

Abstract
Type II Bartter syndrome is caused by mutations in the renal outer medullary potassium (ROMK) channel, but the molecular mechanisms underlying this disease are poorly defined. To rapidly screen for ROMK function, we developed a yeast expression system and discovered that yeast cells lacking endogenous potassium channels could be rescued by WT ROMK but not by ROMK proteins containing any one of four Bartter mutations. We also found that the mutant proteins were significantly less stable than WT ROMK. However, their degradation was slowed in the presence of a proteasome inhibitor or when yeast cells contained mutations in the CDC48 or SSA1 gene, which is required for endoplasmic reticulum (ER)-associated degradation (ERAD). Consistent with these data, sucrose gradient centrifugation and indirect immunofluorescence microscopy indicated that most ROMK protein was ER-localized. To translate these findings to a more relevant cell type, we measured the stabilities of WT ROMK and the ROMK Bartter mutants in HEK293 cells. As in yeast, the Bartter mutant proteins were less stable than the WT protein, and their degradation was slowed in the presence of a proteasome inhibitor. Finally, we discovered that low-temperature incubation increased the steady-state levels of a Bartter mutant, suggesting that the disease-causing mutation traps the protein in a folding-deficient conformation. These findings indicate that the underlying pathology for at least a subset of patients with type II Bartter syndrome is linked to the ERAD pathway and that future therapeutic strategies should focus on correcting deficiencies in ROMK folding.
Keywords: 70-kilodalton heat shock protein (Hsp70), endoplasmic reticulum–associated protein degradation (ERAD), ER quality control, proteasome, Saccharomyces cerevisiae, yeast, Cdc48
Introduction
Approximately one-third of all proteins in eukaryotes are targeted to the secretory pathway. Among these substrates, secreted and membrane proteins play a pivotal role in intercellular communication and the maintenance of cellular homeostasis. Protein folding in the secretory pathway must therefore be meticulously controlled. However, many proteins fold slowly or inefficiently. This can lead to the accumulation of misfolded proteins and subsequent delivery to cellular quality control machineries that target aberrant polypeptides for degradation or for refolding with the assistance of molecular chaperones (1, 2). When a misfolded protein in the secretory pathway is targeted for degradation, it can follow one of several routes: 1) chaperone-mediated selection in the endoplasmic reticulum (ER),2 retrotranslocation to the cytoplasm, and degradation by the cytoplasmic proteasome; 2) aggregation and delivery to the lysosome for destruction via autophagy; 3) entrapment within early compartments in the secretory pathway; or 4) trafficking to the Golgi apparatus or plasma membrane, where post-ER quality control pathways select the protein for degradation in the lysosome. In the first of these routes, proteasome-dependent degradation proceeds via a process known as ER-associated degradation (ERAD). Over 70 human diseases have been linked to ERAD, either through overzealous degradation of modestly misfolded proteins or failure to recognize toxic proteins (3).
ERAD is composed of four main steps: substrate recognition mediated by an extensive network of molecular chaperones and chaperone-like lectins (4, 5); ubiquitination at the ER membrane via cytoplasmic or ER-localized E3 ligases (6, 7); retrotranslocation from the ER to the cytosol via the AAA+ ATPase p97/Cdc48 complex, which provides the mechanical force for substrate removal (8–11); and proteasome-dependent degradation, ultimately resulting in protein cleavage into peptide fragments (12). Prolonged cellular stress in which ERAD fails to keep pace with misfolded protein production triggers the unfolded protein response, which has also been linked to numerous disease states (13–18). Conversely, as noted above, excessive degradation can give rise to other diseases. Many of these diseases are associated with defects in ion channel maturation, such as cystic fibrosis (the cystic fibrosis transmembrane conductance regulator (CFTR) (19, 20)), Liddle's syndrome (the epithelial sodium channel (ENaC) (21–23)), and Anderson-Tawil syndrome (the inward-rectifying potassium channel, Kir2.1 (24, 25)). In some of these cases, the disease phenotype can arise from the ERAD pathway prematurely destroying an immature form of the ion channel.
A key player in the regulation of salt and potassium homeostasis in the kidney is ROMK (KCNJ1, Kir1.1), which is a member of the inwardly rectifying family of potassium channels (Kir) (26). ROMK is expressed at the apical surface of epithelial cells in the distal nephron, including the thick ascending limb of the loop of Henle and the cortical collecting duct (27, 28). In the thick ascending limb, ROMK participates in potassium recycling across the apical membrane, a process vital to support the function of the NKCC2 co-transporter that represents the rate-limiting step for salt reabsorption in this nephron segment (29). In the cortical collecting duct, ROMK contributes to a major potassium excretory pathway in concert with the high-conductance, calcium-activated BK potassium channels (30). The known factors influencing the activity of ROMK channels in these locations within the nephron include PKA-dependent phosphorylation (31, 32), phosphatidylinositol 4,5-bisphosphate interaction (33, 34), ATP binding (35), and intracellular pH (36, 37). Likewise, regulation of the surface density of ROMK via endocytosis, which involves the Src (38, 39) and WNK kinases (40), has also been characterized. Molecular defects in ROMK channel trafficking that give rise to type II Bartter syndrome have also been described (41, 42); however, the mechanisms that cause impaired ROMK surface expression are poorly defined (43), and it is unknown whether any of the disease-causing mutations enhance the selection of ROMK for ERAD.
ROMK exists as three isoforms (ROMK1–3), each with different N-terminal amino acid sequences that are generated via alternative splicing and promoter usage (44–46). All three isoforms exhibit the same biophysical properties, but they are differentially expressed along the nephron. Mutations in any of the isoforms lead to a constellation of deleterious effects associated with type II Bartter syndrome, and to date ∼60 disease-causing mutations in the gene encoding ROMK (KCNJ1) have been identified (47, 48) (Human Gene Mutation Database; OMIM number 600359). Type II Bartter syndrome is evident in the antenatal period and includes polyhydramnios, prematurity, polyuria, nephrocalcinosis, osteopenia, and transient hyperkalemic metabolic acidosis followed by lifelong hypokalemic alkalosis (49). Bartter mutants caused by N-terminal nonsense or frameshift mutations have clear deleterious consequences on protein expression. The other mutations can generally be grouped into two broad categories: those that disrupt the biophysical properties of the channel, such as gating, ion selectivity, or ligand binding, and those that disrupt trafficking and reduce ROMK channel surface density at the plasma membrane (50, 51). Among the ∼60 known type II Bartter mutations, four mutations known to affect protein trafficking, A198T, R212P, H270Y, and Y314C, are clustered in the β-sheet–rich immunoglobulin-like domain in the cytosolic C-terminal region of ROMK (Fig. 1). Previous studies demonstrated that some of these mutations impede post-ER trafficking in Xenopus oocytes or HEK 293 cells (51, 52). Because these Bartter mutants are retained in the ER and may exhibit folding defects, the goal of the current study was to test the hypothesis that the mutations destabilize ROMK so that it is instead targeted for ERAD.
Figure 1.
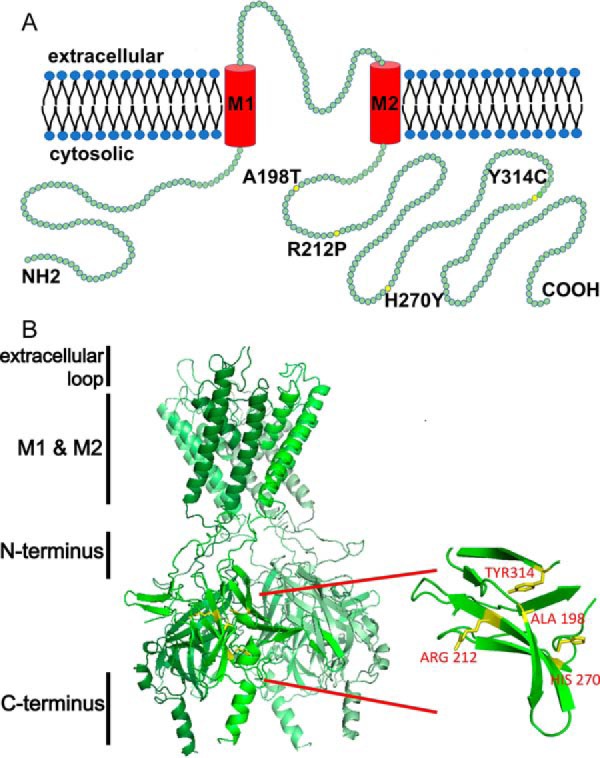
ROMK structure highlighting select mutations associated with type II Bartter syndrome. A, a linear structural model of ROMK. Each green circle represents a single amino acid. Yellow circles indicate mutations described in this study. ROMK shares a common structure with other inwardly rectifying Kir channels: two transmembrane domains (M1 and M2), a conserved potassium selectivity filter, and cytoplasmic N and C-terminal domains. Four subunits tetramerize to form the functional channel. The molecular mass of ROMK is ∼44 kDa. The cytoplasmic N and C termini represent amino acids 1–82 and 181–391, respectively. M1 represents amino acids 83–105, the extracellular loop is amino acids 106–155, and M2 represents amino acids 156–180 (70). B, four type II Bartter syndrome mutations, A198T, R212P, H270Y, and Y314C, reside in an immunoglobulin-like domain, which is assembled from β-sheets packed face-to-face, creating a core populated by highly conserved side chains. The homology model was built and images were rendered using PyMOL software. The amino acid sequence of this protein can be accessed through UniProtKB (UniProt accession number P48048-1).
To this end, we developed a new ROMK1 yeast expression system that allows for a facile and quantitative read-out for ROMK1 function and plasma membrane residence. We discovered that yeast cells expressing the Bartter mutants grow poorly on low-K+ media, suggesting that the mutant proteins are unstable and/or fail to traffic to the plasma membrane. As hypothesized, degradation was proteasome-dependent, relied on the AAA+ ATPase Cdc48, and was mediated by the Hsp70 molecular chaperone, which plays a role in the selection of other unstable ion channels (53–56). Sucrose gradient centrifugation and indirect immunofluorescence microscopy indicated that the majority of ROMK1 was ER-localized, consistent with the mutant channels being subjected to ERAD. We then validated these findings in human cells and showed that the Bartter syndrome-associated ROMK1 alleles are similarly targeted for ERAD. This study demonstrates for the first time that Bartter syndrome is one of a growing number of diseases linked to the ERAD pathway and that the yeast ROMK1 expression system can be co-opted to elucidate the molecular defects associated with other ROMK1 mutants.
Results
Construction and characterization of a screenable ROMK variant
To determine the molecular basis of Bartter disease-causing mutations in ROMK1, we initially took advantage of a well-established phenotypic assay in yeast. Budding yeast have a robust potassium uptake system, enabling them to grow on micromolar concentrations of the ion. The high-affinity potassium transporters Trk1 and Trk2 are central to this system, but yeast lacking these genes fail to propagate on relatively low (∼3–25 mm) potassium. However, heterologous expression of potassium channels from other organisms rescues the potassium-sensitive phenotype of trk1Δtrk2Δ yeast (57), and numerous studies have exploited this phenomenon to probe genetic interactions, identify small molecule modulators, and establish structure-function characteristics of diverse potassium channels (25, 58–63). Therefore, we expressed rat ROMK1 in trk1Δtrk2Δ yeast to assess whether growth on low potassium could be restored.
Initial attempts to express ROMK1 in trk1Δtrk2Δ yeast failed to reveal growth rescue on low potassium media (see below and Fig. 2). This contrasts with the strong rescue phenotype observed in yeast expressing the inwardly rectifying channel Kir2.1 (25), which shares ∼40% sequence identity with ROMK1 (64). To remedy this problem, we made two targeted mutations in the ROMK1-encoding gene. First, ROMK1 contains an intracellular acid-sensitive gate, which keeps the channel closed under conditions of cellular acidification (65) but is absent in Kir2.1. We hypothesized that this pH gate closes ROMK because the yeast cytosol is acidic (pH 5.5–6.8) (66). Therefore, Lys-80, which is crucial for pH gating (67), was mutated to Met (K80M). Second, ROMK1 primarily resides in the ER in mammalian cells due to the presence of multiple RXR motifs at both the N- and C-terminal domains. Aldosterone-regulated phosphorylation at Ser-44 by SGK-1 overrides ER retention, promoting anterograde traffic (68, 69). To favor ER exit in yeast, Ser-44 was mutated to Asp (S44D). Next, vectors engineered for the constitutive expression of ROMK1, each of the single mutants as well as the double mutant, were constructed and introduced into trk1Δtrk2Δ yeast as well as into a strain that our group has previously identified as deficient in a negative regulator of Kir2.1, trk1Δtrk2Δvps23Δ (25). As shown in Fig. 2A, ROMKS44D/K80M rescued the growth of cells on low potassium almost as well as Kir2.1. Although the ROMKS44D species displayed only minimal growth rescue, this mutation augmented growth when combined with the K80M mutation. Importantly, the expression of any of these species did not retard the growth of trk1Δtrk2Δ yeast (data not shown). Therefore, we chose to use the double mutant (ROMKS44D/K80M) for subsequent studies, and for simplicity we will refer to this protein as “ROMK” in the following experiments that utilize the yeast model.
Figure 2.

ROMKS44D/K80M improves the growth of yeast lacking the endogenous Trk1 and Trk2 potassium transporters on low-potassium medium. A, yeast containing an empty expression vector or plasmids engineered for the expression of Kir2.1, wild-type ROMK, or the S44D and K80M mutations alone or in combination were spotted onto SC medium containing the indicated added concentration of KCl. Note that “0 mm KCl” contains residual potassium from the growth medium. As observed with the Kir2.1 potassium channel, deletion of the gene encoding the ESCRT subunit Vps23 greatly increases the ability of ROMKK80M or ROMKS44D/K80M to rescue the growth of trk1Δtrk2Δ yeast on low-potassium medium. B, a Western blot analysis for HA-tagged ROMK1 and Kir2.1 and G6PD (as a loading control) is shown from yeast patches shown in A. C, the ROMKS44D/K80M protein is more stable and less sensitive to proteasome degradation compared with wild-type ROMK. Yeast expressing wild-type ROMK or ROMKS44D/K80M were treated with either 100 μm MG132 (closed squares) or with an equal volume of DMSO (open squares) for 30 min before the addition of cycloheximide. Cycloheximide was added at the zero time point, and aliquots were withdrawn at 0, 30, 60, and 90 min. Protein levels were assessed by Western blot analysis and normalized to the initial time point. Representative blots are shown, and G6PD is used as a loading control. Data represent the means of three independent determinations for all time points ± S.E. D, ROMKS44D/K80M (open circles) is less susceptible to proteolytic cleavage in vitro than wild-type ROMK1 (closed circles). ER-enriched microsomes prepared from wild-type yeast expressing wild-type ROMK or ROMKS44D/K80M were treated with 10 μg/ml Proteinase K for the indicated times. Top, note the more rapid disappearance of the full-length protein and the major proteolytic products when the wild type protein was analyzed. Bottom, protein levels were assessed by Western blot analysis, and the levels of the protein are normalized to the initial time point. Representative blots are shown, and protein-disulfide isomerase was used as a loading control because it is entirely encapsulated within the ER lumen. Data represent the means of four independent experiments ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Curiously, the steady-state expression levels of ROMKK80M and ROMKS44D/K80M were higher than that of ROMK1 (Fig. 2B). Cycloheximide chase analysis revealed that ROMKS44D/K80Mwas also more resistant to proteasomal degradation than ROMK1, because the degradation profile was affected less when ROMKS44D/K80M-expressing cells versus ROMK1-expressing cells were treated with a proteasome inhibitor, MG132 (Fig. 2C). Based on its effects on channel gating, we reasoned that the K80M mutation might stabilize the channel because it would be “locked” into an active conformation. To test this hypothesis, ER-derived microsomes were prepared from yeast expressing wild type ROMK1 and ROMKS44D/K80M. As proposed, we discovered that ROMKS44D/K80M was more resistant to in vitro degradation by Proteinase K as compared to the wild type channel (Fig. 2D).
ROMK is a substrate for ER-associated degradation in yeast
As noted above, the growth phenotype of trk1Δtrk2Δ yeast can be rescued by expressing an exogenous potassium channel that acts in place of the Trk1 and Trk2 transporters. Cell growth is dependent on the potassium channel acting at the plasma membrane, which therefore provides a readout for both channel residence and function. To determine whether the select group of four Bartter mutants are defective for rescue on low potassium, we grew trk1Δtrk2Δ yeast transformed with vectors engineered for the expression of ROMK, Kir2.1 (as a positive control (25)), and the four Bartter mutants with substitutions that reside in the immunoglobulin-like domain (i.e. A198T, R212P, H270Y, and Y314C; Fig. 1). We then performed serial dilution assays on media supplemented with a range of potassium (Fig. 3A). Although the Bartter mutants exhibited growth defects under several of these conditions when compared with cells expressing ROMK, the defect was most clearly evident on the 25 mm potassium plates (Fig. 3A). The growth defects of the Bartter mutants were also pronounced in liquid media (Fig. 3B). Interestingly, immunoblot analysis indicated that the expression of the A198T, R212P, H270Y, and Y314C mutant proteins was reduced by ∼5-fold relative to ROMK (Fig. 3, C and D), suggesting that these proteins are subject to a protein quality control pathway.
Figure 3.

Yeast expressing type II Bartter syndrome mutants exhibit growth defects on low potassium medium. A, yeast lacking the Trk1 and Trk2 potassium channel proteins were transformed with vectors engineered for the expression of ROMK1, Kir2.1 (as a positive control), or ROMK containing the indicated Bartter mutant. Cells were serially diluted and plated onto medium supplemented with 100, 25, 10, or 0 mm potassium. Note that all four mutants were made in the ROMK DM background (see Fig. 2). B, yeast lacking the Trk1 and Trk2 potassium channel proteins were transformed with ROMK or the indicated Bartter mutant and were grown in liquid cultures for ∼12 h and diluted back to an A600 of 0.2. The A600 was measured over the indicated time period in medium containing 100 mm (left) or 25 mm (right) potassium, as indicated. C, Western blot analyses were performed on the indicated transformed cells, as in A. Note that Kir2.1, ROMK, and the Bartter mutant proteins all contain an HA epitope. G6PD serves as a loading control. D, scatter plot depicts the relative protein expression of each Bartter mutant compared with ROMK. Averages and S.E. are shown in red. Data represent the means of four independent colonies ± S.E. (error bars).
The ROMK channel probably encounters multiple layers of protein quality control as it travels through the secretory pathway en route to the plasma membrane. To gain a more comprehensive understanding of the ROMK trafficking and degradation pathways, we next examined ROMK localization by indirect immunofluorescence microscopy. Our results indicated that ROMK primarily resides in a perinuclear compartment that co-localized with an ER-resident chaperone, Kar2 (BiP; Fig. 4A). Sucrose density gradient centrifugation analysis indicated that most ROMK also co-migrated with the ER-resident protein, Sec61 (Fig. 4B, lanes 6–15). However, a fraction of ROMK co-migrated with the plasma membrane protein, Pma1, and this pattern was most evident when the immunoblots were overexposed (Fig. 4B, lanes 16–22). These data suggest that the majority of ROMK is ER-localized, with a small pool advancing to the plasma membrane. Similar results were evident when the trafficking of Kir2.1 was analyzed in yeast (25), and indeed the plasma membrane fraction of both channels appeared to be sufficient to support growth on low potassium because the open probabilities of Kir2.1 and ROMK are high (70) and/or because of the high membrane potential across the yeast plasma membrane. In contrast to results with ROMK, plasma membrane residence of the A198T Bartter mutant was somewhat reduced, especially when longer exposures of ROMK and A198T ROMK were compared (Fig. 4B, ROMK, lanes 19–22). These data are in accordance with the proposal that this Bartter mutant is less stable or traffics poorly (51). These data also indicate that the yeast expression system provides a rapid and facile model to explore the protein quality control pathways encountered by ROMK as it transits the secretory pathway.
Figure 4.
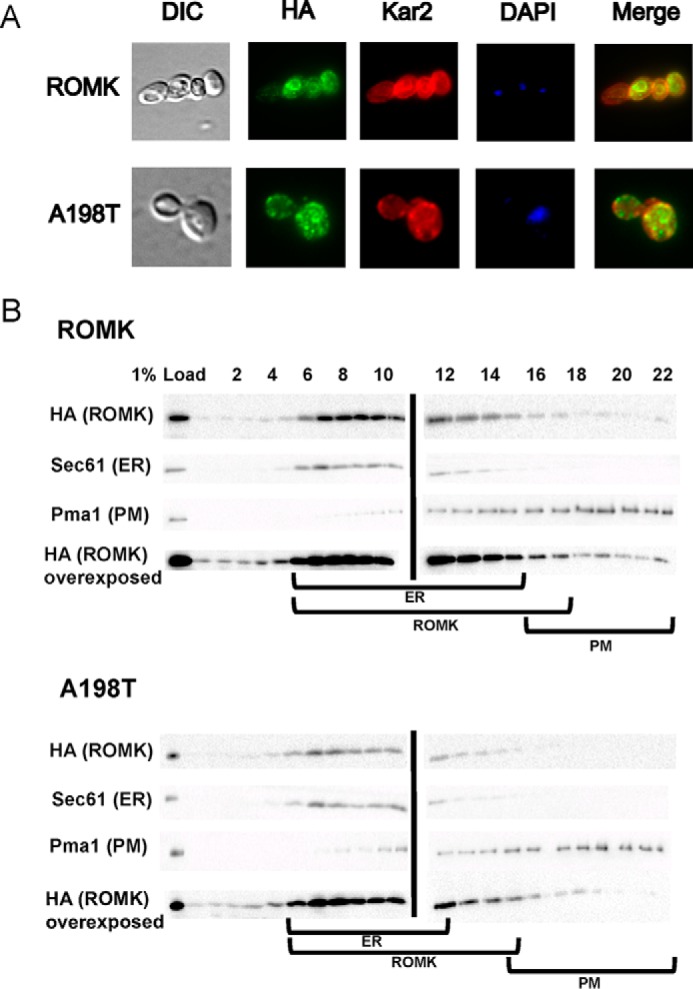
ROMK resides primarily in the yeast ER. A, indirect immunofluorescence microscopy of yeast expressing ROMK. Fixed cells were probed with antibodies against the ER chaperone Kar2 and the HA tag to visualize ROMK or the A198T mutant. DAPI corresponds to the nucleus. Right, merge showing co-localization. B, lysates from cells expressing ROMK or the A198T mutant were analyzed by centrifugation in a 30–70% sucrose gradient under conditions to maximize ER and plasma membrane (PM) isolation. The gradient was fractionated from the top (fraction 1) to the bottom of the tubes (fraction 22). The migrations of ROMK (HA), the ER chaperone Kar2, and the plasma membrane protein Pma1 were evaluated by Western blot analysis. When overexposed, the A198T mutant is absent from the Pma1 peak (i.e. the plasma membrane), in contrast to ROMK. Brackets represent ER and plasma membrane fractions containing ROMK.
Because several ion channels fold or assemble inefficiently in the ER (3, 71), we next asked whether ROMK is targeted for ERAD and whether the Bartter mutants might be more susceptible to degradation. To determine whether ROMK degradation is proteasome-dependent, which is a hallmark of ERAD, we first introduced the ROMK expression vectors into a pdr5Δ strain, which lacks a multidrug pump so that the uptake of the proteasome-specific inhibitor MG132 is facilitated (72). As shown in Fig. 5, ROMK was stabilized after cycloheximide addition in cells treated with MG132 when compared with cells treated with the vehicle, DMSO. In contrast, the stabilities of the A198T, R212P, H270Y, and Y314C mutants were significantly lower compared with ROMK (compare signals corresponding to DMSO treatment in Fig. 5). Moreover, proteasome inhibition led to a more pronounced relative stabilization of the Bartter mutants. Combined with the data provided above, these results indicate that the ROMK Bartter mutants are more strongly targeted to the ERAD pathway than ROMK lacking these mutations.
Figure 5.

Bartter mutant degradation is proteasome-dependent in yeast. A–E, cycloheximide chase reactions were performed, and lysates were blotted with anti-HA antibody to measure the stability of ROMK and the indicated mutant proteins over 90 min. In all cases, a pdr5Δ yeast strain was pretreated for 30 min at 37 ºC with 100 μm MG132 (closed squares) or DMSO (open squares). Data represent the means of six independent experiments ± S.E. (error bars). In each experiment, a representative Western blot is shown at the bottom. G6PD was used as a loading control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We also compared the growth of yeast expressing ROMK versus A198T Bartter mutant in strains lacking DOA10 and HRD1, which are the E3 ubiquitin ligases required for ERAD (73). However, we observed no difference in their growth rates (data not shown). These data suggest that eliminating ERAD does not necessarily increase the plasma membrane pool of ROMK.
The degradation of Bartter mutants is facilitated by the Cdc48 AAA+ ATPase and the Hsp70 molecular chaperone in yeast
ERAD substrates undergo retrotranslocation, a process by which proteins are extracted from the ER lumen or membrane concurrent with or following polyubiquitination in order to be processed by the cytoplasmic 26S proteasome (74, 75). For nearly all ERAD substrates, this event requires the AAA+ ATPase Cdc48 (also known as p97 or VCP in mammals), which provides the mechanical force required for extraction (10, 11, 76–79). To examine whether the degradation of ROMK was Cdc48-dependent, which would provide further evidence that ROMK and especially the Bartter mutants are ERAD substrates, cycloheximide chases were performed in a strain containing a temperature-sensitive mutation in the gene encoding Cdc48, cdc48-2. Following a 2-h shift to a non-permissive temperature, which inactivates Cdc48, we found that ROMK was again significantly more stable than the Bartter mutants and that the modest turnover of this protein was Cdc48-independent. In contrast, the degradation of the Bartter mutant proteins was Cdc48-dependent (Fig. 6).
Figure 6.

Bartter mutant degradation is facilitated by the AAA+ ATPase Cdc48 in yeast. A–E, cycloheximide chase reactions were performed, and lysates were blotted with anti-HA antibody to measure the stability of ROMK and the indicated mutant proteins over 90 min. The degradation of ROMK was measured in CDC48 (open squares) or cdc48-2 (closed squares) mutant yeast, which were incubated at 39 °C for 2 h before the start of the chase, and the incubation was continued at 39 °C. Data represent the means of six independent experiments ± S.E. (error bars). In each experiment, a representative Western blot is shown at the bottom. G6PD was used as a loading control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ERAD substrate recognition is mediated by molecular chaperones and chaperone-like proteins (4–7, 10). Some of the most extensively studied proteins in this family are the Hsp70s (67). In yeast, the major cytoplasmic Hsp70 is Ssa1. Previous data indicated that this chaperone is required for the selection of other ERAD substrates, and especially ion channels, expressed in yeast (25, 54, 55, 80). To examine whether ROMK degradation is chaperone-dependent, we utilized a yeast strain containing a temperature-sensitive mutation in the gene encoding Ssa1. Specifically, in the ssa1-45 strain, a Pro to Leu mutation at residue 417 renders the resulting protein unstable at higher temperatures (81). When expressed in a strain that lacks the genes encoding the paralogous cytosolic Hsp70 proteins Ssa2, Ssa3, and Ssa4, defects in growth, protein translocation, and ERAD were evident at 37 °C (82). As anticipated, we discovered that Bartter mutant protein degradation was highly dependent on Ssa1, as shown by the nearly complete stabilization of the mutant proteins in the ssa1-45 strain at the non-permissive temperature (Fig. 7). The mutants were again also considerably less stable in the isogenic SSA1 wild-type strain when compared with ROMK. Together, these experiments indicate that the ERAD of the A198T, R212P, H270Y, and Y314C ROMK variants is Hsp70- and Cdc48-dependent, which is in stark contrast to wild-type ROMK.
Figure 7.

Bartter mutant degradation is mediated by the Hsp70 chaperone in yeast. A–E, cycloheximide chase reactions were performed, and lysates were blotted with anti-HA antibody to measure the stability of ROMK and the indicated mutant proteins over 90 min. The degradation of ROMK was measured in SSA1 (open squares) or ssa1-45 (closed squares) mutant yeast, which were incubated at 37 °C for 20 min before the start of the chase, and the incubation was continued at 37 °C. Data represent means of six independent experiments ± S.E. (error bars). In each experiment, a representative Western blot is shown at the bottom. G6PD was used as a loading control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ROMK degradation is proteasome- and p97/VCP-dependent in human cells
The data above show that the underlying defect for select cases of Bartter disease arises from an unstable protein that is targeted for ERAD, at least in yeast. To validate this hypothesis in a more biologically relevant system, we performed cycloheximide chase reactions in the presence or absence of MG132 in HEK293 cells transiently expressing either wild-type ROMK1 or the same protein containing either the A198T, R212P, H270Y, or Y314C substitutions. As shown in Fig. 8 (A–E), the results from this human cell line mirror those obtained in the yeast model; the relative instability of the mutant proteins is greater than that of the wild-type protein, but proteasome inhibition slows the degradation of only the disease-causing mutants. In addition, treatment with the Cdc48/VCP inhibitor, CB-5083, increased the steady-state expression of both ROMK and the A198T Bartter mutant in HEK293 cells (Fig. 8, F and G). Taken together, these data indicate that mutations in the immunoglobulin-like domain target ROMK for ERAD, thus linking type II Bartter syndrome to this pathway.
Figure 8.

The degradation of type II Bartter syndrome mutant proteins is proteasome-dependent in HEK293H cells. A–E, cycloheximide chase reactions were performed, and lysates were blotted with anti-HA antibody to measure the stability of ROMK and the indicated mutant proteins over 3 h. The HEK293H cells were pretreated for 4 h with 20 μm MG132 (closed squares) or dimethyl sulfoxide (open squares). Data represent the means of three independent experiments ± S.E. (error bars) Actin served as a loading control. *, p < 0.05; **, p < 0.01; ***, p < 0.001. F, Western blotting of ROMK and A198T in the presence or absence of CB-5083 treatment. G, scatter plot of the data in F, representing the results from three independent experiments. Averages and S.E. are shown in red. H, steady-state levels of HEK293H cells expressing ROMK or the A198T Bartter mutant grown at 26 °C versus 37 °C at the depicted time points. *, nonspecific band. I, scatter plot of the data in H, representing the results from three independent experiments. Data were standardized to ROMK at 24 h and 37 ºC. Averages and S.E. are shown in red.
Expression of a Bartter mutant is temperature-sensitive
In cystic fibrosis, the ΔF508 CFTR mutation fails to fold properly in the ER and is rapidly degraded by ERAD (83). The mutation appears to be temperature-sensitive, because culturing cells expressing ΔF508 CFTR at 26–30 °C for 24–48 h stabilizes the protein and results in the delivery of some ΔF508 CFTR to the cell surface (84). Although the effect of low-temperature correction is quite complex, low-temperature incubation augments domain–domain contacts that are otherwise recognized as being misfolded. To test whether the expression of the A198T mutant can similarly be increased by low-temperature correction, we cultured HEK293 cells expressing this protein at 26 or 37 °C for the indicated times. For comparison, HEK293 cells expressing the wild-type protein were also propagated at 37 °C. As shown in Fig. 8 (H and I), low-temperature incubation significantly increased the steady-state levels of the Bartter mutant. These data are consistent with the notion that the disease-associated protein is misfolded but that thermodynamic and/or kinetic barriers in the folding pathway can be overcome by low-temperature incubation.
Discussion
Type II Bartter syndrome is an autosomal recessive renal salt wasting disorder caused by loss-of-function mutations in the gene encoding ROMK (85, 86). Severe forms of the disease manifest during gestation and cause polyhydramnios, increasing the risk of premature delivery. Postnatally, infants with severe forms of type II Bartter syndrome develop life-threatening volume losses, hypokalemic metabolic alkalosis, hypercalciuria, and nephrocalcinosis (86). A number of these disease-causing mutations have been studied in vitro and have been found to affect different aspects of ROMK functional status, including its gating and pH-sensing characteristics (87, 88). In most cases, however, type II Bartter mutations lead to ROMK mistrafficking and reduced channel surface density (51). Although investigators have speculated that the trafficking mutations may reduce ROMK plasma membrane residence through activation of cellular protein quality control mechanisms, the details underlying this process have not been explored.
We show in this report that select disease-causing mutations in ROMK compromise ER folding, which targets the protein for ERAD in both a new yeast model and a human cell culture system. In the yeast system, the A198T, R212P, H270Y, and Y314C Bartter mutant proteins, which represent substitutions in the cytosolic C-terminal immunoglobulin-like domain, fail to rescue growth in potassium transporter-deficient yeast. These mutants also undergo accelerated degradation that is proteasome-dependent, relies on the Cdc48 AAA+ ATPase, and is mediated by cytosolic Hsp70. Sucrose gradient centrifugation and indirect immunofluorescence microscopy indicate that the majority of Bartter ROMK is ER-localized, consistent with it being subjected to ERAD. Nevertheless, a small but noticeable fraction is found at the plasma membrane, which is sufficient to rescue growth in the potassium transporter-deficient cells. Similar observations were evident when the related Kir2.1 channel was examined in a yeast expression system (25). In accordance with data obtained in the yeast model, the A198T, R212P, H270Y, or Y314C Bartter mutant proteins were also more readily degraded by the proteasome than ROMK in HEK293 cells. Additionally, the steady-state levels of a Bartter mutant rise after incubation at low temperature. These data suggest that future therapies to correct disease-associated mutations in ROMK should target mechanisms to stabilize the protein in the ER. Pharmacological chaperones that stabilize other disease-relevant proteins in the ER have entered clinical trials or have been approved by the Food and Drug Administration (89, 90).
Elucidating the precise biophysical consequences of the four Bartter mutations in the immunoglobulin-like domain is beyond the scope of this study, but based on their location and our molecular model, we propose working hypotheses for why the corresponding proteins are unstable and targeted for ERAD. Notably, all four mutations are predicted to fall within β-strands in the immunoglobulin-like domain. Whereas the side chains at positions 198 and 314 face the hydrophobic core of the β-sandwich, the side chains at positions 212 and 270 face away from the core, although they are still predicted to have low solvent exposure. Mutations of side chains in the hydrophobic core (positions 198 and 314) most likely compromise domain folding, because early events during protein folding pathways require the collapse of a hydrophobic core (91, 92). In contrast, given that the substitutions at positions 212 and 270 affect non-solvent-exposed charged residues, it is possible that the R212P and H270Y substitutions disrupt electrostatic interactions within this domain or possibly that they interfere with the assembly of ROMK monomers into the functional tetrameric species. Until high-resolution structural data for ROMK become available, such observations remain fraught with uncertainty. We do, however, note that Y314C is the least stable protein in HEK293 cells (Fig. 8, compare E with A–D). This observation concurs with previous work suggesting that the Tyr is essential for maintaining the structural integrity of the immunoglobulin-like domain and that even more conservative mutations than Tyr → Cys (such as Tyr → Phe or Tyr → Leu) are poorly tolerated (52).
The development of a yeast model for ROMK function and type II Bartter syndrome represents a gateway to identify factors that control the biogenesis and function of this potassium channel. Yeast genetic approaches to identify factors that affect quality control decisions for other mammalian ERAD substrates have also been used: CFTR (55, 93–95), apolipoprotein B (96, 97), antitrypsin-Z (98–100), and the NaCl co-transporter (54). In these previous studies, the yeast model has aided the discovery of conserved, human homologs of chaperones and chaperone-like proteins that influence ERAD and trafficking in higher eukaryotes. Yeast models have also been applied to define the nature of the toxicity associated with myriad neurological disorders, including Parkinson's disease, Huntington's disease, and ALS (101–105).
In sum, the continued characterization of factors required for the degradation of ROMK mutant alleles will yield additional insights into the multiple pathways by which ERAD substrates are degraded and potentially identify novel therapeutic strategies to treat type II Bartter syndrome. Our approach can also be applied to further characterize other mutations in ROMK associated with type II Bartter syndrome, a pursuit that will allow us to define specific domains within ROMK that play important roles during channel trafficking to the plasma membrane and in ER stability. Ultimately, one hopes that the full characterization of each of the mutations associated with this disease will lead to the development of personalized therapies for patients. This would eliminate the need for lifelong multidrug and supplement regimens currently required for type II Bartter syndrome patients.
Experimental procedures
Plasmid construction
Rat ROMK1, modified with an extracellular HA tag flanked with diglycines, was a generous gift from Paul Welling (University of Maryland School of Medicine) (70). In yeast, ROMK mutants A198T, R212P, H270Y, and Y314C were made by two-stage PCR mutagenesis (106). The ROMK-HA insert was cloned into digested pRS415TEF and pRS416TEF vectors (107) containing different promoters and genes for auxotrophic selection (108) using BamHI and XhoI. The cut vector was treated with Antarctic phosphatase (New England BioLabs, Ipswich, MA), and the digested vectors were run on a 1% agarose gel and purified with the GeneJET Gel Extraction and DNA Cleanup Kit (Thermo Scientific). The cut vectors and inserts were ligated using T4 DNA ligase (Thermo Scientific). All isolated inserts were subject to DNA sequence analysis (GENEWIZ). The same ROMK mutants were cloned into a pcDNA3.1 vector via site-directed mutagenesis using the same protocol as stated above. The yeast Kir2.1 expression vector was obtained and used as described previously (25).
To build the ROMKS44D/K80M construct, two-stage PCR mutagenesis was again used with the exception that SmaI was targeted in the upstream restriction site, located next to BamHI in the pRS polylinker. All primers used for these constructs can be found in supplemental Table S1. The point mutations in the gene encoding ROMK were confirmed by DNA sequence analysis (GENEWIZ).
Yeast strains, growth conditions, and viability assays
A summary of the Saccharomyces cerevisiae strains used in this study is provided in supplemental Table S2. All strains were grown at 30 °C unless otherwise indicated. Standard procedures were followed for propagation and transformation of yeast (109). For growth assays, 200 μl of cultures were grown overnight in 96-well dishes in SC-Leu liquid medium supplemented with 100 mm KCl. Once all cultures reached stationary phase, they were diluted 5-fold in SC-Leu liquid medium lacking additional KCl. Six 5-fold dilutions were made and transferred to SC-Leu solid medium supplemented with 100, 25, 10, or 0 mm KCl using a 48-pin manifold (Sigma-Aldrich). Plates were incubated for 4 days and imaged on days 2, 3, and 4 using a Bio-Rad image station. To assay protein expression, 2 A600 equivalents of yeast were harvested from the plates and lysed with 0.3 m NaOH, 1% β-mercaptoethanol, and protease inhibitors (leupeptin (1:1,000), PepA (1:10,000), and PMSF (1:100)). Total protein was precipitated from the lysate with 10% TCA and analyzed by SDS-PAGE and Western blotting. ROMK was detected using an anti-HA-HRP–conjugated antibody, and blots were probed with anti-glucose-6-phosphate dehydrogenase (G6PD) antiserum as a loading control. Donkey HRP-conjugated anti-rabbit immunoglobulin G secondary antibody was applied. The Supersignal chemiluminescent substrate (Pierce) was utilized to develop the blots, and the signals were quantified with a Bio-Rad ChemiDoc XRS+ image station and Image Lab version 5.2.1 software.
Yeast cycloheximide chase assays
Yeast cultures transformed with a vector engineered for the constitutive expression of the indicated potassium channel under the control of the TEF1 promoter (see above) were grown overnight to saturation in SC-Leu medium. The cultures were diluted into the same medium at an initial A600 of 0.25 and allowed to grow to mid-log phase (A600 = 0.8–1.0). Each culture was dosed with cycloheximide (Sigma-Aldrich) to a final concentration of 150 μg/ml and incubated with shaking in a water bath at either 30 or 37 °C (for assays using temperature-sensitive mutant strains). After the addition of cycloheximide, 1-ml aliquots were removed at 0, 30, 60, and 90 min, added to 35 μl of 0.5 m NaN3 on ice, harvested, flash-frozen in liquid nitrogen, and stored at −80 °C. The cell pellets were thawed on ice, and total protein was TCA-precipitated as described (55). The protein samples were analyzed by SDS-PAGE and immunoblot analysis as described above. To determine whether substrate degradation was proteasome-dependent, MG132 was added to a final concentration of 100 μm (Selleck Chemicals) in DMSO or in the equivalent volume of DMSO and allowed to incubate for 30 min at 37 °C prior to the addition of cycloheximide.
Yeast protein localization studies
The intracellular residence of ROMK was determined by sedimentation in a sucrose gradient as described previously (110). In brief, ∼100 A600 equivalents of log phase wild-type (BY4742) cells expressing HA-tagged ROMK were grown in SC-Leu liquid medium. Cells were pelleted for 5 min at 4,500 rpm in a clinical centrifuge at room temperature, flash-frozen in liquid nitrogen, and stored at −80 °C. A 70% sucrose solution was prepared in 10 mm Tris, pH 8.0, 1 mm EDTA, and 1 mm DTT and subsequently diluted to create a 10–70% layered sucrose gradient. The cells were thawed, resuspended in 400 μl of 10% sucrose solution plus protease inhibitors (1 mm PMSF, 1 μg/ml leupeptin, and 0.05 μg/ml pepstatin A), and lysed by agitation using glass beads on a Vortex mixer three times for 1 min with a 1-min rest on ice between each round. A non-continuous sucrose gradient was created in an SW41 high-speed 14 × 89-mm Polyallomer ultracentrifuge tube (Beckman Instruments, Inc.). A total of 2 ml of each percentage was layered on top of one another, with 70% on the bottom to 10% at the top, although only 1.5 ml each of the 30 and 20% sucrose solutions was used. Finally, 400 μl of the homogenate was loaded onto the gradient, and 20 μl (5%) of the homogenate was retained at −20 °C as the load fraction. The gradient was spun in an SW41 rotor for 18 h at 28,500 rpm (∼140,000 × g) at 4 °C. After the spin, 500-μl fractions were carefully removed from the top of the gradient until the entire gradient was aliquoted. A 20-μl aliquot from each fraction was removed and combined with 5 μl of 5× sample buffer (0.325 m Tris, pH 6.8, 10% SDS, 50% glycerol, 25 mg/ml bromphenol blue, 5% β-mercaptoethanol), and after incubation at room temperature for 10 min, the samples were loaded onto a 10% denaturing polyacrylamide gel. The resolved proteins were transferred to nitrocellulose, and anti-HA-HRP was used to visualize ROMK (see above). Antibodies for the following organelle markers were also used: anti-Anp1p (provided by Hugh Pelham, Medical Research Council Laboratory of Molecular Biology) for the Golgi, anti-Pma1p (AbCam) for the plasma membrane, and anti-Sec61p (111) for the ER fraction.
Indirect immunofluorescence was also applied to determine ROMK residence, as described previously with slight modifications (109). Yeast cells expressing ROMK were grown to an A600 of 0.5–0.8 and fixed with 3.7% formaldehyde for 1 h at 30 °C. The cells were then collected by centrifugation, washed with solution A (1.2 m sorbitol, 40 mm KPO4, pH 6.5), resuspended in 30 μl of solution A supplemented with 10 mg/ml 20T Zymolyase (U.S. Biological), and incubated at 37 °C for 25 min. The cells were pelleted at 2,000 rpm in a clinical centrifuge for 2 min, washed with solution A, and resuspended in 200 μl of solution A. An aliquot of the cell suspension (20 μl) was added to a microscope slide well, which was pretreated with 1 mg/ml polylysine, and incubated at room temperature for 10 min. The wells were then washed with PBS, 0.5% BSA, 0.5% ovalbumin and stained with mouse anti-HA (HA-11, Covance) and rabbit anti-Kar2p (112) primary antibodies at 4 °C in a 1:250 dilution. The primary antibodies were decorated with Alexa Fluor 488 goat anti-mouse and Alexa Fluor 568 goat anti-rabbit, both at 1:500, and nuclei were stained with DAPI. The slides were mounted with Prolong Antifade Gold (Invitrogen) and imaged with a Nikon TiE Eclipse inverted microscope using a ×100 oil immersion objective (1.45 numeric aperture) fitted with a Hamamatsu Orca Flash 4.0 Sci cMos camera. Final images were obtained through the use of Nikon Elements software.
Limited proteolysis
Microsomes were prepared from wild-type (BY4742) yeast expressing the protein of interest under the control of the TEF1 promoter via the medium-scale protocol (113). Approximately 10 μg of microsomes were treated with 10 μg/ml Proteinase K (Sigma) on ice for the times indicated. Reactions were then quenched with 100% TCA added to a final concentration of 33%. After centrifugation at 15,000 × g for 10 min at 4 °C, the precipitate was washed with acetone and resuspended in sample buffer for subsequent SDS-PAGE and immunoblot analysis, as described above.
HEK293H cell culture, transfection, and cycloheximide chase assays
HEK293H cells (Thermo Fisher) were cultured in high glucose Dulbecco's modified Eagle's medium supplemented with 10% FBS at 37 °C. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) per the instructions of the manufacturer, and cells were subjected to analysis 18–20 h post-transfection. Medium containing 50 μg/ml cycloheximide was applied to the cells, and time points were collected at 0, 1, 2, and 3 h. Cells were lysed with 300 μl of TNT buffer (50 mm Tris, 150 mm NaCl, 1% Triton X-100) supplemented with protease inhibitors (1 tablet/6 ml of TNT buffer; Roche Diagnostics, Mannheim, Germany). Next, the cells were incubated on ice with gentle intermittent rocking for 30 min. After centrifugation at 13,000 × g for 15 min at 4 °C, lysate was collected, and proteins were analyzed by SDS-PAGE and immunoblot analysis as described above. When the effect of proteasome inhibition was examined, MG132 (20 μm) or an equivalent amount of DMSO was added 4 h before the addition of cycloheximide. When the effect of cold exposure was examined, the cells were placed at 26 or 37 °C within 4 h after the application of Lipofectamine. When the effect of p97/VCP inhibition was examined, CB-5083 (Selleck Chemicals) (1 μm) or an equivalent amount of DMSO was added 4 h before harvest. The cells were lysed, as described above, and protein was detected by immunoblot analysis at the indicated time points.
Author contributions
B. M. O., T. D. M., A. R. S., and J. L. B. developed and coordinated the study. B. M. O. and T. D. M. performed all of the experiments and wrote a draft of the manuscript, which was edited by J. L. B. and A. R. S. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Paul Welling, Teresa Buck, Allyson O'Donnell, Chris Guerriero, Annette Chiang, Jennifer Goeckeler-Fried, Patrick Needham, Lynley Doonan, George Preston, Sara Sannino, Alex Kolb, Alexandra Socovich, Yang Liu, and Kathleen Makielski for technical assistance, helpful discussion, plasmids, antibodies, and reagents.
This work was supported by National Institutes of Health Grants GM75061 and DK79307 (University of Pittsburgh George O'Brien Kidney Research Center) (to J. L. B.), DK098145 (to A. R. S.), and 5T32 DK71492-10 and 4K12HD052892-10 (to B. M. O.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2.
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- Kir
- inwardly rectifying family of potassium channels
- ROMK
- renal outer medullary potassium channel
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ENaC
- epithelial sodium channel
- G6PD
- glucose-6-phosphate dehydrogenase.
References
- 1. Brandvold K. R., and Morimoto R. I. (2015) The chemical biology of molecular chaperones: implications for modulation of proteostasis. J. Mol. Biol. 427, 2931–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vendruscolo M., Knowles T. P., and Dobson C. M. (2011) Protein solubility and protein homeostasis: a generic view of protein misfolding disorders. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a010454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guerriero C. J., and Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brodsky J. L. (2007) The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 404, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Molinari M., and Hebert D. N. (2015) Glycoprotein maturation and quality control. Semin. Cell Dev. Biol. 41, 70. [DOI] [PubMed] [Google Scholar]
- 6. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bar-Nun S. (2005) The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr. Top. Microbiol. Immunol. 300, 95–125 [DOI] [PubMed] [Google Scholar]
- 9. Hampton R. Y. (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell. Biol. 14, 476–482 [DOI] [PubMed] [Google Scholar]
- 10. Rabinovich E., Kerem A., Fröhlich K. U., Diamant N., and Bar-Nun S. (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22, 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ye Y., Meyer H. H., and Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 12. Lecker S. H., Goldberg A. L., and Mitch W. E. (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819 [DOI] [PubMed] [Google Scholar]
- 13. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 14. Perri E. R., Thomas C. J., Parakh S., Spencer D. M., and Atkin J. D. (2015) The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma Y., and Hendershot L. M. (2004) The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977 [DOI] [PubMed] [Google Scholar]
- 16. Xiao X., Jones G., Sevilla W. A., Stolz D. B., Magee K. E., Haughney M., Mukherjee A., Wang Y., and Lowe M. E. (2017) A carboxyl ester lipase (CEL) mutant causes chronic pancreatitis by forming intracellular aggregates that activate apoptosis. J. Biol. Chem. 292, 7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J., Yang X., and Zhang J. (2016) Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell. Signal. 28, 1099–1104 [DOI] [PubMed] [Google Scholar]
- 18. Dickhout J. G., Carlisle R. E., and Austin R. C. (2011) Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ. Res. 108, 629–642 [DOI] [PubMed] [Google Scholar]
- 19. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 20. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 21. Malik B., Schlanger L., Al-Khalili O., Bao H. F., Yue G., Price S. R., Mitch W. E., and Eaton D. C. (2001) Enac degradation in A6 cells by the ubiquitin-proteosome proteolytic pathway. J. Biol. Chem. 276, 12903–12910 [DOI] [PubMed] [Google Scholar]
- 22. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., and Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valentijn J. A., Fyfe G. K., and Canessa C. M. (1998) Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J. Biol. Chem. 273, 30344–30351 [DOI] [PubMed] [Google Scholar]
- 24. Ma D., Taneja T. K., Hagen B. M., Kim B. Y., Ortega B., Lederer W. J., and Welling P. A. (2011) Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 145, 1102–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolb A. R., Needham P. G., Rothenberg C., Guerriero C. J., Welling P. A., and Brodsky J. L. (2014) ESCRT regulates surface expression of the Kir2.1 potassium channel. Mol. Biol. Cell 25, 276–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nichols C. G., and Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191 [DOI] [PubMed] [Google Scholar]
- 27. Wang W. (2004) Renal potassium channels: recent developments. Curr. Opin. Nephrol. Hypertens. 13, 549–555 [DOI] [PubMed] [Google Scholar]
- 28. Hebert S. C., Desir G., Giebisch G., and Wang W. (2005) Molecular diversity and regulation of renal potassium channels. Physiol. Rev. 85, 319–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hebert S. C., and Andreoli T.E. (1984) Control of NaCl transport in the thick ascending limb. Am. J. Physiol. 246, F745–F756 [DOI] [PubMed] [Google Scholar]
- 30. Rieg T., Vallon V., Sausbier M., Sausbier U., Kaissling B., Ruth P., and Osswald H. (2007) The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 72, 566–573 [DOI] [PubMed] [Google Scholar]
- 31. McNicholas C. M., Wang W., Ho K., Hebert S. C., and Giebisch G. (1994) Regulation of ROMK1 K+ channel activity involves phosphorylation processes. Proc. Natl. Acad. Sci. U.S.A. 91, 8077–8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu Z. C., Yang Y., and Hebert S. C. (1996) Phosphorylation of the ATP-sensitive, inwardly rectifying K+ channel, ROMK, by cyclic AMP-dependent protein kinase. J. Biol. Chem. 271, 9313–9319 [DOI] [PubMed] [Google Scholar]
- 33. Huang C. L. (2007) Complex roles of PIP2 in the regulation of ion channels and transporters. Am. J. Physiol. Renal Physiol. 293, F1761–F1765 [DOI] [PubMed] [Google Scholar]
- 34. Huang C. L., Feng S., and Hilgemann D. W. (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391, 803–806 [DOI] [PubMed] [Google Scholar]
- 35. McNicholas C. M., Yang Y., Giebisch G., and Hebert S. C. (1996) Molecular site for nucleotide binding on an ATP-sensitive renal K+ channel (ROMK2). Am. J. Physiol. 271, F275–F285 [DOI] [PubMed] [Google Scholar]
- 36. Choe H., Zhou H., Palmer L. G., and Sackin H. (1997) A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am. J. Physiol. 273, F516–F529 [DOI] [PubMed] [Google Scholar]
- 37. McNicholas C. M., MacGregor G. G., Islas L. D., Yang Y., Hebert S. C., and Giebisch G. (1998) pH-dependent modulation of the cloned renal K+ channel, ROMK. Am. J. Physiol. 275, F972–F981 [DOI] [PubMed] [Google Scholar]
- 38. Lin D., Sterling H., Lerea K. M., Giebisch G., and Wang W. H. (2002) Protein kinase C (PKC)-induced phosphorylation of ROMK1 is essential for the surface expression of ROMK1 channels. J. Biol. Chem. 277, 44278–44284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin D. H., Sterling H., Lerea K. M., Welling P., Jin L., Giebisch G., and Wang W. H. (2002) K depletion increases protein tyrosine kinase-mediated phosphorylation of ROMK. Am. J. Physiol. Renal Physiol. 283, F671–F677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kahle K. T., Ring A. M., and Lifton R. P. (2008) Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 70, 329–355 [DOI] [PubMed] [Google Scholar]
- 41. Schwalbe R. A., Bianchi L., Accili E. A., and Brown A. M. (1998) Functional consequences of ROMK mutants linked to antenatal Bartter's syndrome and implications for treatment. Hum. Mol. Genet. 7, 975–980 [DOI] [PubMed] [Google Scholar]
- 42. Starremans P. G., van der Kemp A. W., Knoers N. V., van den Heuvel L. P., Bindels R. J. (2002) Functional implications of mutations in the human renal outer medullary potassium channel (ROMK2) identified in Bartter syndrome. Pflugers Arch. 443, 466–472 [DOI] [PubMed] [Google Scholar]
- 43. Kleta R., and Bockenhauer D. (2006) Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol. 104, 73–80 [DOI] [PubMed] [Google Scholar]
- 44. Boim M. A., Ho K., Shuck M. E., Bienkowski M. J., Block J. H., Slightom J. L., Yang Y., Brenner B. M., and Hebert S. C. (1995) ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am. J. Physiol. 268, F1132–F1140 [DOI] [PubMed] [Google Scholar]
- 45. Shuck M. E., Piser T. M., Bock J. H., Slightom J. L., Lee K. S., and Bienkowski M. J. (1997) Cloning and characterization of two K+ inward rectifier (Kir) 1.1 potassium channel homologs from human kidney (Kir1.2 and Kir1.3). J. Biol. Chem. 272, 586–593 [DOI] [PubMed] [Google Scholar]
- 46. Zhou H., Tate S. S., and Palmer L. G. (1994) Primary structure and functional properties of an epithelial K channel. Am. J. Physiol. 266, C809–C824 [DOI] [PubMed] [Google Scholar]
- 47. Welling P. (2016) ROMK and Bartter Syndrome Type 2. In Ion Channels and Transporters of Epithelia in Health and Disease (Hamilton K. L., and Devor D. C.) pp. 643–658, Springer, New York [Google Scholar]
- 48. Ji W., Foo J. N., O'Roak B. J., Zhao H., Larson M. G., Simon D. B., Newton-Cheh C., State M. W., Levy D., and Lifton R. P. (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 40, 592–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seyberth H. W., and Schlingmann K. P. (2011) Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr. Nephrol. 26, 1789–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fang L., Li D., and Welling P. A. (2010) Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am. J. Physiol. Renal Physiol. 299, F1359–F1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peters M., Ermert S., Jeck N., Derst C., Pechmann U., Weber S., Schlingmann K. P., Seyberth H. W., Waldegger S., and Konrad M. (2003) Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 64, 923–932 [DOI] [PubMed] [Google Scholar]
- 52. Fallen K., Banerjee S., Sheehan J., Addison D., Lewis L. M., Meiler J., and Denton J. S. (2009) The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels 3, 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grove D. E., Fan C. Y., Ren H. Y., and Cyr D. M. (2011) The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 22, 301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Needham P. G., Mikoluk K., Dhakarwal P., Khadem S., Snyder A. C., Subramanya A. R., and Brodsky J. L. (2011) The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J. Biol. Chem. 286, 43611–43621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y. M., Nijbroek G., Sullivan M. L., McCracken A. A., Watkins S. C., Michaelis S., and Brodsky J. L. (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ficker E., Dennis A. T., Wang L., and Brown A. M. (2003) Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 92, e87–e100 [DOI] [PubMed] [Google Scholar]
- 57. Nakamura R. L., and Gaber R. F. (1998) Studying ion channels using yeast genetics. Methods Enzymol. 293, 89–104 [DOI] [PubMed] [Google Scholar]
- 58. Haass F. A., Jonikas M., Walter P., Weissman J. S., Jan Y. N., Jan L. Y., and Schuldiner M. (2007) Identification of yeast proteins necessary for cell-surface function of a potassium channel. Proc. Natl. Acad. Sci. U.S.A. 104, 18079–18084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bichet D., Lin Y. F., Ibarra C. A., Huang C. S., Yi B. A., Jan Y. N., and Jan L. Y. (2004) Evolving potassium channels by means of yeast selection reveals structural elements important for selectivity. Proc. Natl. Acad. Sci. U.S.A. 101, 4441–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Minor D. L. Jr., Masseling S. J., Jan Y. N., and Jan L. Y. (1999) Transmembrane structure of an inwardly rectifying potassium channel. Cell 96, 879–891 [DOI] [PubMed] [Google Scholar]
- 61. Paynter J. J., Shang L., Bollepalli M. K., Baukrowitz T., and Tucker S. J. (2010) Random mutagenesis screening indicates the absence of a separate H+-sensor in the pH-sensitive Kir channels. Channels 4, 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zaks-Makhina E., Kim Y., Aizenman E., and Levitan E. S. (2004) Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol. Pharmacol. 65, 214–219 [DOI] [PubMed] [Google Scholar]
- 63. Zaks-Makhina E., Li H., Grishin A., Salvador-Recatala V., and Levitan E. S. (2009) Specific and slow inhibition of the kir2.1 K+ channel by gambogic acid. J. Biol. Chem. 284, 15432–15438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rapedius M., Fowler P. W., Shang L., Sansom M. S., Tucker S. J., and Baukrowitz T. (2007) H bonding at the helix-bundle crossing controls gating in Kir potassium channels. Neuron 55, 602–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Imai T., and Ohno T. (1995) Measurement of yeast intracellular pH by image processing and the change it undergoes during growth phase. J Biotechnol. 38, 165–172 [DOI] [PubMed] [Google Scholar]
- 67. Sackin H., Nanazashvili M., Palmer L. G., and Li H. (2006) Role of conserved glycines in pH gating of Kir1.1 (ROMK). Biophys. J. 90, 3582–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoo D., Fang L., Mason A., Kim B. Y., and Welling P. A. (2005) A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J. Biol. Chem. 280, 35281–35289 [DOI] [PubMed] [Google Scholar]
- 69. Yoo D., Kim B. Y., Campo C., Nance L., King A., Maouyo D., and Welling P. A. (2003) Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J. Biol. Chem. 278, 23066–23075 [DOI] [PubMed] [Google Scholar]
- 70. Ho K., Nichols C. G., Lederer W. J., Lytton J., Vassilev P. M., Kanazirska M. V., and Hebert S. C. (1993) Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362, 31–38 [DOI] [PubMed] [Google Scholar]
- 71. Li K., Jiang Q., Bai X., Yang Y. F., Ruan M. Y., and Cai S. Q. (2017) Tetrameric assembly of K+ channels requires ER-located chaperone proteins. Mol. Cell 65, 52–65 [DOI] [PubMed] [Google Scholar]
- 72. Lee D. H., and Goldberg A. L. (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 271, 27280–27284 [DOI] [PubMed] [Google Scholar]
- 73. Claessen J. H., Kundrat L., and Ploegh H. L. (2012) Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 22, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wolf D. H., and Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 75. Meyer H., Bug M., and Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 76. Bays N. W., and Hampton R. Y. (2002) Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr. Biol. 12, R366–R371 [DOI] [PubMed] [Google Scholar]
- 77. Braun S., Matuschewski K., Rape M., Thoms S., and Jentsch S. (2002) Role of the ubiquitin-selective CDC48 (UFD1/NPL4) chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 21, 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D. H., an Sommer T. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4, 134–139 [DOI] [PubMed] [Google Scholar]
- 79. Hitchcock A. L., Krebber H., Frietze S., Lin A., Latterich M., and Silver P. A. (2001) The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol. Biol. Cell 12, 3226–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kolb A. R., Buck T. M., and Brodsky J. L. (2011) Saccharomyces cerivisiae as a model system for kidney disease: what can yeast tell us about renal function? Am. J. Physiol. Renal Physiol. 301, F1–F11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Needham P. G., Patel H. J., Chiosis G., Thibodeau P. H., and Brodsky J. L. (2015) Mutations in the yeast Hsp70, Ssa1, at P417 alter ATP cycling, interdomain coupling, and specific chaperone functions. J. Mol. Biol. 427, 2948–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Becker J., Walter W., Yan W., and Craig E. A. (1996) Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol. Cell. Biol. 16, 4378–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., and Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 84. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., and Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 85. Welling P. A., and Ho K. (2009) A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am. J. Physiol. Renal Physiol. 297, F849–F863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fremont O. T., and Chan J. C. (2012) Understanding Bartter syndrome and Gitelman syndrome. World J. Pediatr. 8, 25–30 [DOI] [PubMed] [Google Scholar]
- 87. Flagg T. P., Tate M., Merot J., and Welling P. A. (1999) A mutation linked with Bartter's syndrome locks Kir 1.1a (ROMK1) channels in a closed state. J. Gen. Physiol. 114, 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Srivastava S., Li D., Edwards N., Hynes A. M., Wood K., Al-Hamed M., Wroe A. C., Reaich D., Moochhala S. H., Welling P. A., and Sayer J. A. (2013) Identification of compound heterozygous KCNJ1 mutations (encoding ROMK) in a kindred with Bartter's syndrome and a functional analysis of their pathogenicity. Physiol. Rep. 1, e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Coelho T., Maia L. F., Martins da Silva A., Waddington Cruz M., Planté-Bordeneuve V., Lozeron P., Suhr O. B., Campistol J. M., Conceição I. M., Schmidt H. H., Trigo P., Kelly J. W., Labaudinière R., Chan J., Packman J., et al. (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 79, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Stack J. H., Straley K. S., Decker C. J., Miller M., McCartney J., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., and Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Clerico E. M., Tilitsky J. M., Meng W., and Gierasch L. M. (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 427, 1575–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Baldwin R. L. (1989) How does protein folding get started? Trends Biochem. Sci. 14, 291–294 [DOI] [PubMed] [Google Scholar]
- 93. Youker R. T., Walsh P., Beilharz T., Lithgow T., and Brodsky J. L. (2004) Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell 15, 4787–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., and Brodsky J. L. (2007) Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 18, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hutt D. M., Roth D. M., Chalfant M. A., Youker R. T., Matteson J., Brodsky J. L., and Balch W. E. (2012) FK506 binding protein 8 peptidylprolyl isomerase activity manages a late stage of cystic fibrosis transmembrane conductance regulator (CFTR) folding and stability. J. Biol. Chem. 287, 21914–21925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grubb S., Guo L., Fisher E. A., and Brodsky J. L. (2012) Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 23, 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hrizo S. L., Gusarova V., Habiel D. M., Goeckeler J. L., Fisher E. A., and Brodsky J. L. (2007) The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 282, 32665–32675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Werner E. D., Brodsky J. L., and McCracken A. A. (1996) Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. U.S.A. 93, 13797–13801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Scott C. M., Kruse K. B., Schmidt B. Z., Perlmutter D. H., McCracken A. A., and Brodsky J. L. (2007) ADD66, a gene involved in the endoplasmic reticulum-associated degradation of α-1-antitrypsin-Z in yeast, facilitates proteasome activity and assembly. Mol. Biol. Cell 18, 3776–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gelling C. L., Dawes I. W., Perlmutter D. H., Fisher E. A., and Brodsky J. L. (2012) The endosomal protein-sorting receptor sortilin has a role in trafficking α-1 antitrypsin. Genetics 192, 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Narayan P., Ehsani S., and Lindquist S. (2014) Combating neurodegenerative disease with chemical probes and model systems. Nat. Chem. Biol. 10, 911–920 [DOI] [PubMed] [Google Scholar]
- 102. Khurana V., and Lindquist S. (2010) Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat. Rev. Neurosci. 11, 436–449 [DOI] [PubMed] [Google Scholar]
- 103. Tardiff D. F., Jui N. T., Khurana V., Tambe M. A., Thompson M. L., Chung C. Y., Kamadurai H. B., Kim H. T., Lancaster A. K., Caldwell K. A., Caldwell G. A., Rochet J. C., Buchwald S. L., and Lindquist S. (2013) Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 342, 979–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chung C. Y., Khurana V., Auluck P. K., Tardiff D. F., Mazzulli J. R., Soldner F., Baru V., Lou Y., Freyzon Y., Cho S., Mungenast A. E., Muffat J., Mitalipova M., Pluth M. D., Jui N. T., et al. (2013) Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gitler A. D., Chesi A., Geddie M. L., Strathearn K. E., Hamamichi S., Hill K. J., Caldwell K. A., Caldwell G. A., Cooper A. A., Rochet J. C., and Lindquist S. (2009) α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Vallejo A. N., Pogulis R. J., and Pease L. R. (1994) In vitro synthesis of novel genes: mutagenesis and recombination by PCR. PCR Methods Appl. 4, S123–S130 [DOI] [PubMed] [Google Scholar]
- 107. Mumberg D., Müller R., and Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 [DOI] [PubMed] [Google Scholar]
- 108. Mumberg D., Müller R., and Funk M. (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22, 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Adams A., and Kaiser C. (1998) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 110. Sullivan M. L., Youker R. T., Watkins S. C., and Brodsky J. L. (2003) Localization of the BiP molecular chaperone with respect to endoplasmic reticulum foci containing the cystic fibrosis transmembrane conductance regulator in yeast. J. Histochem. Cytochem. 51, 545–548 [DOI] [PubMed] [Google Scholar]
- 111. Stirling C. J., Rothblatt J., Hosobuchi M., Deshaies R., and Schekman R. (1992) Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell 3, 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Brodsky J. L., and Schekman R. (1993) A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nakatsukasa K., and Brodsky J. L. (2010) In vitro reconstitution of the selection, ubiquitination, and membrane extraction of a polytopic ERAD substrate. Methods Mol. Biol. 619, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.