

Abstract
An increased cardiovascular morbidity and mortality, including the risk of sudden cardiac death (SCD), has been shown in patients with rheumatoid arthritis (RA). Abnormalities in autonomic markers such as heart rate variability and ventricular repolarization parameters, such as QTc interval and QT dispersion, have been associated with sudden death in patients with RA. The interplay between these parameters and inflammation that is known to exist with RA is of growing interest. In this article, we review the prevalence and predictors of SCD in patients with RA and describe the potential underlying mechanisms, which may contribute to this. We also review the impact of biologic agents on arrhythmic risk as well as cardiovascular morbidity and mortality.
Keywords: Sudden death, Rheumatoid arthritis, Cardiovascular, QT, Autonomic nervous system, Arrhythmia
Core tip: Patients with rheumatoid arthritis are twice as likely to experience sudden cardiac death (SCD). This excess risk can only partially be explained by the higher rates of heart failure and ischaemic heart disease. Abnormalities of the autonomic nervous system, such as decreased heart rate variability, and abnormalities of ventricular repolarization parameters, such as QTc interval and QT dispersion, have also been implicated. In this article we review the interplay between these parameters and inflammation, exploring whether biologic agents and disease modifying anti-rheumatic drugs may have a role in reducing the burden of SCD.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory condition which affects 0.8% of the adult population[1]. It causes significant morbidity as a result of synovial inflammation, joint destruction, and associated disability. In addition to articular manifestations, there is substantial data demonstrating excess cardiovascular morbidity and mortality in RA[2,3]. Studies as early as the 1950s have shown that RA is associated with premature death[4], with 50% of excess deaths being attributed to cardiovascular disease[2,5]. Sudden cardiac death (SCD) is estimated to account for 50% of cardiovascular deaths in the general population[6,7]. Patients with RA are twice as likely to experience SCD[8], a figure comparable to patients with diabetes mellitus[9]. This review will seek to explore the risk factors that make SCD more prevalent in RA, including coronary artery disease (CAD), structural heart disease, electrophysiological abnormalities including myocardial repolarization (QTc interval, QT dispersion) and autonomic dysfunction [heart rate variability (HRV) analysis], and the interplay of inflammation on all these factors[10]. We will also review the effect that the new biologic agents may have on the incidence of cardiovascular events and SCD.
DEFINING SCD AND ITS PREVALENCE IN RA
Sudden death is defined as “non-traumatic, unexpected fatal event occurring within 1 h of the onset of symptoms in an apparently healthy subject. If death is not witnessed, the definition applies when the victim was in good health 24 h before the event”[11]. The term SCD is used when a congenital, or acquired, potentially fatal cardiac condition was known to be present during life, or an autopsy has identified a cardiac or vascular anomaly as the probable cause of the event, or no obvious extra-cardiac causes have been identified by post-mortem examination, and therefore an arrhythmic event is a likely cause of death[11]. SCD is largely thought to result from fatal arrhythmias, in particular, ventricular tachycardia degenerating to ventricular fibrillation[12,13]. Bradyarrhythmia or electromechanical dissociation can also be associated with SCD but these tend to occur in patients with advanced cardiac disease[12,14].
In the general population sudden death accounts for 60% of cardiovascular deaths in those with known CAD[15], and is the first presentation of CAD in 15% of cases[16]. Over 70% of fatal arrhythmias are thought to be secondary to CAD[12]. These arrhythmias can occur acutely secondary to direct repolarisation changes occurring during ischaemia, or remotely, typically due to initiation of re-entry circuits within areas of electrically unexcitable scar tissue or diseased myocardium in patients with established myocardial infarction (MI)[17]. Dilated and hypertrophic cardiomyopathies account for the second largest number of sudden deaths followed by valvular, congenital, infiltrative, ion- channel disorders which only account for the small remainder[12,13].
Within the RA population, there is a wealth of data regarding the rates of cardiovascular death, however few studies have looked specifically at the epidemiology of SCD. In an inception cohort of 1010 RA patients, Goodson et al[18] found an excess of cardiovascular mortality without a corresponding increase in cardiovascular admission rates as compared to controls, suggesting that cardiovascular disease has a higher case fatality in the RA population, or that it often goes unrecognized before the fatal event. Indeed, Solomon et al[19] found that the rate ratio for cardiovascular death was highest in young RA adults and those with no known prior cardiovascular events. Whilst Van Doornum et al[20] demonstrated that RA patients have a higher 30-d case fatality following MI as compared to controls, 17.6% vs 10.8%. In a large population cohort study of 603 RA patients followed up for 15 years, Maradit-Kremers et al[5] demonstrated that RA patients were twice as likely to experience SCD (hazard ratio 1.94, 95%CI: 1.06-3.55)[8], a figure similar to the risk of SCD amongst patients with diabetes mellitus[21]. The authors also noted a higher risk of unrecognized MIs and a lower likelihood of angina symptoms, suggesting that CAD manifests differently in RA and is more likely to manifest as cardiovascular death[8]. Similarly Mantel et al[22] demonstrated that RA is associated with higher risk acute coronary syndromes, higher cumulative incidence of SCD (0.2% vs 0.13% over 3 years), and higher short term case fatality rate at 7 and 30 d. Indeed, certain RA genetic polymorphisms have been linked to premature cardiovascular disease and mortality[23-26], although none with a strong clinical implication[27,28].
RISK FACTORS FOR SCD IN RA
Accelerated CAD, congestive cardiac failure and inflammation
Whilst there is a higher incidence of ischaemic heart disease (IHD) in RA, several authors have shown that this increased incidence cannot be explained by traditional risk factors alone[5,29], as such there has been growing interest in the role of inflammation as novel risk factor for atherosclerosis[30]. Indeed, in the general population, modest increases in C-reactive protein (CRP) have been associated with increased cardiovascular events[31], and RA has been likened to diabetes as a risk factor for CAD[32].
Studies have also suggested different patterns of CAD in RA with chronic inflammation leading to early endothelial dysfunction[33,34], and a higher incidence of unstable plaques attributed to inflammatory cytokines[35]. Indeed tumour necrosis factor alpha (TNF-α) has been implicated in all stages of atherosclerosis including endothelial dysfunction, plaque formation, rupture and promotion of the clotting cascade[36,37]. Systemic inflammation has also been associated with dyslipidaemia, impaired glucose metabolism, platelet activation and increased clotting factors[36]. However, despite the evidence linking inflammation to accelerated atherosclerosis and IHD, Maradit-Kremers et al[8] demonstrated that the two-fold risk of SCD seen in the RA population persisted after adjustments for history of hospitalized, or unrecognized, MI, revascularization procedures and cardiovascular risk factors. This suggests that the increased risk of SCD in RA cannot be explained by increased rates of IHD alone[10,38].
In two studies[39,40] the excess risk of congestive cardiac failure (CCF) among RA subjects could not be explained by the increased frequency, or effect of, either cardiovascular risk factors, or IHD. In the same cohort, Gabriel et al[41] demonstrated that whilst 80% of CCF in the general population is attributed to classical CVD risk factors, in RA, classical risk factors only explained 40% of the incident heart failure.
Amongst RA patients experiencing new-onset heart failure, ESR levels were highest in the 6 mo immediately preceding diagnosis, suggesting that ESR may signal the onset of heart failure in patients with RA[42]. However, the relationship between SCD and severity of CCF is not as clear-cut as that seen with SCD and IHD, and less is known about RA and CCF. Data from the general population suggests that as left ventricular (LV) systolic function deteriorates, all-cause mortality and the absolute number of sudden deaths increases, but the proportion of deaths due to arrhythmias decreases[14,43]. Thus, the degree of LV systolic dysfunction lacks specificity as a predictor of death secondary to cardiac arrhythmias, because it is also powerful measure of the risk of death[12]. In line with these results, Nicola et al[44] found CCF contributed to the excess cardiovascular mortality in RA, primarily through the increased incidence of CCF in RA rather than increased case fatality. Studies have also shown that patients with RA have higher rates of diastolic dysfunction[45], and heart failure with preserved ejection fraction[46].
Abnormal ventricular repolarization, autonomic dysfunction and inflammation
Inflammation, as an independent predictor of cardiovascular mortality and sudden death, has been the focus of recent research[30,47,48]. Indicators of abnormal ventricular repolarization such as QTc prolongation, QT interval dispersion, and autonomic dysfunction have been implicated in the aetiopathogenesis of SCD. The QT interval represents the time from onset of ventricular depolarization (beginning of the Q wave) to completion of repolarization (end of T wave). The corrected QT interval (QTc) estimates the QT at a standardized heart rate of 60 bpm, while QT interval dispersion (QTd) is measure of the dispersion of ventricular repolarization (maximum QT interval - minimum QT interval). In the general population both prolongation of QTc and increased QTd are known risk factors for SCD[49,50], and there is data linking both CRP to prolongation of QTc[51] and to SCD[47]. In animal models, prolonged QTc is associated with depolarization during phases 2 and 3 of the action potential prior to completion of repolarization[52]. These premature action potentials also known as early after depolarizations (EADs) can generate fatal ventricular arrhythmias, such as torsade de pointes, which can progress to ventricular fibrillation and SCD[12,53].
Several studies have also shown an association between RA and prolonged QTc or increased QT dispersion variables, as well as an association between RA disease activity and QTc length[54-62] (Table 1), with the strongest evidence available for CRP as a marker of disease activity, compared with clinical scoring systems and ESR[10]. Moreover, there is growing evidence from basic science studies demonstrating that pro-inflammatory cytokines, particularly TNF-α, directly prolong cardiomyocyte action potential duration (APD) by regulating ion channels involved in ventricular repolarization[10]. In particular, several experimental studies have shown that TNF-α prolongs APD, triggering re-entrant ventricular arrhythmias[63,64]. TNF-α prolongs APD by inhibiting both the transient outward potassium current[65], and the rapid delayed-rectifier potassium current[10,66]. Similarly, animal studies have shown that the pro inflammatory cytokines IL-1 and IL-6, prolong APD in ventricular myocytes via their effects on calcium channels[67,68]. Interestingly both levels of CRP[48] and levels of soluble TNF-α receptors (sTNFR) are strong and independent predictors of cardiovascular death amongst RA patients[69].
Table 1.
Studies demonstrating associations between rheumatoid arthritis and QT parameters, inflammation and mortality
| Ref. | Design | RA patients | Controls | Impact of RA and QT dispersion (QTd) and QTc | Association between QT parameter, and disease activity/duration (1), arrhythmia (2), autonomic dysfunction (3), mortality (4) |
| [54] | Cross-sectional | 42 | 42 | ↑ QTd variables (QTd, QTcD, JTD, JTcD) vs controls No difference in QTc vs controls | (1) ESR, CRP (2) Complex premature ventricular beats |
| [55] | Cross-sectional | 40 | 48 | ↑ QTd variables (QTd, QTcD) vs controls | (1) Disease duration |
| [56] | Cross-sectional | 40 | 40 | ↑ QTd vs controls | (1) Extra-articular manifestations, erosive disease |
| [57] | Cross-sectional | 58 | 29 | ↑ QT vs controls | (1) Secondary Sjörgen’s syndrome |
| [58] | Cross-sectional | 100 | 100 | ↑ QTd vs controls | (1) Disease duration, DAS28, ESR, number of joints involved |
| [59] | Cross-sectional | 25 | 21 controls 76 with spondylarthopathy | ↑ QTc vs controls and those with spondyloarthopathies Infliximab therapy duration inversely correlated to QTc (P < 0.01) | (1) CRP (3) ↑ QTC associated with ↑ HR, autonomic dysfunction, particularly sympathetic dysfunction as assessed by spectral parameters of heart rate variability |
| [60] | Prospective cohort | 357 | _ | ↑ QTc 10% males (QTc ≥ 450 ms) and 5.6% of females (QTc ≥ 460 ms) | (1) CRP (4) Doubled risk of all-cause mortality per 50 ms increase in QTc, (lost after adjustment for CRP) HR = 2.17 (95%CI: 1.21-3.90) |
| [61] | Retrospective cohort | 417 | 422 | ↑ % of RA patients with QTc prolongation (> 450 ms males, > 460 ms females) vs controls 20 yr after disease onset (48% vs 38%, P = 0.004) | (1) ESR (4) Any cause QTc prolongation was associated with ↑ all-cause mortality HR = 2.99 (95%CI: 1.93-4.65) |
| [62] | Prospective cohort | 17 | _ | ↑ QTc (> 440 ms) in 76% of patients Toclizumab associated with 47% ↓ in No. patients with QTc prolongation (P = 0.006) | (1) CRP and TNF-α |
| [70] | Cross-sectional | 117 | - | (1) CRP, TNF-α, IL-1β and IL 10 ( QTc BAZ) (1) IL-1β and IL 10, trend towards TNF-α (QTc FHS) |
RA: Rheumatoid arthritis; QTd: QT interval dispersion; QTc: Heart-rate corrected QT interval; QTcD: Heart-rate corrected QT interval dispersion; JTD: JT interval dispersion; JTcD: Heart-rate corrected JT interval dispersion; DAS28: Disease activity score in 28 joints; ESR: Erythrocyte sedimentation rate; CRP: C-reactive protein; ms: Milliseconds; QTc BAZ: QTc calculated using Bazett formula; QTc FHS: QTc calculated using Framingham formula.
As early as 1998, a cross-sectional study by Göldeli et al[54], demonstrated a significant increase in QT dispersion variables when comparing RA patients with matched controls, as well as an increase in complex premature ventricular beats. More recently a large prospective study of 357 RA patients demonstrated that prolonged QTc was a strong predictor of death, with a 50 ms increase in QTc being associated with a doubling of the hazard for all-cause mortality[60]. The authors also showed that QTc prolongation was independently associated with CRP levels, and that the association between QTc and mortality was lost after adjustment for CRP, further supporting the role of inflammation in the increased rates SCD seen in this patient group[60]. No association was found between QTc and the presence of cardiovascular disease at baseline, ECG abnormalities suggestive of myocardial ischemia, LV hypertrophy, or the use of common cardiovascular medications[60]. Similarly, Chauhan et al[61] found a higher incidence of idiopathic QTc prolongation amongst RA patients and demonstrated that any cause QTc prolongation was significantly associated with all-cause mortality (HR = 2.99, 95%CI: 1.93-4.65) but only marginally associated with cardiovascular mortality (HR = 2.68, 95%CI: 0.84-5.6, P = 0.09). The authors used a cut off of ≥ 450 ms in males and ≥ 460 ms in females to define QTc prolongation[61], but interestingly, amongst the general over 55 population even a borderline increased QTc interval, defined as 451 to 470 ms in women, and 431 to 450 ms in men, was associated with a two-fold increase in the risk of SCD[53]. Following this, Lazzerini et al[62] showed that treating RA patients with 3 mo of the anti-IL 6-receptor antibody, tocilizumab, was associated with a significant reduction in the QTc interval. The fact improvement was seen in such a short time frame suggests that QTc prolongation is driven by an inflammatory process rather than subclinical coronary atherosclerosis[62].
There is also emerging data about the role of pro and anti-inflammatory cytokines in QTc prolongation amongst patients with RA. Lazzerini et al[62] recently demonstrated a strong association between QTc and circulating TNF-α levels, more so than CRP, although sample sizes were small. Adlan et al[70] performed a cross-sectional study of 112 patients with RA examining the relationship between QTc and cross-sectional sampling of several pro and anti-inflammatory cytokines. The authors demonstrated an association between QTc prolongation and CRP, TNF-α, IL-1β and the anti-inflammatory cytokine IL-10. A surge of IL-10 often follows the release of pro-inflammatory cytokines and its release is stimulated by adrenaline[70,71].
Sympathetic excitation has been associated with prolongation of the QTc[72], while cholinergic stimulation with pyridostigmine shortens QTc interval in patients with CAD[73]. RA has been associated with both reduced parasympathetic tone and increased sympathetic tone, with a recent systematic review demonstrating a 60% prevalence of autonomic dysfunction amongst patients with RA[52]. The majority of studies assessed autonomic dysfunction using clinical cardiovascular tests (CCTs) or by measuring HRV parameters[52]. CCTs include blood pressure and heart rate response to orthostasis, deep breathing and Valsalva manoeuvres, with many studies using Ewing’s battery of CCTs[74]. HRV analysis attempts to assess cardiac autonomic regulation through quantification of variation in sino-atrial activity with rapid variations reflecting vagal modulation and slower variations reflecting a combination of both parasympathetic and sympathetic modulation and non-autonomic factors. HRV can be measured using time domain methods or frequency domain methods. Examples of time domain measures include; mean heart rate, AVNN (Average of all the NN intervals, with “NN” used in place of “RR” to emphasize that these are normal sinus beats), and the difference between the longest and shortest NN interval[75]. More complex statistically derived time domain measures include either those, derived from direct measurements of the NN intervals, such as SDNN (standard deviation of all NN intervals), SDANN (standard deviation of the average of NN intervals in all 5-min segments of a 24 h recording) or those derived from the differences between NN intervals such as RMSSD (square root of the mean of the squares of the differences between adjacent NN intervals) and pNN50 (percentage of differences between adjacent NN intervals that are > 50 ms)[75]. Conversely, frequency domain methods assign bands of frequency, and through fast Fourier transformation quantify the NN interval in each band[75]. The bands are typically high frequency (HF) from 0.15 to 0.4 Hz, low frequency (LF) from 0.04 to 0.15 Hz, and very low frequency from 0.0033 to 0.04 Hz. Vagal activity is the major contributor to the HF component with a combination of both sympathetic and parasympathetic activity contributing to the LF and LF/HF ratio[75].
In the general population reduced HRV has been associated with a significantly increased risk of death post MI[76], although as yet there are no studies demonstrating the association between autonomic dysfunction and mortality amongst RA patients. This said, there is data to show that autonomic dysfunction, namely reduced HRV is associated with prolongation of the QTc in patients with RA[59]. A study of 100 patients with chronic inflammatory arthritis (CIA) demonstrated that while CRP was independently associated with HRV, there was no association between CRP and QTc in the multivariate model, with HRV parameters and RR interval playing a predominant role in producing differences in QTc among the subjects[59]. These findings, together with the evidence that, even after sex-adjustment, QTc was correlated with heart rate and all HRV parameters suggests that the association between CRP and QTc prolongation is most likely an indirect consequence of the autonomic dysfunction and specifically increased sympathetic tone[59]. Indeed, there is growing evidence to show that the release of pro inflammatory cytokines in diseases such as RA increases sympathetic outflow activation via autonomic centres in brain[10]. This represents an adaptive response which dampens immuno-inflammatory activation and inhibits the release of further cytokines via stimulation of β2-adrenoceptors in circulating lymphocytes and monocytes[77,78]. This negative feedback loop is known as the inflammatory reflex. However, sympathetic activation does not only affect the immune system, but all the systems throughout the body, and its effects may either directly[79], or indirectly (by prolonging QT interval parameters via modification in calcium and/or potassium conductance) trigger the onset of arrhythmias and SCD[10].
IMPACT OF DISEASE-MODIFYING ANTI-RHEUMATIC DRUGS AND BIOLOGICS ON CARDIOVASCULAR OUTCOMES?
Disease-modifying anti-rheumatic drugs (DMARDs) are a category of otherwise unrelated drugs defined by their use in RA to slow down disease progression. Throughout this review the term DMARD will be used to refer to synthetic DMARDs such as methotrexate, whereas biologic DMARDs will be simply referred to as biologics.
Whilst currently there are no studies specifically evaluating the impact of synthetic DMARDS and biologics on the incidence of arrhythmias and SCD in RA, the European League Against Rheumatism advocate early aggressive treatment with these agents, for the purpose of reducing cardiovascular morbidity and mortality[3]. These agents are expected to exert their benefits via their direct effect on reducing inflammation, but also by improving joint inflammation and function they will potentially lead to increased levels of physical activity and reduce the incidence of other risk factors such as diabetes mellitus and hypertension[3] (Figure 1). Indeed, a prospective cohort study and concurrent literature review conducted by Meek et al[80] showed a trend towards reducing cardiovascular case fatality since the advent of DMARDS and biologics. However, no comparison was made to a control population, to ensure that the findings were not just tagging the observed reduction in cardiovascular disease burden, in the general population.
Figure 1.
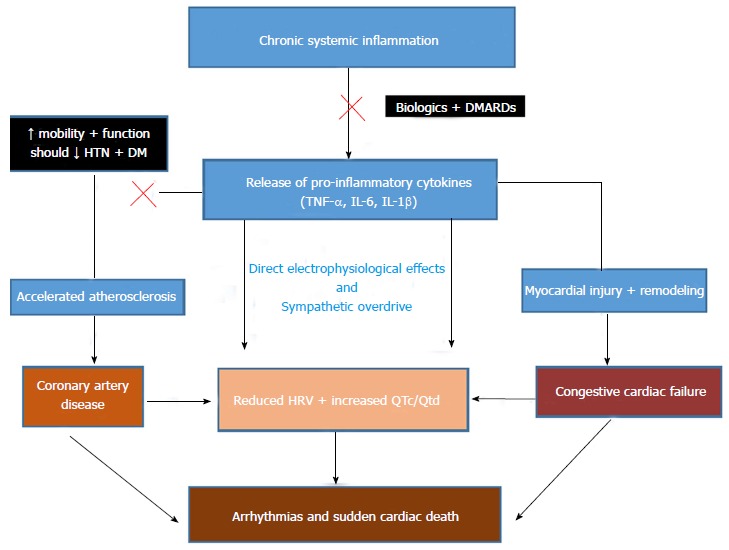
Proposed Mechanisms by which biologics and disease modifying anti-rheumatic drugs will reduce the risk of sudden cardiac death in patients with rheumatoid arthritis. DMARD: Disease modifying anti-rheumatic drugs; HTN: Hypertension; DM: Diabetes; TNF: Tumour necrosis factor; IL: Interleukin; HRV: Heart rate variability; QTc: Corrected QT interval; QTd: QT interval dispersion.
In this section, we will explore whether the advent of DMARDs and particularly Biologics, has indeed reduced cardiovascular mortality and morbidity. We will look closely at the data supporting both the anti-arrhythmic and pro-arrhythmic effects of biologics, as well as their influence on cardiac autonomic dysfunction and repolarisation parameters such as the QTc.
There is growing evidence from observational studies, including observational meta-analyses, to suggest that DMARDs in particular methotrexate[81,82] and biologics, particularly TNF inhibitors (TNFi)[37,83,84] are associated with decreased cardiovascular mortality and morbidity. To date, the largest meta-analysis has been conducted by Roubille et al[84], and included 28 observational studies, over 200000 RA patients and over 5000 cardiovascular adverse events. The authors found that TNFi were associated with a significant reduction in the risk of all cardiovascular events (CVE) (RR = 0.70, P = 0.005), as well as MIs, strokes and major adverse cardiac events. Methotrexate was also associated with a reduction in the risk of all CVEs (RR = 0.72, P = 0.007) and MIs. Moreover, a large observational study by Dixon et al[85] suggested that patients who responded to TNFi experienced a 70% lower risk of MI than their non-responding counterparts. In addition, a recent retrospective post hoc analysis involving approximately 4000 patients with moderate to severe RA, demonstrated that during tocilizumab treatment, greater reductions in disease activity were inversely associated with future major cardiovascular events, including MIs and cardiovascular death[86].
The lack of randomized controlled trials (RCTs) specifically examining the impact of biologics and DMARDS on CVE is perhaps explained by the number of patients that would be required to adequately power the studies, and the duration of follow-up that would be needed to detect an effect. Some studies have examined the impact of biologics on surrogate markers of IHD such as carotid intimal thickness and brachial artery flow mediated dilatation with conflicting results[87]. A single center RCT conducted by Hsue et al[88] demonstrated that depletion of B-cells with rituximab in RA patients improved both macrovascular (brachial artery flow-mediated dilation) and microvascular (reactive hyperemia) endothelial function, despite modest elevation in lipids. There is also an ongoing prospective imaging study, CADERA, bolted onto the single-center VEDERA RCT (very early vs delayed etanercept in RA, NCT 02433184), which will use cardiac MRI to explore the prevalence and change of cardiovascular abnormalities in patients receiving TNF inhibitors vs standard therapy over a 12-mo period[89]. The evidence for a link between inflammation and cardiovascular disease is so compelling that 2 RCTS, the Cardiovascular Inflammation Reduction Trial (CIRT) and the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS), have been commenced. CIRT will investigate whether low-dose methotrexate will reduce rates of recurrent MI, stroke, and cardiovascular death among stable post MI patients with type 2 diabetes or metabolic syndrome, two conditions that are associated with an enhanced inflammatory response (NCT01594333). CANTOS will evaluate whether interleukin-1β inhibition as compared to placebo can reduce rates of recurrent MI, stroke, and cardiovascular death among stable CAD patients who remain at high vascular risk due to persistent elevations of CRP (NCT01327846). In addition, tocilizumab and anakirna have been investigated as treatment for acute MIs with RCTs demonstrating an attenuation in CRP levels, CCF and cardiac remodeling following ST elevation MI[90,91], and attenuation in CRP levels and post percutaneous intervention troponin release following non ST elevation MI[92]. There is a further ongoing trial TOCRIVA, which will assess the effects of tocilizumab on cardiovascular risk in RA patients (NCT 01752335).
The relationship between biologics and CCF is more complex and the data is conflicting. In the general population TNF-α levels are increased in CCF[93] and associated with severity of clinical signs and symptoms[94]. Experimental heart failure models have suggested that TNF inhibitors may improve ventricular dysfunction[95]; however, a large clinical trial assessing etanercept in the treatment of congestive heart failure showed no benefit[96], while another one found that high-dose infliximab 10 mg/kg worsened heart failure in patients with moderate-to-severe chronic heart failure[97]. Consequently, a “black box” warning was introduced not to use these medications in patients with pre-existing heart failure[98]. The most recent data from the United States, which compared 8656 new users of synthetic DMARDs with 11587 new users of TNFi, suggested that TNFi were not associated with an increased risk of hospital admissions due to heart failure (HR = 0.85, 95%CI: 0.63 to 1.14), but identified that such a difference may well have existed prior to the introduction of the black box warning in 2002[99]. This study also noted a dose-dependent association between glucocorticoid use and heart failure. Importantly, the authors acknowledged that they were unable to adjust for potential differences in baseline disease severity between the TNFi and synthetic DMARD groups as this information was not collected[98]. The German RABBIT registry also reported similar results, with similar rates of heart failure reported in those receiving TNFi compared to those receiving combined synthetic DMARDs as well as a dose-dependent association with glucocorticoids[100]. The authors of that study suggested that the overall effect of TNFi is more beneficial than harmful, through improved control of disease activity and reduced need for glucocorticoid. Glucocorticoid use in RA patients has also been associated with a dose dependent increase in all-cause and cardiovascular mortality, at a threshold of 8 mg per day[101]. There is less data about the other biologics and CCF but a recent pilot study has shown that 12 mo of treatment with the anti-IL6 tocilizumab significantly increases LV ejection fraction and reduces LV mass index with a concomitant reduction in disease activity[102]. As aforementioned anakinra has also been associated with reduced heart failure following ST elevation MIs[91].
It has been suggested that by suppressing inflammation, biologics may attenuate the autonomic and electrophysiological disturbances that have been linked with SCD and arrhythmic risk amongst patients with RA. Indeed, in a small interventional study of 17 patients with active RA, 76% of which had a prolonged QTc, Lazzerini et al[62] demonstrated that tocilizumab was associated with significant shortening of the QTc within a 3 to 6 mo period. This was also correlated with a decrease in both CRP and TNF-α levels. Similarly, Senel et al[103] showed a reduction in both QTc interval, and inflammatory markers, amongst patients with Ankylosing spondylitis (AS), following 6 mo of treatment with infliximab. Amongst patient with CIA, infliximab therapy duration has also been shown to be inversely and independently associated with QTc duration[59]. Conversely, DI Franco et al[104] found no significant difference in QTc interval duration amongst CIA patients treated with TNFi and rituximab. However, the authors did not report on disease activity and thus there may be have been a large proportion of patients who did not achieve adequate disease control[105]. In the Diana study, 12 wk of treatment with combination synthetic DMARDs or biologics significantly improved cardiac autonomic dysfunction (P < 0.05) in both RA and AS patients[106]. Inflammatory markers (ESR and CRP) correlated with variables of autonomic neuropathy before and after biologic treatment, suggesting that inflammatory markers may both predict the occurrence of autonomic neuropathy and response to treatment, especially with biologics[106]. Infliximab has also been associated with acute changes in HRV consistent with a decrease in sympathetic tone and shift towards relative vagal predominance[107]. Additionally, duration of treatment was also found to be correlated to increased HRV and improved cardiac autonomic function[59].
However, the use of biologics is not without risk, and beyond the well-recognized risks of malignancy and infection, there have been reports of cardiac arrhythmias, in some cases life threatening, particularly following the use of anti-TNF monoclonal antibodies and rituximab[105]. Case reports have described ventricular arrhythmias[108,109], supraventricular arrhythmias[110,111], and various degrees of heart block[112-114], associated with the use of infliximab, although in one case, the complete heart block did in fact resolve spontaneously[112]. It is thought that biologics and in particular TNF inhibiting monoclonal antibodies may be acutely unmasking the inflammation driven myocardial instability that characterizes RA, and other inflammatory conditions[105]. They could be doing this in one of three ways; firstly by worsening LV function, secondly by reducing coronary blood flow[115], and thirdly, a mechanism which would be exclusive to monoclonal antibodies, via complement-mediated cytotoxic or inflammatory damage to the myocardium[105,116]. This is supported by two interesting studies in the literature. The first, is a case report of 42-year-old AS patient who developed ventricular tachycardia requiring defibrillation following 3 doses of infliximab[117]. Despite this, the patient was retreated with infliximab given that there was no evidence of CAD on angiography and his inflammatory markers remained high. Over 2 mo a marked reduction in ventricular arrhythmias was noted with an associated attenuation of inflammatory response. The second study, a prospective, single-blind, crossover study of 75 patients with CIA, demonstrated that during acute infliximab infusion, there was 8% incidence of new-onset ventricular tachyarrhythmia vs 2.7% with placebo (OR = 3.17, 95%CI: 0.61-16.26)[107]. Although the difference was not statistically significant, the study was likely underpowered to detect this. Interestingly, those patients that experienced new onset ventricular arrhythmia showed baseline QTc and HRV values that were significantly prolonged and depressed, respectively, as compared to the patients who did not develop ventricular arrhythmia. Translating this into clinical practice, rituximab and monoclonal TNFi should be avoided in patients with significant CV risk factors, known structural heart disease or ECG abnormalities (conduction disease, QTc prolongation and HRV depression). If these drugs are still required, careful ECG monitoring should be performed in the early phases of administration, until disease activity is adequately controlled, to detect and treat any complications[105].
CONCLUSION
RA is a chronic inflammatory condition, which is associated with significant cardiovascular mortality and morbidity. Data suggests that RA patients are twice as likely to experience SCD compared to the general population[8], a figure comparable to diabetics[9]. Whilst some of this excess risk can be explained by the higher rates of heart failure and IHD, thought to be partially trigged and mediated by inflammation, direct influence of inflammatory cytokines on electrophysiological parameters has been implicated in arrhythmogenesis leading to SCD in RA. Interest is growing in examining the interplay between QTc prolongation, autonomic dysfunction and the risk of SCD. Both autonomic dysfunction and QTc prolongation have been shown to be correlated to inflammation, with the best evidence in place for CRP[10]. The advent of DMARDS and biologics has improved cardiovascular morbidity and mortality[37,82], with novel evidence demonstrating a direct normalisation effect on repolarization parameters such as QTc[62], as well as improvement in parameters of autonomic dysfunction[106]. Randomised controlled trials assessing the impact of DMARDs and biologics on autonomic dysfunction and repolarization parameters, such as QTc or QT dispersion, are urgently required to demonstrate the potential for reduction of SCD in this patient population.
Key messages
Patients with RA are twice as likely to experience SCD compared to the general population; This excess risk can be partially explained by the higher rates of heart failure and IHD, thought to be partially trigged and mediated by inflammation; Direct influence of inflammatory cytokines and autonomic dysfunction on electrophysiological parameters has been implicated in arrhythmogenesis in this patient group; Biologic agents and disease modifying anti-rheumatic drugs may have a role in reducing the burden of SCD by controlling inflammation.
Footnotes
Conflict-of-interest statement: All authors declare no conflict of interest
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: United Kingdom
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: December 28, 2016
First decision: February 17, 2017
Article in press: April 19, 2017
P- Reviewer: Pocar M, Salemi VMC S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Symmons D, Turner G, Webb R, Asten P, Barrett E, Lunt M, Scott D, Silman A. The prevalence of rheumatoid arthritis in the United Kingdom: new estimates for a new century. Rheumatology (Oxford) 2002;41:793–800. doi: 10.1093/rheumatology/41.7.793. [DOI] [PubMed] [Google Scholar]
- 2.Aviña-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690–1697. doi: 10.1002/art.24092. [DOI] [PubMed] [Google Scholar]
- 3.Peters MJ, Symmons DP, McCarey D, Dijkmans BA, Nicola P, Kvien TK, McInnes IB, Haentzschel H, Gonzalez-Gay MA, Provan S, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–331. doi: 10.1136/ard.2009.113696. [DOI] [PubMed] [Google Scholar]
- 4.Cobb S, Anderson F, Bauer W. Length of life and cause of death in rheumatoid arthritis. N Engl J Med. 1953;249:553–556. doi: 10.1056/NEJM195310012491402. [DOI] [PubMed] [Google Scholar]
- 5.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005;52:722–732. doi: 10.1002/art.20878. [DOI] [PubMed] [Google Scholar]
- 6.Myerburg RJ, Kessler KM, Castellanos A. Sudden cardiac death: epidemiology, transient risk, and intervention assessment. Ann Intern Med. 1993;119:1187–1197. doi: 10.7326/0003-4819-119-12-199312150-00006. [DOI] [PubMed] [Google Scholar]
- 7.Myerburg RJ, Interian A, Mitrani RM, Kessler KM, Castellanos A. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol. 1997;80:10F–19F. doi: 10.1016/s0002-9149(97)00477-3. [DOI] [PubMed] [Google Scholar]
- 8.Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52:402–411. doi: 10.1002/art.20853. [DOI] [PubMed] [Google Scholar]
- 9.Siscovick DS, Sotoodehnia N, Rea TD, Raghunathan TE, Jouven X, Lemaitre RN. Type 2 diabetes mellitus and the risk of sudden cardiac arrest in the community. Rev Endocr Metab Disord. 2010;11:53–59. doi: 10.1007/s11154-010-9133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazzerini PE, Capecchi PL, Acampa M, Galeazzi M, Laghi-Pasini F. Arrhythmic risk in rheumatoid arthritis: the driving role of systemic inflammation. Autoimmun Rev. 2014;13:936–944. doi: 10.1016/j.autrev.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC) Europace. 2015;17:1601–1687. doi: 10.1093/europace/euv319. [DOI] [PubMed] [Google Scholar]
- 12.Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. doi: 10.1056/NEJMra000650. [DOI] [PubMed] [Google Scholar]
- 13.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 14.Luu M, Stevenson WG, Stevenson LW, Baron K, Walden J. Diverse mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation. 1989;80:1675–1680. doi: 10.1161/01.cir.80.6.1675. [DOI] [PubMed] [Google Scholar]
- 15.Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104:2158–2163. doi: 10.1161/hc4301.098254. [DOI] [PubMed] [Google Scholar]
- 16.Kannel WB, Doyle JT, McNamara PM, Quickenton P, Gordon T. Precursors of sudden coronary death. Factors related to the incidence of sudden death. Circulation. 1975;51:606–613. doi: 10.1161/01.cir.51.4.606. [DOI] [PubMed] [Google Scholar]
- 17.Bunch TJ, Hohnloser SH, Gersh BJ. Mechanisms of sudden cardiac death in myocardial infarction survivors: insights from the randomized trials of implantable cardioverter-defibrillators. Circulation. 2007;115:2451–2457. doi: 10.1161/CIRCULATIONAHA.106.683235. [DOI] [PubMed] [Google Scholar]
- 18.Goodson N, Marks J, Lunt M, Symmons D. Cardiovascular admissions and mortality in an inception cohort of patients with rheumatoid arthritis with onset in the 1980s and 1990s. Ann Rheum Dis. 2005;64:1595–1601. doi: 10.1136/ard.2004.034777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solomon DH, Goodson NJ, Katz JN, Weinblatt ME, Avorn J, Setoguchi S, Canning C, Schneeweiss S. Patterns of cardiovascular risk in rheumatoid arthritis. Ann Rheum Dis. 2006;65:1608–1612. doi: 10.1136/ard.2005.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Doornum S, Brand C, King B, Sundararajan V. Increased case fatality rates following a first acute cardiovascular event in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:2061–2068. doi: 10.1002/art.21932. [DOI] [PubMed] [Google Scholar]
- 21.Bergner DW, Goldberger JJ. Diabetes mellitus and sudden cardiac death: what are the data? Cardiol J. 2010;17:117–129. [PubMed] [Google Scholar]
- 22.Mantel Ä, Holmqvist M, Jernberg T, Wållberg-Jonsson S, Askling J. Rheumatoid arthritis is associated with a more severe presentation of acute coronary syndrome and worse short-term outcome. Eur Heart J. 2015;36:3413–3422. doi: 10.1093/eurheartj/ehv461. [DOI] [PubMed] [Google Scholar]
- 23.López-Mejías R, García-Bermúdez M, González-Juanatey C, Castañeda S, Miranda-Filloy JA, Gómez-Vaquero C, Fernández-Gutiérrez B, Balsa A, Pascual-Salcedo D, Blanco R, et al. NFKB1-94ATTG ins/del polymorphism (rs28362491) is associated with cardiovascular disease in patients with rheumatoid arthritis. Atherosclerosis. 2012;224:426–429. doi: 10.1016/j.atherosclerosis.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Rodríguez-Rodríguez L, González-Juanatey C, Palomino-Morales R, Vázquez-Rodríguez TR, Miranda-Filloy JA, Fernández-Gutiérrez B, Llorca J, Martin J, González-Gay MA. TNFA -308 (rs1800629) polymorphism is associated with a higher risk of cardiovascular disease in patients with rheumatoid arthritis. Atherosclerosis. 2011;216:125–130. doi: 10.1016/j.atherosclerosis.2010.10.052. [DOI] [PubMed] [Google Scholar]
- 25.Panoulas VF, Nikas SN, Smith JP, Douglas KM, Nightingale P, Milionis HJ, Treharne GJ, Toms TE, Kita MD, Kitas GD. Lymphotoxin 252A& gt; G polymorphism is common and associates with myocardial infarction in patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:1550–1556. doi: 10.1136/ard.2007.082594. [DOI] [PubMed] [Google Scholar]
- 26.Panoulas VF, Stavropoulos-Kalinoglou A, Metsios GS, Smith JP, Milionis HJ, Douglas KM, Nightingale P, Kitas GD. Association of interleukin-6 (IL-6)-174G/C gene polymorphism with cardiovascular disease in patients with rheumatoid arthritis: the role of obesity and smoking. Atherosclerosis. 2009;204:178–183. doi: 10.1016/j.atherosclerosis.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 27.Ibrahim I, McAllister K, Plant D, Symmons D, Marshall T, Barton A, Eyre S. Investigation of an interleukin-6 receptor gene polymorphism (rs2228145) as a predictor of cardiovascular mortality in inflammatory polyarthritis: results from the Norfolk Arthritis Register. Ann Rheum Dis. 2014;73:787–788. doi: 10.1136/annrheumdis-2013-204330. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahim I, Plant D, Lunt M, Verstappen S, Flynn E, Symmons D, Barton A. Investigation of C reactive protein gene polymorphisms as predictors of cardiovascular mortality in inflammatory polyarthritis: results from the Norfolk Arthritis Register. Ann Rheum Dis. 2013;72:1429–1430. doi: 10.1136/annrheumdis-2012-202920. [DOI] [PubMed] [Google Scholar]
- 29.del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–2745. doi: 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- 30.John H, Kitas G. Inflammatory arthritis as a novel risk factor for cardiovascular disease. Eur J Intern Med. 2012;23:575–579. doi: 10.1016/j.ejim.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 31.Casas JP, Shah T, Hingorani AD, Danesh J, Pepys MB. C-reactive protein and coronary heart disease: a critical review. J Intern Med. 2008;264:295–314. doi: 10.1111/j.1365-2796.2008.02015.x. [DOI] [PubMed] [Google Scholar]
- 32.Stamatelopoulos KS, Kitas GD, Papamichael CM, Chryssohoou E, Kyrkou K, Georgiopoulos G, Protogerou A, Panoulas VF, Sandoo A, Tentolouris N, et al. Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler Thromb Vasc Biol. 2009;29:1702–1708. doi: 10.1161/ATVBAHA.109.190108. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Gay MA, Gonzalez-Juanatey C, Martin J. Inflammation and endothelial dysfunction in rheumatoid arthritis. Clin Exp Rheumatol. 2006;24:115–117. [PubMed] [Google Scholar]
- 34.Vaudo G, Marchesi S, Gerli R, Allegrucci R, Giordano A, Siepi D, Pirro M, Shoenfeld Y, Schillaci G, Mannarino E. Endothelial dysfunction in young patients with rheumatoid arthritis and low disease activity. Ann Rheum Dis. 2004;63:31–35. doi: 10.1136/ard.2003.007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aubry MC, Maradit-Kremers H, Reinalda MS, Crowson CS, Edwards WD, Gabriel SE. Differences in atherosclerotic coronary heart disease between subjects with and without rheumatoid arthritis. J Rheumatol. 2007;34:937–942. [PubMed] [Google Scholar]
- 36.van Leuven SI, Franssen R, Kastelein JJ, Levi M, Stroes ES, Tak PP. Systemic inflammation as a risk factor for atherothrombosis. Rheumatology (Oxford) 2008;47:3–7. doi: 10.1093/rheumatology/kem202. [DOI] [PubMed] [Google Scholar]
- 37.Westlake SL, Colebatch AN, Baird J, Curzen N, Kiely P, Quinn M, Choy E, Ostor AJ, Edwards CJ. Tumour necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology (Oxford) 2011;50:518–531. doi: 10.1093/rheumatology/keq316. [DOI] [PubMed] [Google Scholar]
- 38.Lazzerini PE, Capecchi PL, Laghi-Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J. 2017;38:1717–1727. doi: 10.1093/eurheartj/ehw208. [DOI] [PubMed] [Google Scholar]
- 39.Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med. 2004;116:305–311. doi: 10.1016/j.amjmed.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 40.Crowson CS, Nicola PJ, Kremers HM, O’Fallon WM, Therneau TM, Jacobsen SJ, Roger VL, Ballman KV, Gabriel SE. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52:3039–3044. doi: 10.1002/art.21349. [DOI] [PubMed] [Google Scholar]
- 41.Gabriel SE. Heart disease and rheumatoid arthritis: understanding the risks. Ann Rheum Dis. 2010;69 Suppl 1:i61–i64. doi: 10.1136/ard.2009.119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Jacobsen SJ, Roger VL, Gabriel SE. Raised erythrocyte sedimentation rate signals heart failure in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:76–80. doi: 10.1136/ard.2006.053710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bigger JT, Fleiss JL, Kleiger R, Miller JP, Rolnitzky LM. The relationships among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation. 1984;69:250–258. doi: 10.1161/01.cir.69.2.250. [DOI] [PubMed] [Google Scholar]
- 44.Nicola PJ, Crowson CS, Maradit-Kremers H, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Contribution of congestive heart failure and ischemic heart disease to excess mortality in rheumatoid arthritis. Arthritis Rheum. 2006;54:60–67. doi: 10.1002/art.21560. [DOI] [PubMed] [Google Scholar]
- 45.Liang KP, Myasoedova E, Crowson CS, Davis JM, Roger VL, Karon BL, Borgeson DD, Therneau TM, Rodeheffer RJ, Gabriel SE. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1665–1670. doi: 10.1136/ard.2009.124362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis JM, Roger VL, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58:2603–2611. doi: 10.1002/art.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albert CM, Ma J, Rifai N, Stampfer MJ, Ridker PM. Prospective study of C-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation. 2002;105:2595–2599. doi: 10.1161/01.cir.0000017493.03108.1c. [DOI] [PubMed] [Google Scholar]
- 48.Goodson NJ, Symmons DP, Scott DG, Bunn D, Lunt M, Silman AJ. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: a ten-year followup study of a primary care-based inception cohort. Arthritis Rheum. 2005;52:2293–2299. doi: 10.1002/art.21204. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Post WS, Blasco-Colmenares E, Dalal D, Tomaselli GF, Guallar E. Electrocardiographic QT interval and mortality: a meta-analysis. Epidemiology. 2011;22:660–670. doi: 10.1097/EDE.0b013e318225768b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niemeijer MN, van den Berg ME, Eijgelsheim M, van Herpen G, Stricker BH, Kors JA, Rijnbeek PR. Short-term QT variability markers for the prediction of ventricular arrhythmias and sudden cardiac death: a systematic review. Heart. 2014;100:1831–1836. doi: 10.1136/heartjnl-2014-305671. [DOI] [PubMed] [Google Scholar]
- 51.Kazumi T, Kawaguchi A, Hirano T, Yoshino G. C-reactive protein in young, apparently healthy men: associations with serum leptin, QTc interval, and high-density lipoprotein-cholesterol. Metabolism. 2003;52:1113–1116. doi: 10.1016/s0026-0495(03)00184-7. [DOI] [PubMed] [Google Scholar]
- 52.Adlan AM, Lip GY, Paton JF, Kitas GD, Fisher JP. Autonomic function and rheumatoid arthritis: a systematic review. Semin Arthritis Rheum. 2014;44:283–304. doi: 10.1016/j.semarthrit.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, Deckers JW, Kingma JH, Sturkenboom MC, Stricker BH, et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362–367. doi: 10.1016/j.jacc.2005.08.067. [DOI] [PubMed] [Google Scholar]
- 54.Göldeli O, Dursun E, Komsuoglu B. Dispersion of ventricular repolarization: a new marker of ventricular arrhythmias in patients with rheumatoid arthritis. J Rheumatol. 1998;25:447–450. [PubMed] [Google Scholar]
- 55.Cindas A, Gökçe-Kutsal Y, Tokgözoglu L, Karanfil A. QT dispersion and cardiac involvement in patients with rheumatoid arthritis. Scand J Rheumatol. 2002;31:22–26. doi: 10.1080/030097402317255327. [DOI] [PubMed] [Google Scholar]
- 56.Alkaabi JK, Ho M, Levison R, Pullar T, Belch JJ. Rheumatoid arthritis and macrovascular disease. Rheumatology (Oxford) 2003;42:292–297. doi: 10.1093/rheumatology/keg083. [DOI] [PubMed] [Google Scholar]
- 57.Pirildar T, Sekuri C, Utük O, Tezcan UK. QT dispersion in rheumatoid arthritis patients with and without Sjögren’s syndrome. Clin Rheumatol. 2003;22:225–228. doi: 10.1007/s10067-003-0707-4. [DOI] [PubMed] [Google Scholar]
- 58.Ahmad S, Garg S, Dhar M, Srivastava S, Biswas D, Barthwal SP, Shirazi N, Srivastava R. Predictors of atherosclerosis in rheumatoid arthritis. Vasa. 2012;41:353–359. doi: 10.1024/0301-1526/a000221. [DOI] [PubMed] [Google Scholar]
- 59.Lazzerini PE, Acampa M, Capecchi PL, Hammoud M, Maffei S, Bisogno S, Barreca C, Galeazzi M, Laghi-Pasini F. Association between high sensitivity C-reactive protein, heart rate variability and corrected QT interval in patients with chronic inflammatory arthritis. Eur J Intern Med. 2013;24:368–374. doi: 10.1016/j.ejim.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Panoulas VF, Toms TE, Douglas KM, Sandoo A, Metsios GS, Stavropoulos-Kalinoglou A, Kitas GD. Prolonged QTc interval predicts all-cause mortality in patients with rheumatoid arthritis: an association driven by high inflammatory burden. Rheumatology (Oxford) 2014;53:131–137. doi: 10.1093/rheumatology/ket338. [DOI] [PubMed] [Google Scholar]
- 61.Chauhan K, Ackerman MJ, Crowson CS, Matteson EL, Gabriel SE. Population-based study of QT interval prolongation in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2015;33:84–89. [PMC free article] [PubMed] [Google Scholar]
- 62.Lazzerini PE, Acampa M, Capecchi PL, Fineschi I, Selvi E, Moscadelli V, Zimbone S, Gentile D, Galeazzi M, Laghi-Pasini F. Antiarrhythmic potential of anticytokine therapy in rheumatoid arthritis: tocilizumab reduces corrected QT interval by controlling systemic inflammation. Arthritis Care Res (Hoboken) 2015;67:332–339. doi: 10.1002/acr.22455. [DOI] [PubMed] [Google Scholar]
- 63.London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, McTiernan CF, Feldman AM, Salama G. Calcium-dependent arrhythmias in transgenic mice with heart failure. Am J Physiol Heart Circ Physiol. 2003;284:H431–H441. doi: 10.1152/ajpheart.00431.2002. [DOI] [PubMed] [Google Scholar]
- 64.Petkova-Kirova PS, Gursoy E, Mehdi H, McTiernan CF, London B, Salama G. Electrical remodeling of cardiac myocytes from mice with heart failure due to the overexpression of tumor necrosis factor-alpha. Am J Physiol Heart Circ Physiol. 2006;290:H2098–H2107. doi: 10.1152/ajpheart.00097.2005. [DOI] [PubMed] [Google Scholar]
- 65.Fernández-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MA, Delgado C. TNF-alpha downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Heart Circ Physiol. 2007;293:H238–H245. doi: 10.1152/ajpheart.01122.2006. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K(+) channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem. 2004;279:13289–13292. doi: 10.1074/jbc.C400025200. [DOI] [PubMed] [Google Scholar]
- 67.Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, Murata M, Takahashi E, Shimoda K, Hirano T, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. 2007;43:710–716. doi: 10.1016/j.yjmcc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 68.Li YH, Rozanski GJ. Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc Res. 1993;27:525–530. doi: 10.1093/cvr/27.3.525. [DOI] [PubMed] [Google Scholar]
- 69.Mattey DL, Glossop JR, Nixon NB, Dawes PT. Circulating levels of tumor necrosis factor receptors are highly predictive of mortality in patients with rheumatoid arthritis. Arthritis Rheum. 2007;56:3940–3948. doi: 10.1002/art.23075. [DOI] [PubMed] [Google Scholar]
- 70.Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol. 2015;42:421–428. doi: 10.3899/jrheum.140861. [DOI] [PubMed] [Google Scholar]
- 71.Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–344. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 72.Li C, Hu D, Shang L, Ma S, Liu W, Li Y, Ma Z, Tang C, Mei Y, Wang L. Surgical left cardiac sympathetic denervation for long QT syndrome: effects on QT interval and heart rate. Heart Vessels. 2005;20:137–141. doi: 10.1007/s00380-005-0820-1. [DOI] [PubMed] [Google Scholar]
- 73.Castro RR, Porphirio G, Serra SM, Nóbrega AC. Cholinergic stimulation with pyridostigmine reduces the QTc interval in coronary artery disease. Braz J Med Biol Res. 2002;35:685–689. doi: 10.1590/s0100-879x2002000600008. [DOI] [PubMed] [Google Scholar]
- 74.Ewing DJ, Martyn CN, Young RJ, Clarke BF. The value of cardiovascular autonomic function tests: 10 years experience in diabetes. Diabetes Care. 1985;8:491–498. doi: 10.2337/diacare.8.5.491. [DOI] [PubMed] [Google Scholar]
- 75.Malik M. Heart rate variability. Ann Noninvas Electro. 1996;1:151–181. [Google Scholar]
- 76.La Rovere MT, Bigger JT, Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet. 1998;351:478–484. doi: 10.1016/s0140-6736(97)11144-8. [DOI] [PubMed] [Google Scholar]
- 77.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 78.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 79.Verrier RL, Antzelevitch C. Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr Opin Cardiol. 2004;19:2–11. doi: 10.1097/00001573-200401000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meek IL, Vonkeman HE, van de Laar MA. Cardiovascular case fatality in rheumatoid arthritis is decreasing; first prospective analysis of a current low disease activity rheumatoid arthritis cohort and review of the literature. BMC Musculoskelet Disord. 2014;15:142. doi: 10.1186/1471-2474-15-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westlake SL, Colebatch AN, Baird J, Kiely P, Quinn M, Choy E, Ostor AJ, Edwards CJ. The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology (Oxford) 2010;49:295–307. doi: 10.1093/rheumatology/kep366. [DOI] [PubMed] [Google Scholar]
- 82.Micha R, Imamura F, Wyler von Ballmoos M, Solomon DH, Hernán MA, Ridker PM, Mozaffarian D. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108:1362–1370. doi: 10.1016/j.amjcard.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barnabe C, Martin BJ, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken) 2011;63:522–529. doi: 10.1002/acr.20371. [DOI] [PubMed] [Google Scholar]
- 84.Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, Siu S, Kraft J, Lynde C, Pope J, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74:480–489. doi: 10.1136/annrheumdis-2014-206624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007;56:2905–2912. doi: 10.1002/art.22809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rao VU, Pavlov A, Klearman M, Musselman D, Giles JT, Bathon JM, Sattar N, Lee JS. An evaluation of risk factors for major adverse cardiovascular events during tocilizumab therapy. Arthritis Rheumatol. 2015;67:372–380. doi: 10.1002/art.38920. [DOI] [PubMed] [Google Scholar]
- 87.Lim DT, Cannella AC, Michaud KD, Mikuls TR. Cardiovascular risk and the use of biologic agents in rheumatoid arthritis. Curr Rheumatol Rep. 2014;16:459. doi: 10.1007/s11926-014-0459-y. [DOI] [PubMed] [Google Scholar]
- 88.Hsue PY, Scherzer R, Grunfeld C, Imboden J, Wu Y, Del Puerto G, Nitta E, Shigenaga J, Schnell Heringer A, Ganz P, et al. Depletion of B-cells with rituximab improves endothelial function and reduces inflammation among individuals with rheumatoid arthritis. J Am Heart Assoc. 2014;3:e001267. doi: 10.1161/JAHA.114.001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Erhayiem B, Pavitt S, Baxter P, Andrews J, Greenwood JP, Buch MH, Plein S. Coronary Artery Disease Evaluation in Rheumatoid Arthritis (CADERA): study protocol for a randomized controlled trial. Trials. 2014;15:436. doi: 10.1186/1745-6215-15-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW, Oddi C, Carbone S, Trankle CR, Roberts CS, et al. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies) Am J Cardiol. 2015;115:288–292. doi: 10.1016/j.amjcard.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E, Michelsen AE, Bendz B, Amundsen BH, Espevik T, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J. 2016;37:2406–2413. doi: 10.1093/eurheartj/ehw171. [DOI] [PubMed] [Google Scholar]
- 93.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 94.McMurray J, Abdullah I, Dargie HJ, Shapiro D. Increased concentrations of tumour necrosis factor in “cachectic” patients with severe chronic heart failure. Br Heart J. 1991;66:356–358. doi: 10.1136/hrt.66.5.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kadokami T, Frye C, Lemster B, Wagner CL, Feldman AM, McTiernan CF. Anti-tumor necrosis factor-alpha antibody limits heart failure in a transgenic model. Circulation. 2001;104:1094–1097. doi: 10.1161/hc3501.096063. [DOI] [PubMed] [Google Scholar]
- 96.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 97.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 98.Humphreys J, Hyrich K, Symmons D. What is the impact of biologic therapies on common co-morbidities in patients with rheumatoid arthritis? Arthritis Res Ther. 2016;18:282. doi: 10.1186/s13075-016-1176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Solomon DH, Rassen JA, Kuriya B, Chen L, Harrold LR, Graham DJ, Lewis JD, Lii J, Liu L, Griffin MR, et al. Heart failure risk among patients with rheumatoid arthritis starting a TNF antagonist. Ann Rheum Dis. 2013;72:1813–1818. doi: 10.1136/annrheumdis-2012-202136. [DOI] [PubMed] [Google Scholar]
- 100.Listing J, Strangfeld A, Kekow J, Schneider M, Kapelle A, Wassenberg S, Zink A. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum. 2008;58:667–677. doi: 10.1002/art.23281. [DOI] [PubMed] [Google Scholar]
- 101.del Rincón I, Battafarano DF, Restrepo JF, Erikson JM, Escalante A. Glucocorticoid dose thresholds associated with all-cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:264–272. doi: 10.1002/art.38210. [DOI] [PubMed] [Google Scholar]
- 102.Kobayashi H, Kobayashi Y, Giles JT, Yoneyama K, Nakajima Y, Takei M. Tocilizumab treatment increases left ventricular ejection fraction and decreases left ventricular mass index in patients with rheumatoid arthritis without cardiac symptoms: assessed using 3.0 tesla cardiac magnetic resonance imaging. J Rheumatol. 2014;41:1916–1921. doi: 10.3899/jrheum.131540. [DOI] [PubMed] [Google Scholar]
- 103.Senel S, Cobankara V, Taskoylu O, Guclu A, Evrengul H, Kaya MG. Effect of infliximab treatment on QT intervals in patients with ankylosing spondylitis. J Investig Med. 2011;59:1273–1275. doi: 10.2130/JIM.0b013e3182330720. [DOI] [PubMed] [Google Scholar]
- 104.DI Franco M, Paradiso M, Ceccarelli F, Scrivo R, Spinelli FR, Iannuccelli C, Valesini G. Biological drug treatment of rheumatoid arthritis and spondyloarthritis: effects on QT interval and QT dispersion. J Rheumatol. 2012;39:41–45. doi: 10.3899/jrheum.110158. [DOI] [PubMed] [Google Scholar]
- 105.Lazzerini PE, Capecchi PL, Galeazzi M, Laghi-Pasini F. Biologic drugs and arrhythmic risk in chronic inflammatory arthritis: the good and the bad. Immunol Res. 2016:1–14. doi: 10.1007/s12026-016-8833-7. [DOI] [PubMed] [Google Scholar]
- 106.Syngle A, Verma I, Krishan P, Garg N, Syngle V. Disease-modifying anti-rheumatic drugs improve autonomic neuropathy in arthritis: DIANA study. Clin Rheumatol. 2015;34:1233–1241. doi: 10.1007/s10067-014-2716-x. [DOI] [PubMed] [Google Scholar]
- 107.Lazzerini PE, Acampa M, Hammoud M, Maffei S, Capecchi PL, Selvi E, Bisogno S, Guideri F, Galeazzi M, Pasini FL. Arrhythmic risk during acute infusion of infliximab: a prospective, single-blind, placebo-controlled, crossover study in patients with chronic arthritis. J Rheumatol. 2008;35:1958–1965. [PubMed] [Google Scholar]
- 108.de’ Clari F, Salani I, Safwan E, Giannacco A. Sudden death in a patient without heart failure after a single infusion of 200 mg infliximab: does TNF-alpha have protective effects on the failing heart, or does infliximab have direct harmful cardiovascular effects? Circulation. 2002;105:E183. doi: 10.1161/01.cir.0000017216.41471.df. [DOI] [PubMed] [Google Scholar]
- 109.Boyer JF, Jamard B, El Mahou S, Laroche M, Mazières B, Cantagrel A, Constantin A. New-onset acute heart failure and ventricular tachycardia after therapy with a tumor necrosis factor antagonist. Clin Exp Rheumatol. 2004;23:274–275. [PubMed] [Google Scholar]
- 110.Choi JW, Tae HJ, Oh IH, Lee MK, Shin JH, Kim TH, Jun JB, Uhm WS. A case of atrial fibrillation induced by infliximab in a patient with rheumatoid arthritis. J Rheum Dis. 2011;18:302–305. [Google Scholar]
- 111.Singh M, Diwan MM, Patel KC. A rare case of supraventricular tachycardia induced by Infliximab: a case report. Cases J. 2009;2:147. doi: 10.1186/1757-1626-2-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sofos S, Savoye G, Ramirez S, Bauer F, Lerebours E. Transient type III atrioventricular block after infliximab infusion in a fistulizing perianal Crohn’s disease patient. Am J Gastroenterol. 2007;102:217–219. [PubMed] [Google Scholar]
- 113.Sote Y, Green S, Maddison P. Complete heart block after infliximab therapy. Rheumatology (Oxford) 2008;47:227–228. doi: 10.1093/rheumatology/kem336. [DOI] [PubMed] [Google Scholar]
- 114.Anand CP, Al-Juburi A, Sandeep B. Heart block occurring during infliximab infusion: A report of two cases. Am J Gastroenterol. 2003;98(s9):S144. [Google Scholar]
- 115.Di Diego JM, Antzelevitch C. Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm. 2011;8:1963–1968. doi: 10.1016/j.hrthm.2011.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sarzi-Puttini P, Atzeni F, Shoenfeld Y, Ferraccioli G. TNF-alpha, rheumatoid arthritis, and heart failure: a rheumatological dilemma. Autoimmun Rev. 2005;4:153–161. doi: 10.1016/j.autrev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 117.Özcan ÖU, Özcan DS, Polat CS, Özyüncü N, Erol Ç. Ventricular arrhythmia with cessation of infliximab in a patient with ankylosing spondylitis. Joural Ankara University Faculty Medicine. 2015;68:77–79. [Google Scholar]