

Abstract
High-grade gliomas such as glioblastoma (GBM) and diffuse intrinsic pontine glioma (DIPG) are characterized by an aggressive phenotype with nearly universal local disease progression despite multimodal treatment, which typically includes chemotherapy, radiation therapy (RT), and possibly surgery. Radiosensitizers that have improved the effects of RT for extracranial tumors have been ineffective for the treatment of GBM and DIPG, in part due to poor blood brain barrier penetration and rapid intracranial clearance of small molecules. Here, we demonstrate that nanoparticles can provide sustained drug release and minimal toxicity. When administered locally, these nanoparticles conferred radiosensitization in vitro and improved survival in rats with intracranial gliomas when delivered concurrently with a 5-day course of fractionated RT. Compared to previous work using locally-delivered radiosensitizers and cranial radiation, our approach – based on rational selection of agents and a clinically-relevant radiation dosing schedule – produces the strongest synergistic effects between chemo- and radio-therapy approaches to the treatment of high-grade gliomas.
Keywords: Glioblastoma, Diffuse intrinsic pontine glioma, Nanoparticles, Ataxia Telangiectasia and Rad3-related protein inhibitor, Convection-enhanced delivery, Fractionated radiation therapy
Introduction
High-grade gliomas are devastating intracranial tumors that occur in both adults and children, and there has been limited progress in the development of more effective therapies for these tumors over the last several decades. In the most malignant form of the disease, glioblastoma (GBM), median survival has stagnated at approximately 14.5 months in adults (1). Similarly, children who develop a particularly aggressive type of glioma in their brainstem, diffuse intrinsic pontine glioma (DIPG), typically live no longer than 12 months (2). The greatest barrier to treatment efficacy in nearly all gliomas is local recurrence, despite treatments that include surgical resections followed by high doses of radiation therapy (RT) and chemotherapy. For example, nearly 80% of GBMs (3) and 100% of DIPGs recur after standard treatment at the primary site (2). This has prompted a number of clinical studies which have been focused on combining novel targeted agents with RT and chemotherapy, as means to sensitize tumor cells to the effects of these potent DNA damaging agents (4, 5). To date, these studies have largely been unsuccessful (6).
The highly protective nature of blood-brain barrier (BBB) is a formidable obstacle to achieving therapeutic levels of many drugs in glioma tumor tissue (7). While some tumors such as high grade glioma (including glioblastoma) demonstrate tumor enhancement (reflecting a disruption of the normal BBB in regions of the tumor), DIPG often does not enhance with contrast. For this reason, it is thought that the blood-brain barrier and blood-tumor barrier may be more intact than in GBM which prevents most drugs and all nanoparticles from crossing as readily as might be seen in tumors with significant enhancement. While the BBB may be broken down in some areas of the bulk tumor, all GBMs contain regions of intact BBB (8), which is a major factor underlying the universal recurrence after systemic drug therapy. Tight junctions within the BBB endothelium limit the entry of over 98% of all small molecules, and essentially 100% of all macromolecular agents (9). In addition, efflux pumps are expressed in the BBB and have been shown to actively exclude a number of small molecules tested in glioma clinical trials, including gefitinib (10), dasatinib (11), cediranib (12), and vemurafenib (13). Several approaches have been developed to either enhance or circumvent the BBB, including transient disruption of the BBB using ultrasound or hypertonic intravascular solutions, nanoparticle carriers, and direct intratumoral delivery in the brain (8). Nanoparticles (NPs) are attractive drug delivery vehicles because they can be created with biodegradable materials that have been approved by the FDA in other settings; we and others have shown that they can be engineered for sustained drug release over many weeks (14). Convection-enhanced delivery (CED) is a direct intratumoral delivery method that uses a mild hydrostatic pressure gradient to distribute particles through brain tissue via trans-cranial catheters (15, 16). The use of CED in humans with intracranial tumors has been shown to be safe in both adults and children with gliomas (17–19). Although CED provides enhanced drug penetration, most small molecules administered directly to the brain are prone to rapid clearance from the brain parenchyma, which usually leads to undetectable drug levels 24 hours after the end of a course of CED (20). CED typically is only administered once because of the complexity and potential morbidity of the procedure. As chemotherapy and RT are delivered over periods of weeks to months, CED of chemo- and radio-sensitizers thus is likely to be ineffective without a method for sustained drug release.
In this study, we sought to develop and validate a method for the sustained release of radiosensitizers, encapsulated in nanoparticles and administered via CED, as a novel approach to treat gliomas. Overall, we found that CED of radiosensitizer-loaded NPs is feasible, effective, and represents a promising new therapeutic strategy for the treatment gliomas.
Materials and Methods
Materials
Polyethylene glycol-b-Polylactic acid diblock polymer (Mw PEG = 5kDa, Mw PLA = 10kDa) was purchased from Polysciences, Inc. (Warrington, PA, USA). Anhydrous paraformaldehyde and Tween 80, were obtained from Sigma-Aldrich (St. Louis, MO, USA). Acetonitrile and dimethylsulfoxide were obtained from J.T. Baker (Avantor Performance Materials, Central Valley, PA, USA). KU60648 was purchased from Axon MedChem (Groningen, Netherlands) and V822 was purchased from Selleck (Houston, TX, USA). All other small molecule DNA repair inhibitors were purchase from Tocris Bioscience (Avonmouth, Bristol, UK). All dyes were purchase from ThermoFisher Scientific (Waltham, MA, USA). All animals were obtained from Charles River Laboratories (Wilmington, MA, USA)
NPs preparation
PLA-PEG NPs
PLA-PEG NPs were synthesized using a nanoprecipitation technique. Briefly, polymer was dissolved in DMSO at 20mg/ml. The polymer solution was added drop-wise to at a 1:5 volume ratio to diH2O under vortex. This suspension was then diluted in diH2O and washed x2 via centrifugation filtration with an Amicon Ultracell 100k centrifugal filter unit. The final NP suspension was then either immediately used for in vivo or in vitro experiments, or snap-frozen at −80°C until use. Unloaded (blank) NPs were fabricated exactly as described above. For dye-loaded NPs, Didyes dissolved in the polymer solution at 0.2% by weight to the polymer. For drug-loaded NPs, all drugs were dissolved in DMSO and added to the polymer solution at given weight ratios before nanoprecipitation.
NPs characterization
Size and zeta potential measurements
The hydrodynamic diameter of a 0.05mg/ml solution of NPs in 1×PBS was measured by Dynamic Light Scattering (DLS). Particle hydrodynamic diameter was reported as the mean of the diameter distribution.
Zeta potential of a 0.5mg/ml solution of NPs in diH2O was measured using a Malvern Nano-ZS (Malvern Instruments, UK).
TEM imaging
For TEM imaging, particle solutions were placed on a CF400-CU TEM grid (Electron Microscopy Sciences, Hatfield, PA). Grids were stained with a 0.2% uranyl acetate solution for 15 s and washed three times in DI water and then mounted for imaging with a Tecnai T12 TEM microscope (FEI, Hillsboro, OR).
Particle stability in aCSF
Particles were measured using Malvern Nano-ZS in artificial cerebrospinal fluid (aCSF; Harvard Apparatus, Holliston, MA) at 37°C with a standard operating procedure taking measurements at given time points up to 21 d.
Particle loading
Dye loading was determined by suspending 10 mg of NPs in a diH2O and comparing against a standard curve using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) at 456/590 (nm). Drug loading was determined by dissolving NPs in acetonitrile (ACN) and analyzing the filtered solution using a Shimadzu HPLC System (SpectraLab Scientific, Markham, ON, Canada), with comparison against standard curves for each agent.
Particle Release
The release of radiosensitizers from nanoparticles was measured for up to 14 days after fabrication. NPs were dispersed in 1× PBS with 0.5% tween 80 at 37°C. At predetermined time points, this suspension was removed, filtered, and the filtrate was collected for HPLC analysis. The same volume of solution removed was added back to the suspension for continued release.
Particle Release Analysis
Drug release results for each agent were fit to a two-phase decay curve to determine the percent of burst release. The results from the linear release phase were then fit to a linear regression curve, where the slope of the fit line corresponded to the rate of drug release.
Cell culture
RG2 (rat glioma) cell lines were obtained from ATCC (Manassas, VA). SF188 (human pediatric glioma) were obtained from Daphne Hass-Kogan at the University of California, San Francisco. KNS42 cell lines were obtained from the Ryken Cell Bank in Japan. Primary human DIPG spheroids were obtained from Dr. Michelle Monje of Stanford University. Normal Human Astrocytes were obtained from Tim Chan at Memorial Sloan Kettering Cancer Center. All cells were cultured in 5% CO2 and air humidified in at 37°C incubator. Each cell line was stocked at early passages and kept in culture up to passage number 20. All cell lines used were tested via PCR and confirmed negative for mycoplasma contamination.
NP and TEM Uptake kinetic studies
U87 and SF188 cells were plated at a density of 10,000 cells/well in 96 well plates. 24 h after, cells were treated with either fluorescent particles at a concentration of 100 µg/mL or left untreated as a control. At different time points cells were harvested, washed 3× and re-suspending cells in a cold 1% BSA solution on ice. Flow cytometry was performed using Attune NxT (Invitrogen) and at least 10,000 iterations were acquired, then the data was analyzed using FlowJo v.10.0.8r1. The mean fluorescence intensity (MFI) in the DiA channel was then recorded and divided by background MFIs from control cell populations for each time point to yield a normalized fold increase in MFI in this channel for each of the different cell types. As NPs from a singular batch preparation and thus with the same dye loading per weight ratio were used on all of these experiments, the normalized MFI can directly be translated to relative particle uptake.
For TEM, the same procedure was repeated as above except only in U87 cells plated at a density of 200,000 cells/well in 6 well plates. After harvest the cells were fixed in 4% paraformaldehyde then prepared for TEM imaging using a Tecnai T12 TEM microscope (FEI, Hillsboro, OR).
In vitro uptake imaging
DIPG spheroids and SF188 cells were grown in 12-well plates and exposed to NP containing C6 dye. Hoechst dye (Thermo Fisher) was added to cells at given timepoints and cells were imaged using a plate reader capable of fluorescent detection and imaging (Cytation3, Cytek).
DNA Repair Assay
This U2OS reporter (EJ-DR) cell assay developed by Bindra et al. was performed as previously described (21). Free drug results were normalized to an equivalent volume of DMSO in the cell media and nanoparticle-drug formulations were normalized to an equivalent weight of unloaded nanoparticles in the media
Western Blot
SF188 cells were cultured under sterile conditions and exposed to 1 µM NP-VE822 or free VE822. Length of time of drug exposure varied from 24 h to 72 h. As indicated, cells were irradiated with 10 Gy using an XRAD 320 Cabinet X-ray irradiator (Precision Xray, CT, USA). Cells were harvested 60 min after RT and prepared for western blot analysis. The following primary antibodies were used: rabbit polyclonal anti-SMC1 (Bethyl), rabbit monoclonal anti-pChk1 (Ser345) (133D3, Cell Signaling), and mouse monoclonal anti-Chk1 (2G1D5, Cell Signaling).
Clonogenic survival assays
KNS42 cells were incubated with test compounds for 20 h before irradiation on an XRAD 320 Cabinet X-ray irradiator at multiple doses, and the cells were then trypsinized after a 4-hour incubation post-IR, seeded at decreasing plating densities and grown for 14 d as previously described (22). Colonies (>50 cells) were visualized by fixing with methanol and staining with crystal violet. Surviving fractions were then calculated by normalizing to the plating efficiency for each experiment (colony formation after 0 Gy dose).
Cell Viability assays
Normal Human Astrocytes and SF188 cells were plated in 96 well plates (5,000 cells/well) and exposed to varying concentration of NP-VE822, free VE-822 or DMSO control for 24 to 96 h. Cell viability was evaluated using the CellTiter 96 AQ One Solution Cell Proliferation Assay (Promega) per Promega’s published protocol. Cell viability was calculated as a percentage of the absorbance at the target dose divided by the absorbance at nominal dose.
In vivo experiments
All animal husbandry and procedures were performed in accordance with the guidelines and policies of the Yale Animal Resource Center (YARC) and approved by the Institutional Animal Care and Use Committee (IACUC) per protocol # 2015-20060. Male Fischer 344 rats (200–220 g) obtained from Charles River Laboratories were used. Surgical procedures were performed using standard sterile surgical techniques.
Convection enhanced delivery
CED of all cells and therapeutic or therapeutic controls was performed as previously described (10). CED in tumor bearing rats was conducted following the exact same procedure as for the healthy rats, by reopening the burr hole used for tumor implantation. 2.5 × 105 RG2 cells/animal were used. Tumors were grown for 4 d before randomization into treatment groups with either free drug, NPs, or controls. All groups had 8 animals per treatment, per group. In order to avoid confounding effects of toxicity and to isolate the therapeutic benefit conferred by specifically-radiosensitizing effects, we desired the maximum average local concentration to be approximately 5uM. In vivo, we demonstrated that the volume of distribution of nanoparticles when delivered locally via convection-enhanced delivery was ~2× the volume infused. Thus, if a solution at a given concentration was infused into the tumor, the resulting average local concentration (assuming homogenous distribution) was estimated to be ½ of the original solution concentration (i.e. if a 10uM solution was infused, this would result in a distributed solution with an average concentration of 5uM). Thus to achieve an initial average local concentration of 5uM in vivo using local administration via convection-enhanced delivery, we infused a solution with a concentration of 10uM.
Volume of distribution
Volume of distribution was measured as previously described (23) using DiI-loaded PLAPEG NPs.
Intracranial drug retention studies
20 µL of drug-loaded NPs, free drug, unloaded NPs, or 1× PBS was delivered into the R caudate via CED exactly as described above. Brains were harvested at certain time points after injection ranging from 0 h to 10 d and immediately divided into L and R hemispheres. Each hemisphere was processed separately using centrifugation and ACN to extract small hydrophobic compounds. Samples were analyzed for VE822 levels using Agilent LCMS 6120B (Agilent Technologies, Santa Clara, CA) compared against previously established standards.
Intracranial NP TEM
20 µL of Dye-loaded NPs were infused into the caudate exactly as described above. At given time points animals were sacrificed and their tissue was processed for TEM imaging using a Tecnai T12 TEM microscope (FEI, Hillsboro, OR).
Radiation Therapy and Survival Endpoints
Animals in the single-fraction groups were irradiated 24 h after CED of the drug, while animals in the 5-fraction RT groups were irradiated starting 48 h after drug delivery. All animals were anesthetized prior to RT using a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg), injected intraperitoneally. Animals were placed in a custom plexiglass holder with molded lead shielding to collimate the beam, allowing for selective cranial irradiation. The animals were irradiated in a XRAD 320 Cabinet X-ray irradiator (Precision Xray, CT, USA). In-depth dosimetry was performed using thermoluminescent dosimeter to ensure that the entire cranium was treated with 10 Gy per fraction for the single-dose groups or 3 Gy per fraction of radiation for the 5-fraction groups. Animals in the 5-fraction groups were treated on consecutive days, spaced 24 h apart for a total of 5 d. The animals’ health status were monitored daily, and decision to euthanize was made after either a 15% loss in body weight or when it was humanely necessary due to clinical symptoms from tumor progression.
Graphing and Statistical Analysis
Prism 6 was used for all graphing and statistical analysis, unless otherwise noted. Flow cytometry data analysis was done with FlowJo v.10.0.8r1 and Microsoft Office Excel 2011. ImageJ and Matlab were used for all image analysis, unless otherwise noted. When two groups were being compared, the significance of data was assessed by the two-tailed Student’s unpaired t test All error bars represent S.D. unless otherwise noted. Differences in survival curves were determined by Log-Rank test. P values are denoted in figure legends when applicable.
For more details please see Supplementary Methods
Results
Encapsulation and characterization of candidate DNA repair inhibitors in PLA-PEG NPs
We selected six small molecule inhibitors that selectively target molecules involved in key DNA damage response pathways for our study (Table 1) (24–29). We first sought to encapsulate the collection of drugs into PLA-PEG NPs, followed by an assessment of each compound’s encapsulation efficiency and the biological properties of the agent after formulation into NPs. We found that the encapsulation efficiency of candidate molecules was highly dependent on the agent’s solubility in the organic solvent used for fabrication. The candidate molecule solubilities ranged from 1mM (BEZ235) to 140mM (AZD7762) in DMSO (Table 1). The drug encapsulation efficiencies as measured using high-performance liquid chromatography trended in a similar manner, ranging from 4.2 to 52% for BEZ235 and AZD7762, respectively (Table 2, Supplemental Fig. 1). None of the agents, when encapsulated into NPs, had a significant effect on either the diameter or surface potential of the NPs (Table 2). Dynamic light scattering analysis revealed a homogenous size distribution for each formulation, with mean diameters ranging from 65 to 82 nm (Fig. 1a, Table 2). These values are well below the previously established size cutoff of 100 nm diameter required for effective penetration through the brain parenchyma (30). In addition, all formulations were shown to have slightly negative surface potentials (ranging from −2 to −9 mV), which are well within the range of 0 to −15mV that we and others previously have shown to be optimal for preventing aggregation of particles, and for minimizing toxicity to cell membranes (31, 32).
Table 1.
Candidate DNA repair inhibitors for NP encapsulation.
| Candidate DNA Repair Inhibitors for nanoparticle Encapsulation | |||||
|---|---|---|---|---|---|
| Drug Name |
Drug Target(s) |
Chemical Structure | Active dose range |
Solubility in DMSO (mM) |
LogP |
| NU7441 | DNA-PK |
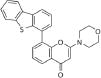
|
0.3–500 uM | 5 | 4.73 |
| KU60648 | DNA-PK |
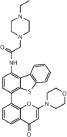
|
200–500 nM | 10 | 3.55 |
| BEZ235 | DNA-PK/ATM |
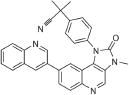
|
0.5–1 uM | 1 | 5.71 |
| KU60019 | ATM |
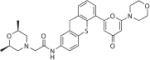
|
1–10 uM | 100 | 2.7 |
| VE822 | ATR |
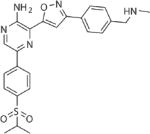
|
20–200 nM | 80 | 2.89 |
| AZD7762 | Chk1 |
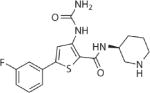
|
0.2–12 uM | 140 | −0.47 |
Table 2.
NP characteristics for each optimized drug-NP formulation
| Characteristics for each Optimized Nanoparticle Formulation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Candidate Molecule |
NP diam (nM) |
NP zeta (surface ) potentia l (mV) |
Drug-NP encapsulation (nmol drug/mg NP) |
Encapsulation efficiency (%)* |
Release during burst phase (%) |
Duration of linear phase (d) |
Release Rate during linear phase (nmol/d/mg NP) |
Release Rate during linear phase (%) |
| NU7441 | 80 ± 3 | −4 ± 1 | 110 ± 10 | 21 ± 9 | 33 ± 3 | 4 | 1.0 | 0.91 |
| KU60648 | 82 ± 2 | −6 ± 3 | 240 ± 10 | 44 ± 6 | 31 ± 1 | 5 | 2.6 | 3.4 |
| BEZ235 | 81 ± 5 | −3 ± 2 | 1.8 ± 0.1 | 4.2 ± 2.0 | 30 ± 3 | >14 | 0.011 | 0.60 |
| KU60019 | 78 ± 3 | −5 ± 2 | 320 ± 10 | 45 ± 8 | 45 ± 5 | >14 | 3.9 | 1.0 |
| VE822 | 65 ± 3 | −9 ± 4 | 180 ± 10 | 49 ± 4 | 36 ± 2 | >14 | 1.4 | 0.80 |
| AZD7762 | 76 ± 5 | −5 ± 1 | 640 ± 10 | 52 ± 1 | 31 ± 1 | 0 | 0 | 0 |
| Unloaded (blank) | 62 ± 3 | −2 ± 1 | n/a | n/a | n/a | n/a | n/a | n/a |
| DiD (dye) | 66 ± 4 | −7 ± 1 | 0.2% by weight | 100 | ~0 | n/a | n/a | n/a |
Drug loaded/theoretical drug encapsulated
Figure 1. in vitro uptake characteristics and uptake of PLA-PEG NPs.

a. Transmission electron micrography (top row) and diffusion light scattering of hydrodynamic diameter (bottom row) of drug-encapsulated PLAPEG nanoparticles suspended in diH2O b. Release of encapsulated drugs from PLA-PEG NPs at 37°C suspended at 0.5mg NP/ml in 1×PBS spiked with 0.5% TWEEN80. c. in vitro transmission electron micrography imaging of cumarin6-loaded PLA-PEG NP uptake into U87 cells after incubation with PLA-PEG NPs suspended in cell culture media at 100mg/ml for 30 min, 1h, 2h, 4h. NPs indicated with red arrow. d., e. Fluorescence imaging of DAPI-stained SF188 cells (d.) and patient-derived DIPG spheroids (e.) after 4h incubation with coumarin6-loaded PLA-PEG NPs suspended in cell culture media at 100mg/ml. DAPI channel (nuclei) in blue, coumarin6 channel (NPs) in green.
Next, we characterized the release of each molecule from NPs during continuous incubation in conditions mimicking CSF (Fig. 1b). The percent release during the burst phase (i.e. during the initial 24 hr) ranged between 30% and 45% (Table 2). The duration and pattern of sustained release of the compounds (i.e. after the initial 24 hours) varied significantly: AZD7762 demonstrated minimal additional release beyond the burst period; NU7441 and KU60648 demonstrated linear release for less than 5 d; BEZ235, VE822, and KU60019 demonstrated linear sustained release for at least 14 d (Fig. 1b). For the three agents that exhibited >14 d of linear sustained release, VE822 and KU60019 were released at substantial levels, 1.4 and 3.9 nmol/d/mg NP, respectively (Table 2). Interestingly, we found that neither the percent released during burst phase nor the rate of release during the linear phase, trended with the solubilities, partition coefficients (logP), molecular weight (MW), or total polar surface area (tPSA) of each compound. Linear regression analysis found a R2 < 0.25 for all potential correlations and F-testing for non-zero slope found no significance, with p>0.1, for all potential correlations (Supplementary Table 1). PLA-PEG NPs were found to be stable in size and surface potential when incubated in a surfactant-spiked isotonic solution at 37°C over the course of 28 days (Supplementary Fig. 2a) consistent with a slow degradation profile and accounting for the prolonged release of several of the encapsulated agents.
One advantage of NPs is internalization into cells, which should enhance the activity of agents that act intracellularly, and promote retention in the brain. But a potential concern about PEGylated NPs is that internalization is hindered. To assess this in tumor cells, we produced dye-loaded NPs that were comparable in size and surface potential to the radiosensitizer-loaded NPs (Table 2) and measured their internalization in cultured glioma cells in vitro. Substantial cellular uptake was evident within 24 hours of exposure in both an adult and pediatric glioma cell line (U87 and SF188 cells, respectively), as measured using flow cytometry (Supplementary Figs. 2b–d). NPs were associated with cell membranes within 30 minutes of exposure to U87 cells and were internalized by 4 hours, as confirmed by TEM (Fig. 1c). Live fluorescence imaging of DAPI-stained SF188 cells revealed substantial perinuclear localization 4h after incubation with NPs (Fig. 1d). We also detected robust, homogenous uptake of dye-loaded NPs in cultured primary, patient-derived DIPG spheroids as large as 200 µm in diameter (Fig. 1e).
Taken together, these data indicate that a diverse collection of DNA repair inhibitors can be encapsulated stably into NPs. These NPs have characteristics that are favorable for brain tissue penetration and robust cell uptake in both established and primary, patient-derived glioma models in vitro. Only some of the DNA repair inhibitors demonstrate slow-release kinetics that match clinical courses of fractionated RT.
Comparison of the functional activities of free versus encapsulated forms of DNA repair inhibitors
To determine the utility of NPs for DNA repair inhibitors, we compared their functional activity as free drugs and as NPs on HR and NHEJ repair. We utilized a live-cell, dual HR and NHEJ repair developed by our group, which measures repair activity at two site-specific, intrachromosomally-integrated DSBs (schematic shown in Fig.2a) (21). This cell-based assay is ideal for the comparison of DSB repair inhibition by free drugs versus NPs, because the DSB kinetics can be precisely controlled during exposure to slow-release NPs, and the assay can be miniaturized to test a range of drugs, variable doses, and formulations in a single experiment (33, 34). The HR repair pathway is active in the S and G2 phases of the cell cycle, and thus is the primary form of DNA repair in mitotically active cells such as tumor cells, while the canonical NHEJ pathway is most active in G0 and G1 phases and is the dominant mode of DNA repair in quiescent cells such as neurons (35). The NHEJ component of our assay measures the more mutagenic, non-canonical NHEJ repair pathway; it is now well-established that the non-canonical pathway actively competes with canonical NHEJ repair (36–39). Two examples of the dynamic responsiveness of this assay are shown in Fig. 2b: (1) treatment with the DNA-PK inhibitor, NU7441, induced a marked shift towards HR and mutagenic NHEJ repair, and (2) treatment with the ATR inhibitor, VE822, selectively suppressed HR, with minimal effects on mutagenic NHEJ repair, which we and others have reported previously (21, 40, 41).
Figure 2. dsDNA repair pathway activity using a U2OS reporter assays.

a. Assay schematic showing induction of dsDNA breaks using I-SceI with repair to functional gene for RFP (L) and GFP (R), using either the m-NHEJ repair mechanism or the HR repair mechanism, respectively. b., c. examples of dose-dependent response of HR and mNHEJ pathway repair in the U2OS reporter assay to treatment with the DNA-PK inhibitor, NU7441 (b.) and the ATR inhibitor, VE822 (c.). Relative Free drug levels normalized to equivalent volume of DMSO (free drug solvent). Full dose-response curves in Supplementary Figure 3. d. Upregulation of the alternative mNHEJ pathway by inhibition of the canonical NHEJ pathway at a single dose for each drug. e. Inhibition of HR pathway at a single dose for each drug. (p-values indicated as: *<0.05, **<0.01, ***<0.001, ****<0.0001). Doses chosen for each drug based on dose-response curves (Supplementary Figure 3). Relative Free drug levels normalized to equivalent volume of DMSO (free drug solvent), drug-NP levels normalized to equivalent weight of blank NP.
Free- and NP-drug formulations were tested in parallel at a range of drug doses and in multiple replicates, and HR/NHEJ activity was normalized to appropriate vehicle controls (either DMSO or blank NPs). In each case, agent-loaded NP produced the expected phenotype on DSB repair, which was comparable to its free drug counterpart (Fig. 2c,d, Supplementary Fig 3). The DNA-PK inhibitors, NU7441 and KU60648, induced both HR and mutagenic NHEJ repair when delivered as free drugs or in NPs. Interestingly, NU7441 induced higher rates of mutagenic NHEJ repair than KU60648 specifically when given in NPs; this effect was statistically significant and reproducible across increasing drug doses (Supplementary Fig. 3a). BEZ235 induced moderate mutagenic NHEJ and suppressed HR when given as free drug and as NPs, which is consistent with its known suppression of both DNA-PK and mTOR pathways that appear to stimulate HR (42). VE822 selectively blocked HR with minimal effects on mutagenic NHEJ as a free drug and in NPs, which is an expected phenotype for ATR inhibitors. The ATM inhibitor, KU60019, induced mutagenic NHEJ repair as both a free drug and in NPs, with less consistent effects on HR (see also the dose-response curves in Supplementary Fig. 3e). Finally, the dual Chk1/Chk2 inhibitor, AZD7762, suppressed both HR and mutagenic NHEJ as a free drug and as NPs, which is an expected effect because of the central role of these two proteins in the proximal DNA damage response (43). The NP-encapsulated form of AZD7762 was slightly less potent than the free drug form, but nonetheless it suppressed DSB repair at levels similar to the free drug at high doses (Supplemental Fig. 3f). Collectively, these data indicate that each of the encapsulated molecules retain robust activity as DNA repair inhibitors.
Further testing and validation of free drug versus NP-VE822 as a tumor cell radiosensitizer
Our selection of the best NP agent for testing was guided by several key characteristics (Supplementary Table 2). We chose to focus on NPs containing VE822 (NP-VE822) as a potential glioma radiosensitizer for the following reasons: (1) it was efficiently encapsulated at high levels (49%) with a nmol drug/mg NP value that is within the range of that required to reach concentrations for functional DSB repair inhibition, (2) it demonstrated excellent slow-release kinetics (Table 2, Fig. 1b) which would be suitable for combination with fractionated RT, and (3) it demonstrated a dose-responsive, selective suppression of HR which was comparable to that observed with free drug. Importantly, emerging data suggests that ATR is a viable target for both radio- and chemo-sensitization in gliomas(44–46). In addition, as ATR activity and HR repair are most critical in actively replicating cells (47), there is an obvious therapeutic index, with the potential for reduced toxicity in surrounding quiescent brain tissue.
Further in vitro testing of free versus NP-VE822 revealed robust and reproducible, statistically significant HR suppression, compared to blank NPs (Supplementary Fig. 4a). HR suppression was comparable to that observed with free drug, and minimal effects were seen on mutagenic NHEJ repair (Supplemental Fig. 4b). We tested for sustained effects of VE822 in our DSB repair reporter assay by varying the time between treatment and induction of DSBs in the assay. While the relative HR pathway inhibition by free VE822 was constant for pre-treatment times of 0, 24, and 48h, the effect of the NP-VE822s increased with pre-exposure time (Supplementary Fig. 4c). Additionally, at the 48h pre-exposure time, the effect of the NP was significantly greater than free drug. These data suggest that slow-release of the drug in a NP formulation, in contrast to the free drug alone, has the potential for sustained suppression of HR repair.
We tested the biochemical effects of VE822 versus NP-VE822 directly on the known IR-induced phosphorylation targets of ATR, including Chk1. Both VE822 and NP-VE822 substantially prevented IR-induced phosphorylation of Chk1 at Serine 345 in SF188 glioma cells (Fig. 3a). ATR-dependent phosphorylation of this site on Chk1 leading to HR stimulation is well-established (48), and VE822 has been shown to block Chk1 phosphorylation at this site previously (49). Similar results were obtained in the primary, patient-derived DIPG spheroids.
Figure 3. VE822 protein regulation, radiosensitization, and toxicity.

a. Western blot of phospho-Chk1 levels in non-radiated (left three columns) and irradiated (right three columns) SF188 cells (top 2 rows) and DIPG spheroids (bottom two rows) with 24h pre-treatment with free VE822 (columns 2 and 5), VE822-NP (columns 3 and 6), or no treatment (columns 1 and 4). Smc1 used to normalize for protein levels. b. Clonogenic survival assay in colony-forming pediatric glioma cells, KNS42 using either VE822-NPs (red) or DMSO control (black) c. Dose-dependent SF188 cell viability at 72h following treatment with free VE822 (red), VE822-NPs (green), or DMSO control (blue).
NP-VE822 sensitized glioma cells to IR in vitro. Tumor cell killing was significantly enhanced with for cells treated with NP-VE822 and IR in clonogenic survival assays, which is a standard approach to assess for radiosensitivity (Fig. 3b; (50)). Importantly, NP-VE822 was minimally toxic to cell lines in culture over the range of therapeutically relevant doses, in comparison to free VE822 (Fig. 3c). Taken together, these data indicate that NP-VE822 effectively blocks ATR function leading to substantial glioma cell radiosensitization in vitro.
Distribution and retention of NPs in vivo after CED
To examine retention and distribution in the brain, we synthesized fluorescent NPs that were identical in size and surface potential to NP-VE822 (Table 2), and we administered them by intracranial CED in rats. We found that the particles distributed beyond the injection site with a Vd/Vi ratio of ~2, covering a volume of 40 mm3 (Fig. 4a,b) which provides good coverage of a typical xenografted tumor in this model (23, 30). Since the tumors are very small at the time of the CED delivery, we did not expect that relative levels of drug would vary significantly in the tumor-bearing brain, compared to brains without tumors, nor would the overall volume of distribution. Indeed, as shown in Fig. 4c, we observed good coverage of the tumor volume using our optimized CED conditions. NPs were observed in cells by TEM (Fig. 4d); NP size increased, although not significantly over the 5 days after CED, which might suggest accelerated degradation in this setting (Fig. 4d,e). The average density of nanoparticles (number per field of view) declined with an exponential decay pattern demonstrating a half-life of 10h, suggesting a relatively rapid clearance of a large portion of the NPs (Fig. 4f).
Figure 4. in vivo intracranial CED of PLA-PEG NPs into the R caudate of immunocompetent Fisher344 rats.

a.,b., and c.. NP volume of distribution immediately following CED of DiI-loaded PLA-PEG NPs. a. 3D distribution of DiI-loaded NPs superimposed on rat brain. The volume of distribution was reconstructed from serial 50um-thick coronal slices through the injection site. b. Representative fluorescently-imaged 2D coronal section at the site of injection. DiI channel (NPs) in red. c. Comparison of DiI-loaded NP distribution in normal rat brain (left) and rat brain with tumor xenograft (right). d. in vivo TEM imaging of caudate tissue samples from healthy rat brains harvested 4, 24, and 120 h after CED of DiD-loaded PLA-PEG NPs. NP indicated by red arrow. e. Average NP diameter over time as measured via TEM image analysis f. Average number of NPs per wide (2um scale) field of view as measured via TEM image analysis. g. and h. VE822 tissue levels following CED of either free VE822 (squares) or VE822-NPs (circles). Data analyzed using ex vivo standards of brains spiked with drug. Saline with equivalent volume of DMSO (drug solvent) and equivalent weight of blank NPs, respectively, were subtracted out as background in data analysis. Free VE822 was fit to a single-phase exponential decay model (R2 = 0.99) with t1/2 = 10.5h. VE822-NP was fit to a two-phase exponential decay model (R2 = 0.99) with 42.3% being rapidly cleared during the fast phase, with t1/2 = 2.2h followed by a slow phase with t1/2 = 8.2d. g. full data through 10d following CED h. close-up of initial 24h following CED
VE822 or NP-VE822 were administered by CED into the right hemisphere of rats, and drug concentrations were measured in brain tissue by LC/MS. After CED infusion of free drug, VE822 levels in the right hemisphere declined continuously over ~24 hr (Fig. 4g,h); this decline is well represented by an exponential decay with a half-life of 11 h. No drug was detected in the left hemisphere at any time. In contrast, after CED infusion of NP-VE822, VE822 concentrations in the right hemisphere were sustained over a period of 10 d: ~35% of the injected dose was present in the hemisphere after 5 days and ~ 10% was present 10 days after CED (Fig. 4g). VE822 was not detectable in the left hemisphere. Disappearance of VE822 after NP-VE822 CED was well represented by a biexponential decay: ~45% of the drug was cleared with a half-life of 2 h, and the remainder of the drug was cleared more slowly with a half-life of 8.3 d. These data indicate that we can achieve sustained concentrations of VE822, a potent and selective DNA repair inhibitor, in the brain by CED of NP-VE822. In addition, these findings confirm previous literature findings demonstrating rapid clearance of small molecules from the brain parenchyma (20).
Enhanced radiosensitization with CED of VE822-loaded NPs in a rat intracranial xenograft model in vivo
Finally, we tested whether our approach to administer VE822-loaded NPs directly into the brain via CED could lead to enhanced radiosensitization in vivo. We chose the rat glioma model, RG2, because the resulting tumors present a highly aggressive and invasive pattern of growth (as compared to the U87 and 9L, which are highly encapsulated) (50). These intracranial tumors can be established in syngeneic rats and the average time for survival without treatment is ~14 d. Rats with xenografted RG2 intracranial tumors were treated via CED with either free VE822, NP-VE822, or blank NPs. We tested three different strategies: CED of free VE822, VE822-loaded NPs, or blank NPs, versus no treatment (NT), and all without RT (Group 1); the same conditions as Group 1 but with a single fraction of RT (10 Gy; Group 2), or with fractionated RT (3 Gy×5; Group 3) (Fig. 5a). In each group, the CED procedure was performed 4 days after tumor implants, to allow time for initial tumor formation based on our prior work (23, 51). When indicated, RT was initiated 1 or 2 days after CED. For the RT, rats where placed in a cradle fitted with lead shielding for a focused cranial RT field, which consisted of a single posterior-anterior (PA) field generated by a kilovoltage irradiator. Dose was prescribed to the skull base and confirmatory dosimetry was performed using calibrated micro-cube thermoluminescent dosimeters.
Figure 5. in vivo survival study using RG2 rat glioma model in immunocompetent Fisher344 rats.

a. Treatment schematic: Large dashed line indicates infusion of RG2 tumor cells into the R caudate at day 0. Small dashed line indicates intratumoral CED delivery of either free VE822, VE822-NPs, or blank NPs, or no treatment at day 4. Light gray lines indicate administration of focal cranial radiation of either a single dose of 10 Gy (group 2) or five consecutive doses of 3 Gy (group 3). Black line indicates recorded survival day for each animal. Animals were euthanized prior to death at using pre-set and standardized symptomatic criteria from the tumors. b., c., and d. Kaplan-Meyer survival curves with CED treatment groups indicated: No treatment (NT, black), blank NPs (blue), free VE822 at 10uM (yellow), VE822-NPs at 10uM (green). b. Group 1 (no RT) survival curves c. Group 2 (Single fraction RT) survival curves with dotted line indicating NT (no RT) group d. Group 3 (Fractionated RT) survival curves with dotted line indicating NT (no RT) group and dashed line indicating NT (with RT) group. e., f. Median OS survival advantage calculated as median OS minus median OS of NT (no RT) group (14.5d). Comparison of median OS survival advantage between CED of free VE822 v. VE822-NPs treatment groups for each RT treatment group (e.) and between RT treatment groups for either free VE822 or VE822-NPs treatment group (f.). p values calculated as pair-wise log-rank comparisons from the survival curves in b.–d. and indicated as: *<0.05, **<0.01, ***<0.005, ****<0.001)
As expected, survival analysis showed no significant difference between any of the non-radiated cohorts in Group 1 (log-rank p > 0.05) (Fig. 5b). In the single-fraction radiated groups (Group 2), there was a moderate advantage over the no treatment group conferred by treatment with blank NP (log-rank p<0.01) and NP-VE822 (log-rank p<0.01), with no statistically significant advantage conferred by treatment with free VE822 (Fig. 5c). Additionally, in the single-dose RT groups, there was no statistically significant survival difference between the free VE822 and NP-VE822 treatment groups (Fig. 5e). However, in the animals treated with fractionated RT (Group 3), NP-VE822 provided a significant survival advantage over both control groups (log-rank p<0.001), as well as over the free VE822 group (log-rank p<0.05) (Figs. 5d and 5e). This corresponds to a ~25% increase in median overall survival in rats treated with fractionated RT plus NP-VE822 versus fractionated RT alone (mOS 22.5 vs 17.5 days). No significant difference in survival was observed between animals receiving blank NP versus no treatment in the fractionated RT group (Fig. 5d). Free VE822 did confer a slight survival advantage (log-rank p<0.05) compared to the no treatment control, but did not confer a survival advantage over the blank NP control (Fig. 5d).
Analysis of potential synergy between the CED treatment and radiation dosing schedules revealed that while the combination of NP-VE822 and single-dose RT showed neither synergistic nor antagonistic effects (SF = 1.0), the combination of NP-VE822 and fractionated RT demonstrated strong synergistic effects (SF = 1.8). This synergism is highlighted by the improvement in median overall survival seen in rats treated with NP-VE822 and fractionated RT compared to that of rats treated with NP-VE822 and single-dose RT (Fig. 5f). We did not observe a similar advantage of fractionated RT over single-dose RT in the free VE822 groups (Fig. 5f), which suggests that the NP-VE822 formulation is more suitable for use during a prolonged and more clinically relevant course of radiation. Overall these results demonstrate that NP-VE822 combined with fractionated RT can significantly enhance glioma radiosensitization in vivo.
Discussion
Despite dozens of clinical trials over the past decades, the prognoses for both GBM and DIPG remain grim. Currently, fractionated RT is the only treatment that has been shown to improve median survival of patients with DIPG (2). Both GBM and DIPG are characterized by nearly universal local recurrence despite aggressive local therapy, which highlights the need for new therapies. We believe that the lack of survival improvements for this disease can be attributed to three major issues: (1) systemically-delivered drug penetration into high-grade gliomas is severely limited by the highly-protective BBB (7, 52–54); (2) even when therapies are able to penetrate the tumor volume, they are rarely able to achieve therapeutic intratumoral levels, because agents are rapidly cleared from the brain and repeated systemic doses are toxic (20, 55); and finally, (3) even when present at therapeutically-relevant concentrations, the agents that have been tested for combination with RT and chemotherapy do not have sufficient pre-clinical data to justify their use as sensitizers (6).
To overcome these issues, we developed a new approach to radiosensitize gliomas via CED of DNA repair inhibitors loaded into NPs that provide sustained release. We encapsulated our agents in biocompatible nanoparticles formed from PLA-PEG, which slowly degrade after intracranial delivery and provide continuous drug levels that are sustained over the period of radiotherapy. We selected PLA-PEG because of its use in other applications that have reached advanced clinical trials demonstrating its safety (56). VE822 was encapsulated with moderate efficiency, 49%. We confirmed that NP-VE822 blocked HR repair at levels similar to that observed with the free drug, and we confirmed it could radiosensitize glioma cells, in vitro. We then extended these findings to intracranial tumors in animals, by demonstrating a 9 d (64%) increase in median survival using the combination of NP-VE822 + a 5 d course of fractionated RT when compared to untreated controls.
Sustained release of VE822 from the infused NP-VE822 is essential for enhancement in survival with fractionated radiation therapy. We showed that the NP formulation was highly effective at increasing the intracranial half-life of VE822 when delivered via CED: NP-VE822 extended the intracranial half-life to 8.3 d, compared to 0.45 d (11 h) for free VE822. Based on its in vitro properties, we believe that NP-VE822 releases VE822 in a burst (accounting for ~50% of the administered drug), followed by a long period of linear release of ~1 nmol/mg/day. Previous work studying drug release patterns from NPs has demonstrated a typical bi-phasic release pattern, which we observed for all six of the drugs (57, 58). Prior studies have shown that polymer hydrolysis and degradation rates play a significant role in the second, slower release phase; whereas the method of NP fabrication and drug encapsulation, the crystallinity of the polymer, and thus degree of initial water penetration into the polymer matrix, as well as specific drug properties are more important in determining this first burst release phase. We believe that drug is released through multiple mechanisms including passive diffusion, as well as endocytosis and degradation of the nanoparticle in the intracellular space.
Our measurements suggest two mechanisms for clearance of VE822 from the brain when administered as NP-VE822: elimination of the free drug (with a half-life of 0.45 d) and elimination of NP-VE822 in the nanoparticle form. The contribution of this second mechanism is difficult for us to quantify at present, but our preliminary results suggest that a substantial fraction of the NPs are cleared from the brain over the first few hours (Fig. 4f). These observations are consistent with the profile of VE822 clearance observed after NP-VE822 infusion in the brain (Fig. 4g). The period of rapid elimination of VE822 (half-life of 2 hr, more rapid than the clearance rate of free drug) is due to elimination of NPs containing VE822, whereas the sustained levels are the result of a competition between slow VE822 release from the remaining intracellular NPs, elimination of free VE822, and slow elimination/degradation of the remaining NPs. We still have work to do in determining the relative contributions of these multiple routes for elimination in the tumor microenvironment, but recent experiments suggest that some of these mechanisms can be manipulated by changing properties of the NP, such as surface chemistry (59–61). Still, even with our present formulation, NP-VE822 was able to maintain intracranial drug levels above the IC50 of VE822 (0.125uM) for at least 10 d after administration.
Other DNA repair pathway inhibitors, such as DNA-PK inhibitors have received attention as popular candidates for radiosensitization (62–64). However, more selective agents such as ATM and ATR inhibitors have started to emerge as promising agents (35, 65, 66). These agents have the potential to promote selective tumor cell radiosensitization while minimizing toxicity in healthy quiescent cells (67) – an approach particularly applicable to the brain environment. Additionally, the overlap between the enzymatic structures of ATM and DNA-PK allows for substantial off-target effects of small molecule ATM inhibitors (68, 69) and renders an ATR inhibitor relatively more specific to HR inhibition. In this context, we selected a molecule with minimal effect on NHEJ in an effort to maximize the therapeutic ratio. Additionally, the passive targeting of NP-formulations that allows enhanced uptake in mitotically active cells such as tumor cells seems to supplement this selective tumor targeting and even further widen the gap of relative toxicity between healthy and tumor cells (59).
We believe that the ability to confer sustained radiosensitization both in vitro and in vivo is key in establishing true, clinically-relevant radiosensitization and survival advantage. Most pre-clinical, particularly in vitro, screens of candidate radiosensitizing molecules involve single time point read-outs of drug activity (70). Additionally, many in vivo studies of radiosensitizers involve only one or two doses of RT (51, 71–73), often at both drug and radiation doses much too high to be clinically relevant (51, 71–75). Finally, as most radiation dosing studies use only one or two doses of RT, they do not account for the temporal profile of drug levels in the brain throughout a prolonged course of radiation therapy. However, particularly in the setting of rapid intracranial clearance of small molecules such as VE822, which we measured has a half-life of 11 h, a more thorough evaluation of the sustainability of the drug’s radiosensitizing capabilities is necessary. Because local delivery methods such as CED are invasive, it is important to minimize the number of repeat treatments required to achieve maximal clinical efficacy. To this extent, drug-NP formulations that have the potential to retain relevant intracranial drug levels for up to weeks—such as those we created here that were able to extended the intracranial half-life from <12h to >8d—is an important goal, with both in vitro and in vivo assays designed to study this property of candidate formulations.
Here, we designed our approach with the objective of developing a therapeutic strategy to improve local control for aggressive gliomas by using a clinically-relevant fractionated dosing schedule, and with a focus on sustained radiosensitization through slowly-releasing NPs. We demonstrated the feasibility of this new multi-modal approach to treatment of high-grade gliomas both in vitro, where we showed selective and sustained radiosensitization, and in vivo, where we demonstrated a synergistic effect of our radiosensitizing strategy superior to similar approaches reported in literature. Additionally, this work suggests a broader application for the novel combination of local delivery of radiosensitizer-loaded NPs for sustained radiosensitization during fractionated RT that can potentially be applied to tumors of the brain, spine, liver and beyond.
Supplementary Material
Acknowledgments
The authors thank Dr. Terence Wu for help with LC-MS and Marc Llaguno and Xinran Liu for help with TEM. We especially thank Kerry McLaughlin for invaluable technical assistance. We also thank Dr. Kevin Becker (Yale School of Medicine, Department of Neuro-Oncology) for his insights and review of this manuscript.
Financial Support: National Institutes of Health (R01 CA149128 – W.M. Saltzman and F30 CA206386 – A.R. King), Brain Research Foundation (SIA-2014-02 – R.S. Bindra), the DIPG Collaborative (R.S. Bindra), the Curesearch Foundation (R.S. Bindra), and a Yale Cancer Center Collaborative Co-Pilot Award (R.S. Bindra and W.M. Saltzman).
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Khatua S, Moore KR, Vats TS, Kestle JR. Diffuse intrinsic pontine glioma-current status and future strategies. Childs Nerv Syst. 2011;27(9):1391–7. doi: 10.1007/s00381-011-1468-z. [DOI] [PubMed] [Google Scholar]
- 3.Milano MT, Okunieff P, Donatello RS, Mohile NA, Sul J, Walter KA, et al. Patterns and timing of recurrence after temozolomide-based chemoradiation for glioblastoma. Int J Radiat Oncol Biol Phys. 2010;78(4):1147–55. doi: 10.1016/j.ijrobp.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Seiwert TY, Salama JK, Vokes EE. The concurrent chemoradiation paradigm - general principles. Nat Clin Pract Oncol. 2007;4(2):86–100. doi: 10.1038/ncponc0714. [DOI] [PubMed] [Google Scholar]
- 5.Xu R, Shimizu F, Hovinga K, Beal K, Karimi S, Droms L, et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin Cancer Res. 2016;22(19):4786–96. doi: 10.1158/1078-0432.CCR-16-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corso CD, Bindra RS. Success and Failures of Combined Modalities in Glioblastoma Multiforme: Old Problems and New Directions. Seminars in radiation oncology. 2016;26(4):281–98. doi: 10.1016/j.semradonc.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Laquintana V, Trapani A, Denora N, Wang F, Gallo JM, Trapani G. New strategies to deliver anticancer drugs to brain tumors. Expert Opin Drug Del. 2009;6(10):1017–32. doi: 10.1517/17425240903167942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. 2016;18(1):27–36. doi: 10.1093/neuonc/nov164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aiken R. Molecular neuro-oncology and the challenge of the blood-brain barrier. Seminars in oncology. 2014;41(4):438–45. doi: 10.1053/j.seminoncol.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. The Journal of pharmacology and experimental therapeutics. 2010;334(1):147–55. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. The Journal of pharmacology and experimental therapeutics. 2009;330(3):956–63. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 12.Wang T, Agarwal S, Elmquist WF. Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. The Journal of pharmacology and experimental therapeutics. 2012;341(2):386–95. doi: 10.1124/jpet.111.190488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032) The Journal of pharmacology and experimental therapeutics. 2012;342(1):33–40. doi: 10.1124/jpet.112.192195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ediriwickrema A, Saltzman WM. Nanotherapy for Cancer: Targeting and Multifunctionality in the Future of Cancer Therapies. ACS Biomater Sci Eng. 2015;1(2):64–78. doi: 10.1021/ab500084g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):2076–80. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson SD, Foster K, Yamini B. Convection-enhanced delivery for treatment of brain tumors. Expert review of anticancer therapy. 2007;7(12 Suppl):S79–85. doi: 10.1586/14737140.7.12s.S79. [DOI] [PubMed] [Google Scholar]
- 17.Barua NU, Lowis SP, Woolley M, O'Sullivan S, Harrison R, Gill SS. Robot-guided convection-enhanced delivery of carboplatin for advanced brainstem glioma. Acta Neurochir (Wien) 2013;155(8):1459–65. doi: 10.1007/s00701-013-1700-6. [DOI] [PubMed] [Google Scholar]
- 18.Guisado DI, Singh R, Minkowitz S, Zhou Z, Haque S, Peck KK, et al. A Novel Methodology for Applying Multivoxel MR Spectroscopy to Evaluate Convection-Enhanced Drug Delivery in Diffuse Intrinsic Pontine Gliomas. AJNR Am J Neuroradiol. 2016 doi: 10.3174/ajnr.A4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12(8):871–81. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vecchio D, Daga A, Carra E, Marubbi D, Raso A, Mascelli S, et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int J Cancer. 2015;136(6):1445–57. doi: 10.1002/ijc.29121. [DOI] [PubMed] [Google Scholar]
- 21.Bindra RS, Goglia AG, Jasin M, Powell SN. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013;41(11):e115. doi: 10.1093/nar/gkt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods Mol Med. 2005;110:21–8. doi: 10.1385/1-59259-869-2:021. [DOI] [PubMed] [Google Scholar]
- 23.Saucier-Sawyer JK, Seo YE, Gaudin A, Quijano E, Song E, Sawyer AJ, et al. Distribution of polymer nanoparticles by convection-enhanced delivery to brain tumors. Journal of controlled release : official journal of the Controlled Release Society. 2016;232:103–12. doi: 10.1016/j.jconrel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14(24):6083–7. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 25.Dumont F, Altmeyer A, Bischoff P. Radiosensitising agents for the radiotherapy of cancer: novel molecularly targeted approaches. Expert Opin Ther Pat. 2009;19(6):775–99. doi: 10.1517/13543770902967666. [DOI] [PubMed] [Google Scholar]
- 26.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 27.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8(10):2894–902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7(9):2955–66. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 29.Fokas E, Prevo R, Hammond EM, Brunner TB, McKenna WG, Muschel RJ. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev. 2014;40(1):109–17. doi: 10.1016/j.ctrv.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J, Patel TR, Sirianni RW, Strohbehn G, Zheng MQ, Duong N, et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):11751–6. doi: 10.1073/pnas.1304504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–91. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mura S, Hillaireau H, Nicolas J, Le Droumaguet B, Gueutin C, Zanna S, et al. Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int J Nanomedicine. 2011;6:2591–605. doi: 10.2147/IJN.S24552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surovtseva YV, Jairam V, Salem AF, Sundaram RK, Bindra RS, Herzon SB. Characterization of Cardiac Glycoside Natural Products as Potent Inhibitors of DNA Double-Strand Break Repair by a Whole-Cell Double Immunofluorescence Assay. Journal of the American Chemical Society. 2016;138(11):3844–55. doi: 10.1021/jacs.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goglia AG, Delsite R, Luz AN, Shahbazian D, Salem AF, Sundaram RK, et al. Identification of novel radiosensitizers in a high-throughput, cell-based screen for DSB repair inhibitors. Mol Cancer Ther. 2015;14(2):326–42. doi: 10.1158/1535-7163.MCT-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 36.Soong CP, Breuer GA, Hannon RA, Kim SD, Salem AF, Wang G, et al. Development of a novel method to create double-strand break repair fingerprints using next-generation sequencing. DNA repair. 2015;26:44–53. doi: 10.1016/j.dnarep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter AC, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016;44(12):5743–57. doi: 10.1093/nar/gkw326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutation research Genetic toxicology and environmental mutagenesis. 2015;793:166–75. doi: 10.1016/j.mrgentox.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. The Journal of biological chemistry. 2011;286(49):42470–82. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goglia AG, Delsite R, Luz AN, Shahbazian D, Salem AF, Sundaram RK, et al. Identification of Novel Radiosensitizers in a High-Throughput, Cell-Based Screen for DSB Repair Inhibitors. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gil del Alcazar CR, Hardebeck MC, Mukherjee B, Tomimatsu N, Gao X, Yan J, et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin Cancer Res. 2014;20(5):1235–48. doi: 10.1158/1078-0432.CCR-13-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benada J, Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules. 2015;5(3):1912–37. doi: 10.3390/biom5031912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Middleton FK, Patterson MJ, Elstob CJ, Fordham S, Herriott A, Wade MA, et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget. 2015;6(32):32396–409. doi: 10.18632/oncotarget.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer research. 2015;75(20):4416–28. doi: 10.1158/0008-5472.CAN-14-3790. [DOI] [PubMed] [Google Scholar]
- 46.Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347(6219):273–7. doi: 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3 doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes & development. 2000;14(12):1448–59. [PMC free article] [PubMed] [Google Scholar]
- 49.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protocols. 2006;1(5):2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 51.Gaudin A, Song E, King AR, Saucier-Sawyer JK, Bindra R, Desmaele D, et al. PEGylated squalenoyl-gemcitabine nanoparticles for the treatment of glioblastoma. Biomaterials. 2016;105:136–44. doi: 10.1016/j.biomaterials.2016.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.WM P. Blood-brain barrier delivery. Drug Discovery Today. 2007;12(1–2):54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Merkus P, Guchelaar HJ, Bosch DA, Merkus FW. Direct access of drugs to the human brain after intranasal drug administration? Neurology. 2003;60(10):1669–71. doi: 10.1212/01.wnl.0000067993.60735.77. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Dalwadi G, Benson HA. Drug delivery across the blood-brain barrier. Curr Drug Deliv. 2004;1(4):361–76. doi: 10.2174/1567201043334542. [DOI] [PubMed] [Google Scholar]
- 55.Noble CO, Krauze MT, Drummond DC, Yamashita Y, Saito R, Berger MS, et al. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer research. 2006;66(5):2801–6. doi: 10.1158/0008-5472.CAN-05-3535. [DOI] [PubMed] [Google Scholar]
- 56.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Science translational medicine. 2012;4(128):128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 57.Allison SD. Analysis of initial burst in PLGA microparticles. Expert Opin Drug Deliv. 2008;5(6):615–28. doi: 10.1517/17425247.5.6.615. [DOI] [PubMed] [Google Scholar]
- 58.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel) 2011;3(3):1377–97. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song E, Gaudin A, King AR, Seo Y, Won P, Suh H, et al. Abstract B46: Surface chemistry governs cellular tropism of nanoparticles in the brain. Cancer research. 2017;77(2 Supplement):B46–B. doi: 10.1038/ncomms15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agarwal R, Roy K. Intracellular delivery of polymeric nanocarriers: a matter of size, shape, charge, elasticity and surface composition. Ther Deliv. 2013;4(6):705–23. doi: 10.4155/tde.13.37. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Liu Y, Sen S, Kral P, Gemeinhart RA. Charged group surface accessibility determines micelleplexes formation and cellular interaction. Nanoscale. 2015;7(17):7559–64. doi: 10.1039/c5nr00095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell death and differentiation. 2014;21(2):258–69. doi: 10.1038/cdd.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget. 2015;6(16):14005–25. doi: 10.18632/oncotarget.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Novotna E, Tichy A, Pejchal J, Lukasova E, Salovska B, Vavrova J. DNA-dependent protein kinase and its inhibition in support of radiotherapy. Int J Radiat Biol. 2013;89(6):416–23. doi: 10.3109/09553002.2013.767993. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Lai J, Du Z, Gao J, Yang S, Gorityala S, et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget. 2016 doi: 10.18632/oncotarget.9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Llona-Minguez S, Hoglund A, Jacques SA, Koolmeister T, Helleday T. Chemical strategies for development of ATR inhibitors. Expert Rev Mol Med. 2014;16:e10. doi: 10.1017/erm.2014.10. [DOI] [PubMed] [Google Scholar]
- 67.Dal Pra A, Locke JA, Borst G, Supiot S, Bristow RG. Mechanistic Insights into Molecular Targeting and Combined Modality Therapy for Aggressive, Localized Prostate Cancer. Front Oncol. 2016;6 doi: 10.3389/fonc.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee BS, Gapud EJ, Zhang S, Dorsett Y, Bredemeyer A, George R, et al. Functional intersection of ATM and DNA-dependent protein kinase catalytic subunit in coding end joining during V(D)J recombination. Mol Cell Biol. 2013;33(18):3568–79. doi: 10.1128/MCB.00308-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalvass JC, Olson ER, Cassidy MP, Selley DE, Pollack GM. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50,u and correlation of in vitro, preclinical, and clinical data. The Journal of pharmacology and experimental therapeutics. 2007;323(1):346–55. doi: 10.1124/jpet.107.119560. [DOI] [PubMed] [Google Scholar]
- 70.Nahas SA, Davies R, Fike F, Nakamura K, Du LT, Kayali R, et al. Comprehensive Profiling of Radiosensitive Human Cell Lines with DNA Damage Response Assays Identifies the Neutral Comet Assay as a Potential Surrogate for Clonogenic Survival. Radiat Res. 2012;177(2):176–86. doi: 10.1667/rr2580.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang W, Barth RF, Huo T, Nakkula RJ, Weldon M, Gupta N, et al. Radiation therapy combined with intracerebral administration of carboplatin for the treatment of brain tumors. Radiat Oncol. 2014;9:25. doi: 10.1186/1748-717X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi MH, Fortin D, Sanche L, Paquette B. Convection-enhancement delivery of platinum-based drugs and Lipoplatin(TM) to optimize the concomitant effect with radiotherapy in F98 glioma rat model. Invest New Drug. 2015;33(3):555–63. doi: 10.1007/s10637-015-0228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Recinos VT, BM, Bekelis K, Sunshine SB, Vellimana A, Li KW, Brem H. Combination of intracranial temozolomide with intracranial carmustine improves survival when compared with either treatment alone in a rodent glioma model. Neurosurgery. 2010;66(3):530–7. doi: 10.1227/01.NEU.0000365263.14725.39. [DOI] [PubMed] [Google Scholar]
- 74.Rousseau J, Boudou C, Barth RF, Balosso J, Esteve F, Elleaume H. Enhanced survival and cure of F98 glioma-bearing rats following intracerebral delivery of carboplatin in combination with photon irradiation. Clin Cancer Res. 2007;13(17):5195–201. doi: 10.1158/1078-0432.CCR-07-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robbins ME, Payne V, Tommasi E, Diz DI, Hsu FC, Brown WR, et al. The AT1 receptor antagonist, L-158,809, prevents or ameliorates fractionated whole-brain irradiation-induced cognitive impairment. Int J Radiat Oncol Biol Phys. 2009;73(2):499–505. doi: 10.1016/j.ijrobp.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.