

Abstract
The mechanisms of action of three C-10 non-acetal trioxane dimers (TDs) were examined in human (LNCaP) and mouse (TRAMP-C1A and -C2H) prostate cancer cell lines. 1(AJM3/23), 2(GHP-TM-III-07w, 3(GHP-KB-06) inhibited cell growth with 3 being the most potent in C1A (GI50 = 18.0 nM), C2H (GI50 = 17.0 nM) and LNCaP (GI50 = 17.9 nM) cells. In comparison to a standard cytotoxic agent such as doxorubicin (GI50 = 45.3 nM), 3 (GI50 = 17.9 nM) inhibited LNCaP cell growth more potently. TDs induced G0/G1 cell cycle arrest in LNCaP cells and decreased cells in the S phase. These changes correlated with modulation of G1 phase cell cycle proteins including decreased cyclin D1, cyclin E, cdk2; and increased p21waf1 and p27Kip1. TDs also promoted apoptosis in LNCaP cells with increased expression of pro-apoptotic bax. These results demonstrate that TDs are potentially useful agents that warrant further preclinical development for treatment of prostate cancer.
Keywords: Trioxane dimers, Artemisinin derivatives, Prostate cancer, TRAMP, LNCaP, Cell cycle, Apoptosis, Doxorubicin
Graphical abstract
Introduction
It is estimated that prostate cancer is the most prevalent cancer and the third leading cause of cancer death in American men.1 Current treatment options for localized, early stage prostate cancer include surgery, radiation therapy and watchful waiting.2 However, most patients relapse with advanced metastatic disease, that when treated with androgen deprivation therapy eventually becomes hormone refractory.2 Clinical trials have been conducted with chemotherapeutic agents such as docetaxel, mitoxantrone and doxorubicin in men with hormone refractory prostate cancer, however, these agents have limited efficacy and significant adverse side effects.3 The poor clinical outcome of advanced metastatic prostate cancer highlights the urgent need to develop effective novel therapeutic agents for prevention and treatment of the disease.
Trioxane dimers (TDs) are semi-synthetic derivatives of the Chinese antimalarial compound, 1,2,4-trioxane artemisinin (Artemesia annua L., qinghaosu).4 The instability of first generation TDs has prompted synthesis of more potent, orally active, thermally and hydrolytically stable second generation C-10 non-acetal TDs.5–7 These compounds display potent antimalarial, antiproliferative and anticancer activities.5–7 They inhibit proliferation of chloroquine-sensitive Plasmodium falciparum,5–7 murine keratinocytes5,6 and several malignant human cancer cell lines in the National Cancer Institute’s (NCI) Developmental and Therapeutics Program.5–7 Newly synthesized TDs are selectively cytotoxic in HeLa, human cervical cancer cells compared to normal cervical cells.8 In vivo, TDs decrease tumor mass in the NCI mouse hollow fiber assay.5 Toxicity studies in antimalarial mouse models indicate that up to 125 mg/kg of TDs can be safely administered.9 Although the antitumor modes of action of TDs have not been extensively characterized, the mechanisms of action of the parent compound (artemisinin) and its semi-synthetic derivatives have been studied. Artemisinin derivatives inhibit proliferation, induce G1 phase cell cycle arrest and increase apoptosis in P388 leukemia cells.10 mRNA expression profiling studies indicate that the antineoplastic activity of artemisinin derivatives in a diverse panel of tumor cell lines from the NCI correlates with changes in expression of genes involved in cell proliferation.11 Further studies demonstrated that the cytotoxicity of an artemisinin derivative (artesunate), in a panel of 55 cancer cell lines results in induction of G0/G1 cell cycle arrest, inhibition of S phase progression and changes in expression of cell cycle regulatory proteins.12
Cyclin/cyclin dependent kinase (cdk) complexes including cyclin D-cdk4/cdk6 and cyclinE-cdk2 complexes are important for progression of cells through the G1 phase of the cell cycle and initiation of DNA replication.13 Overexpression of cyclin D1,14 and cdk415,16 appear to be important for oncogenic transformation. The Cip/Kip family of cdk inhibitors, p21Waf1 (p21) and p27Kip1 (p27), impede cell cycle progression by inhibiting cyclin E-cdk2 complexes.17 p21 induces G1 or G2 cell cycle arrest and inhibits DNA synthesis via inhibition of cdk activity and proliferating cell nuclear antigen (PCNA).18
We previously demonstrated that 1(AJM3/23) inhibits the growth of prostate cancer clonal cell lines (C1A, C2D, C2G and C2H).9 These clonal cell lines were derived from a poorly differentiated tumor that developed in the transgenic adenocarcinoma of mouse prostate (TRAMP) model.19 Probasin promoter-driven expression of SV40 early genes (T and t) in the prostatic epithelium leads to progressive development of prostate cancer in the TRAMP mouse model.20 The mechanisms of action of three C-10 non-acetal TDs that were synthesized by chemical substitution at the C-10 position (Fig. 1), were evaluated using human prostate cancer cells (LNCaP) and two clonal cell lines derived from the TRAMP mouse model (C1A and C2H). The molecular effects of 1, 2(GHP-TM-III-07w) and 3(GHP-KB-06) in prostate cancer cells include inhibition of cell proliferation, regulation of cell cycle progression and induction of apoptosis.
Figure 1.
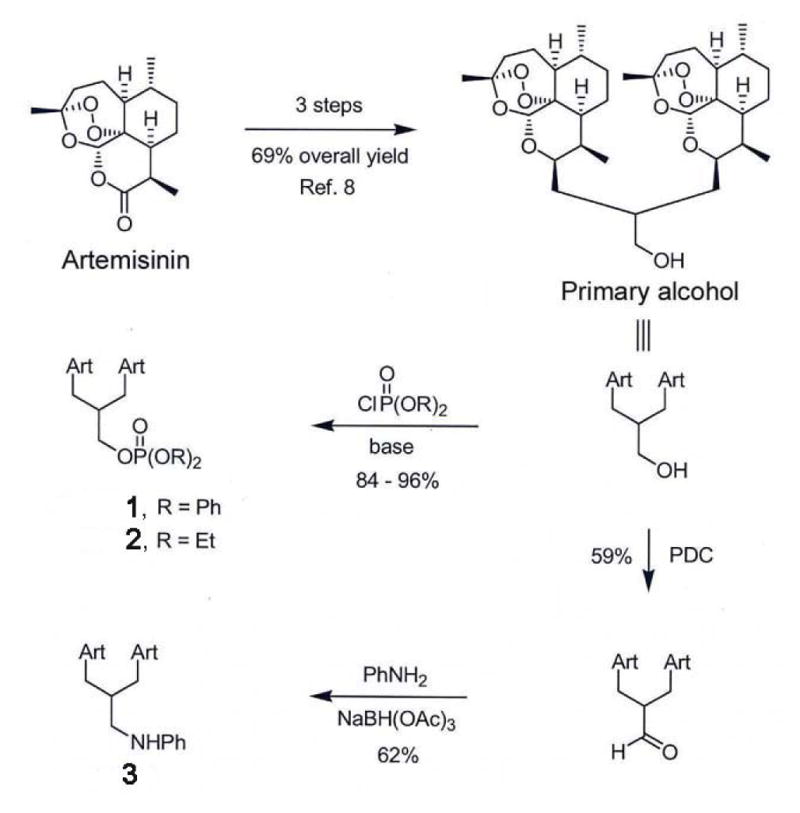
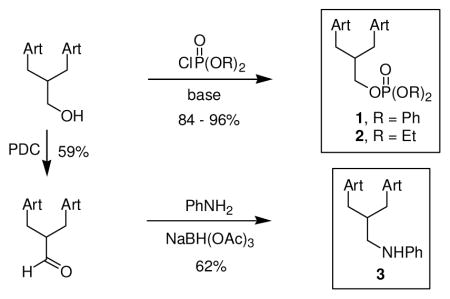
Chemical synthesis of C-10 non-acetal trioxane dimers. 1, 2 and 3, were synthesized from trioxane dimer primary alcohol.
Results
TDs inhibit proliferation of prostate cancer cells
To assess the effect of TDs on cell growth, C1A, C2H and LNCaP cells were treated with TDs or DOX for 72 h. Relative to other TDs, 3 was the most potent at inhibiting cell growth in C1A (GI50 = 18.0 nM), C2H (GI50 = 17.0 nM) and LNCaP (GI50 = 17.9 nM) cells (Figs. 2A–C & Table 1). The efficacy of TDs was compared to doxorubicin (DOX), an anthracycline and standard chemotherapeutic agent that is used clinically to treat advanced prostate cancer.21,22 DOX was used in these studies because antimalarial artemisinin derivatives share similar properties with DOX such as generation of reactive oxygen species.23 3 (GI50 = 17.9 nM) inhibited the growth of LNCaP cells more potently than DOX (GI50 = 45.3 nM, Table 1). At the highest dose tested, DOX (100 nM) inhibited LNCaP cell growth by 69%, however, 3 inhibited cell growth by 97% (Fig. 2C). Interestingly, 3 (GI50 =17.0 nM, Table 1) was also as effective as DOX (GI50 =15.8 nM, Table 1) in C2H cells that are aggressive and metastatic.
Figure 2.

Inhibition of prostate cancer cell growth by trioxane dimers. (A) C1A, (B) C2H, and (C) LNCaP cells were treated with different concentrations of 1, 2, 3 or DOX for 72 h. Cell viability was determined by MTT assay and results are reported as mean cell growth (%) of at least three replicate wells ± SD. Results are normalized to cell growth at timepoint zero.
Table 1.
GI50 of trioxane dimers in prostate cancer cell linesa
|
|
|||
|---|---|---|---|
| GI50 (nM) | |||
|
| |||
| Compounds | C1A | C2H | LNCaP |
| 2 | 42.2 | 39.4 | 36.3 |
| 3 | 18.0 | 17.0 | 17.9 |
| 1 | 31.9 | 44.8 | 34.4 |
| DOX | 20.4 | 15.8 | 45.3 |
Summary of GI50s of trioxane dimers in prostate cancer cells. Cells were treated with 1, 2, 3 or DOX for 72 h. Cell viability was determined by MTT assay. GI50, concentration that inhibits 50% cell growth. These results are representative of at least 3 independent experiments.
In subsequent studies, cells were treated with 50 nM of TDs or DOX for 72 h because maximal inhibition was observed with all compounds at this dose (Figs. 2A–C). The effect of TDs on DNA synthesis was examined by BrdU incorporation assay. TDs (50 nM) inhibited BrdU incorporation in C1A, C2H and LNCaP cells (P < 0.0001, Fig. 3) after 72h of treatment. Compared to other TDs, 3 strongly inhibited DNA synthesis (>90%) in C2H and LNCaP cells. Although cell growth inhibition studies indicated that 3 was more potent than DOX in LNCaP cells, both compounds appear to be equally effective at inhibiting DNA synthesis. These differences may be attributable to the variability in the sensitivity of the assays used. LNCaP cells were used to further investigate the mechanisms of action of TDs.
Figure 3.

Inhibition of DNA synthesis in C1A, C2H and LNCaP cells. Cells were treated with vehicle (control) or 50 nM of either 1, 2, 3 or DOX for 72 h. DNA synthesis was measured by BrdU incorporation assay. Results are reported as mean BrdU incorporation (%) ± SEM normalized to control group. *, P < 0.0001 by ANOVA.
TDs regulate cell cycle progression in LNCaP cells
The effect of TDs on the cell cycle profile of LNCaP cells was examined by PI staining and flow cytometry. Compared to control, TDs induced G0/G1 cell cycle arrest in LNCaP cells (Figs. 4A–B). Modulation of G1 phase cell cycle regulators may be a key mechanism of action of TDs in LNCaP cells. Consistent with earlier findings that TDs inhibited DNA synthesis in LNCaP cells (Fig. 3), a dramatic reduction in the S phase population was observed (Figs. 4A–B), indicating a block in G1-S transition. DOX dramatically promoted accumulation of LNCaP cells in the G2/M phase and decreased the G1 and S phase populations (Figs. 4A–B), consistent with previous findings by Martinez et al. 24 The effects of DOX in the cell cycle include induction of G2/M phase cell cycle arrest.
Figure 4.
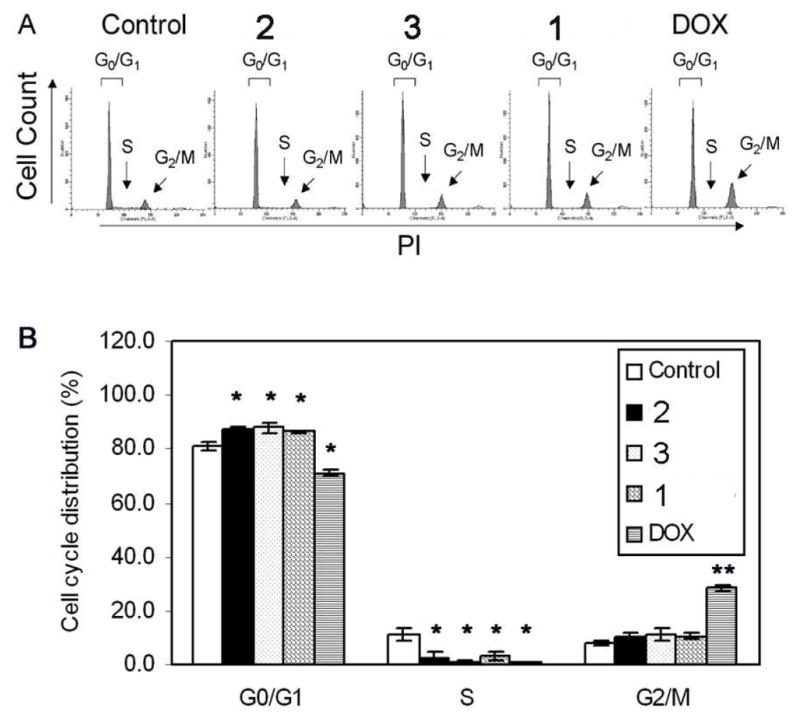
Induction of G0/G1 cell cycle arrest in LNCaP cells. Cells were treated with vehicle (control) or 50 nM of either 1, 2, 3 or DOX for 72 h. Cell cycle distribution was determined by PI staining and flow cytometry. (A) Histograms of cell cycle distribution, and (B) mean cell cycle distribution (%) in the G0/G1, S and G2/M phases of three independent experiments ± SEM. *, P < 0.05; **, P < 0.0001 by ANOVA.
Deregulation of G1 phase cell cycle regulators is critical for tumor progression, and genetic alterations of cell cycle proteins occurs frequently in human cancers.25 The effect of TDs on protein expression of G1 phase cell cycle regulatory proteins was examined in LNCaP cells by Western blot analysis following treatment for 72h. Cyclin D1-cdk4/6 and cyclin E-cdk2 complexes promote G1 phase cell cycle progression.13 TDs had minimal effects on cdk4 expression as evidenced by a 10–30% decrease (Fig. 5A). However, 1 and 3 increased cdk6 expression (Fig. 5A). This coincided with dramatic inhibition of cyclin D1 expression by TDs (Fig. 5A). The effect of TDs on the cyclin E-cdk2 complex was also examined. TDs markedly inhibited cdk2 protein expression and kinase activity as demonstrated by decreased phosphorylation of histone H1, a cdk2 substrate (Fig. 5B). These changes correlated with decreased expression of cyclin E (Fig. 5B) that forms a complex with cdk2.
Figure 5.
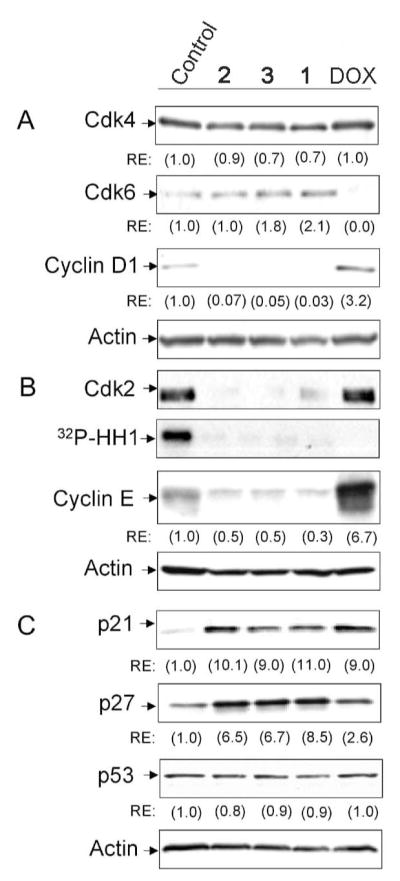
Regulation of G1 phase cell cycle regulators in LNCaP cells. Cell lysates were collected following treatment with vehicle (control) or 50 nM of either 1, 2, 3 or DOX for 72 h. (A) Western blot analysis with anti-cdk4, -cdk6 and -cyclin D1, (B) Western blot analysis with anti- cdk2 and -cyclin E, and cdk2 kinase assay; 32P-HH1, γ-32P-histone H1. (C) Western blot analysis with anti-p21, -p27 and -p53. Actin was used as loading control. RE, relative expression of proteins was determined by densitometry and normalized to actin.
The effect of TDs on expression of the Cip/Kip family of cdk inhibitors (p21 and p27) was evaluated. Compared to control, TDs increased expression of p21 (9–11 fold) and p27 (6.5–8.5 fold) (Fig. 5C). Induction of p21 expression may be mediated by p53-dependent or -independent pathways.26 TDs did not increase p53 expression relative to control (Fig. 5C). This suggests that at the doses tested, induction of p21 expression by TDs in LNCaP cells does not correlate with increased p53 levels.
TDs increase apoptosis in LNCaP cells
To further examine the mechanisms of action of TDs in LNCaP cells, apoptosis was measured by annexin V staining. Cells in early stages of apoptosis stain positive for annexin V only, while late stage apoptotic and necrotic cells stain positive for annexin V and 7-AAD.27 TDs increased early apoptosis by ~4–6 fold compared to control (Fig. 6A), with 3 having the greatest effect. In contrast, DOX induced late apoptosis and necrosis in LNCaP cells (Fig. 6A). The apoptotic effect of TDs in LNCaP cells was accompanied by increased expression of pro-apoptotic bax (Fig. 6B) and a decrease in anti-apoptotic Bcl-2 by 1 and 3 (Fig. 6B).
Figure 6.
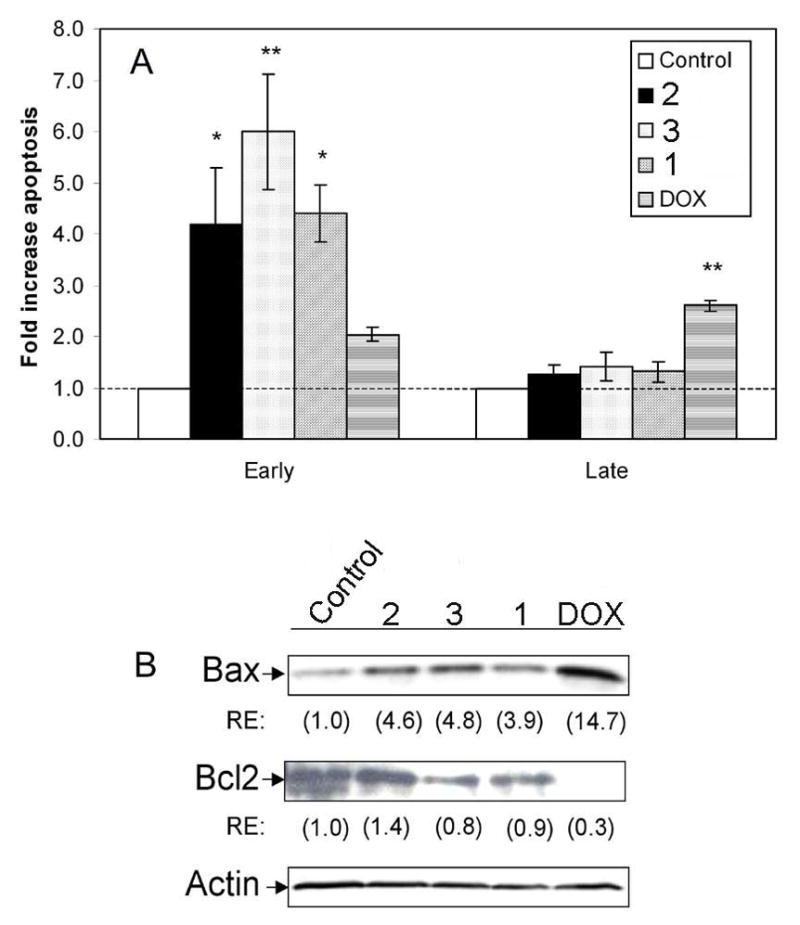
Induction of apoptosis in LNCaP cells. Cells were treated with vehicle (control) or 50 nM of either 1, 2, 3 or DOX for 72 h. Apoptosis was measured by annexin V-PE/7-AAD staining. (A) Mean fold increase in early stage (annexin V positive cells) and late stage (annexin V and 7-AAD positive cells) apoptosis ± SEM normalized to control group (fold increase = 1.0). *, P < 0.05; **, P < 0.001 by ANOVA. (B) Western blot analysis was performed with anti-bax and -Bcl-2. Actin was used as loading control. RE, relative expression of proteins was determined by densitometry and normalized to actin.
Discussion
The antiproliferative effects of artemisinin-derived C-10 non-acetal TDs in prostate cancer cells include inhibition of cell proliferation, induction of G0/G1 cell cycle arrest and promotion of apoptosis. These effects were accompanied by changes in expression of key proteins in the G1 phase of the cell cycle and increased expression of pro-apoptotic bax. 3 was the most potent TD tested, as it elicited the most profound growth inhibition in C1A and LNCaP cells. Interestingly, compared to the other agents, 3 has different chemical substituents in the linker position (Fig. 1). The linkers of 1 and 2 contain phosphate esters,28 however, in 3, the linker has a basic aniline functionality that can hydrogen bond with various biological hydrogen-bond donors. We speculate that this chemical difference may account for the increased efficacy of 3 in prostate cancer cell lines.
Uncontrolled cell cycle progression and evasion of apoptosis are hallmarks of cancer.29 Therefore, therapeutic agents such as TDs that can simultaneously impede cell cycle progression and promote apoptosis in cancer cells are highly desirable. TDs inhibited DNA synthesis, induced G0/G1 cell cycle arrest and regulated expression of key G1 phase cell cycle proteins in LNCaP cells. Aberrant proliferation of cancer cells involves deregulation of key G1 phase cell cycle regulators25 and overexpression of cyclins and cdks provides a selective growth advantage to tumor cells.25,30 Therefore, targeting cyclin/cdk complexes that promote tumor progression is therapeutically relevant for treatment of cancer. D-type cyclins are considered to be required for cell cycle entry and early G1 phase cell cycle progression,13 as inhibition of cyclin D1 specifically triggers G1 phase cell cycle arrest31. Cyclin D1 is overexpressed in many human cancers,32,33 including prostate cancer.34 Cyclin D1 forms a complex with either of its catalytic partners, cdk4 or cdk6, to promote G1 phase cell cycle progression.30 Induction of G0/G1 cell cycle arrest by TDs in LNCaP cells coincided with inhibition of cyclin D1, thus inactivating the cyclin D1-cdk4/6 complex. This suggests that inhibition of pro-growth signals such as cyclin D1 is critical for the anticancer effects of TDs in prostate cancer.
p21 and p27 impede cell cycle progression by inhibiting cyclin E-cdk2 complexes, that promote G1-S phase cell cycle progression.17 The role of p21 expression in prostate cancer remains controversial. Overexpression of p21 in the nucleus has been associated with poor prognosis in prostate cancer,35 however, other studies have indicated otherwise.36 Decreased p27 expression is associated with poor prognosis in prostate cancer.36 Interestingly, TDs strongly upregulated p21 and p27 expression in LNCaP cells. These effects coincided with inhibition of cyclin E-cdk2, induction of G0/G1 cell cycle arrest and inhibition of DNA synthesis in the S phase. p53, is a tumor suppressor gene that regulates expression of p21 following DNA damage.37 However, p53-independent upregulation of p21 expression has been reported.38 Induction of p21 expression by TDs in LNCaP cells was not accompanied by increased p53 expression. Past studies by Efferth et al. indicate that artesunate (an artemisinin derivative) increases p21 expression in a p53-dependent and -independent manner in HCT-116 colon cancer cells.12
TDs promoted early apoptosis and increased expression of bax, a pro-apoptotic protein.39 Wang et al. previously demonstrated that artesunate induces apoptosis in hepatic carcinoma in vivo by upregulating bax.40 Although TDs increased apoptosis in LNCaP cells, it appears that their major mechanism of action involves modulation of cell cycle progression. The cytostatic and cytotoxic properties of TDs in prostate cancer indicates that the biological mechanisms of action of these agents are diverse, thus rendering them more attractive for preclinical and clinical development for prostate cancer.
The efficacy of TDs was compared to DOX, a standard chemotherapeutic agent that is used clinically to treat several cancers, including prostate cancer.22 DOX inhibited the growth of prostate cancer cells, decreased DNA synthesis, induced cell cycle arrest, modulated cell cycle regulatory proteins and promoted apoptosis. These findings are consistent with previous findings that the antitumor effects of DOX are mediated by inhibition of DNA synthesis,41 induction of cell cycle arrest24 and promotion of apoptosis.42 Interestingly, 3 was more potent than DOX in human LNCaP cells. However, while cardiotoxicity is a major side effect of DOX,43 no serious adverse side effects have been observed clinically with artemisinin derivatives.44 In vivo studies with TDs9 and other artemisinin derivatives45 indicate that these compounds can be safely administered. The effect of TDs on cell cycle progression appears to differ from DOX. TDs induced G1 phase cell cycle arrest, while DOX largely promoted accumulation of cells in the G2/M phase. These findings are consistent with previous reports that DOX induces G2/M arrest in LNCaP24 and DU14546 prostate cancer cell lines. DOX increased cyclin D1 expression, while cdk4 levels were unchanged. Although DOX reduced cdk6 levels, cyclin D1 can complex with cdk4 to facilitate G1 phase cell cycle progression. The strong induction of G2/M arrest by DOX may be an early event. It appears that DOX later induces G1 cell cycle arrest as evidenced by increased expression of G1 phase cell cycle inhibitors, p21 and p27; decreased cdk2 kinase activity, and inhibition of DNA synthesis in LNCaP cells. At the dose of DOX utilized in these studies, increased p21 expression did not correlate with upregulation of p53 levels in LNCaP cells. These results appear to be contrary to studies by Martinez et al. that demonstrated that high doses of DOX (0.2 μg/mL, 345 nM) increase p21 and p53 expression in LNCaP cells.24 In the current studies, LNCaP cells were treated with much lower doses of DOX (50 nM) that increased p21 levels with no change in p53 expression.
These are the first studies to specifically explore the anticancer effects and biological mechanisms of action of TDs in prostate cancer. The novel TDs tested in these studies inhibit the growth of prostate cancer cells by inducing G0/G1 cell cycle arrest, inhibiting DNA synthesis, modulating expression of G1 phase cell cycle regulators and promoting apoptosis. We have identified a highly potent dimer (3) that was more effective than DOX in human LNCaP cells. In contrast to DOX which causes cardiotoxicity, there are no serious adverse side effects associated with clinically used artemisinin derivatives.47 Based on these results, further preclinical studies are warranted to develop TDs such as 3 for prostate cancer treatment.
Experimental Section
Semi-synthetic trioxane dimers
The trioxane dimer primary alcohol9 in figure 1 was phosphorylated to give diphenyl phosphate ester 1 (96%) and diethyl phosphate ester 2 (84%). Separately, this dimer primary alcohol was oxidized into the corresponding aldehyde (59%) that was reductively aminated to produce aniline 3 (62%). Each of these new trioxane dimers is stable in the absence of solvent at 60 °C for at least 24 h and is hydrolytically stable in a solution of pH 7.4 H2O/DMSO (1/4) at 25 °C for at least 12 h.
1
To a solution of the dimer primary alcohol9 (60 mg, 0.099 mmol) in CH2Cl2 (1 mL) at 0 °C was added pyridine (0.5 mL), 4-(dimethylamino)pyridine (1.2 mg, 9.8 μmol) and diphenyl phosphoryl chloride (61 μL, 0.29 mmol). The solution was warmed to rt and stirred for 16 h. The reaction was diluted with EtOAc (5 mL) and quenched by addition of aq. citric acid (1 N, 10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 3 mL). The combined organic solution was washed with aq. citric acid (0.1 N, 2 mL), dried (MgSO4), filtered and concentrated. The residue was purified by flash column chromatography (elution with EtOAc:hexanes = 1:3) to provide 1 (80 mg, 96%) as a colorless oil: [α]D24 +68 (c 0.26, CHCl3); IR (neat) 2919, 2861, 1589, 1485, 1455, 1376, 1290,1220, 1190, 1108, 1008, 944, 767, 686 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.33 (t, 4H, J = 7.7 Hz), 7.28-7.22 (m, 4H), 7.17 (t, 2H, J = 7.7 Hz), 5.30 (s, 1H), 5.29 (s, 1H), 4.58-4.51 (m, 1H), 4.50-4.37 (m, 2H), 4.25 (dd, 1H, J = 10.1, 5.9 Hz), 2.68 (q, 1H, J = 6.6 Hz), 2.52 (q, 1H, J = 6.6 Hz), 2.37-2.24 (m, 3H), 2.05-1.97 (m, 2H), 1.95-1.79 (m, 4H), 1.79-1.33 (m, 16H, including s at 1.40 and 1.37), 1.32-1.17 (m, 6H), 0.98-0.78 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 150.7 (d, 2C, J = 7.2 Hz), 129.7 (4C), 125.1 (2C), 120.2 (d, 2C, J = 3.9 Hz), 120.1 (d, 2C, J = 3.9 Hz), 103.2, 102.8, 89.5, 88.7, 81.14, 81.10, 73.8, 71.9 (d, J = 7.0 Hz), 70.8, 52.4, 52.1, 44.5, 44.1, 37.4, 37.3, 36.64, 36.58, 35.3 (d, J = 7.7 Hz), 34.5, 34.4, 30.5, 30.4, 30.2, 29.4, 26.11, 26.05, 24.9, 24.8, 24.7, 24.6, 20.2, 20.1, 13.2, 12.6; 31P NMR (162 MHz, CDCl3) δ −11.8; HRMS (ESI) calculated for C46H63O12PNa [(M + Na)+] 861.3955, found 861.3879.
2
To a solution of the dimer primary alcohol9 (103 mg, 0.170 mmol) in CH2Cl2 (1 mL) at 0 °C was added pyridine (42 μL, 0.52 mmol) and diethyl phosphoryl chloride (74 μL, 0.51 mmol). The solution was stirred for 4 h at rt, diluted with EtOAc (3 mL) and quenched with aq. citric acid (0.1 N, 5 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 3 mL). The combined organic solution was dried (MgSO4) and concentrated. The resulting oil was purified by flash column chromatography (elution with EtOAc:hexanes = 1:1) to yield 2 (106 mg, 84%) as an oily solid: [α]D24 +13 (c 0.30, CHCl3); IR (thin film) 2941, 2880, 1454, 1373, 1271, 1103, 1037, 1011, 878, 843, 665 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.31 (s, 1H), 5.28 (s, 1H), 4.38 (m, 1H), 4.23 (q, J = 5.2 Hz, 2H), 4.18-4.05 (m, 5H), 2.68 (sextet, J = 6.8 Hz, 1H), 2.56 (sextet, J = 6.8 Hz, 1H), 2.30 (m, 2H), 2.1 (m, 1H), 2.0 (dm, J = 14.4 Hz, 2H), 1.94-1.18 (m, 32H including s at 1.38 and 1.37, and dt at 1.32 with J = 0.8, 7.0 Hz), 0.95-0.85 (m, 14H including d at 0.94 with J = 6.0 Hz and dd at 0.85 with J = 4.2, 7.4 Hz); 13C NMR (100 MHz, CDCl3) δ 103.1, 102.8, 89.4, 88.7, 81.2, 81.1, 74.0, 71.1, 69.8, 63.6, 63.5, 52.4, 52.2, 44.5, 44.2, 37.4, 37.4, 36.7, 36.6, 35.5, 35.5, 34.5, 34.4, 30.6, 30.5, 30.1, 29.5, 26.1, 26.0, 24.8, 24.8, 24.7, 24.7, 20.2, 20.1, 16.2, 16.1, 13.2, 12.7; HRMS (FAB) calculated for C38H64O12P [(M + H)+] 743.4135, found 743.4144.
3
A 100 mL round bottom flask was charged with the dimer primary alcohol9 (230 mg, 0.38 mmol), CH2Cl2 (40 mL) and pyridinium dichromate (PDC) (260 mg, 0.68 mmol). The reaction was stirred at rt overnight, filtered through a pad of celite and the filtrate was concentrated under reduced pressure. The crude mixture was purified by flash column chromatography (elution with EtOAc) to give the dimer aldehyde (130 mg, 59%) as a sticky white solid: 1H NMR (400 MHz, CDCl3) δ 9.86 (d, 1H, J = 1.2 Hz), 5.26 (s, 1H), 5.23 (s, 1H), 4.38-4.26 (m, 2H), 2.82 (m, 1H), 2.67 (m, 1H), 2.56 (m, 1H), 2.36-2.24 (m, 2H), 2.18-2.06 (m, 2H), 2.04-1.96 (m, 2H), 1.92-1.84 (m, 4H), 1.82-1.74 (m, 2H), 1.70-1.58 (m, 4H), 1.46-1.20 (m, 14H including s at 1.39 and 1.38), 0.96-0.86 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 205.6, 103.8, 103.1, 89.7, 89.2, 81.5, 81.4, 73.4, 70.9, 52.5, 52.3, 47.8, 44.5, 44.2, 37.72, 37.65, 36.8, 34.70, 34.65, 30.7, 30.5, 30.0, 28.9, 26.3, 26.2, 25.13, 25.11, 25.03, 24.95, 20.43, 20.37, 13.3, 12.7; HRMS (ESI) calculated for C34H52O9Na [(M + Na)+] 627.3504, found 627.3509. A 25 mL round bottom flask was charged with the dimer aldehyde (93 mg, 0.15 mmol) and CH2Cl2 (10 mL) at rt. To this was added aniline (70 μL, 0.78 mmol) and sodium triacetoxy borohydride (69 mg, 0.32 mmol). The reaction was stirred at rt for 5 h until complete consumption of starting material was observed by TLC analysis. The reaction was quenched with saturated aq. NaHCO3. The aqueous layer was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried (MgSO4) and concentrated to give a pale yellow oil. The crude product was purified by silica gel flash column chromatography (10–20% EtOAc in PE) to yield 3 (64 mg, 62%) as a white fluffy solid: mp 84–87 °C; IR (thin film) 3584, 3378, 3051, 2925, 2874, 2359, 1716, 1602, 1508, 1453, 1376, 1322, 1253, 1222, 1186, 1103, 1053, 1008, 940, 909, 878, 846, 827 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.16-7.11 (m, 2H), 6.70-6.60 (m, 3H), 5.34 (s, 1H), 5.31 (s, 1H), 4.46-4.26 (m, 2H), 3.29-3.15 (m, 2H), 2.72-2.56 (m, 2H), 2.36-2.27 (m, 2H), 2.22-2.16 (m, 1H), 2.10-1.99 (m, 2H), 1.93-1.86 (m, 2H), 1.79-1.71 (m, 5H), 1.65-1.55 (m, 5H), 1.50-1.21 (m, 14 H including s at 1.41 and 1.38), 0.96-0.84 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 149.1, 129.1, 116.3, 112.7, 103.2, 103.0, 89.4, 88.9, 81.2, 81.2, 73.9, 71.5, 52.3, 52.2, 47.3, 44.4, 44.2, 37.4, 37.3, 36.6, 36.6, 34.8, 34.4, 31.5, 31.1, 30.7, 30.5, 26.1, 26.0, 24.8, 24.8, 24.7, 24.7, 20.2, 20.1, 13.1, 12.8; HRMS (ESI) calculated for C40H59NO8Na [(M + Na)+] 704.4133, found 704.4136.
Chemicals
Trioxane dimers (1, 2, and 3) were dissolved in pure ethanol and stored at −80 °C. Doxorubicin (DOX, a gift from Dr. Enrico Mihich’s laboratory) was dissolved in sterile water, protected from light exposure and stored at −20 °C.
Cell culture conditions
TRAMP cells (C1A and C2H) were cultured in DMEM media (Invitrogen, Frederick, MD) supplemented with 10% FBS (Hyclone, Logan, UT), 10−8 M dihydrotestosterone (Sigma-Aldrich, St. Louis, MO), 5 μg/mL insulin (Sigma) and 25 U/mL penicillin/streptomycin (Invitrogen) as previously described.9,19 LNCaP cells were cultured in RPMI media (Invitrogen) supplemented with 10% FBS, 1% L-glutamine (Invitrogen), 10 mM HEPES buffer (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 100 U/mL penicillin/streptomycin and 2.4 mg/mL glucose (Sigma). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Ethanol (≤ 0.002%) was used as vehicle control for all experiments
MTT assay
Cell viability was measured as previously described.9 Briefly, C1A (2500 cells/well), C2H (1500 cells/well) and LNCaP (10,000 cells/well) were seeded in 96-well plates overnight. Cells were treated with media containing vehicle or different concentrations of TDs or DOX for an additional 72 h. 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma) solution was added to each well for 4 h. Cells were solubilized with 100 μL 20% SDS/0.02 N HCl and absorbance measured at 570 nm using an ELISA plate reader (Molecular Devices, Sunnyvale, CA). The concentration of test compounds required to inhibit 50% of cell growth (GI50) was determined using CalcuSyn (Biosoft, Ferguson, MO).
Propidium iodide (PI) staining
Sub-confluent LNCaP cells were treated with vehicle or 50 nM of TDs and DOX for 72 h. Cells were harvested, washed with ice-cold PBS and fixed with ice-cold 70% ethanol for at least 30 min at 4 °C. About 106 cells were resuspended in PBS containing PI (10 μg/mL, Sigma) and RNase A (100 μg/mL, Sigma) for 30 min in the dark. Cells were sorted by Florescence Activated Cell Sorting (FACS) analysis at the RPCI Laboratory of Flow Cytometry.
Bromodeoxyuridine (BrdU) incorporation assay
DNA synthesis was measured in CIA and C2H cells using Cell Proliferation ELISA BrdU Colorimetric Kit (Roche, Indianapolis, IN) according to manufacturer’s instructions. BrdU is a brominated analog of thymidine that is incorporated into actively replicating DNA of cells in the S phase of the cell cycle.48 Briefly, sub-confluent cells were treated with vehicle or 50 nM of either TDs or DOX for 72 h. Cells were labeled with BrdU for 4 h and BrdU incorporation was assessed by measuring absorbances at 370 nm using an ELISA plate reader.
DNA synthesis was measured in LNCaP cells by flow cytometry. Sub-confluent cells were treated with vehicle or 50 nM of either TDs or DOX for 72 h. Cells were labeled for 30 min with 100 μg/mL BrdU (Sigma) and 1 μg/mL 5-fluoro-2′-deoxyuridine (FdU, Sigma). FdU enhances BrdU incorporation by inhibiting thymidylate synthetase.49 Cells were harvested and fixed for flow cytometry as described previously. Approximately 106 cells were denatured with 2 N HCl, neutralized with 0.1 M sodium borate (pH 8.5), and incubated with mouse anti-BrdU antibody (BD PharMingen, San Diego, CA) in 0.5% BSA/PBS overnight at 4 °C. Cells were labeled with Alexa Fluor® 488 goat anti-mouse secondary antibody (Molecular Probes, Eugene, OR) for 1 h at 4 °C. Samples were stained with PI and sorted by FACS analysis as previously described.
Annexin V staining
Apoptotic cells were detected using the Annexin V-PE Apoptosis Detection Kit (BD PharMingen) according to manufacturer’s instructions. Annexin V detects cells in the earliest stages of apoptosis27 and 7-AAD is a vital dye which is excluded by viable cells. Briefly, sub-confluent LNCaP cells were treated with vehicle or 50 nM of TDs or DOX for 72 h. Cells were trypsinized, washed twice with ice-cold PBS and stained with annexin V-PE and 7-AAD for 15 min in the dark. Apoptotic cells were detected by FACS analysis as previously described.
Western blot analysis
Sub-confluent LNCaP cells were treated with vehicle or 50 nM of TDs or DOX for 72 h. Cells were lysed with Triton X-100/SDS lysis buffer (10% Triton X-100, 10% SDS, 1.0 M Tris-Cl, pH 8.0 and 5.0 M NaCl) containing 1X Protease Inhibitor Cocktail (BD PharMingen), 2 mM sodium orthovanadate, 2 mM EDTA, 12 mM beta-glycerol phosphate and 10 mM sodium fluoride. Proteins were quantified using the Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA) according to manufacturer’s instructions. Proteins were resolved by SDS-PAGE and transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford, MA) overnight at 4 °C by electrophoresis. Membranes were blocked at room temperature for a minimum of 1 h using 5% (w/v) blotto (Santa Cruz Biotechnology, Santa Cruz, CA) in TBST (25 mM Tris-HCl, pH 7.4, 137 mM NaCl, 2.7 mM KCl and 0.05% Tween 20). Western blots were incubated overnight at 4 °C with these primary antibodies: cdk4 (1:1000, sc-260, Santa Cruz); cdk6 (1:1000, sc-177, Santa Cruz); cyclin D1 (1:1000, sc-753, Santa Cruz); cyclin E (1:1000, sc-247, Santa Cruz), p21 (1:1000, sc-397, Santa Cruz); p27 (1:1000, sc-528, Santa Cruz), p53 (1:1000, sc-126, Santa Cruz), bax (1:1000, sc-493, Santa Cruz), Bcl-2 (1:200, sc-492, Santa Cruz) and actin (1:6000, CP01, Calbiochem, San Diego, CA). Membranes were washed at least 3 times with TBST followed by incubation with HRP-conjugated secondary antibodies for a minimum of 1 h. Three additional washes were performed, and proteins were detected using Western Lightning Chemiluminescence Reagent (Perkin Elmer Life Sciences, Boston, MA). Protein expression was quantified using the GS-800 calibrated densitometer and Quantity One software (Bio-Rad).
Kinase assay
LNCaP cells were treated with vehicle, TDs or DOX as described above. Cells were lysed with NP40 lysis buffer (10 mM HEPES, pH 7.9, 1 mM EDTA, 150 mM NaCl, 1% NP40) containing 1X Protease Inhibitor Cocktail and phosphatase inhibitors (2 mM sodium orthovanadate, 2 mM EGTA, 10 mM sodium fluoride). Protein concentration was determined as described above. Lysates (100 μg) were immunoprecipitated with 2 μg cdk2 antibody (sc-163-G, Santa Cruz) and protein A/G agarose (Santa Cruz) overnight at 4 °C with rocking. Immunoprecipitated proteins were washed twice with NP40 lysis buffer and once with kinase buffer (20 mM HEPES, pH 7.8, 10 mM MgCl2, 20 mM beta-glycerol phosphate, 1 mM DTT, 50 μM active sodium orthovanadate). Kinase activity was assayed by resuspending beads in kinase buffer containing 2 μg histone H1 (cdk2 substrate), 50 μM active sodium orthovanadate, 20 μM ATP and 5 μCi (γ-32P) ATP, for 30 min at 30 °C. Kinase reactions were terminated with equal volume 2X Laemmli buffer (Bio-Rad) and boiled for 5 min. Samples were resolved on a SDS-PAGE gel and transferred to a PVDF membrane as described above. (γ-32P)-Histone H1 was detected by autoradigraphy. Cdk2 protein expression was confirmed by immunoblot analysis with anti-cdk2 antibody (1:500).
Supplementary Material
Acknowledgments
This work was supported by NIH/NCI Grant 5R01CA95367-4 (to B.A.F), DOD Pre-doctoral Fellowship Grant 57-8046-01 (to A.A.A.), and NIH AI Grant 34885 (to G.H.P.). We thank Drs. Jennifer Black and Adrian Black of the Department of Pharmacology and Therapeutics at Roswell Park Cancer Institute for providing antibodies for cell cycle studies.
Abbreviations
- TDs
Trioxane Dimers
- GI50
Concentration That Inhibits 50% Cell Growth
- TRAMP
Transgenic Adenocarcinoma of Mouse Prostate
- DOX
Doxorubicin
- PI
Propidium Iodide
Footnotes
Supporting Information Available: HPLC traces and NMR spectra for trioxane dimers (1, 2 and 3).
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society (ACS) website. http://www.cancer.org.
- 3.Sonpavde G, Hutson TE, Berry WR. Hormone refractory prostate cancer: Management and advances. Cancer Treatment Reviews. 2006;32:90–100. doi: 10.1016/j.ctrv.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Posner GH, Ploypradith P, Hapangama W, Wang D, Cumming JN, Dolan P, Kensler TW, Klinedinst D, Shapiro TA, Zheng QY, Murray CK, Pilkington LG, Jayasinghe LR, Bray JF, Daughenbaugh R. Trioxane dimers have potent antimalarial, antiproliferative and antitumor activities in vitro. Bioorg Med Chem. 1997;5:1257–1265. doi: 10.1016/s0968-0896(97)00079-5. [DOI] [PubMed] [Google Scholar]
- 5.Posner GH, Ploypradith P, Parker MH, O’Dowd H, Woo SH, Northrop J, Krasavin M, Dolan P, Kensler TW, Xie S, Shapiro TA. Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J Med Chem. 1999;42:4275–4280. doi: 10.1021/jm990363d. [DOI] [PubMed] [Google Scholar]
- 6.Posner GH, Northrop J, Paik IH, Borstnik K, Dolan P, Kensler TW, Xie S, Shapiro TA. New chemical and biological aspects of artemisinin-derived trioxane dimers. Bioorg Med Chem. 2002;10:227–232. doi: 10.1016/s0968-0896(01)00270-x. [DOI] [PubMed] [Google Scholar]
- 7.Posner GH, Paik IH, Sur S, McRiner AJ, Borstnik K, Xie S, Shapiro TA. Orally active, antimalarial, anticancer, artemisinin-derived trioxane dimers with high stability and efficacy. J Med Chem. 2003;46:1060–1065. doi: 10.1021/jm020461q. [DOI] [PubMed] [Google Scholar]
- 8.Paik IH, Xie S, Shapiro TA, Labonte T, Narducci Sarjeant AA, Baege AC, Posner GH. Second Generation, Orally Active, Antimalarial, Artemisinin-Derived Trioxane Dimers with High Stability, Efficacy, and Anticancer Activity. J Med Chem. 2006;49:2731–2734. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 9.Posner GH, McRiner AJ, Paik IH, Sur S, Borstnik K, Xie S, Shapiro TA, Alagbala A, Foster BA. Anticancer and antimalarial efficacy and safety of artemisinin-derived trioxane dimers in rodents. J Med Chem. 2004;47:1299–1301. doi: 10.1021/jm0303711. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Shan F, Wu JM, Wu GS, Ding J, Xiao D, Yang WY, Atassi G, Leonce S, Caignard DH, Renard P. Novel antitumor artemisinin derivatives targeting G1 phase of the cell cycle. Bioorg Med Chem Lett. 2001;11:5–8. doi: 10.1016/s0960-894x(00)00578-3. [DOI] [PubMed] [Google Scholar]
- 11.Efferth T, Olbrich A, Bauer R. mRNA expression profiles for the response of human tumor cell lines to the antimalarial drugs artesunate, arteether, and artemether. Biochem Pharmacol. 2002;64:617–623. doi: 10.1016/s0006-2952(02)01221-2. [DOI] [PubMed] [Google Scholar]
- 12.Efferth T, Sauerbrey A, Olbrich A, Gebhart E, Rauch P, Weber HO, Hengstler JG, Halatsch ME, Volm M, Tew KD, Ross DD, Funk JO. Molecular modes of action of artesunate in tumor cell lines. Mol Pharmacol. 2003;64:382–394. doi: 10.1124/mol.64.2.382. [DOI] [PubMed] [Google Scholar]
- 13.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 14.Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, Russell R, Pestell RG. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–7611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Puebla ML, Miliani de Marval PL, LaCava M, Moons DS, Kiyokawa H, Conti CJ. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol. 2002;161:405–411. doi: 10.1016/S0002-9440(10)64196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miliani de Marval PL, Macias E, Rounbehler R, Sicinski P, Kiyokawa H, Johnson DG, Conti CJ, Rodriguez-Puebla ML. Lack of cyclin-dependent kinase 4 inhibits c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol. 2004;24:7538–7547. doi: 10.1128/MCB.24.17.7538-7547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 19.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- 20.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Culine S, Kattan J, Zanetta S, Theodore C, Fizazi K, Droz JP. Evaluation of estramustine phosphate combined with weekly doxorubicin in patients with androgen-independent prostate cancer. Am J Clin Oncol. 1998;21:470–474. doi: 10.1097/00000421-199810000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Harris KA, Harney E, Small EJ. Liposomal doxorubicin for the treatment of hormone-refractory prostate cancer. Clin Prostate Cancer. 2002;1:37–41. doi: 10.3816/cgc.2002.n.005. [DOI] [PubMed] [Google Scholar]
- 23.Posner GH, Oh CH. A regiospecifically O-18 labeled 1,2,4-trioxane-a simple chemical model system to probe the mechanism(s) for the antimalarial activity of artemisinin (qinghaosu) J Am Chem Soc. 1992;114:8328–8329. [Google Scholar]
- 24.Martinez LA, Yang J, Vazquez ES, del Rodriguez-Vargas MC, Olive M, Hsieh JT, Logothetis CJ, Navone NM. p21 modulates threshold of apoptosis induced by DNA-damage and growth factor withdrawal in prostate cancer cells. Carcinogenesis. 2002;23:1289–1296. doi: 10.1093/carcin/23.8.1289. [DOI] [PubMed] [Google Scholar]
- 25.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 26.Gartel AL, Tyner AL. Transcriptional regulation of the p21 ((WAF1/CIP1)) gene. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 27.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 28.Jeyadevan JP, Bray PG, Chadwick J, Mercer AE, Byrne A, Ward SA, Park BK, Williams DP, Cosstick R, Davies J, Higson AP, Irving E, Posner GH, O’Neill PM. Antimalarial and antitumor evaluation of novel C-10 non-acetal dimers of 10beta-(2-hydroxyethyl) deoxoartemisinin. J Med Chem. 2004;47:1290–1298. doi: 10.1021/jm030974c. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 30.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 31.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 32.Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- 33.Williams ME, Swerdlow SH, Meeker TC. Chromosome t(11; 14)(q13; q32) breakpoints in centrocytic lymphoma are highly localized at the bcl-1 major translocation cluster. Leukemia. 1993;7:1437–1440. [PubMed] [Google Scholar]
- 34.Wang L, Habuchi T, Mitsumori K, Li Z, Kamoto T, Kinoshita H, Tsuchiya N, Sato K, Ohyama C, Nakamura A, Ogawa O, Kato T. Increased risk of prostate cancer associated with AA genotype of cyclin D1 gene A870G polymorphism. Int J Cancer. 2003;103:116–120. doi: 10.1002/ijc.10793. [DOI] [PubMed] [Google Scholar]
- 35.Aaltomaa S, Lipponen P, Eskelinen M, Ala-Opas M, Kosma VM. Prognostic value and expression of p21 (waf1/cip1) protein in prostate cancer. Prostate. 1999;39:8–15. doi: 10.1002/(sici)1097-0045(19990401)39:1<8::aid-pros2>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 36.Kuczyk MA, Bokemeyer C, Hartmann J, Schubach J, Walter C, Machtens S, Knuchel R, Kollmannsberger C, Jonas U, Serth J. Predictive value of altered p27Kip1 and p21WAF/Cip1 protein expression for the clinical prognosis of patients with localized prostate cancer. Oncol Rep. 2001;8:1401–1407. doi: 10.3892/or.8.6.1401. [DOI] [PubMed] [Google Scholar]
- 37.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 38.Macleod K, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 39.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Wu LM, Zhao Y, Zhang XL, Wang NP. The anticancer effect of artesunate and its mechanism. Yao Xue Xue Bao. 2002;37:477–478. [PubMed] [Google Scholar]
- 41.Kim SH, Kim JH. Lethal effect of adriamycin on the division cycle of HeLa cells. Cancer Res. 1972;32:323–325. [PubMed] [Google Scholar]
- 42.Ling YH, Priebe W, Perez-Soler R. Apoptosis induced by anthracycline antibiotics in P388 parent and multidrug-resistant cells. Cancer Res. 1993;53:1845–1852. [PubMed] [Google Scholar]
- 43.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 44.Gordi T, Lepist EI. Artemisinin derivatives: toxic for laboratory animals, safe for humans? Toxicology Letters. 2004;147:99–107. doi: 10.1016/j.toxlet.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Moore JC, Lai H, Li JR, Ren RL, McDougall JA, Singh NP, Chou CK. Oral administration of dihydroartemisinin and ferrous sulfate retarded implanted fibrosarcoma growth in the rat. Cancer Lett. 1995;98:83–87. [PubMed] [Google Scholar]
- 46.Tyagi AK, Singh RP, Agarwal C, Chan DC, Agarwal R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth Inhibition, G2-M arrest, and apoptosis. Clin Cancer Res. 2002;8:3512–3519. [PubMed] [Google Scholar]
- 47.Ribeiro IR, Olliaro P. Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Medecine Tropicale: Revue Du Corps De Sante Colonial. 1998;58:50–53. [PubMed] [Google Scholar]
- 48.Dolbeare F, Gratzner H, Pallavicini MG, Gray JW. Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad Sci U S A. 1983;80:5573–5577. doi: 10.1073/pnas.80.18.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for biochemical research. 3. Oxford University Press; New York: 1986. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.