

Abstract
Inflammatory responses are elicited through lipid products of phospholipase A2 activity that acts on the membrane phospholipids, including the phosphoinositides, to form the proinflammatory arachidonic acid and, in parallel, the glycerophosphoinositols. Here, we investigate the role of the glycerophosphoinositol in the inflammatory response. We show that it is part of a negative feedback loop that limits proinflammatory and prothrombotic responses in human monocytes stimulated with lipopolysaccharide. This inhibition is exerted both on the signaling cascade initiated by the lipopolysaccharide with the glycerophosphoinositol-dependent decrease in IκB kinase α/β, p38, JNK, and Erk1/2 kinase phosphorylation and at the nuclear level with decreased NF-κB translocation and binding to inflammatory gene promoters. In a model of endotoxemia in the mouse, treatment with glycerophosphoinositol reduced TNF-α synthesis, which supports the concept that glycerophosphoinositol inhibits the de novo synthesis of proinflammatory and prothrombotic compounds and might thus have a role as an endogenous mediator in the resolution of inflammation. As indicated, this effect of glycerophosphoinositol can also be exploited in the treatment of manifestations of severe inflammation by exogenous administration of the compound.
Keywords: inflammation, lipid signaling, lipopolysaccharide (LPS), monocyte, NF-κB (NF-KB), pharmacology, second messenger, sepsis, tissue factor, transcription regulation
Introduction
Inflammation is a beneficial host response to foreign pathogens and tissue injury that ultimately leads to bacterial clearance and restoration of tissue structure and function (1). The host response involves different cell types and signaling molecules, such as cytokines, chemokines, and bioactive lipids (2). These bioactive lipids include prostaglandins, leukotrienes, and endoperoxides, which originate enzymatically from the phospholipase A2/arachidonic acid cascade and are formed and act in a cell-specific fashion (3). More recently, other compounds that intervene in the resolution of inflammatory responses have been characterized; these include lipoxins, resolvins, protectins, and maresins (all of which are biosynthesized from essential polyunsaturated fatty acids), the receptors for the bioactive lipids mentioned above, and various microRNAs (4). It is now evident that the control of the initiation of inflammation and its natural resolution involve several essential components that need to be finely modulated in space and time to restore tissue homeostasis. Indeed, prolonged or uncontrolled inflammation represents the pathogenetic basis of chronic inflammatory diseases (5). As a prolonged activation of the cells of the immune system is the driving force behind inflammatory diseases, the identification of anti-inflammatory compounds that can switch off proinflammatory responses at one of the crucial steps and thus restore immunological homeostasis remains of great interest. Recent data obtained in our laboratory have addressed some aspects of this need as they show the potential of glycerophosphoinositol (GroPIns)6 as an anti-inflammatory compound that can prevent endotoxin shock in the mouse.
GroPIns is one of the naturally occurring phosphoinositide metabolites, the glycerophosphoinositols, that are produced through the activity of phospholipase A2IVα (PLA2IVα), an enzyme that preferentially hydrolyzes phospholipids carrying arachidonic acid in position sn-2 of the glycerol backbone (6, 7). The formation of GroPIns requires two deacylation steps, both carried out by PLA2IVα. PLA2IVα has intrinsic phospholipase A2 and lysolipase activities; thus, it hydrolyzes the membrane phosphoinositide (carrying the arachidonic acid in the sn-2 position), producing in parallel arachidonic acid and the lysoderivative lysophosphatidylinositol, and then in sequence, PLA2IVα forms GroPIns by deacylating lysophosphatidylinositol (6, 8). Noticeably, this PLA2IVα-dependent metabolism gives rise to three biologically active metabolites that start independent cascades, i.e. G-protein–coupled receptor-dependent signaling (by the GPR55 ligand lysophosphatidylinositol), cyclooxygenase/lipoxygenase metabolism (by producing their substrate, arachidonic acid), and glycerophosphoinositol-modulated processes (which include cell proliferation and actin cytoskeleton organization) (6, 8–10).
The glycerophosphoinositols are ubiquitous, freely diffusible molecules, and they can be detected both within cells and in the extracellular space (11, 12). Their intracellular concentrations are cell type-dependent, and their production can vary upon oncogenic transformation, cell differentiation, and hormonal stimulation (6, 11). They are present in different forms that can be either unphosphorylated (GroPIns) or phosphorylated (e.g. glycerophosphoinositol 4-phosphate (GroPIns4P), glycerophosphoinositol 4,5-phosphate, and glycerophosphoinositol 3-phosphate) with different cellular activities (6, 13). Hematopoietic cells, such as monocytes and macrophages, have a strictly regulated PLA2IVα activity that provides a fine modulation of the intracellular glycerophosphoinositol levels in response to environmental stimuli, including proinflammatory agents such as bacterial lipopolysaccharide (LPS) (7). To further clarify the potential roles of these lipid-derived mediators in inflammation, we have investigated the effect of GroPIns in a well characterized in vitro model of inflammatory and procoagulant responses mimicked by human blood monocytes challenged with LPS.
Here we show that GroPIns can inhibit the expression of prothrombotic and proinflammatory genes that are induced by Escherichia coli LPS through inhibition of the signaling cascade downstream of the toll-like receptor 4 (TLR4) and of the nuclear activity of nuclear factor-κB (NF-κB). Of note, the measurement of inflammatory markers in mice subjected to LPS-induced endotoxemia showed that treatment with GroPIns is associated with reduced plasma levels of tumor necrosis factor-α (TNF-α) and decreased expression of αMβ2 integrin (Mac-1) on the surface of circulating neutrophils, which strengthens the potential pharmacological relevance of this lipid-derived mediator. Thus, the present study provides evidence that GroPIns, which is produced endogenously by activated inflammatory cells, can act as a resolution signal to control the proinflammatory and prothrombotic responses associated with particularly exuberant inflammatory states (14).
Results
GroPIns inhibits endotoxin-induced prothrombotic and inflammatory responses in human monocytes
Increases of the intracellular GroPIns levels have been observed in response to inflammatory stimuli, such as LPS, in macrophages (7). Based on this, we investigated the possibility that GroPIns can regulate the inflammatory reactions in a simple and well characterized in vitro model system: the production of inflammatory cytokines in human blood monocytes upon challenge with endotoxin.
Human monocytes have a major role in the innate immune response to bacterial infection. Bacterial LPS binds TLR4 on the monocyte membrane and triggers the expression of a variety of inflammatory mediators, which include cytokines such as TNF-α and interleukin (IL)-1β, and enzymes, such as cyclooxygenase (COX)-2. These in turn produce inflammatory prostanoids, such as prostaglandin E2 and thromboxane A2 (TxA2) (15), which act locally to elicit the cardinal signs of acute inflammation (16).
We first tested the effects of GroPIns on mRNA levels of some of the major inflammatory mediators in freshly isolated human monocytes challenged with LPS (0.1 μg/ml). As expected, LPS induced a pronounced increase in the expression of IL-1β, TNF-α, COX-2, and the anti-inflammatory mediator IL-10 (Fig. 1, A–D). Preincubation with 300 μm GroPIns reduced the levels of IL-1β, TNF-α, and COX-2 mRNAs although to different extents (by 50–75% at 24 h; Fig. 1, A–C). Of note, treatment with GroPIns considerably reduced the expression of the inflammatory genes at both early (1.5 h) and long times (24 h) of stimulation with very consistent effects at long times (24 h; Fig. 1, A–C). However, GroPIns did not affect the expression of the anti-inflammatory cytokine IL-10 (Fig. 1D) or the suppressor of cytokine signaling 3, a transcriptional target of the signal transducer and activator of transcription 3 (STAT3) that is downstream of IL-10 signaling (Ref. 17 and supplemental Fig. S1). In parallel experiments, monocytes preincubated with 300 μm GroPIns were challenged with TNF-α (50 ng/ml) for 1.5 h; no effect on gene expression was observed under these conditions (Fig. 1, E and F), indicating that GroPIns specifically interferes with the LPS signaling cascade. Accordingly, GroPIns also affected the expression of cytokines when monocytes were challenged with agonists of TLR7/8 and TLR9 that share a common downstream pathway with TLR4 (i.e. the myeloid differentiation primary response gene 88 (MyD88)-dependent pathway (Ref. 18 and supplemental Fig. S1).
Figure 1.

GroPIns reduces the LPS-induced proinflammatory response in human monocytes. A–C, GroPIns inhibits the LPS-mediated transcription of TNF-α, IL-1β, and COX-2 at 1.5 and 24 h. D, GroPIns does not inhibit the LPS-mediated expression of IL-10. E and F, GroPIns does not inhibit the TNF-α-mediated expression of proinflammatory genes (TNF-α and IL-1β). Human monocytes were purified from peripheral blood of healthy donors and incubated at 37 °C for 20 min without or with 300 μm GroPIns and for an additional 1.5 or 24 h in the absence or presence of 0.1 μg/ml LPS or 50 ng/ml TNF-α. At each time point, the RNA was extracted and converted to cDNA for real-time PCR analysis. Data were calculated using the ΔΔ method (2−ΔΔCT). Transcription of the housekeeping gene GAPDH was used to normalize the data. Measurements of mRNA levels are expressed as -fold increase over the respective control (unstimulated) cells, and are means ± S.D. of triplicates (n = 3) from two experiments performed with cells from two different donors (A-C) and means ± S.D. of three independent experiments (n = 3) performed in duplicate (D–F). Asterisks indicate statistically significant differences (*, p < 0.05; **, p < 0.01). ns, not significant. G, GroPIns inhibits LPS-induced protein expression levels of TNF-α and IL-1β and reduces COX-derived TxB2. Monocytes were preincubated for 20 min at 37 °C in the absence or presence of 300 μm GroPIns and then incubated for a further 5 and 24 h without or with 0.1 μg/ml LPS. At the end of this incubation, the supernatants were prepared, and the levels of TNF-α, IL-1β, and TxB2 in the medium were assessed using ELISA. In untreated monocytes, the protein concentrations were under the assay detection limits at both times. Data show the percentage of inhibition induced by GroPIns treatment on LPS-induced protein release. Data represent three independent experiments performed with cells from three different donors. Error bars represent S.D.
In line with the effects on gene expression, 300 μm GroPIns reduced the LPS-stimulated release of both TNF-α and IL-1β cytokines at 24 h (with up to 60% inhibition) with negligible effects at earlier times (Fig. 1G). Moreover, GroPIns reduced the accumulation of TxB2 by 60%. TxB2 is the stable metabolite of TxA2, which is a major product of the metabolic activity of COX-2 in monocytes (Fig. 1G), thus confirming that these decreased mRNA levels result in reduced metabolic activity.
Monocytes also have a major role in thrombotic events through the de novo synthesis of tissue factor (TF), a transmembrane glycoprotein that initiates blood coagulation through its binding to factors VII and VIIa, which leads to fibrin clot formation (19). Treatment with 300 μm GroPIns reduced the LPS-induced expression of TF gene by 50% at 24 h of stimulation (Fig. 2A). Following these observations, we investigated whether modulation of TF gene expression also translates into a reduced procoagulant activity of LPS-challenged monocytes. Procoagulant activity is practically undetectable in freshly isolated monocytes, and it is strongly up-regulated by exposure to LPS (Fig. 2B). Treatment of monocytes with 50–300 μm GroPIns before LPS stimulation resulted in a dose-dependent reduction in TF activity with up to 60% inhibition (Fig. 2B). Similar to what was observed with the cytokines, the efficacy of GroPIns in reducing TF activity became significant after 24 h of stimulation (Fig. 2B). Moreover, a series of experiments were conducted at 24 h of GroPIns stimulation where the cells were challenged with 0.1–1000 ng/ml LPS after pretreatment with 300 μm GroPIns; the inhibitory effect of GroPIns on the procoagulant activity of TF was evident across this whole range of LPS concentrations (Fig. 2C).
Figure 2.

GroPIns inhibits LPS-induced tissue factor expression and activity in human monocytes. A, human monocytes were purified from peripheral blood of healthy donors and incubated at 37 °C for 20 min without or with GroPIns 300 μm and for an additional 1.5 or 24 h in the absence or presence of 0.1 μg/ml LPS. The RNA was extracted and converted to cDNA for real-time PCR analysis. Each sample was measured in triplicate, and the data were calculated using the ΔΔ method (2−ΔΔCT). Transcription of the housekeeping gene GAPDH was used to normalize the data. Measurements of mRNA levels are expressed as -fold increase over the respective control (unstimulated) cells and are means ± S.D. of triplicates from two experiments performed with cells from two different donors. Asterisks indicate statistically significant differences (*, p < 0.05). B, human monocytes were purified from peripheral blood of healthy donors and incubated at 37 °C for 20 min without or with increasing concentrations (50–300 μm) of GroPIns and for a further 5 or 24 h in the presence of 0.1 μg/ml LPS. At these time points, the cells were lysed, and the procoagulant activity of tissue factor was assessed using one-stage clotting time (see “Experimental procedures”). For each experiment, monocytes that were not activated with LPS were analyzed as the negative control. The data are means ± S.D. of triplicates from two experiments performed on cells from two independent donors. Asterisks indicate statistically significant differences (**, p < 0.01; ***, p < 0.001). C, monocytes pretreated with 300 μm GroPIns or left untreated were stimulated for 24 h with increasing concentrations of LPS. At the end of incubation, the cells were lysed, and procoagulant activity of tissue factor was assessed using one-stage clotting time (see “Experimental procedures”). The data are representative of more than 10 experiments performed with cells from different donors. Error bars represent S.D.
Thus, GroPIns can suppress proinflammatory and prothrombotic responses in human monocytes stimulated with LPS. This activity was consistently observed in numerous assays with samples obtained from different donors.
The action of GroPIns involves the inhibition of the extracellular signal-regulated kinase 1/2 (Erk1/2), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38) kinase activities
To investigate the mechanism through which GroPIns affects LPS-induced monocyte responses, we analyzed their effects on the phosphorylation of p38, JNK, and Erk1/2, three kinases that are known to transduce the LPS signal downstream of TLR4 (20). In these cells, LPS (0.1 μg/ml) activated Erk1/2, JNK, and p38, and treatment with GroPIns (300 μm) for different times reduced their phosphorylation by about 60% (Fig. 3A), indicating that GroPIns acts by modulating the activity of these kinases. Similar experiments were carried out in monocytes pretreated with 300 μm GroPIns4P, and no difference was observed between control and treated cells (Fig. 3A). This phosphorylated derivative, besides the already reported immunomodulatory activity in T lymphocytes (21), has also been shown to affect the LPS-induced transcription of proinflammatory genes in monocytes.7 Therefore, GroPIns and GroPIns4P are able to exert similar anti-inflammatory activity acting at different levels of the signaling cascade. These results are in line with current knowledge that both GroPIns and GroPIns4P act intracellularly as there is so far no evidence of a membrane receptor or of specific activity at the membrane level (6, 11).
Figure 3.

GroPIns affects the LPS-induced activation of Erk1/2, JNK, p38, and the IKKα–β complex. A, GroPIns affects the phosphorylation of Erk1/2, JNK, and p38. B, GroPIns affects both the phosphorylation of the IKK complex and the degradation of IκBα. C, GroPIns does not affect the TNF-α-induced phosphorylation of the IKK complex and degradation of IκBα. A–C, blots, left side of the panel, monocytes were treated without or with either GroPIns or GroPIns4P (300 μm) for 20 min and for an additional 5, 10, 20, and 30 min in the presence of 0.1 μg/ml LPS (A and B) or 50 ng/ml TNF-α (C). Representative Western blots resolved using 10% SDS-PAGE and revealing phosphorylated (p-) Erk1/2 (Thr-202/Tyr-204), phosphorylated JNK (Thr-183/Tyr-185), phosphorylated p38 (Thr-180/Tyr-182), and total levels of Erk1/2, JNK, and p38 are shown. The phosphorylated IKKα–β complex (Ser-176/180 on IKKα and Ser-177/181 on IKKβ), the phosphorylated IκBα (Ser-32/36), and the total levels of IκBα are also shown. These blots are representative of three independent experiments performed with cells purified from three independent donors. A–C, bar graphs, right side of the panel, densitometric analysis of the bands was performed with ImageJ software. The protein abundance has been expressed relative to the housekeeping signal (vinculin or GAPDH).
GroPIns inhibits the LPS-dependent signaling leading to the nuclear translocation of NF-κB
To evaluate the consequence of the reduced phosphorylation of Erk1/2, JNK, and p38, we have analyzed the well documented LPS-dependent control of transcription. LPS-responsive, cis-acting DNA promoter elements have been characterized in the 5′-flanking regions of several prothrombotic and inflammatory genes, including TF, COX-2, IL-1β, and TNF-α (22). The transcription factors that bind to these LPS-responsive elements include NF-κB, activator protein-1, and cAMP response element-binding protein, which all cooperate to produce various cytokines, the levels of which are barely detectable in resting cells. NF-κB, however, is the only transcription factor that is required for the induction of all of the LPS-inducible genes so far analyzed (23); therefore, it was selected for our investigation. The activation signaling to NF-κB involves its release from the cytosolic inhibitor IκBα followed by its translocation into the nucleus. This event is tightly dependent on the phosphorylation and proteasomal degradation of IκBα, which relies directly on the phosphorylation-dependent activation of the upstream IκB kinase (IKK) complex (24). We therefore investigated the activation of the IKK kinases and the phosphorylation and total levels of IκBα. In monocytes, LPS (0.1 μg/ml) elicited a rapid phosphorylation of the IKKα–β complex that was completely prevented when cells were exposed to 300 μm GroPIns (10-min LPS treatment). Consistent with this observation, the LPS-induced degradation kinetics of IκBα was delayed by GroPIns, reaching a maximum at 10 min (Fig. 3B). The inhibitory effect of GroPIns was not observed when monocytes were treated with TNF-α (50 ng/ml) (Fig. 3C). These results support the observation that GroPIns action is exerted on the signaling events triggered by LPS treatment and that its inhibitory effect is specific for the LPS signaling.
GroPIns reduces LPS-induced nuclear translocation of NF-κB and the binding to the promoters of target genes
As indicated above, IκBα determines the cell localization, and thus activity, of NF-κB. Because GroPIns was able to affect both the activation and degradation of IκBα, we evaluated the nuclear translocation of NF-κB in the presence of GroPIns by using antibodies specific for p65 and p50 proteins (the subunits forming the heterodimer mainly involved in transcriptional regulation; see also Ref. 24 and “Experimental procedures”). The LPS-induced nuclear translocation of NF-κB became evident after 15 min and maximal after 30 min (Fig. 4). Within 30 min of the GroPIns addition, the nuclear translocation of both p65 and p50 was reduced by 45 and 50%, respectively (Fig. 4B). This impairment in the nuclear translocation of NF-κB after LPS stimulation is in line with, and corroborates, the rapid effect of the GroPIns on IKKα–β complex phosphorylation (see above).
Figure 4.

GroPIns reduces the LPS-induced nuclear translocation of NF-κB. Representative confocal microscopy images of NF-κB intracellular localization in human peripheral blood monocytes are shown. A, cells were incubated at 37 °C for 20 min in the presence or absence of 300 μm GroPIns and then treated for 15 and 30 min (′) with 0.1 μg/ml LPS. Cells were then fixed and processed for immunofluorescence (see “Experimental procedures”). The intracellular localization of NF-κB was detected using antibodies specific for p50 and p65 proteins revealed with Alexa Fluor 488-labeled (green) and Alexa Fluor 568-lebeled (gray) secondary antibodies, respectively. Nuclei were stained with DAPI (blue). The samples were analyzed on a laser scanning confocal microscope (LSM710) equipped with a 63× objective. B, the nuclear translocation of NF-κΒ on cells left untreated or treated with LPS in the presence or absence of GroPIns has been quantified by randomly counting 100 cells/sample and expressing the percentage of cells with nuclear staining of p50 or p65. Quantification of nuclear events is presented as the mean ± S.D. of three independent experiments. Statistical analysis was performed using Student's t test (*, p < 0.05; **, p < 0.01). Error bars represent S.D. Scale bars, 5 μm.
Following these observations, we performed an electrophoretic mobility shift assay (EMSA) on nuclear extracts of human monocytes treated with LPS (0.1 μg/ml) in the absence and presence of GroPIns (50 and 300 μm). Because the LPS-induced nuclear translocation of NF-κB becomes evident after 15 min of treatment and maximal after 30 min and the GroPIns inhibition was detected within this time frame, we analyzed these nuclear extracts after 30 min of stimulation. As shown in Fig. 5A, GroPIns consistently reduced the LPS-induced binding of NF-κB to a radiolabeled oligo probe containing the NF-κB-binding site sequence (see “Experimental procedures”); of note, GroPIns was effective at both 50 and 300 μm. Under the same experimental conditions, GroPIns did not affect the binding to DNA of either cAMP response element-binding protein or activator protein-1, implying that the GroPIns modulation is specific for the LPS-induced activation of NF-κB (supplemental Fig. S3).
Figure 5.

GroPIns reduces the LPS-induced binding of NF-κB to promoters of target genes. A, GroPIns reduces the LPS-triggered binding of NF-κB to DNA. For the EMSA, monocytes were incubated at 37 °C for 20 min without or with the indicated concentrations of GroPIns and then incubated for a further 30 min in the presence of 0.1 μg/ml LPS. Unstimulated monocytes were incubated in parallel as a control (T0). At the end of the incubation, the cells were lysed, and the nuclear extracts were incubated with radiolabeled oligonucleotide probes that contained the NF-κB-binding site (see “Experimental procedures”). Protein–DNA complexes were separated using 5% non-denaturing acrylamide gels and visualized by autoradiography. Data are representative of two independent experiments. B, GroPIns directly displaces NF-κB from DNA. For the EMSA competition assay, GroPIns (at the indicated concentrations) was added to the binding mixture containing the radiolabeled probe and the nuclear lysates from monocytes treated with 0.1 μg/ml LPS for 60 min. The EMSA was performed as reported above. Data are representative of three independent experiments. C, left side of the blot, both p65–p50 heterodimer and p50–p50 homodimer participate in the binding to the radiolabeled probe. For the EMSA supershift, antibody specific for p65 or p50 protein was added to the binding mixture containing the radiolabeled probe and the nuclear lysates from monocytes treated with 0.1 μg/ml LPS for 60 min; an antibody specific for tubulin was used as a negative control. C, right side of the blot, HeLa cells treated with 20 ng/ml TNF-α for 20 min were used as a positive control. The EMSA was performed as reported above. D, GroPIns reduces the recruitment of p65 to promoters of tissue factor and TNF-α genes. For the ChIP assay, the binding of p65 on chromatin was evaluated by a specific rabbit polyclonal antibody to p65 protein; a normal rabbit IgG was used as a background control. The enriched promoters were quantified by real-time PCR using primers that specifically amplify a region containing the κB site. The intensity of the PCR signal is proportional to the occupancy on the binding site. The amount of p65-immunoprecipitated chromatin is represented as signal relative to the total amount of input chromatin (percentage of binding relative to input). Data are mean ± S.D. of three independent experiments with cells from three independent donors. Significance was tested by analysis of variance with Dunnett post hoc analysis (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Error bars represent S.D. UNTD, untreated.
To better clarify whether GroPIns could interfere with NF-κB activity by directly displacing it from the promoter, we performed in vitro competition assays. Nuclear extracts of LPS-treated monocytes were incubated with the radiolabeled probe and increasing concentrations of GroPIns; these were able to displace NF-κB from DNA (Fig. 5B).
Next, we examined which of the NF-κB subunits was involved in the binding to the radiolabeled probe by supershift assays (see “Experimental procedures”). The preincubation of nuclear lysates with an antibody specific either for p65 or for p50 induced a supershift that was more pronounced in the case of the p50 antibody (Fig. 5C). This observation is in line with the reported high expression of p50 homodimer in fresh monocytes (25).
The specificity of the antibodies used was validated in nuclear extracts of HeLa cells treated with TNF-α (20 ng/ml) for 20 min. Both antibodies supershifted the same complex, indicating that in this cellular model the heterodimer p50–p65 is mainly involved in the binding.
According to the data obtained, GroPIns may participate in the regulation of transcription by modulating the nuclear-cytoplasmic trafficking of NF-κB as well as by affecting the binding to the promoters of target genes. Therefore, we investigated the recruitment of NF-κB to the promoters of those genes. To this end, we performed a chromatin immunoprecipitation (ChIP) assay (see “Experimental procedures”) using a p65-specific antibody. The binding of p65 to promoters was quantified by real-time PCR performed on immunoprecipitated chromatin using primers that specifically amplify a genomic region containing the sequence bound by NF-κB (κB site) (26–28). As shown in Fig. 5D, treatment of monocytes with LPS (0.1 μg/ml; 60 min) elicited a significant binding of p65 on the promoters of both TF and TNF-α genes, whereas the concomitant addition of GroPIns (300 μm) decreased the LPS-induced binding by about 60 and 40%, respectively. In summary, the GroPIns inhibitory activity may be exerted both at the cytosolic level by regulating the kinase activities necessary for NF-κB translocation and at the nuclear level by reducing the binding of the NF-κB subunits to the promoter region.
GroPIns reduces LPS-induced endotoxin shock in the mouse
The actions of GroPIns as anti-inflammatory agent indicated by the in vitro data reported above prompted us to analyze its potential efficacy in an in vivo model of endotoxin shock in mice (29). The efficacy of GroPIns on inflammatory responses triggered by LPS in vivo was evaluated by measuring the levels of the TNF-α cytokine in the plasma of placebo-treated and GroPIns (10 mg/kg)-treated mice 2 h after challenge with LPS (1 mg/kg). TNF-α was practically undetectable in healthy, placebo-treated mice (Fig. 6A). In contrast, TNF-α significantly increased in the plasma of the LPS-treated mice, and it was significantly reduced by GroPIns treatment (Fig. 6A).
Figure 6.
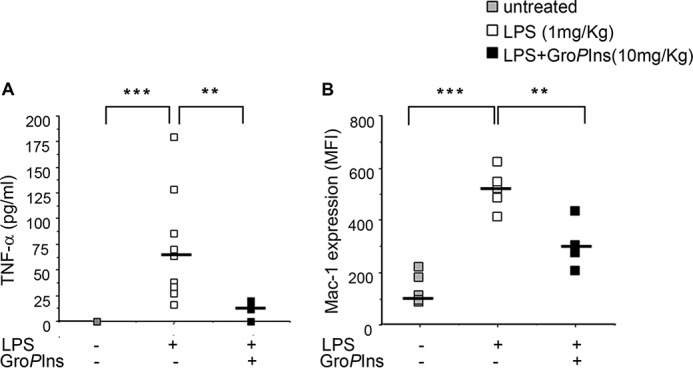
GroPIns reduces the endotoxin shock in LPS-treated mice. Effects on plasma levels of TNF-α cytokine and on Mac-1 expression in circulating neutrophils in LPS-treated mice are shown. The treatment protocols were described under “Experimental procedures.” Citrated whole blood from vehicle- or GroPIns-treated mice (nine mice per group) was collected 2 h after LPS challenge. A, aliquots of whole blood were immediately centrifuged, and the platelet-poor plasma was stored at −80 °C for measurements of TNF-α cytokine using specific immunoassays. B, aliquots were incubated for 15 min with phycoerythrin-conjugated rat anti-mouse Mac-1 antibody (clone M1/70). Nonspecific phycoerythrin-conjugated rat IgG was used as a negative control staining. After staining, the red blood cells were lysed by addition of lysing-fixing solution according to the manufacturer's instructions. Flow cytometric analysis was performed with a FACStar flow cytometer as follows. Events showing side light scatter (granularity) and forward light scatter (dimension) characteristic of polymorphonuclear leukocytes were analyzed for Mac-1 expression levels. A and B, data are for single animals, and the horizontal lines indicate the median values. **, p < 0.01; ***, p < 0.001. MFI, mean fluorescence intensity.
As an additional marker of inflammatory cell activation, we evaluated the up-regulation of Mac-1 in circulating neutrophils using flow cytometry. LPS induces the delivery of the intracellularly stored Mac-1 to the plasma membrane of neutrophils. The increased cell surface expression favors leukocyte adhesion to the activated endothelium and crawling into blood vessels, contributing to transmigration toward inflamed tissues (30). Once there, Mac-1 also mediates inflammatory effector functions, such as phagocytosis and release of bactericidal products, that are responsible for tissue damage during acute inflammation (31). As shown in Fig. 6B, LPS significantly up-regulated Mac-1 expression, and this was significantly affected by the treatment with GroPIns.
Discussion
The present study provides new information on the physiological role and pharmacological potential of the PLA2IVα metabolite GroPIns as a novel mediator in the resolution of inflammation. The data thus pertain to the line of studies that have addressed the possibility of using lipid derivatives for the treatment of immune/inflammatory diseases (4).
Although lipid mediators that originate from PLA2/arachidonic acid metabolism, such as the prostaglandins and leukotrienes, are essential for mounting an inflammatory reaction, other lipid metabolites derived from arachidonic, eicosapentaenoic, and docosahexaenoic acids, such as lipoxins, resolvins, protectins, and maresins, mediate the resolution phase of inflammation (32). From a physiological point of view, GroPIns can be considered as the counterpart of the arachidonic acid cascade as it is formed in the same enzymatic pathway when the PLA2 acts preferentially on the membrane phosphoinositides (7, 8), and importantly, it parallels some of the anti-inflammatory effects of the arachidonic acid metabolites (see above). GroPIns can therefore be thought of as an adjuvant/cooperator in anti-inflammatory strategies.
The GroPIns anti-inflammatory potential is also involved in the ability of this compound to counteract blood-brain barrier failure, replicating in this way the effects of dexamethasone as analyzed in an in vitro model based on a co-culture of endothelial cells and glia (33). Indeed, GroPIns improved blood-brain barrier functions and repair in a dose-dependent manner without the cytotoxic effect that was observed with high doses of dexamethasone (33). The authors have thus proposed this natural compound as a powerful alternative to steroidal drugs to be further validated in in vivo studies (33).
As indicated, GroPIns counteracts the LPS-induced proinflammatory and prothrombotic responses in human blood monocytes, inhibiting the signaling of TLR4 and leading to a decrease in the nuclear translocation and binding of the transcription factor NF-κB to promoters. This reduces the transcription of inflammatory genes.
The activity of GroPIns is specific because it was ineffective when cells were treated with TNF-α. It is worth noting that both TNF-α and LPS share a common pathway downstream of the IKK complex, but they differ in regard to the components linking the receptor to the activation of the IKK complex (24). Thus, GroPIns appears to act on this early part of the LPS pathway, which is independent of the TNF-α signaling. Accordingly, we observed a specific inhibitory effect of GroPIns in the TLR-activated pathway involving MyD88 but not in that involving toll/interleukin-1 receptor (TIR) domain-containing adapter inducing interferon-β (supplemental Figs. S1 and S2). This is also in line with our observation that GroPIns directly binds to a component of the TLR pathway, the protein-tyrosine phosphatase Shp1 (34), as indicated by our proteomic analysis of coimmunoprecipitated complexes, with consequences on its activity.8
The reported competition assays also suggest a direct nuclear activity of GroPIns. NF-κB binding to promoters and target gene expression rely on a number of nuclear events, such as the correct recruitment of remodeling complexes that act on the chromatin architecture (35), balance among the regulators of NF-κB stability (36), and relocalization of the monomer p65 from κB sites to specific nuclear compartments (37).
We have analyzed canonical and validated κB sites within the promoters (26) because it is common knowledge that functional NF-κB-binding sites are present inside enhancer/intronic regions (38). Indeed, NF-κB is a well recognized master regulator of the inflammatory response and a valuable therapeutic target. However, we cannot rule out an indirect effect of GroPIns involving its binding and/or action on other cofactors that cooperate with NF-κB in the transcriptional output (39). This possibility is supported by the observation that under the conditions tested there is no evidence of a direct binding of GroPIns to members belonging to the NF-κB family. At the same time, more than 50 different GroPIns potential targets with different activities have been identified in a proteomic study.9 These include several nuclear proteins that could play a role in the process reported, but their characterization and validation are still in progress.9
These actions of GroPIns have been studied using a pharmacological approach (i.e. exogenous addition); under physiological conditions, this process starts from the production and release of GroPIns upon activation of PLA2IVα in monocytes (7). Indeed, the stimulated intracellular levels of GroPIns fall well within the range of concentrations that can elicit the effects described (40); however, a direct demonstration that these phenomena can take place in co-culture of cells or tissue is hampered by the lack of a means to fully inhibit GroPIns production. Actually, GroPIns is still produced in PLA2IVα−/− cells, potentially by a rescuing activity of other cytosolic PLA2s in the cells (11, 41).
A possible physiological role of endogenously formed GroPIns can be envisioned in the context of transcription. The nucleus is considered a functionally distinct compartment for inositol lipid metabolism (42), and because PLA2IVα is able to translocate to the nuclear envelope and locally hydrolyze phosphoinositides (43), it could form GroPIns from the nuclear membrane phosphoinositides and be responsible for its reported nuclear activity. Alternatively, the GroPIns formed in the cytosol (40, 44) could diffuse in the nucleus to exert its function as also proposed for the inositol phosphates (45). The diverse nuclear roles of these compounds have long been known, but a clear quantification or visualization has been hampered by their ability to diffuse and by the lack of specific probes to follow their dynamic distribution (45). In either case, in parallel to the pharmacological applications reported in this study, endogenous GroPIns could exert a similar function intracellularly during the spontaneous resolution of inflammation. In support of this hypothesis, we have observed a pronounced increase (about 20-fold) in GroPIns levels (as measured by liquid chromatography-mass spectrometry (LC-MS/MS) (46)) in isolated human monocytes challenged with LPS; this may reflect the increase occurring in all cell compartments, including the nucleus (Fig. 7; the time course of the LPS-induced GroPIns production is also indicated; see supplemental Fig. S4).
Figure 7.
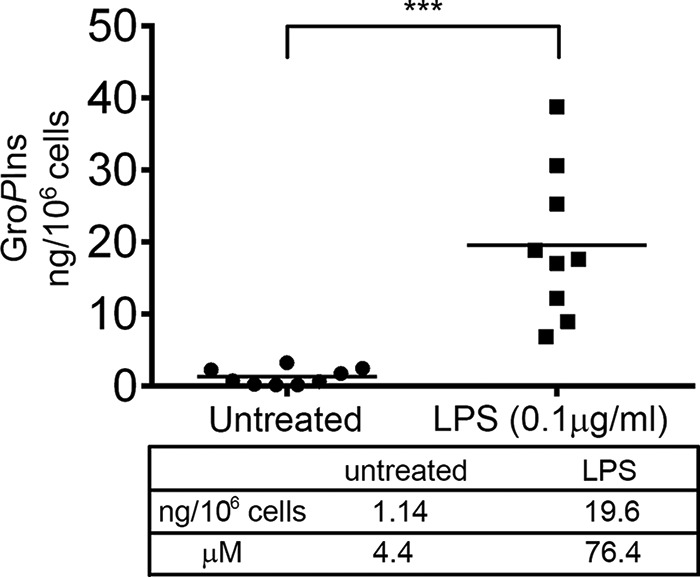
LPS treatment induces a 20-fold increase in the GroPIns levels in human monocytes. Human peripheral blood monocytes purified from nine donors were treated with LPS (0.1 μg/ml) for 45 min or left untreated as a control, and the intracellular GroPIns was collected by acidified Bligh and Dyer (56) extraction (54) following the modifications reported previously (46). The quantification of the intracellular GroPIns was performed by an LC-MS/MS method recently reported in detail (46). Dots represent single donors, and the horizontal lines indicate the mean values. Statistical analysis was performed using Student's t test (***, p < 0.001).
In addition to the ability to modulate the inflammatory response, GroPIns and the other glycerophosphoinositols have been demonstrated to be active in different contexts (13, 41). For example, in human leukemia T-lymphocytes and in peripheral blood lymphocytes, GroPIns4P was shown to synergize with the chemotactic factor stromal cell-derived factor 1α (CXCL12) by increasing cytokine-dependent migration of T-lymphocytes, thus revealing an immunomodulatory effect of this compound (21). Although this effect of GroPIns4P was observed upon exogenous/pharmacological addition, its potential physiological relevance is consistent with the fact that a number of hematopoietic cells produce significant levels of glycerophosphoinositols upon proinflammatory stimuli (6, 11). In particular, antigen-presenting cells have been shown to produce and release glycerophosphoinositols in the extracellular medium where they could function as paracrine factors for lymphocytes (13, 21, 41).
It should be pointed out that, based on the information collected so far, the different glycerophosphoinositols exert separate functions based on the specific intracellular target/receptor to which they can bind.8 This is not surprising and in fact parallels the behavior of the other inositol-containing molecules, such as the phosphoinositide and inositol phosphate families, which participate in different mechanisms according to the number of phosphate residues bound to the inositol ring (6). In addition, the metabolism of the inositol-containing molecules needs to be thought of as a whole in the context of cell homeostasis, and it depends on the available inositol pool in the different cell systems and conditions (resting versus receptor stimulation and transformation (40)).
Here we have focused on the pharmacological exploitation of GroPIns. The administration of GroPIns might act on monocytes to decrease the expression of cytokines and TF gene. This would favor the resolution of inflammation and control potentially harmful prothrombotic activities. The same mechanism might be involved in the GroPIns-mediated blood-brain barrier repair mentioned above (33).
It is conceivable that the capability of GroPIns to regulate TF expression might be exploited to control disseminated intravascular coagulation, the most devastating complication of sepsis (14, 47), which remains untouched by specific therapeutic treatments. The advantages of using this compound in the resolution of inflammation are severalfold. Because of its chemical nature, GroPIns is easily manageable: it is water-soluble, and it crosses the plasma membrane, rapidly reaching its intracellular targets (48). As it is a natural compound that is present in virtually all cell types, it could be administered (e.g. intravenously) in cases of disseminated intravascular coagulation that do not respond to therapy (i.e. septic shock) (47). These are proposals that require further evaluation in in vivo models of inflammatory disease. Nevertheless, GroPIns is a very promising compound as indicated by the reduced plasma levels of TNF-α it induces in a model of endotoxin shock in mice (see “Results”). Moreover, GroPIns reduces the surface expression of Mac-1, which is involved in both chemokine-guided leukocyte infiltration and detrimental inflammatory effector functions; indeed, the inhibition of Mac-1 has been shown to reduce both neutrophil transmigration and tissue injury in vivo (49). Although it is widely accepted to consider leukocyte infiltration as a common marker of inflammation, it is worth mentioning that neutrophils can also perform active healing functions during resolution (50); indeed, the inhibition of their trafficking toward injured sites is a common and effective mechanism shared by several proresolving lipid mediators (4).
We conclude that GroPIns has the potential to regulate the inflammatory response. When produced by monocytes/macrophages, GroPIns can act in a potential negative feedback loop that signals the switching off of the inflammatory response and, in parallel, the inhibition of both expression10 and activity (51) of PLA2. In summary, by targeting proinflammatory and prothrombotic mediators and thereby affecting inflammatory-related transcription factors and deactivating inflammatory cells, GroPIns would shape an anti-inflammatory microenvironment favoring the restoration of tissue homeostasis. In view of this scenario, our data supportive the pharmacological exploitation of this compound (55).
Experimental procedures
Cell culture
Monocytes were isolated from whole blood obtained from healthy donors who gave their informed consent to participate in the study and who did not take any form of medication for at least 10 days before blood donation. Approval was obtained from the independent ethics committee, and buffy coats were provided by Istituto Nazionale per lo Studio e il Trattamento dei Tumori, Fondazione Giovanni Pascale (Naples, Italy). Initial experiments were carried out on human monocytes obtained with approval from the Consorzio Mario Negri Sud Review Board for these studies. The human monocytes were taken to more than 95% purity through two centrifugation steps on Lymphoprep (Axis-Shield, Oslo, Norway) followed by a Percoll (GE Healthcare) gradient as described previously (52) and finally resuspended in serum-free RPMI 1640 medium (Gibco, Life Technologies). These monocytes were left unstimulated or were challenged with 0.1 μg/ml bacterial LPS (E. coli serotype 055:B5; Sigma-Aldrich) or TNF-α (210-TA-010/CF, R&D Systems, Minneapolis, MN) with incubations for different times at 37 °C in 5% CO2. GroPIns and GroPIns4P were provided by Echelon Biosciences Inc. HeLa cells were from American Tissue Type Collection (ATTC) and were grown in minimum Eagle's medium supplemented with 10% fetal calf serum (Biochrom, Cambridge, UK). Cells were grown under a controlled atmosphere in the presence of 5% CO2 at 37 °C.
Procoagulant activity
The procoagulant activity of TF was measured in cell lysates according to one-stage clotting time. Briefly, 100 μl of cell lysate was added to a tube containing 100 μl of prewarmed, pooled normal human plasma. After addition of 100 μl (20 mm) CaCl2, the clotting time was determined using a KC4A Amellung coagulometer (Mascia Brunelli, Milan, Italy). The clotting times were converted to arbitrary units by interpolation with a standard curve generated with serial dilutions of human recombinant thromboplastin (Hemoliance RecombiPlasTin, Instrumentation Laboratory Co., Pleasantville, NY). As the concentration of recombinant TF (Hemoliance RecombiPlasTin) used to produce the standard curve of procoagulant activity is not reported by the manufacturer, we measured the TF concentrations in the serial dilutions used to produce the standard activity curve using an IMUBIND enzyme-linked immunosorbent assay (ELISA) (International Laboratory Co.). The correlations between TF activity and the concentration of the TF antigen in the recombinant TF preparations have been reported (52).
Western blotting
The levels of phosphorylation of p38, Erk1/2, and JNK were analyzed by Western blotting using the following phosphospecific antibodies: phospho-p38 MAPK (Thr-180/Tyr-182, catalog number 9211, rabbit polyclonal, Cell Signaling Technology, Beverly, MA; 1:3000), phospho-JNK (Thr-183/Tyr-185, catalog number 4668, rabbit polyclonal, Cell Signaling Technology; 1:3000), and phospho-p44/42 MAPK (Erk1/2) (Thr-202/Tyr-204, catalog number 4370, rabbit monoclonal, Cell Signaling Technology; 1:3000). The level of phosphorylated IκBα was analyzed using an antibody that specifically recognizes phosphoserine 32/36 on IκBα (phospho-IκBα, catalog number 9246, mouse monoclonal, Cell Signaling Technology; 1:3000). The levels of IKKα–β phosphorylation were analyzed using an antibody that recognizes phosphoserine 176/180 on IKKα and phosphoserine 177/181 on IKKβ (phospho-IKKα–β, catalog number 2697, rabbit monoclonal, Cell Signaling Technology; 1:3000). In parallel, total proteins were monitored with the following specific antibodies: p38 (p38α mitogen-activated protein kinase, catalog number 9218, rabbit polyclonal, Cell Signaling Technology; 1:3000), JNK (SAPK/JNK, catalog number 9258, rabbit monoclonal, Cell Signaling Technology; 1:3000), Erk1/2 (Erk1, sc-94, rabbit polyclonal, Santa Cruz Biotechnology, Inc., San Diego, CA; 1:3000), IKKα (catalog number 11930, mouse monoclonal, Cell Signaling Technology; 1:2000), and IκBα (catalog number 4812, rabbit polyclonal, Cell Signaling Technology; 1:3000). The levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog number 4699-9555, mouse monoclonal, AbD Serotec, Kidlington, Oxford, UK; 1:10,000) and vinculin (hVin-1, catalog number v9131, Sigma-Aldrich) were used as loading controls.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells using the thiocyanate/cesium chloride method. One microgram of total RNA was converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Applied Biosystems). Real-time PCR were performed using 10 ng of cDNA, 50 nm concentration of each primer, and SYBR Green Master Mix (Applied Biosystems) in 20-μl reactions using an Applied Biosystems PRISM 7500 Fast Real-time PCR System. The reverse transcription was primed using random hexamers. The following TF gene sequences were used as validated primers: cag tga ttc cct ccc gaa ca (forward) and tgc ctt tct aca act gtg tag ag (reverse). Preliminary validation assays showed a band on agarose gel electrophoresis of the expected size for amplification of TF cDNA in samples of monocytes/platelets co-incubated for 24 h. The TF cDNA band was present in samples treated with DNase I but not in the samples in which Moloney murine leukemia virus reverse transcriptase was omitted, indicating that the signal is not from contaminated genomic DNA (52). Similarly, real-time PCR measurements of mRNA expression of COX-2, TNF-α, IL-1β, and IL-10 were performed using the following validated primers: COX-2 forward, ttc cag atc cag agc tca tta aa; COX-2 reverse, ccg gag cgg gaa gaa ct; TNF-α forward, cct gta gcc cat gtt gta g; TNF-α reverse, tgg tta tct ctc agc tcc ac; IL-1β forward, gat gca cct gta cga tca ct; IL-1β reverse, gac atg gag aac aac act t; IL-10 forward, ttc ttc cct gtg aaa aca ag; IL-10 reverse, tca aac tca ctc atg gct tt; GAPDH forward, caa ctt tgg tat cgt gga agg ac; and GAPDH reverse, aca gtc ttc ttg gtg gca gtg (GAPDH served as the housekeeping gene). Each sample was measured in triplicate, and the data generated were analyzed with SDS 2.0 software (Applied Biosystems) using the ΔΔ method (2−ΔΔCT) for comparison of relative expression results. Resting monocytes incubated alone were considered as the reference sample.
ELISA
Conditioned medium from monocytes was sedimented by centrifugation at 300 × g at room temperature for 10 min, and the supernatant was recovered. The concentrations of TNF-α, IL-1β, and TxB2 were assessed according to the manufacturer's protocol (Amersham Biosciences).
NF-κB DNA-binding assay
Nuclear extracts for EMSA were prepared as reported previously (53) with modifications. Monocytes, pretreated with GroPIns (300 μm; 20 min) and challenged with LPS (0.1 μg/ml), were harvested in PBS and centrifuged 5 min at 300 × g. Pellets were lysed in buffer A (10 mm Hepes, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT) supplemented with protease and phosphatase inhibitors (Roche Applied Science) and incubated for 15 min at 4 °C. 10% Triton X-100 was added, and extracts were centrifuged for 1 min at 12,000 × g. The supernatants were recovered as the cytosolic fraction, and the pellets containing intact nuclei were lysed in buffer C (20 mm Hepes, pH 7.9, 25% (v/v %) glycerol, 0.4 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm DTT, 0.5 mm PMSF) supplemented with protease and phosphatase inhibitors and incubated for 30 min at 4 °C on a shaker. Lysates were centrifuged for 10 min at maximum speed, and the supernatants were recovered as the nuclear fraction. Nuclear extracts were incubated with 32P-radiolabeled (Amersham Biosciences) double-stranded oligonucleotide probes containing the NF-κB-binding site, 5′-AgTTgAggggATTTCCCAggC-3′ (Sigma-Aldrich). Protein–DNA complexes were separated on a 5% non-denaturing acrylamide gel in 0.5× Tris borate-EDTA. Autoradiography was performed on Kodak XAR film. For competition assays, monocytes were treated with LPS (0.1 μg/ml) or left untreated as a control, and nuclear lysates were obtained as reported above. Different concentrations of GroPIns were added to the nuclear lysates together with the radiolabeled probe. For supershift assays, p65 antibody (NF-κB p65, catalog number 8242, rabbit monoclonal, Cell Signaling Technology), p50 antibody (NF-κB1 p105/p50, catalog number 12540, rabbit monoclonal, Cell Signaling Technology), or tubulin (sc-5286, mouse monoclonal, Santa Cruz Biotechnology, Inc.) was added together with the radiolabeled probe to the nuclear lysates obtained from untreated or LPS-treated monocytes.
ChIP assay
Monocytes were treated with GroPIns (300 μm; 20 min) and then challenged with LPS (0.1 μg/ml) for 60 min. Chromatin for the immunoprecipitation was prepared by fixing the cells by adding 37% formaldehyde (Sigma-Aldrich) into the medium at a final concentration of 1% for 10 min and quenching with 125 mm glycine (Sigma-Aldrich) for 5 min. Cells were collected and lysed in SDS buffer (1.1% SDS, 10 mm EDTA, pH 8, 50 mm Tris, pH 8) with protease inhibitors. Chromatin was sonicated for three cycles of 10-s pulse at 30% of maximum power (Branson Sonifier) to obtain fragments 500–1000 kb in size. Samples were then centrifuged and diluted by 10-fold with dilution buffer (1% Triton X-100, 1.2 mm EDTA, pH 8, 16.7 mm Tris, pH 8, 167 mm NaCl) with protease inhibitors and precleared with 25 μl of 50% salmon sperm/protein A-agarose slurry (EMD Millipore) for 30 min at 4 °C. Precleared chromatin was incubated overnight with 2 μg of p65 polyclonal antibody (7970, chip grade, Abcam, Cambridge, UK) or normal rabbit IgG (Santa Cruz Biotechnology, Inc.). A portion of the precleared chromatin (10% of the sample) was stored at 4 °C as input to further quantitate the amount of immunoprecipitated DNA. The antibody–protein–chromatin complex was isolated with 25 μl of 50% salmon sperm/protein A-agarose slurry (Millipore) for 2 h at 4 °C. After washing, pellets were eluted (1% SDS, 0.1 m NaHCO3), reverse cross-linked with 5 m NaCl at 65 °C overnight, and treated with RNase A (Thermo Fisher Scientific, Waltham, MA) and Proteinase K (Thermo Fisher Scientific). Samples were purified with a commercially available purification kit (Qiagen, Hilden, Germany) and used as templates in real-time PCR analysis. The intensity of the PCR signal is proportional to the occupancy on the binding site. Normalization of data has been performed by using the percent input method: signals obtained by ChIP/PCR have been divided by signals obtained from input/PCR. The validated primers used were: TNF-α forward, GCGATGGAGAAGAAACCGAG; TNF-α reverse, GAGGGCGGGGAAAGAATCA; TF forward, GCAACTAGACCCGCCTGC; and TF reverse, CTCCTCCCGGTAGGAAACTC.
Immunofluorescence
For nuclear trafficking of NF-κB, immunofluorescence experiments were performed as follows. Cells (3 × 105) were seeded on glass coverslips in culture medium and treated the day after according to the experimental protocol. Cells were rinsed with PBS and fixed with 4% (w/v) paraformaldehyde for 10 min at room temperature and permeabilized with blocking solution containing 0.05% (w/v) saponin for 20 min. The intracellular localization of NF-κB was revealed using antibodies specific to p65 (catalog number 7970, Abcam; dilution, 1:30) or p50 (catalog number 12540, Cell Signaling Technology; dilution, 1:50) for 1 h at room temperature. After washes, cells were stained with Alexa Fluor 568-labeled (A10042, Molecular Probes) or Alexa Fluor 488-labeled (A21206, Molecular Probes) secondary antibody and 4,6-diamidino-2-phenylindole (DAPI) dye for 1 h at room temperature. Coverslips were mounted in Mowiol (Calbiochem) and analyzed on a laser scanning confocal microscope (LSM710, Carl Zeiss) equipped with a 63× objective. The nuclear translocation of NF-κB on monocytes treated with LPS in the presence or absence of GroPIns was quantified by randomly counting 100 cells/sample and expressing the percentage of cells labeled with p50 or p65 nuclear staining for each sample.
Analysis of the intracellular GroPIns levels
Peripheral blood monocytes purified from nine healthy donors were treated with LPS (0.1 μg/ml) for 45 min or left untreated, and intracellular GroPIns was recovered by Bligh and Dyer (56) extraction (54) with minor modifications (as detailed under “Sample preparation” in Ref.46). The quantitative analysis of GroPIns was performed by an LC-MS/MS method as reported previously (46).
Mouse experiments
Mouse treatments
C57BL/6J male mice (9–12 weeks old) were obtained from Charles River Laboratories (Calco, Lecco, Italy), fed ad libitum, and housed in a temperature-controlled and light-controlled room. Treatments were approved by the Ethics Committee of the Consorzio Mario Negri Sud. GroPIns (Euticals S.p.A, Lodi, Italy) or saline (vehicle) was given intraperitoneally (i.p.). The mice were treated with three doses of GroPIns (10 mg/kg) administered 15 min before, at the same time as, and 60 min after LPS administration (1 mg/kg; E. coli serotype 055:B5). Two hours after LPS administration, the mice were anesthetized with a mixture of ketamine and xylazine (100 and 20 mg/kg, respectively), and their blood was collected in 0.38% trisodium citrate (final concentration) by cardiac puncture.
Measurement of TNF-α cytokine
Blood was centrifuged for 10 min to obtain platelet-poor plasma. This plasma from each mouse was divided in aliquots and stored at −80 °C before analysis. Mouse TNF-α was measured using ELISA kits (Pierce Biotechnology).
Flow cytometric analysis of Mac-1 expression in neutrophils from mouse whole blood
Citrated whole blood was diluted 1:1 with citrated saline and incubated for 15 min with phycoerythrin-conjugated rat anti-mouse Mac-1 (clone M1/70, BD PharmingenTM). Nonspecific phycoerythrin-conjugated rat IgG was used as the staining negative control. After staining, the red blood cells were lysed by addition of lysing-fixing solution (BD Biosciences) according to the manufacturer's instructions. Flow cytometry analysis was performed with a FACStar flow cytometer as follows. Events showing side light scatter (granularity) and forward light scatter (dimension) characteristic of polymorphonuclear leukocytes were analyzed for Mac-1 expression levels. The data are shown as mean fluorescence intensities for each of the nine mice analyzed.
Statistical analysis
The data are expressed as means ± S.E. Statistical differences between two groups were determined by paired t tests. To test for differences across different treatment groups, repeated-measures analysis of variance and Dunnett tests were used. Statistical significance was defined as p < 0.05.
Author contributions
M. V. and P. Z. designed, performed, and analyzed all the experiments and co-wrote the manuscript. A. V. contributed to the in vitro experiments and to the discussion. S. M. and V. E. designed and analyzed the initial experiments; V. E. also co-wrote the manuscript. M. Z and T. d. C. performed and discussed the EMSA. A. D. S., C. A, and G. D.' E. performed all the in vivo experiments and part of the in vitro experiments. A. C. and A. F. performed and discussed LC-MS/MS analysis. C. C. and G. D. C. provided buffy coats. D. C. conceived and supervised the project, discussed and analyzed the data, and co-wrote the manuscript.
Supplementary Material
Acknowledgments
We thank C. Valente (Institute of Protein Biochemistry (IBP), National Research Council (CNR)) for assistance with the immunofluorescence analyses and discussion, D. Acampora (Institute of Genetics and Biophysics, CNR) for assistance in ChIP analysis, D. Boraschi and A. Luini (IBP, CNR) for discussion and critical reading of the manuscript, L. Grauso for help in the LC-MS/MS analysis, N. Martelli for FACS analysis, and A. Capasso (IBP, CNR) for help with the biochemical assays.
This work was supported in part by Italian Association for Cancer Research (Milan, Italy) Grants IG10341 and IG14675, the Ministry for Education, Universities and Research Project “FaReBio di Qualità,” Programma Operativo Nazionale Project 01-00862, Research Projects of National Interest Project 2012CK5RPF_05, National Research Plan-National Research Council Aging Program 2012–2014, Progetto Bandiera “Epigen,” and the Programma Operativo Regionale Project OcKey. D. Corda, P. Zizza, and S. Mariggiò are authors of United States Patent 9351983, Use of Glycerophosphoinositols for the Treatment of Septic Shock.
This article contains supplemental Figs. S1–S4.
P. Zizza and D. Corda, unpublished observations.
A. Varone, S. Mariggiò, A. Varriale, M. Vessichelli, F. Formiggini, N. Brancati, M. Frucci, S. D'Auria, P. Pucci, A. Flagiello, C. Iannuzzi, C. Valente, and D. Corda, submitted for publication.
A. Varone and D. Corda, unpublished observations.
M. Vessichelli, A. Varone, and D. Corda, unpublished observations.
- GroPIns
- glycerophosphoinositol
- GroPIns4P
- glycerophosphoinositol 4-phosphate
- TLR
- toll-like receptor
- Mac-1
- αMβ2 integrin
- TF
- tissue factor
- COX
- cyclooxygenase
- IκBα
- inhibitor of κB
- IKK
- IκB kinase
- p38
- p38 mitogen-activated protein kinase
- MyD88
- myeloid differentiation primary response gene 88
- PLA2
- phospholipase A2
- Tx
- thromboxane.
References
- 1. Medzhitov R. (2008) Origin and physiological roles of inflammation. Nature 454, 428–435 [DOI] [PubMed] [Google Scholar]
- 2. Dinarello C. (2007) Historical insights into cytokines. Eur. J. Immunol 37, S34–S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dennis E. A., and Norris P. C. (2015) Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 15, 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serhan C. N., Chiang N., Dalli J., and Levy B. D. (2014) Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 7, a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nathan C., and Ding A. (2010) Nonresolving inflammation. Cell 140, 871–882 [DOI] [PubMed] [Google Scholar]
- 6. Corda D., Iurisci C., and Berrie C. P. (2002) Biological activities and metabolism of the lysophosphoinositides and glycerophosphoinositols. Biochim. Biophys. Acta 1582, 52–69 [DOI] [PubMed] [Google Scholar]
- 7. Zizza P., Iurisci C., Bonazzi M., Cossart P., Leslie C. C., Corda D., and Mariggiò S. (2012) Phospholipase A2IVα regulates phagocytosis independent of its enzymatic activity. J. Biol. Chem. 287, 16849–16859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mariggiò S., Sebastià J., Filippi B. M., Iurisci C., Volonté C., Amadio S., De Falco V., Santoro M., and Corda D. (2006) A novel pathway of cell growth regulation mediated by a PLA2α-derived phosphoinositide metabolite. FASEB J. 20, 2567–2569 [DOI] [PubMed] [Google Scholar]
- 9. Filippi B. M., Mariggiò S., Pulvirenti T., and Corda D. (2008) SRC-dependent signalling regulates actin ruffle formation induced by glycerophosphoinositol 4-phosphate. Biochim. Biophys. Acta 1783, 2311–2322 [DOI] [PubMed] [Google Scholar]
- 10. Mancini R., Piccolo E., Mariggiò S., Filippi B. M., Iurisci C., Pertile P., Berrie C. P., and Corda D. (2003) Reorganization of actin cytoskeleton by the phosphoinositide metabolite glycerophosphoinositol 4-phosphate. Mol. Biol. Cell 14, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corda D., Zizza P., Varone A., Filippi B. M., and Mariggiò S. (2009) The glycerophosphoinositols: cellular metabolism and biological functions. Cell. Mol. Life Sci. 66, 3449–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falasca M., Iurisci C., Carvelli A., Sacchetti A., and Corda D. (1998) Release of the mitogen lysophosphatidylinositol from H-Ras-transformed fibroblasts; a possible mechanism of autocrine control of cell proliferation. Oncogene 16, 2357–2365 [DOI] [PubMed] [Google Scholar]
- 13. Corda D., Zizza P., Varone A., Bruzik K. S., and Mariggiò S. (2012) The glycerophosphoinositols and their cellular functions. Biochem. Soc. Trans. 40, 101–107 [DOI] [PubMed] [Google Scholar]
- 14. Levi M. (2014) Diagnosis and treatment of disseminated intravascular coagulation. Int. J. Lab. Hematol. 36, 228–236 [DOI] [PubMed] [Google Scholar]
- 15. Suzuki T., Hashimoto S., Toyoda N., Nagai S., Yamazaki N., Dong H. Y., Sakai J., Yamashita T., Nukiwa T., and Matsushima K. (2000) Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. Blood 96, 2584–2591 [PubMed] [Google Scholar]
- 16. Lawrence T., Willoughby D. A., and Gilroy D. W. (2002) Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2, 787–795 [DOI] [PubMed] [Google Scholar]
- 17. Murray P. J. (2006) Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 6, 379–386 [DOI] [PubMed] [Google Scholar]
- 18. O'Neill L. A., Golenbock D., and Bowie A. G. (2013) The history of Toll-like receptors—redefining innate immunity. Nat. Rev. Immunol. 13, 453–460 [DOI] [PubMed] [Google Scholar]
- 19. Nemerson Y. (1988) Tissue factor and hemostasis. Blood 71, 1–8 [PubMed] [Google Scholar]
- 20. Kim J. H., Na H. K., Pak Y. K., Lee Y. S., Lee S. J., Moon A., and Surh Y. J. (2008) Roles of ERK and p38 mitogen-activated protein kinases in phorbol ester-induced NF-κB activation and COX-2 expression in human breast epithelial cells. Chem. Biol. Interact. 171, 133–141 [DOI] [PubMed] [Google Scholar]
- 21. Patrussi L., Mariggiò S., Paccani S. R., Capitani N., Zizza P., Corda D., and Baldari C. T. (2007) Glycerophosphoinositol-4-phosphate enhances SDF-1α-stimulated T-cell chemotaxis through PTK-dependent activation of Vav. Cell. Signal. 19, 2351–2360 [DOI] [PubMed] [Google Scholar]
- 22. Sweet M. J., and Hume D. A. (1996) Endotoxin signal transduction in macrophages. J. Leukoc. Biol. 60, 8–26 [DOI] [PubMed] [Google Scholar]
- 23. Müller J. M., Ziegler-Heitbrock H. W., and Baeuerle P. A. (1993) Nuclear factor κB, a mediator of lipopolysaccharide effects. Immunobiology 187, 233–256 [DOI] [PubMed] [Google Scholar]
- 24. Karin M. (1999) How NF-κB is activated: the role of the IκB kinase (IKK) complex. Oncogene 18, 6867–6874 [DOI] [PubMed] [Google Scholar]
- 25. Lewin S. R., Lambert P., Deacon N. J., Mills J., and Crowe S. M. (1997) Constitutive expression of p50 homodimer in freshly isolated human monocytes decreases with in vitro and in vivo differentiation: a possible mechanism influencing human immunodeficiency virus replication in monocytes and mature macrophages. J. Virol. 71, 2114–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lim C. A., Yao F., Wong J. J., George J., Xu H., Chiu K. P., Sung W. K., Lipovich L., Vega V. B., Chen J., Shahab A., Zhao X. D., Hibberd M., Wei C. L., Lim B., et al. (2007) Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell 27, 622–635 [DOI] [PubMed] [Google Scholar]
- 27. Oeth P. A., Parry G. C., Kunsch C., Nantermet P., Rosen C. A., and Mackman N. (1994) Lipopolysaccharide induction of tissue factor gene expression in monocytic cells is mediated by binding of c-Rel/p65 heterodimers to a κB-like site. Mol. Cell. Biol. 14, 3772–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Udalova I. A., Knight J. C., Vidal V., Nedospasov S. A., and Kwiatkowski D. (1998) Complex NF-κB interactions at the distal tumor necrosis factor promoter region in human monocytes. J. Biol. Chem. 273, 21178–21186 [DOI] [PubMed] [Google Scholar]
- 29. Lowell C. A., and Berton G. (1998) Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. Proc. Natl. Acad. Sci. U.S.A. 95, 7580–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolaczkowska E., and Kubes P. (2013) Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 [DOI] [PubMed] [Google Scholar]
- 31. Wright H. L., Moots R. J., Bucknall R. C., and Edwards S. W. (2010) Neutrophil function in inflammation and inflammatory diseases. Rheumatology 49, 1618–1631 [DOI] [PubMed] [Google Scholar]
- 32. Serhan C. N. (2011) The resolution of inflammation: the devil in the flask and in the details. FASEB J. 25, 1441–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cucullo L., Hallene K., Dini G., Dal Toso R., and Janigro D. (2004) Glycerophosphoinositol and dexamethasone improve transendothelial electrical resistance in an in vitro study of the blood-brain barrier. Brain Res. 997, 147–151 [DOI] [PubMed] [Google Scholar]
- 34. An H., Hou J., Zhou J., Zhao W., Xu H., Zheng Y., Yu Y., Liu S., and Cao X. (2008) Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat. Immunol. 9, 542–550 [DOI] [PubMed] [Google Scholar]
- 35. Natoli G. (2009) Control of NF-κB-dependent transcriptional responses by chromatin organization. Cold Spring Harb. Perspect. Biol.[ 1, a000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neumann M., and Naumann M. (2007) Beyond IκBs: alternative regulation of NF-κB activity. FASEB J. 21, 2642–2654 [DOI] [PubMed] [Google Scholar]
- 37. Ghosh S., and Hayden M. S. (2008) New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 8, 837–848 [DOI] [PubMed] [Google Scholar]
- 38. Martone R., Euskirchen G., Bertone P., Hartman S., Royce T. E., Luscombe N. M., Rinn J. L., Nelson F. K., Miller P., Gerstein M., Weissman S., and Snyder M. (2003) Distribution of NF-κB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U.S.A. 100, 12247–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krappmann D., Wegener E., Sunami Y., Esen M., Thiel A., Mordmuller B., and Scheidereit C. (2004) The IκB kinase complex and NF-κB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol. Cell. Biol. 24, 6488–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berrie C. P., Dragani L. K., van der Kaay J., Iurisci C., Brancaccio A., Rotilio D., and Corda D. (2002) Maintenance of PtdIns45P2 pools under limiting inositol conditions, as assessed by liquid chromatography-tandem mass spectrometry and PtdIns45P2 mass evaluation in Ras-transformed cells. Eur. J. Cancer 38, 2463–2475 [DOI] [PubMed] [Google Scholar]
- 41. Patrussi L., Mariggiò S., Corda D., and Baldari C. T. (2013) The glycerophosphoinositols: from lipid metabolites to modulators of T-cell signaling. Front. Immunol. 4, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Faenza I., Fiume R., Piazzi M., Colantoni A., and Cocco L. (2013) Nuclear inositide specific phospholipase C signalling—interactions and activity. FEBS J. 280, 6311–6321 [DOI] [PubMed] [Google Scholar]
- 43. Schievella A. R., Regier M. K., Smith W. L., and Lin L. L. (1995) Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J. Biol. Chem. 270, 30749–30754 [DOI] [PubMed] [Google Scholar]
- 44. Dragani L. K., Berrie C. P., Corda D., and Rotilio D. (2004) Analysis of glycerophosphoinositol by liquid chromatography-electrospray ionisation tandem mass spectrometry using a β-cyclodextrin-bonded column. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 802, 283–289 [DOI] [PubMed] [Google Scholar]
- 45. Fisher S. K., Novak J. E., and Agranoff B. W. (2002) Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J. Neurochem. 82, 736–754 [DOI] [PubMed] [Google Scholar]
- 46. Grauso L., Mariggiò S., Corda D., Fontana A., and Cutignano A. (2015) An improved UPLC-MS/MS platform for quantitative analysis of glycerophosphoinositol in mammalian cells. PLoS One 10, e0123198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Angus D. C., and van der Poll T. (2013) Severe sepsis and septic shock. N. Engl. J. Med. 369, 2063. [DOI] [PubMed] [Google Scholar]
- 48. Mariggiò S., Iurisci C., Sebastià J., Patton-Vogt J., and Corda D. (2006) Molecular characterization of a glycerophosphoinositol transporter in mammalian cells. FEBS Lett. 580, 6789–6796 [DOI] [PubMed] [Google Scholar]
- 49. Hirahashi J., Hishikawa K., Kaname S., Tsuboi N., Wang Y., Simon D. I., Stavrakis G., Shimosawa T., Xiao L., Nagahama Y., Suzuki K., Fujita T., and Mayadas T. N. (2009) Mac-1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation 120, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Colgan S. P. (2015) Neutrophils and inflammatory resolution in the mucosa. Semin. Immunol. 27, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Corda D., and Falasca M. (1996) Glycerophosphoinositols as potential markers of ras-induced transformation and novel second messengers. Anticancer Res. 16, 1341–1350 [PubMed] [Google Scholar]
- 52. Di Santo A., Amore C., Dell'Elba G., Manarini S., and Evangelista V. (2011) Glycogen synthase kinase-3 negatively regulates tissue factor expression in monocytes interacting with activated platelets. J. Thromb. Haemost. 9, 1029–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berrie C. P., Iurisci C., Piccolo E., Bagnati R., and Corda D. (2007) Analysis of phosphoinositides and their aqueous metabolites. Methods Enzymol. 434, 187–232 [DOI] [PubMed] [Google Scholar]
- 55. Corda D., Zizza P., Luini A., and Mariggiò S. (May 31, 2016) Use of Glycerophosphoinositols for the Treatment of Septic Shock, U. S. Patent 9351983
- 56. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.