

ABSTRACT
Oncogenic “driver” mutations are theoretically attractive targets for the immunotherapy of lymphoid cancers, yet the proportion that can be recognized by T cells remains poorly defined. To address this issue without any confounding effects of the patient's immune system, we assessed T cells from 19 healthy donors for recognition of three common driver mutations in lymphoma: MYD88L265P, EZH2Y641F, and EZH2Y641N. Donors collectively expressed the 10 most prevalent HLA class I alleles, including HLA-A*02:01. Peripheral blood T cells were primed with peptide-loaded dendritic cells (DC), and reactive T cells were assessed for recognition of naturally processed mutant versus wild type full-length proteins. After screening three driver mutations across 17–26 HLA class I alleles and 3 × 106−3 × 107 T cells per donor, we identified CD4+ T cells against EFISENCGEII from EZH2Y641N (presented by HLA-DRB1*13:02) and CD8+ T cells against RPIPIKYKA from MYD88L265P (presented by HLA-B*07:02). We failed to detect RPIPIKYKA-specific T cells in seven other HLA-B*07:02-positive donors, including two lymphoma patients. Thus, healthy donors harbor T cells specific for common driver mutations in lymphoma. However, such responses appear to be rare due to the combined limitations of antigen processing, HLA restriction, and T cell repertoire size, highlighting the need for highly individualized approaches for selecting targets.
KEYWORDS: Driver mutation, EZH2, immunotherapy, lymphoma, MYD88, neoantigen, next-generation sequencing
Introduction
Next generation sequencing has revealed extensive mutational heterogeneity in tumors; a key clinical challenge is how best to target these mutations for the benefit of patients. An individual tumor can bear tens to thousands of mutations (collectively referred to as the “mutanome”), and these can vary markedly between patients and even between physical locations within a patient.1 “Driver” mutations promote carcinogenesis and disease progression, while “passenger” mutations have minimal influence on tumor behavior or fitness.2 A small number of driver mutations are common to multiple patients (so-called “recurrent” mutations), while others may affect the same pathways but differ with respect to the precise sequence alteration (so-called “private” mutations). A subset of driver mutations, such as BCR-ABL and BRAFV600E, can be targeted pharmacologically;3,4 however, many are difficult to target in this manner because of biochemical impediments or a lack of selective agents.
An alternative approach for targeting mutations is to harness the exquisite sensitivity and specificity of the adaptive immune system.5 Each mutation has the potential to generate a new target, or “neoantigen,” which can be recognized by T cells when processed and presented on the cell surface by major histocompatibility complex (MHC) molecules. Presentation by MHC molecules allows T cell recognition of both intracellular and extracellular proteins. Moreover, T cells can detect even single amino acid changes in proteins.6 Activated T cells are able to kill target cells presenting their cognate epitope, regardless of protein structure or function. Finally, the adaptive immune system provides immunological memory, which may prevent recurrence of tumors expressing the same mutant proteins. In support of these theoretical advantages, T cells specific for neoantigens have recently been identified as an important component of the antitumor T cell response.7 For example, next-generation sequencing has been used to identify mutant proteins recognized by tumor-infiltrating lymphocytes (TIL) in melanoma patients who responded well to adoptive T cell therapy (ACT).8-10
Despite the conceptual advantages of targeting mutant proteins with T cells, a limitation of this approach is that the immune system is able to recognize only a fraction of mutations. Although the size of this fraction remains poorly defined, empirical evidence from melanoma, ovarian cancer, and gastrointestinal cancers indicates that only 0.5–4% of non-synonymous point mutations elicit spontaneous T cell responses in patients.9-13 There are several reasons for this. First, due to the selective nature of antigen processing, not all mutations give rise to bona fide mutation-bearing epitopes that are processed and presented by the MHC class I or II pathways. Second, even when present, not all epitopes spontaneously trigger T cell responses, owing to a lack of cognate T cells, inefficient T cell priming, or other immunological issues. Third, when responses are triggered, the responding T cells can become exhausted or even deleted from the T cell repertoire, as suggested from our work in ovarian cancer.12 Fourth, as with any antigen, tumors can undergo “immune editing” which leads to the outgrowth of tumor cells that no longer express the targeted neoantigen.14
One approach to address the second and third issues (i.e., a lack of responding T cells in patients) is to perform in vitro priming of neoantigen-specific T cells using blood from an MHC-matched healthy donor. In a clinical setting, the resulting neoantigen-specific T cell receptors (TCRs) could then be used to engineer the patient's T cells to create a mutation-specific infusion product. To this end, a recent study interrogated the naive T cell repertoire of healthy donors to identify TCRs specific for predicted epitopes derived from melanoma-specific neoantigens from three patient samples.15 Most, if not all, of these neoantigens were derived from passenger mutations. In total, T cell responses were successfully identified for 10/45 mutations from 2/3 patients, providing proof-of-concept for this approach.
The fourth issue (immune editing) could potentially be addressed by targeting driver mutations rather than passengers. Since drivers are important for the survival and spread of cancer cells, expression is more likely to be maintained even in the face of immunological pressure. Indeed, T cell responses have been demonstrated against driver mutations such as BRAFV600E, KRASG12D, and BCR-ABL.13,16,17 Recently, infusion of a TIL product specific for KRASG12D resulted in regression of metastases in a colorectal cancer patient.13 Furthermore, using an in vitro priming approach, we recently demonstrated that lymphoma patients can harbor CD8+ T cells specific for driver mutations in CREBBP and MEF2B.18 However, such T cell responses were detected in only 23% of cases, raising questions about the widespread applicability of this approach. Given that our prior study used patient-derived T cells, we hypothesized that the third issue from above applied, namely that neoantigen-specific T cells may have become exhausted or deleted from the patient's T cell repertoire, as suggested by previous work with patient samples and animal models.12,19
Bringing these concepts together, the present study explored whether T cells specific for driver mutations can be obtained from blood samples from MHC-matched healthy donors. We focused on three common driver mutations in lymphoma: MYD88L265P, EZH2Y641F, and EZH2Y641N. MYD88 encodes an adaptor protein involved in toll-like receptor and NF-κB signaling.20 MYD88L265P is present in 91% of lymphoplasmacytic lymphomas (LPL), 62% of primary central nervous system lymphomas, 29% of activated B-cell-like (ABC)-diffuse large B-cell lymphomas (DLBCL), and subsets of other lymphomas and leukemias.21-28 EZH2 is involved in histone methylation and subsequent repression of a multitude of genes.29 EZH2Y641 is mutated to one of four residues (F, N, H, or S) in approximately 22% of germinal center B-cell (GCB)-DLBCL and follicular lymphomas (FL).30,31 We found that CD4+ and CD8+ T cells specific for common driver mutations can, indeed, be obtained from MHC-matched healthy donors. However, our results underscore the rarity of such responses owing to the combined limitations of antigen processing, MHC restriction, and the finite size of the human T cell repertoire in individuals.
Materials and methods
Biospecimens
Specimens and clinical data were collected with informed consent under protocols approved by the ethics review boards of the BC Cancer Agency/University of British Columbia or the Dana Farber/Harvard Cancer Center. The average age of healthy donors was 45 y, and the female:male ratio was 13:6. For mutational analysis, CD19+ cells were sorted from bone marrow aspirates of 20 LPL patients. Tumor tissue from the remaining LPL and FL patients was obtained from diagnostic biopsies that were cryopreserved or fixed in formalin. Peripheral blood mononuclear cells (PBMC) from healthy donors and patients were collected into sodium heparin tubes (BD Biosciences), isolated by density centrifugation over Ficoll-Paque PLUS (GE Healthcare) and cryopreserved in nitrogen vapor freezers. DNA was isolated using the QIAGEN AllPrep kit.
DNA sequencing
High-resolution MHC class I typing of patient samples was performed in-house using sequence-based approaches or commercially using PCR-SSOP (ProImmune). Genomic tumor DNA was screened for MYD88 and EZH2 mutations using Sanger sequencing or Illumina-based sequencing after PCR amplification or targeted exon capture (Supplemental materials).
Peptide libraries
We designed libraries comprised of all possible 8-, 9-, 10-, and 11-mer peptides corresponding to mutant or wildtype MYD88 and EZH2 proteins (38 peptides per library, Table S1). Peptides were synthesized commercially (ThinkPeptides and Genscript), reconstituted in 80% DMSO, and stored at −80°C.
Derivation of T cell lines
Monocyte-derived DC were generated by culturing adherent PBMC in AIM-V serum-free media (Life Technologies) with HEPES, L-glutamine, 800 IU/mL GM-CSF (PeproTech), and 800 IU/mL IL-4 (PeproTech). 50 μg/mL poly(I:C) (Sigma-Aldrich) was added on day 6, and DC were used as antigen presenting cells (APC) on day 8.32 DC were pulsed with MYD88L265P, EZH2Y641N, or EZH2Y641F peptide libraries (1 μM/peptide; 38 μM total per library), irradiated, and cultured for 10 d with autologous PBMC to activate antigen-specific CD8+ T cells. Cells were cultured in 96-well plates (15,000 APC plus 150,000 PBMC) in 0.22 μm-filtered CTL media: RPMI-1640 (Hyclone) with HEPES, L-glutamine, penicillin/streptomycin, β-mercaptoethanol, and 10% heat-inactivated human AB serum (Sigma-Aldrich). After 10–11 d, irradiated, peptide-pulsed autologous PBMC (50,000 cells/well) were added, along with 50 μg/mL poly(I:C). Cytokines were added 1 d later: 60 IU/mL IL-2 (National Cancer Institute) and 5 ng/mL each of IL-7 and IL-15 (PeproTech). After two rounds of stimulation, cells were rested in CTL media with 1 IU/mL IL-2 and 10 ng/mL IL-7. Cultures were screened by IFNγ ELISPOT 3 d later. Positive cultures were expanded using an established polyclonal expansion protocol involving allogeneic PBMC, anti-CD3 (clone OKT-3, eBioscience), and IL-2.33
Results
Mapping peripheral T cell responses to shared driver mutations in MYD88 and EZH2
To map the immunogenicity of shared driver mutations across a broad range of human HLA alleles, we designed peptide libraries comprised of all possible 8-, 9-, 10-, and 11-mer mutant peptides (38 peptides per library; Table S1) corresponding to three common driver mutations: MYD88L265P, EZH2Y641N, and EZH2Y641F (Table S2). To avoid any potentially confounding effects associated with the size and functional capacity of the T cell repertoire of cancer patients, experiments were performed with blood samples from MHC-matched healthy donors. We selected individuals who collectively harbored a broad range of MHC alleles (Table S3). Donors for experiments with MYD88L265P, EZH2Y641F, and EZH2Y641N included, respectively, individuals with 10/10, 10/10, and 7/10 of the most prevalent MHC class I alleles in North American Caucasians. Furthermore, for most of the common MHC alleles, we screened T cells from multiple healthy donors expressing the relevant allele, which allowed us to assess inter-patient variability in T cell responses (Table 1). For example, in the case of HLA-A*02:01, each of the three mutations was screened against T cells from 3–6 different HLA-A*02:01-positive donors. DC were loaded with the MYD88L265P, EZH2Y641F, or EZH2Y641N library, lethally irradiated, and cultured with autologous PBMC as a source of T cells. On average, 5.1 × 107 PBMC were screened from each donor (range: 107–108 cells). After two rounds of in vitro stimulation, cultures were screened by interferon gamma (IFNγ) ELISPOT. Positive T cell responses against the MYD88L265P library were observed for 7/10 donors (Table 2). Similarly, 10/14 donors showed positive responses to the EZH2Y641N or EZH2Y641F library (Table 3).
Table 1.
Coverage of the most prevalent MHC class I alleles in healthy donors used for T cell stimulations (number of donors with each allele).
| MHC allele | MYD88L265P | EZH2Y641F | EZH2Y641N |
|---|---|---|---|
| HLA-A*02:01 | 3 | 6 | 4 |
| HLA-C*07:01 | 3 | 3 | 1 |
| HLA-A*01:01 | 2 | 2 | 1 |
| HLA-A*03:01 | 4 | 2 | 1 |
| HLA-C*07:02 | 4 | 4 | 0 |
| HLA-C*04:01 | 3 | 3 | 1 |
| HLA-B*44:02 | 2 | 1 | 0 |
| HLA-B*07:02 | 4 | 4 | 0 |
| HLA-B*08:01 | 2 | 2 | 1 |
| HLA-C*05:01 | 1 | 3 | 1 |
Table 2.
T cells from the majority of healthy donors recognize peptides derived from mutant MYD88.
| Number of MYD88-reactive T cell lines generated |
|||
|---|---|---|---|
| Donor | Total | L265P-reactive | WT-reactive |
| DC038 | 1 | 1 | 0 |
| DC057 | 1 | 1 | 0 |
| DC086 | 1 | 1 | 0 |
| DC042 | 2 | 2 | 1 |
| DC044 | 7 | 7 | 6 |
| DC011 | 1 | 1 | 1 |
| DC060 | 1 | 1 | 1 |
| DC002 | 0 | 0 | 0 |
| DC029 | 0 | 0 | 0 |
| DC049 | 0 | 0 | 0 |
| Total | 14 | 14 | 9 |
Table 3.
T cells from the majority of healthy donors recognize peptides derived from mutant EZH2.
| Number of EZH2-reactive T cell lines generated |
||||
|---|---|---|---|---|
| Donor | Total | Y641F-reactive | Y641N-reactive | WT-reactive |
| DC011 | 7 | 7 | — | 0 |
| DC038 | 1 | 1 | — | 0 |
| DC044 | 9 | 9 | — | 0 |
| DC059 | 1 | 1 | — | 0 |
| DC041 | 6 | 6 | — | 3 |
| DC056 | 4 | 4 | — | 2 |
| DC023 | 30 | 30 | — | 30 |
| DC029 | 0 | 0 | — | 0 |
| DC054 | 0 | 0 | — | 0 |
| DC060 | 0 | 0 | — | 0 |
| DC040 | 4 | — | 4 | 0 |
| DC058 | 1 | — | 1 | 0 |
| DC035 | 4 | — | 4 | 1 |
| DC091 | 0 | — | 0 | 0 |
| Total | 67 | 58 | 9 | 36 |
To assess specificity, T cell lines were tested for recognition of the corresponding wildtype peptide libraries. For each muta-tion, 60–75% of donors yielded T cell lines with specificity for the mutant versus wildtype peptide library (Tables 2 and 3).
MYD88L265P is naturally processed and presented to T cells
To define the minimal peptides underlying each MYD88-spe-cific response, T cell lines were assessed by ELISPOT for reac-tivity to individual peptides from the MYD88L265P library (Table S1). Four minimal peptides were identified, one of which (RPIPIKYKAM) was recognized by 5/6 donors (Fig. 1A and B). In the seventh donor, the minimal peptide could not be identi-fied due to a high level of background reactivity after T cell expansion.
Figure 1.
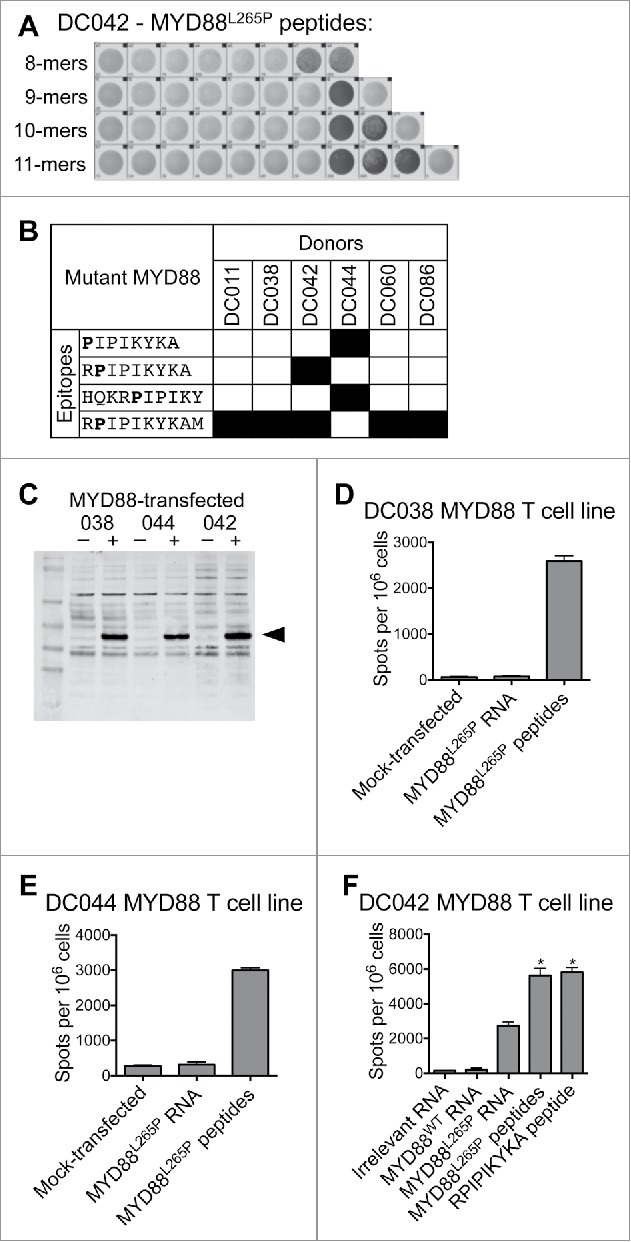
T cells from healthy donors recognize MYD88L265P peptides and full-length MYD88L265P. Healthy donor T cell lines derived from in vitro stimulation with the MYD88L265P peptide library were screened by IFNγ ELISPOT for reactivity against each individual peptide and against B cells expressing full-length MYD88L265P. (A, B) T cell lines were stimulated with each of the 38 peptides in the MYD88L265P library (1 peptide/well). The minimal peptide was defined as the shortest peptide with the strongest response of all peptides tested. (A) ELISPOT wells from one DC042 T cell line showing reactivity to eight closely related peptides derived from MYD88L265P. (B) Summary of minimal epitopes recognized by T cells. The minimal epitope was defined as the shortest peptide with the strongest response of all peptides tested. Mutant residues are indicated in bold font. (C–F) Autologous CD40-activated B cells were transfected with ivtRNA encoding full-length MYD88L265P and used to stimulate T cell lines. (C) Western blot demonstrating expression of MYD88 by transfected B cells. (D–F) ELISPOT results for T cells (1–1.5 × 105 cells/well) incubated with B cells (2 × 105/well) that had been pulsed with peptides or transfected with ivtRNA. T cell lines from (D) DC038 and (E) DC044 did not recognize processed MYD88L265P, but a T cell line from (F) DC042 did recognize processed MYD88L265P. Asterisks refer to conditions that were too numerous to count (TNTC).
We next assessed whether peptides derived from MYD88L265P are processed and presented on MHC class I or II. Autologous tumor samples were not available, as surgery is not part of standard care for lymphoma patients. Instead, we transfected CD40-activated autologous B cells with ivtRNA encoding full-length MYD88L265P. Expression was confirmed by Western blot (Fig. 1C). Autologous transfected B cells were used as APC in IFNγ ELISPOT assays with mutation-reactive T cell lines. We tested seven T cell lines from five healthy donors, which covered all candidate epitopes shown in Fig. 1B. Six of seven T cell lines failed to recognize MYD88L265P ivtRNA-transfected B cells despite showing responses to peptide-pulsed B cells (Fig. 1D and E and data not shown). However, the T cell line specific for RPIPIKYKA (from donor DC042) showed a clear positive response (Fig. 1F).
MYD88L265P is recognized by an HLA-B*07:02-restricted CD8+ T cell line
The T cell response to the MYD88L265P RPIPIKYKA epitope was further characterized. Analysis of CD137-upregulation by MYD88L265P-stimulated cells revealed that the responding T cells were CD8+ (Fig. 2A). According to flow cytometry-based TCR-Vβ spectratyping, the responding T cells expressed Vβ3 (TRBV28) (Fig. 2B). MHC-restriction experiments with peptide-pulsed LCL showed that the processed MYD88L265P peptide, RPIPIKYKA was presented by HLA-B*07:02 (Fig. 2C). Moreover, dose titration experiments demonstrated that T cells responded to the mutant peptide at 100,000-fold lower concentration than the wildtype peptide (Fig. 2D and E). These data were in accord with NetMHC v4.0,36,37 which predicted an IC50 binding score of 613 nM for the mutant epitope (ranked as a weak binder in the top 1.3% of peptides) versus 18,936 nM for the wildtype epitope (ranked as a non-binder in the top 14.0% of peptides). Furthermore, proteasomal cleavage algorithms (NetChop v3.1) predicted the presence of a putative proteasomal cleavage site within the wildtype protein (immediately C-terminal to leucine-265) that was eliminated in the mutant version. Thus, the RPIPIKYKA peptide was naturally processed, presented, and recognized by CD8+ T cells.
Figure 2.
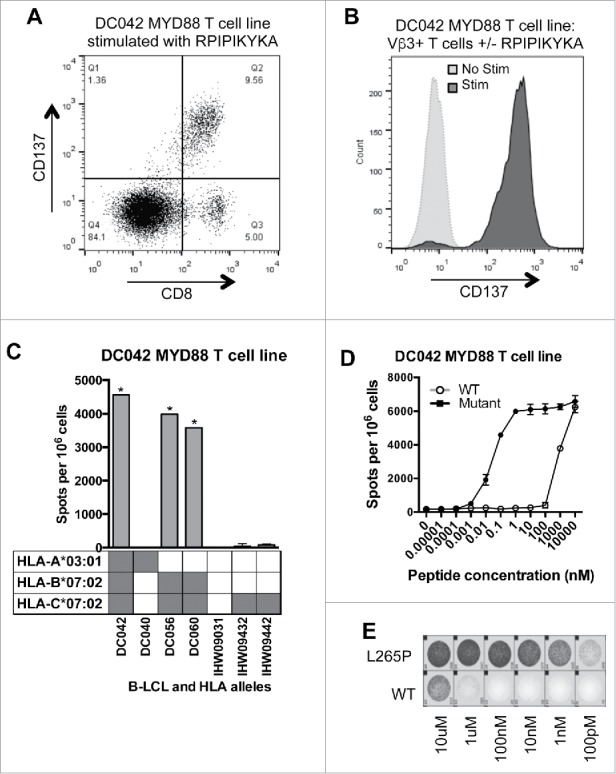
MYD88 L265P is recognized by an HLA-B*07:02-restricted CD8+ T cell line. (A, B) The MYD88L265P RPIPIKYKA-specific T cell line was stimulated overnight with or without peptide (10 μM) and then analyzed by flow cytometry. (A) Peptide stimulated cells were gated on single lymphocytes and assessed for expression of CD8+ and CD137. This showed that activated (CD137+) cells were CD8+. (B) After overnight incubation with or without peptide, cells were gated on single lymphocytes and then on expression of CD8+ and TCR-Vβ3. Stimulated CD8+ TCRVβ3+ cells express CD137, demonstrating that the relevant TCR is from the Vβ3 family. (C) IFNγ ELISPOT results from an HLA restriction experiment showing that the RPIPIKYKA peptide is presented in the context of HLA-B*07:02. T cells (1 × 105 cells) were incubated with a panel of LCL (2 × 105 cells/well) that were matched with the donor T cells at 0–2 alleles and that had been peptide-pulsed with RPIPIKYKA (10 μM). Background spot numbers (T cells plus LCL without peptide pulse) were subtracted. * = TNTC. Gray boxes indicate alleles present in each donor. (D) ELISPOT data showing recognition of MYD88L265P RPIPIKYKA (solid circles) at over 100,000-fold lower peptide concentration than MYD88WT RLIPIKYKA (open circles). Data points at the top of the curve are an underestimate, as spots were TNTC. (E) A subset of ELISPOT wells from the experiment described in Fig. 2D.
To determine whether RPIPIKYKA-reactive T cells were present in additional donors, we stimulated peripheral T cells from two more HLA-B*07:02+ healthy donors using autologous DC loaded with the RPIPIKYKA peptide. We also used the entire MYD88L265P library to stimulate peripheral T cells from two HLA-B*07:02+ lymphoma patients (2.6 × 107–7.6 × 107 PBMC per patient) whose tumors harbored the MYD88L265P mutation. We were unable to isolate T cells specific for RPIPIKYKA from any of these individuals (data not shown), indicating that RPIPIKYKA-specific T cells are rare even among HLA-B*07:02+ individuals, whether they are healthy donors or MYD88L265P-positive patients.
EZH2Y641N is naturally processed and presented to T cells on HLA-DRB1*13:02
To define the minimal peptides underlying each EZH2-specific response, T cell lines were assessed by ELISPOT for reactivity to individual peptides from the appropriate library (Table S1). We found responses to 16 minimal peptides, the most commonly recognized being SENCGEII, SEFCGEII, and NEFISEFCG (Fig. 3A). Individual donors recognized anywhere from 1–5 mutant EZH2 minimal peptides.
Figure 3.
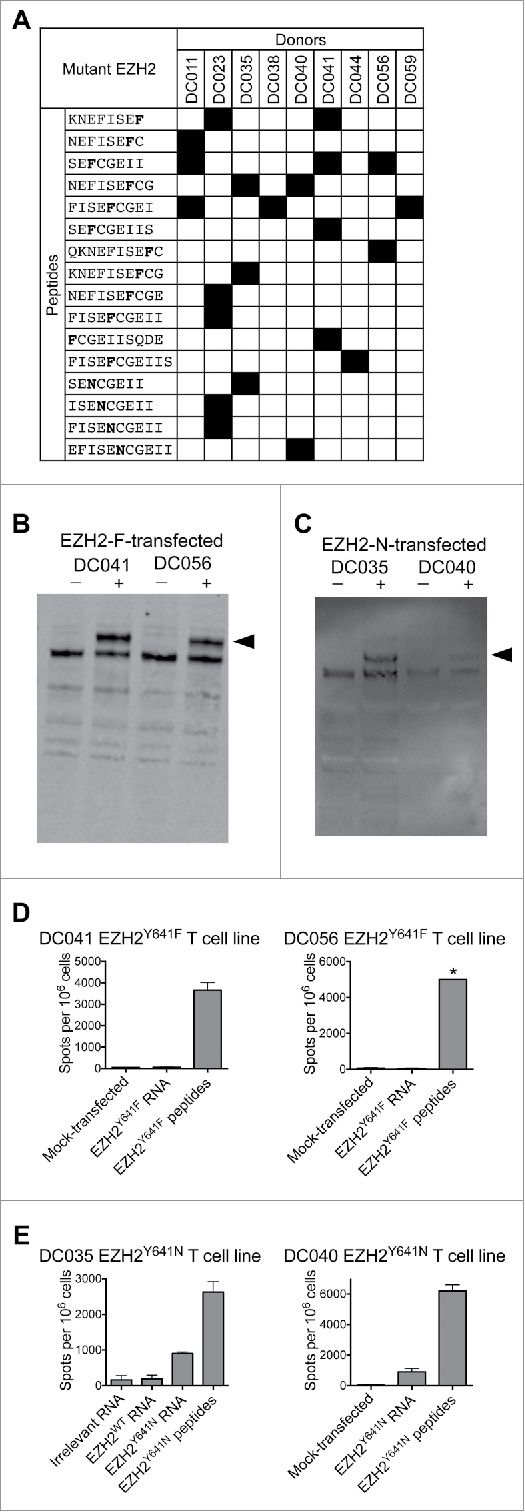
T cells from healthy donors recognize mutant EZH2 peptides and full-length EZH2Y641N. Healthy donor T cell lines derived from in vitro stimulation with the EZH2Y641F or EZH2Y641N peptide libraries were screened by IFNγ ELISPOT for reactivity against each individual peptide and against B cells expressing full-length EZH2. (A) Summary of minimal epitopes recognized by T cells. The minimal epitope was defined as the shortest peptide with the strongest response of all peptides tested. Mutant residues are indicated in bold font. (B–E) Autologous CD40-activated B cells were transfected with ivtRNA encoding full-length EZH2 and used to stimulate T cell lines. (B, C) Western blots demonstrating expression of EZH2 by B cells transfected with (B) EZH2Y641F or (C) EZH2Y641N. Although the staining is weak in panel C, the results were reproducible in independent experiments. (D, E) ELISPOT results for (D) EZH2Y641F or (E) EZH2Y641N-specific T cells (1–1.5 × 105 cells/well) incubated with B cells (2 × 105/well) that had been pulsed with peptides or transfected with ivtRNA. (D) T cell lines from DC041 and DC056 did not recognize processed EZH2Y641F, whereas (E) T cell lines from DC035 and DC040 did recognize processed EZH2Y641N. These ELISPOT results are from the same experiments shown in panels B and C. * = TNTC. All data are representative of at least 2–3 independent experiments.
To assess whether the above epitopes from EZH2Y641F and EZH2Y641N are processed and presented on MHC class I or II, we transfected CD40-activated autologous B cells with ivtRNA encoding full-length EZH2 bearing the relevant mutations. Expression was confirmed by Western blot (Fig. 3B and C). Transfected B cells were used as APC in IFNγ ELISPOT assays with autologous mutation-reactive T cell lines.
For EZH2Y641F, we tested eight T cell lines from four healthy donors, which covered 8/12 candidate EZH2Y641F epitopes shown in Fig. 3A. None of the T cell lines reacted to EZH2Y641F ivtRNA-transfected B cells, despite showing responses to peptide-pulsed cells (Fig. 3D). T cell responses could not be accurately assessed in the remaining T cell lines due to limited sample availability or low T cell reactivity to peptides. Thus, we failed to find evidence that any of the candidate epitopes from EZH2Y641F were naturally processed and presented.
For EZH2Y641N, we tested seven T cell lines from two healthy donors, which covered all candidate EZH2Y641N epitopes shown in Fig. 3A. Four T cell lines from two donors reacted to EZH2Y641N ivtRNA-transfected B cells (Fig. 3E). Collectively, these T cell lines recognized three peptides that shared the core sequence ISENCGEII.
Analysis of CD137-upregulation by EZH2Y641N EFISENCGEII-stimulated cells revealed that the responding T cells from the first donor (DC040) were CD4+ (Fig. 4A and data not shown). Dose titration experiments revealed that the response was specific for the mutant versus wildtype peptide (Fig. 4B). HLA blocking experiments (Fig. 4C) and HLA restriction experiments with peptide-pulsed LCL (Fig. 4D) showed that the response was restricted to HLA-DRB1*13:02. This agreed with the predicted IC50 score (NetMHCII v2.2)38,39 of 29 nM for this MHC:peptide combination. For the second donor (DC035), HLA restrictions could not be determined due to a low frequency of reactive T cells in T cell lines. However, by in silico analysis the cognate peptides (ISENCGEII and FISENCGEII) were also predicted to be presented by HLA-DRB1*13:02. Moreover, these peptides were not predicted to bind to the donor's MHC class I alleles (data not shown), further suggesting they were restricted by MHC class II. Thus, although we did not find any evidence of T cells reactive against full-length EZH2Y641F, we did identify CD4+ T cells specific for a naturally processed and presented HLA-DRB1*13:02-restricted epitope from EZH2Y641N.
Figure 4.
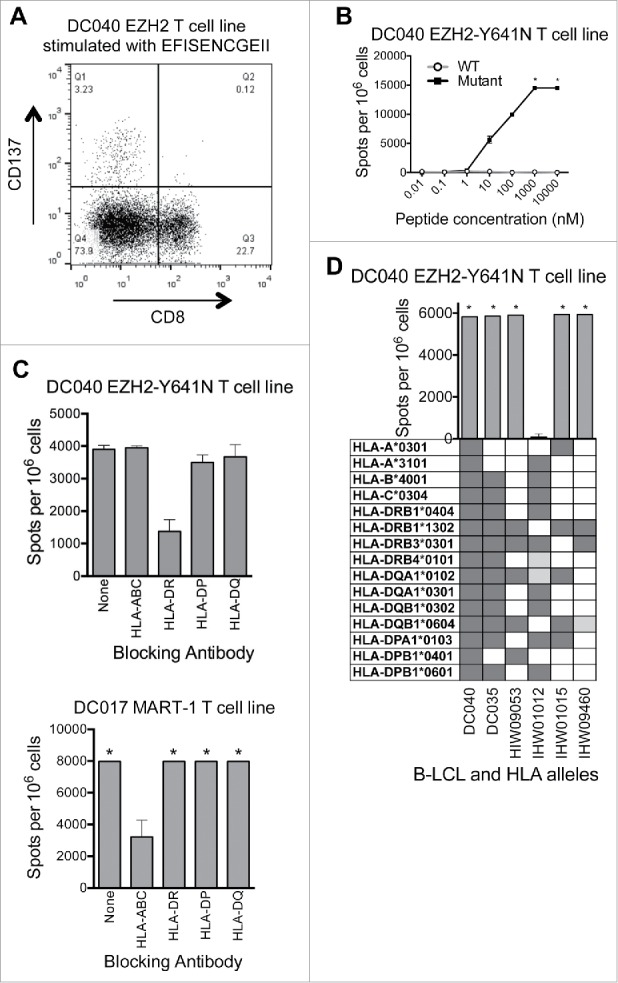
EZH2 Y641N is recognized by an HLA-DRB1*13:02-restricted CD4+ T cell line. (A) The EZH2Y641N EFISENCGEII-specific T cell line was stimulated overnight with peptide (10 μM) and then analyzed by flow cytometry. Peptide stimulated cells were gated on single lymphocytes and assessed for expression of CD8+ and CD137. This showed that activated (CD137+) cells were CD8−. Subsequent analysis confirmed that these cells were CD4+ (data not shown). (B) ELISPOT data showing recognition of EZH2Y641N EFISENCGEII (solid squares) and no recognition of EZH2WT EFISEYCGEII (open circles). Data points at the top of the curve are an underestimate, as this represents only the number of spots that were counted; in some areas, spots were TNTC (asterisks). Negative control (no peptide) counts have been subtracted. (C) IFNγ ELISPOT results from an HLA blocking experiment showing that the EFISENCGEII-specific T cell response from donor DC040 is inhibited in the presence of an antibody against HLA-DR (upper panel). Equal numbers of T cells and autologous B cells were pre-incubated with blocking antibodies (5–10 μg/mL) and then 2 × 105 cells were added per well along with 50 nM peptide and 5–10 μg/mL antibody. Background spot numbers (T cells plus B cells without antibodies or peptides) were subtracted. An HLA-A2-restricted MART-1-reactive T cell line from donor DC017 was used as a negative control (lower panel). * = TNTC. (D) IFNγ ELISPOT results from an HLA restriction experiment showing that the EFISENCGEII peptide is presented in the context of HLA-DRB1*13:02. 1 × 105 T cells were incubated with a panel of LCL (2 × 105 cells/well) that were matched with the donor T cells at 2–12 alleles and that had been peptide-pulsed with EFISENCGEII (10 μM). Background spot numbers (T cells plus LCL without peptide pulse) were subtracted. * = TNTC. Gray boxes indicate alleles present in each donor. Dark gray boxes represent alleles matched to four-digit resolution with DC040. Light gray boxes represent alleles matched to two-digit resolution. This experiment indicates that the T cell response is restricted to either HLA-DRB1*13:02 or HLA-DQB1*06:04.
Discussion
To map the TCR repertoire to shared driver mutations, we stimulated healthy donor T cells with autologous APC presenting three common driver mutations from lymphoma. Specifically, T cells from donors collectively expressing the most common MHC class I alleles were primed in vitro with peptide libraries encompassing MYD88L265P, EZH2Y641F, and EZH2Y641N. We identified T cells specific for bona fide, processed epitopes derived from full-length MYD88L265P and EZH2Y641N. However, these responses were rare, even across individuals with the same MHC alleles. Thus, although targeting driver mutations with T cell-based therapies remains a compelling strategy, individualized approaches will be required to overcome limitations in both the number of bona fide neoantigens and the rarity of cognate T cells in the naïve repertoire.
Our analysis of over 100 mutant peptides across the 10 most common HLA class I alleles using approximately 4 × 108 T cells, followed by validation of processing and presentation of full-length proteins, provides new insights into the accuracy of epitope prediction algorithms in the context of mutanome studies. Current algorithms generally perform well for predicting MHC:peptide binding40,41 but poorly for predicting protein processing.42 The two confirmed epitopes in our study - RPIPIKYKA and EFISENCGEII - have predicted IC50 values of 613 nM and 29 nM, respectively. In this case, empirical analysis was beneficial since the standard threshold of <500 nM36,37,41 would have eliminated HLA-B*07:02/RPIPIKYKA while nominating 23 other false positives from our set (i.e., peptides that were not processed and presented by ivtRNA-transfected cells and HLA/peptide combinations that did not stimulate T cell responses). The binding differential between wild type and mutant peptides may also influence immunogenicity.43 For example, the IC50 values for the wild type versions of the two aforementioned peptides are 18,936 nM and 2,380 nM. Based on these values, these peptides are unlikely to be presented on MHC, making it unlikely that cognate T cells would be eliminated through central tolerance. However, the binding differential between wild type and mutant peptides is not always predictive of immunogenicity, as some point mutations affect TCR binding without altering MHC interactions, as shown in our prior study of lymphoma.18 Thus, our findings highlight the utility of current prediction algorithms for identifying some mutant epitopes (e.g., EFISENCGEII from EZH2Y641N); however, empirical analyses are still required to identify others (e.g., RPIPIKYKA from MYD88L265P).
Another limitation to targeting the mutanome is that T cells specific for a given epitope may not be present, or functionally competent, in a given patient. Cognate T cells simply might not exist in the naive repertoire (e.g., due to central tolerance), or they may become anergic, exhausted or deleted after activation. By screening allogeneic donors, we avoided the latter possibilities. Nevertheless, the presence of “holes” in the naive TCR repertoire remained a considerable problem. Despite extensively screening blood from six HLA-B*07:02-positive healthy donors and two HLA-B*07:02-positive lymphoma patients, which represented a collective total of approximately 5.5 × 107 CD8+ T cells, we identified T cells specific for the MYD88L265P epitope (RPIPIKYKA) in only one individual (Fig. 1 and data not shown). This is in line with a recent study in which T cells reactive against individual melanoma-specific mutations were only detected in a subset of HLA-matched donors.15 Due to a lack of available HLA-DRB1*13:02-positive donors, we were not able to interrogate the naive CD4+ T cell repertoire to the same degree. However, our data suggests that mutation-specific responses, considered as a whole, are rare given that we screened a total of 1.2 × 109 PBMC and identified only five mutation-reactive T cell lines. The issue of individuals having holes in their TCR repertoires in relation to specific epitopes has been underappreciated in mutanome studies to date and underscores the need to screen multiple donors to identify relevant TCRs corresponding to high-value epitopes.
Our study has several limitations. First, our analysis involved only three driver mutations and one malignancy. While this narrow focus allowed us to perform exceptionally deep screening, the extent to which our conclusions apply to other driver mutations and malignancies awaits further study. Second, given that surgery is not part of standard care in lymphoma, we were unable to assess recognition of autologous tumor cells. We addressed this to the extent possible by using autologous B cells transfected with RNA encoding full-length mutant and wildtype genes. Indeed, this approach revealed that 75% of candidate mutant peptides were not naturally processed. Tumor sample availability is a common limitation in this field, so neoantigen-reactive T cells are often characterized using peptide-pulsed or transfected cells, or with peptide:MHC multimers.8,13,15,34,35 Furthermore, neoantigens have even been targeted clinically without first confirming that they are expressed on tumor cells.13 Our findings and those of others highlight the significant “drop out” of candidate neoantigens that occurs between recognition of synthetic peptides versus full-length proteins. One would expect additional losses to be observed when assessing recognition of autologous tumor cells, due to inadequate expression of the target antigen and/or MHC pathway components. Thus, in the setting of clinical trials, we believe there is strong justification to take extra measures to collect autologous tumor samples for T cell recognition assays.
Immune-based targeting of the cancer mutanome has promise, but the limited number of immunogenic epitopes and mutation-specific T cells must be considered when developing therapeutic strategies. Our work and other recent studies have revealed three key points. First, “off-the-shelf” therapeutic strategies such as TCR engineering might be practical in cases where the driver mutation, HLA allele, and cancer type are all sufficiently prevalent, such as HLA-A*02-restricted epitopes from mutant KRAS in pancreatic and colorectal cancers.44,45 Although MYD88L265P and EZH2Y641N are recurrent mutations, they are found in relatively rare malignancies and presented by less common HLA alleles (approximately 30% of North American Caucasians for each of HLA-B*07 and HLA-DRB1*13, respectively).46 Therefore, we estimate that each of these two epitopes will be applicable to fewer than 2,000 patients per year in the United States, which substantially limits the practicality of TCR engineering and similar strategies. In such scenarios, it makes more sense to use an expanded list of target antigens, such as the approximately 40 other genes that frequently harbor putative driver mutations in lymphoma.30,47-51 Second, while most mutations do not spontaneously elicit detectable T cell responses, it is possible to prime responses to a subset of these “ignored” mutations using in vitro stimulation methods such as those described here. Our data indicate that the peripheral TCR repertoires of patients and healthy donors are similar in this regard, so either could be used as a source of T cells (although the use of patient blood would be simpler).18 Finally, both CD4+ and CD8+ T cell responses should be considered. We and others have identified both CD4+ and CD8+ mutation-reactive T cells,8-10,18,52,53 and mutation-specific T cells from both compartments have demonstrated clinical efficacy when used for ACT.11,14,54 Further work addressing these various considerations should help to elucidate the clinical scenarios in which T cell-based targeting of driver mutations is not only practical but offers the most therapeutic benefit to patients.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the study participants; referring clinicians; Kristy Dillon, Bemuluyigza Baraki, Adria Devlieger, and Tim Turcotte for technical assistance; and Drs Marco Marra, Robert Holt, and Richard Moore for helpful advice. Canada's Michael Smith Genome Sciences Center provided sequencing services.
Funding
This work was supported by the British Columbia Cancer Foundation, the Canadian Cancer Society (Grant #701789), Genome British Columbia, the Whittle Family Multiple Myeloma Research Fund, the Waldenstrom's Macroglobulinemia Foundation of Canada, the International Waldenstrom's Macroglobulinemia Foundation (IWMF), and the David and Janet Bingham Research Partners Fund of the IWMF. JSN received funding from the Canadian Institutes of Health Research (CIHR) and the Lymphoma Foundation Canada. ZLB is supported by a scholar award from the Michael Smith Foundation for Health Research. NNK is supported by a CIHR Frederick Banting and Charles Best MSc award.
References
- 1.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. . Signatures of mutational processes in human cancer. Nature 2013; 500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torkamani A, Verkhivker G, Schork NJ. Cancer driver mutations in protein kinase genes. Cancer Lett 2009; 281:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. . Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–16. PMID:21639808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, et al. . Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002; 346:645–52. PMID:11870241. [DOI] [PubMed] [Google Scholar]
- 5.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J 2013; 32:194–203. PMID:23258224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reinbold CJ, Malarkannan S. Recognition of allo-peptide is governed by novel anchor imposition and limited variations in TCR contact residues. Mol Immunol 2008; 45:1318–26. PMID:17981332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 2005; 102:16013–8. PMID:16247014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. . High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015; 21:81–5. PMID:25531942. [DOI] [PubMed] [Google Scholar]
- 9.Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. . Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 2014; 20:3401–10. PMID:24987109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. . Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747–52; PMID:23644516; https://doi.org/ 10.1038/nm.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. . Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641–5; PMID:24812403; https://doi.org/ 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, Castellarin M, Twumasi-Boateng K, Watson PH, Holt RA, et al. . Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res 2014; 20:1125–34; PMID:24323902; https://doi.org/ 10.1158/1078-0432.CCR-13-2147 [DOI] [PubMed] [Google Scholar]
- 13.Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. . Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015; 350:1387–90; PMID:26516200; https://doi.org/ 10.1126/science.aad1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. . T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 2016; 375:2255–62; PMID:27959684; https://doi.org/ 10.1056/NEJMoa1609279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, et al. . Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016; 352:1337–41; PMID:27198675; https://doi.org/ 10.1126/science.aaf2288 [DOI] [PubMed] [Google Scholar]
- 16.Sharkey MS, Lizee G, Gonzales MI, Patel S, Topalian SL. CD4(+) T-cell recognition of mutated B-RAF in melanoma patients harboring the V599E mutation. Cancer Res 2004; 64:1595–9; PMID:14996715; https://doi.org/ 10.1158/0008-5472.CAN-03-3231 [DOI] [PubMed] [Google Scholar]
- 17.Yotnda P, Firat H, Garcia-Pons F, Garcia Z, Gourru G, Vernant JP, Lemonnier FA, Leblond V, Langlade-Demoyen P. Cytotoxic T cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J Clin Invest 1998; 101:2290–6; PMID:9593785; https://doi.org/ 10.1172/JCI488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen JS, Sedgwick CG, Shahid A, Zong Z, Brumme ZL, Yu S, Liu L, Kroeger DR, Treon SP, Connors JM, et al. . Toward Personalized Lymphoma Immunotherapy: Identification of Common Driver Mutations Recognized by Patient CD8+ T Cells. Clin Cancer Res 2016; 22:2226-36; PMID:26631611 https://doi.org/ 10.1158/1078-0432.CCR-15-2023 [DOI] [PubMed] [Google Scholar]
- 19.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 2005; 437:141–6; PMID:16136144; https://doi.org/ 10.1038/nature03954 [DOI] [PubMed] [Google Scholar]
- 20.Watters TM, Kenny EF, O'Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol 2007; 85:411–9; PMID:17667936; https://doi.org/ 10.1038/sj.icb.7100095 [DOI] [PubMed] [Google Scholar]
- 21.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. . Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470:115–9; PMID:21179087; https://doi.org/ 10.1038/nature09671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al. . MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med 2012; 367:826–33; PMID:22931316; https://doi.org/ 10.1056/NEJMoa1200710 [DOI] [PubMed] [Google Scholar]
- 23.Jimenez C, Sebastian E, Chillon MC, Giraldo P, Mariano Hernandez J, Escalante F, Gonzalez-Lopez TJ, Aguilera C, de Coca AG, Murillo I, et al. . MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom's macroglobulinemia. Leukemia 2013; 27:1722–8; PMID:23446312; https://doi.org/ 10.1038/leu.2013.62 [DOI] [PubMed] [Google Scholar]
- 24.Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol 2011; 122:791–2; PMID:22020631; https://doi.org/ 10.1007/s00401-011-0891-2 [DOI] [PubMed] [Google Scholar]
- 25.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, et al. . Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475:101–5; PMID:21642962; https://doi.org/ 10.1038/nature10113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varettoni M, Arcaini L, Zibellini S, Boveri E, Rattotti S, Riboni R, Corso A, Orlandi E, Bonfichi M, Gotti M, et al. . Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood 2013; 121:2522–8; PMID:23355535; https://doi.org/ 10.1182/blood-2012-09-457101 [DOI] [PubMed] [Google Scholar]
- 27.Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, Morra E, Trojani A, Greco A, Arcaini L, et al. . MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 2013; 121:2051–8; PMID:23321251; https://doi.org/ 10.1182/blood-2012-09-454355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, Tanaka K, Fukushima S, Takami H, Arita H, et al. . Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol 2016; 42:279–90; PMID:26111727; https://doi.org/ 10.1111/nan.12259 [DOI] [PubMed] [Google Scholar]
- 29.Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer 2012; 106:243–7; PMID:22187039; https://doi.org/ 10.1038/bjc.2011.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. . Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42:181–5; PMID:20081860; https://doi.org/ 10.1038/ng.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodor C, Grossmann V, Popov N, Okosun J, O'Riain C, Tan K, Marzec J, Araf S, Wang J, Lee AM, et al. . EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013; 122:3165–8; PMID:24052547; https://doi.org/ 10.1182/blood-2013-04-496893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fucikova J, Rozkova D, Ulcova H, Budinsky V, Sochorova K, Pokorna K, Bartunkova J, Spisek R. Poly I: C-activated dendritic cells that were generated in CellGro for use in cancer immunotherapy trials. J Transl Med 2011; 9:223; PMID:22208910; https://doi.org/ 10.1186/1479-5876-9-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 2003; 26:332–42; PMID:12843795; https://doi.org/ 10.1097/00002371-200307000-00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al. . A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; PMID:25837513; https://doi.org/ 10.1126/science.aaa3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB, et al. . Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 2009; 6:520–6; PMID:19543285; https://doi.org/ 10.1038/nmeth.1345 [DOI] [PubMed] [Google Scholar]
- 36.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2016; 32:511–7; PMID:26515819; https://doi.org/ 10.1093/bioinformatics/btv639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003; 12:1007–17; PMID:12717023; https://doi.org/ 10.1110/ps.0239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics 2009; 10:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics 2007; 8:238; PMID:17608956; https://doi.org/ 10.1186/1471-2105-8-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology 2014; 3:e28836; PMID:25083320; https://doi.org/ 10.4161/onci.28836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res 2014; 2:522–9; PMID:24894089; https://doi.org/ 10.1158/2326-6066.CIR-13-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calis JJ, Reinink P, Keller C, Kloetzel PM, Kesmir C. Role of peptide processing predictions in T cell epitope identification: contribution of different prediction programs. Immunogenetics 2015; 67:85–93; PMID:25475908; https://doi.org/ 10.1007/s00251-014-0815-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A, et al. . Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014; 211:2231–48; PMID:25245761; https://doi.org/ 10.1084/jem.20141308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abrams SI, Khleif SN, Bergmann-Leitner ES, Kantor JA, Chung Y, Hamilton JM, Schlom J. Generation of stable CD4+ and CD8+ T cell lines from patients immunized with ras oncogene-derived peptides reflecting codon 12 mutations. Cell Immunol 1997; 182:137–51; PMID:9514698; https://doi.org/ 10.1006/cimm.1997.1224 [DOI] [PubMed] [Google Scholar]
- 45.Bergmann-Leitner ES, Kantor JA, Shupert WL, Schlom J, Abrams SI. Identification of a human CD8+ T lymphocyte neo-epitope created by a ras codon 12 mutation which is restricted by the HLA-A2 allele. Cell Immunol 1998; 187:103–16; PMID:9732698; https://doi.org/ 10.1006/cimm.1998.1325 [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Galarza FF, Christmas S, Middleton D, Jones AR. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res 2011; 39:D913–9; PMID:21062830; https://doi.org/ 10.1093/nar/gkq1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al. . Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319:1676–9; PMID:18323416; https://doi.org/ 10.1126/science.1153629 [DOI] [PubMed] [Google Scholar]
- 48.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. . Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476:298–303; PMID:21796119; https://doi.org/ 10.1038/nature10351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, et al. . Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014; 46:176–81; PMID:24362818; https://doi.org/ 10.1038/ng.2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. . Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011; 471:189–95; PMID:21390126; https://doi.org/ 10.1038/nature09730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S, Saiya-Cork K, Parkin B, Jacobi K, Shedden K, et al. . Activating STAT6 mutations in follicular lymphoma. Blood 2015; 125:668-79; https://doi.org/ 10.1182/blood-2014-06-582650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, et al. . Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692–6; PMID:25901682; https://doi.org/ 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. . Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 2015; 125:3981–91; PMID:26389673; https://doi.org/ 10.1172/JCI82416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. . Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126–9; PMID:16946036; https://doi.org/ 10.1126/science.1129003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.