in the immune response to heterologous rat CII. Using these models, we performed tamoxifen‐inducible deletion of CTLA‐4 in specific T‐cell populations. This allowed us to address at which stage and by which type of T cells the development of autoimmune arthritis is regulated. Interestingly, the results suggest that the immune priming of naive heteroreactive T cells may not be the most important stage for CTLA‐4 regulation. Instead the critical time for CTLA‐4 to control autoreactive T cells is after priming and in connection with the secondary exposure to antigen. Hence, preventing an antigen‐specific attack on target tissue, i.e. the development of arthritis. Moreover, CTLA‐4 had an impact on disease spreading even during chronic stages in which previously unaffected, healthy joints were subjected to an inflammatory attack.
Despite the general immune activation and aggravated disease due to loss of CTLA‐4, we could not detect a major impact on specific T‐ and B‐cell responses to the heterologous CII immunization. This strongly suggests that CTLA‐4 is not involved in the priming of heterologous, naive, CII‐reactive T cells but instead has important regulatory functions at the onset of disease. A similar conclusion was drawn in a study on diabetes38 and it is fully compliant with the notion that CTLA‐4 regulation is not effective in situations where co‐stimulatory signals are in abundance.39 CTLA‐4 regulation is most likely redundant when naive T cells are activated with a heterologous antigen presented by fully stimulating professional APC, as is the case after immunization with heterologous CII. The situation is different in a fully autologous setting, in which already tolerized autoreactive T cells are involved. Indeed, deletion of CTLA‐4 fully broke T‐cell tolerance in mice with the relevant peptide expressed in cartilage CII, i.e. MMC+ mice, irrespective of whether depletion occurred before immunization or at the onset of disease. In this setting, loss of CTLA‐4 resulted in increased CII‐specific T‐cell responses. This clearly shows that CTLA‐4 regulates the priming of autoreactive but not heteroreactive T cells and confirms a previous study demonstrating that CTLA‐4 limits the proliferation of T cells to an antigen if it is expressed as a tissue‐restricted self antigen but not if the same antigen is supplied exogenously.39
CTLA‐4‐mediated regulation of already tolerized autoreactive T cells is a critical issue for understanding not only its protective role in autoimmune diseases but also its role in tumour escape from immune surveillance. A vital step in the development of arthritis, both CIA and RA, is the autoimmune inflammatory attack on joints. T cells involved in the development of arthritis probably need to receive additional stimulation with antigen derived from the cartilage. At this stage, germinal centres leading to the production of autoantibodies are already formed and it is unclear whether the initiation of arthritis is dependent on both antibodies targeting joints and autoreactive T cells activating synovial cells and osteoclasts.1
Our findings indicate that CTLA‐4‐deficient T cells, in particular FOXP3+ Treg cells, facilitate initiation of arthritis. T cells predominantly recognize the galactosylated form of the immunodominant CII peptide and obviously, not all autoreactive T cells have been centrally deleted in MMC mice. It is also known that the dominating post‐translational form of the epitope expressed in cartilage is the galactosylated form that is most likely not presented in the thymus. Hence, the tolerization of these T cells is likely to take place in the periphery by exposure to galactosylated CII presented by APCs in the draining lymph nodes. The expression of CTLA‐4 may dampen T‐cell activation by decreased APC function or by intrinsic mechanisms of the responding T cells.40, 41
To maintain a robust tolerance to self it is usually important that both Tconv and Treg cells express CTLA‐4. However, a previous study had indicated that CTLA‐4 expression on Tconv cells was enough to maintain tolerance to rat CII in MMC+ mice.32 To further sort out which cell types are important in MMC‐mediated tolerance we used inducible CTLA‐4 deletion specifically in FOXP3+ Treg cells. Intriguingly, iCKO MMC+ mice remained protected from CIA if depletion occurred before immunization, confirming previous results,32 but they were no longer protected if depletion occurred during the secondary encounter with antigen at disease onset. This indicates by deduction, that the initial activation of autoreactive T cells is critically dependent on the ability of Tconv cells to express CTLA‐4 themselves. The notion that CTLA‐4 on Tconv cells can regulate T‐cell responses in a cell extrinsic manner is supported by several studies that show that CTLA‐4‐sufficient Tconv cells are sufficient to limit the excessive proliferation of CTLA‐4 KO T cells in trans but are unable to control the development of systemic autoimmunity, which requires CTLA‐4‐sufficient Treg cells.20, 42, 43 Nonetheless, the ultimate proof that CTLA‐4 on Tconv cells is mediating disease protection during priming would require a model that allows for the specific depletion of CTLA‐4 on Tconv cells without affecting Treg cells, which so far is unfortunately not feasible. CTLA‐4‐mediated Treg cell suppression was, however, important for tolerance maintenance at disease onset (Fig. 6d), although absence of CTLA‐4 on Tconv might enhance this effect as iKO mice develop more severe disease than iCKO mice. It is therefore tempting to speculate that CTLA‐4 is highly up‐regulated on tolerized CII‐specific Tconv cells, which would keep them immunologically inert even in the presence of their antigen in adjuvant. It should be noted that no effect was detected on the adaptive response to CII in iCKO mice and that disease severity in WT littermates was lower than would be expected. This is likely linked with the overall genetic makeup of the iCKO mice and the Foxp3 transgene.20, 44 Also, the fact that only iCKO MMC+ mice depleted at the boost and not before immunization became ill, even though both groups were lacking CTLA‐4 at the onset of disease, is difficult to understand. However, one can speculate that the progressive general autoimmunity caused by loss of Treg‐specific CTLA‐4 might overrule any effect on CIA if CTLA‐4 has been absent for prolonged periods. In fact C57BL/10.Q mice with congenital CTLA‐4 deficiency in Treg cells only, are inert to CIA, which is probably due to a disturbed immune homeostasis (unpublished observation, K. Klocke et al.). In any event, this reduces the chance to see significant changes in T‐ and B‐cell responses. It is therefore hard to say if the absence of CTLA‐4 on Treg cells affects antigen‐specific responses directly or has to do with a more general immune activation. Another theoretical possibility is that the response to CII is skewed from the immunodominant CII256‐270 epitope in the WT setting to other CII epitopes, although we have never seen any evidence for alternative epitopes that could both bind the MHC class II Aq molecule and activate T cells. It is likely that the downstream effects of loss of CTLA‐4 on autoreactive T cells are affecting synovial cells expressing MHC class II, but also other cells of importance for development of arthritis such as neutrophils, macrophages and osteoclasts. For example it has been shown that CTLA‐4 directly inhibits osteoclast formation45, 46 and thereby limits bone erosion in CIA.47 CTLA‐4 has also been implicated in the induction of indoleamine 2,3‐dioxygenase (IDO) production by APCs.48 Recently it was suggested that human B cells could be triggered by CTLA‐4‐expressing T cells to become regulatory B cells that produce IDO, IL‐10 and transforming growth factor‐β.49 Indeed synovial T cells from patients with RA have been reported to have a methylated CTLA‐4 promotor and as a consequence reduced CTLA‐4 expression and IDO induction.22 More studies are clearly needed to decipher the intricate network of CTLA‐4 interactions and cross talk between not only adaptive cells but also innate cells. In summary, our data show that CTLA‐4 expressed on Tconv cells controls priming of naive autoreactive T cells whereas CTLA‐4 on Treg cells critically protects the joints from being attacked by a destructive inflammatory response (Fig. 6d).
Author contributions
KK, RH and KW designed the research; KK performed the research and analysed the data and KK, RH and KW wrote the paper.
Disclosures
The authors declare no conflict of interest.
Acknowledgements
We thank Kristina Palstro, Carlos Palestro and Jinlian Zheng for excellent animal care. This work was funded by the Swedish research council [522‐2009‐2548 and 521‐2010‐2894 to RH], Åke Wibergs Stiftelse [367990049, 924563126, and 940219686], Stiftelsen Professor Nanna Svartz Fond, the Swedish Society of Medicine, Ulla och Gustaf af Ugglas Stiftelse, Swedish Rheumatism Association, Stiftelsen Konung Gustaf V:s 80‐års fond and RH, the Swedish Foundation for Strategic Research to RH, Knut and Alice Wallenberg Foundation [KAW2010·0148] to RH, the EU IMI project BTCure and EU FP7 Neurinox projects to RH.
References
- 1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011; 365:2205–19. [DOI] [PubMed] [Google Scholar]
- 2. Bäcklund J, Treschow A, Bockermann R, Holm B, Holm L, Issazadeh‐Navikas S et al Glycosylation of type II collagen is of major importance for T cell tolerance and pathology in collagen‐induced arthritis. Eur J Immunol 2002; 32:3776–84. [DOI] [PubMed] [Google Scholar]
- 3. Clague RB, Morgan K, Reynolds I, Williams HJ. The prevalence of serum IgG antibodies to type‐II collagen in American patients with rheumatoid arthritis. Br J Rheumatol 1994; 33:336–8. [DOI] [PubMed] [Google Scholar]
- 4. Kim WU, Yoo WH, Park W, Kang YM, Kim SI, Park JH et al IgG antibodies to type II collagen reflect inflammatory activity in patients with rheumatoid arthritis. J Rheumatol 2000; 27:575–81. [PubMed] [Google Scholar]
- 5. Mullazehi M, Mathsson L, Lampa J, Ronnelid J. High anti‐collagen type‐II antibody levels and induction of proinflammatory cytokines by anti‐collagen antibodycontaining immune complexes in vitro characterise a distinct rheumatoid arthritis phenotype associated with acute inflammation at the time of disease onset. Ann Rheum Dis 2007; 66:537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holmdahl R, Bockermann R, Bäcklund J, Yamada H. The molecular pathogenesis of collagen‐induced arthritis in mice – a model for rheumatoid arthritis. Ageing Res Rev 2002; 1:135–47. [DOI] [PubMed] [Google Scholar]
- 7. Morgan ME, Sutmuller RPM, Witteveen HJ, van Duivenvoorde LM, Zanelli E, Melief CJM et al CD25+ cell depletion hastens the onset of severe disease in collagen‐induced arthritis. Arthritis Rheum 2003; 48:1452–60. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen LT, Jacobs J, Mathis D, Benoist C. Where FoxP3‐dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum 2007; 56:509–20. [DOI] [PubMed] [Google Scholar]
- 9. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM et al Effective treatment of collagen‐induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum 2005; 52:2212–21. [DOI] [PubMed] [Google Scholar]
- 10. Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev 2011; 10:744–55. [DOI] [PubMed] [Google Scholar]
- 11. Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A et al Defective CD4+CD25+ regulatory T cell functioning in collagen‐induced arthritis: an important factor in pathogenesis, counter‐regulated by endogenous IFN‐γ . Arthritis Res Ther 2005; 7:R402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L et al Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF‐α in rheumatoid arthritis. Nat Med 2013; 19:322–8. [DOI] [PubMed] [Google Scholar]
- 13. Xiao H, Wang S, Miao R, Kan W. TRAIL is associated with impaired regulation of CD4+CD25– T cells by regulatory T cells in patients with rheumatoid arthritis. J Clin Immunol 2011; 31:1112–9. [DOI] [PubMed] [Google Scholar]
- 14. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA et al Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNF‐α therapy. J Exp Med 2004; 200:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol 2005; 140:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J et al Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c‐akt hyperactivation in effector cells. Blood 2011; 118:3538–48. [DOI] [PubMed] [Google Scholar]
- 17. Rudd CE, Taylor A, Schneider H. CD28 and CTLA‐4 coreceptor expression and signal transduction. Immunol Rev 2009; 229:12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP et al Lymphoproliferative disorders with early lethality in mice deficient in Ctla‐4. Science 1995; 270:985–8. [DOI] [PubMed] [Google Scholar]
- 19. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA‐4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA‐4. Immunity 1995; 3:541–7. [DOI] [PubMed] [Google Scholar]
- 20. Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T, Miyara M, Fehervari Z et al CTLA‐4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271–5. [DOI] [PubMed] [Google Scholar]
- 21. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM et al Trans‐endocytosis of CD80 and CD86: a molecular basis for the cell‐extrinsic function of CTLA‐4. Science 2011; 332:600–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cribbs AP, Kennedy A, Penn H, Read JE, Amjadi P, Green P et al Regulatory T cell function in rheumatoid arthritis is compromised by CTLA‐4 promoter methylation resulting in a failure to activate the IDO pathway. Arthritis Rheumatol 2014; 66:2344–54. [DOI] [PubMed] [Google Scholar]
- 23. Flores‐Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA‐4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci 2008; 105:19396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP et al Genome‐wide association study meta‐analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 2010; 42:508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. von Kempis J, Dudler J, Hasler P, Kyburz D, Tyndall A, Zufferey P et al Use of abatacept in rheumatoid arthritis. Swiss Med Wkly 2012; 142:w13581. [DOI] [PubMed] [Google Scholar]
- 26. Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M et al EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2013; 73:492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pentcheva‐Hoang T, Simpson TR, Montalvo‐Ortiz W, Allison JP. Cytotoxic T lymphocyte antigen‐4 (CTLA‐4) blockade enhances anti‐tumor immunity by stimulating melanoma‐specific T cell motility. Cancer Immunol Res 2014; 2:970–80. [DOI] [PubMed] [Google Scholar]
- 28. Kyi C, Carvajal RD, Wolchok JD, Postow MA. Ipilimumab in patients with melanoma and autoimmune disease. J Immunother Cancer 2014; 2:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA‐4 in mice in adulthood. Proc Natl Acad Sci 2016; 113:E2383–E2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malmstrom V, Michaëlsson E, Burkhardt H, Mattsson R, Vuorio E, Holmdahl R. Systemic versus cartilage‐specific expression of a type II collagen‐specific T‐cell epitope determines the level of tolerance and susceptibility to arthritis. Proc Natl Acad Sci USA 1996; 93:4480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Michaëlsson E, Andersson M, Engström A, Holmdahl R. Identification of an immunodominant type‐II collagen peptide recognized by T cells in H‐2q mice: self tolerance at the level of determinant selection. Eur J Immunol 1992; 22:1819–25. [DOI] [PubMed] [Google Scholar]
- 32. Treschow AP, Bäcklund J, Holmdahl R, Issazadeh‐Navikas S. Intrinsic tolerance in autologous collagen‐induced arthritis is generated by CD152‐dependent CD4+ suppressor cells. J Immunol 2005; 174:6742–50. [DOI] [PubMed] [Google Scholar]
- 33. Mori L, Loetscher H, Kakimoto K, Bluethmann H, Steinmetz M. Expression of a transgenic T cell receptor β chain enhances collagen‐induced arthritis. J Exp Med 1992; 176:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holmdahl R, Carlsen S, Mikulowska A, Vestberg M, Brunsberg U., Hansson A‐S et al Genetic analysis of murine models for rheumatoid arthritis In: Adolph KW, ed. Human Genome methods. Chapter 11: CRC press, Boca Raton, New York, 1998:215–38. [Google Scholar]
- 35. Malmstrom V, Backlund J, Jansson L, Kihlberg J, Holmdahl R. T cells that are naturally tolerant to cartilage‐derived type II collagen are involved in the development of collagen‐induced arthritis. Arthritis Res 2000; 2:315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malmstrom V, Kjellen P, Holmdahl R. Type II collagen in cartilage evokes peptide‐specific tolerance and skews the immune response. J Autoimmun 1998; 11:213–21. [DOI] [PubMed] [Google Scholar]
- 37. Bäcklund J, Nandakumar KS, Bockermann R, Mori L, Holmdahl R. Genetic control of tolerance to type II collagen and development of arthritis in an autologous collagen‐induced arthritis model. J Immunol 2003; 171:3493–9. [DOI] [PubMed] [Google Scholar]
- 38. Luhder F, Chambers C, Allison JP, Benoist C, Mathis D. Pinpointing when T cell costimulatory receptor CTLA‐4 must be engaged to dampen diabetogenic T cells. Proc Natl Acad Sci USA 2000; 97:12204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walker LS, Ausubel LJ, Chodos A, Bekarian N, Abbas AK. CTLA‐4 differentially regulates T cell responses to endogenous tissue protein versus exogenous immunogen. J Immunol 2002; 169:6202–9. [DOI] [PubMed] [Google Scholar]
- 40. Wing K, Yamaguchi T, Sakaguchi S. Cell‐autonomous and ‐non‐autonomous roles of CTLA‐4 in immune regulation. Trends Immunol 2011; 32:428–33. [DOI] [PubMed] [Google Scholar]
- 41. Walker LSK, Sansom DM. Confusing signals: recent progress in CTLA‐4 biology. Trends Immunol 2015; 36:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang CJ, Kenefeck R, Wardzinski L, Attridge K, Manzotti C, Schmidt EM et al Cutting edge: cell‐extrinsic immune regulation by CTLA‐4 expressed on conventional T cells. J Immunol 2012; 189:1118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corse E, Allison JP. Cutting edge: CTLA‐4 on effector T cells inhibits in trans. J Immunol 2012; 189:1123–7. [DOI] [PubMed] [Google Scholar]
- 44. Franckaert D, Dooley J, Roos E, Floess S, Huehn J, Luche H et al Promiscuous Foxp3‐cre activity reveals a differential requirement for CD28 in Foxp3⁺ and Foxp3⁻ T cells. Immunol Cell Biol 2015; 93:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J et al CTLA‐4 directly inhibits osteoclast formation. Ann Rheum Dis 2008; 67:1603–9. [DOI] [PubMed] [Google Scholar]
- 46. Bozec A, Zaiss MM, Kagwiria R, Voll R, Rauh M, Chen Z et al T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med 2014; 6:235ra60. [DOI] [PubMed] [Google Scholar]
- 47. Kong N, Lan Q, Chen M, Zheng T, Su W, Wang J et al Induced T regulatory cells suppress osteoclastogenesis and bone erosion in collagen‐induced arthritis better than natural T regulatory cells. Ann Rheum Dis 2012; 71:1567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A et al CTLA‐4‐Ig regulates tryptophan catabolism in vivo . Nat Immunol 2002; 3:1097–101. [DOI] [PubMed] [Google Scholar]
- 49. Nouël A, Pochard P, Simon Q, Ségalen I, Le Meur Y, Pers JO et al B‐Cells induce regulatory T cells through TGF‐β/IDO production in A CTLA‐4 dependent manner. J Autoimmun 2015; 59:53–60. [DOI] [PubMed] [Google Scholar]