

Abstract
Virus-related type 2 diabetes is commonly observed in individuals infected with the hepatitis C virus (HCV); however, the underlying molecular mechanisms remain unknown. Our aim was to unravel these mechanisms using FL-N/35 transgenic mice expressing the full HCV ORF. We observed that these mice displayed glucose intolerance and insulin resistance. We also found that Glut-2 membrane expression was reduced in FL-N/35 mice and that hepatocyte glucose uptake was perturbed, partly accounting for the HCV-induced glucose intolerance in these mice. Early steps of the hepatic insulin signaling pathway, from IRS2 to PDK1 phosphorylation, were constitutively impaired in FL-N/35 primary hepatocytes via deregulation of TNFα/SOCS3. Higher hepatic glucose production was observed in the HCV mice, despite higher fasting insulinemia, concomitant with decreased expression of hepatic gluconeogenic genes. Akt kinase activity was higher in HCV mice than in WT mice, but Akt-dependent phosphorylation of the forkhead transcription factor FoxO1 at serine 256, which triggers its nuclear exclusion, was lower in HCV mouse livers. These findings indicate an uncoupling of the canonical Akt/FoxO1 pathway in HCV protein-expressing hepatocytes. Thus, the expression of HCV proteins in the liver is sufficient to induce insulin resistance by impairing insulin signaling and glucose uptake. In conclusion, we observed a complete set of events leading to a prediabetic state in HCV-transgenic mice, providing a valuable mechanistic explanation for HCV-induced diabetes in humans.
Keywords: glucose metabolism, hepatitis C virus (HCV), insulin resistance, transgenic mice, type 2 diabetes
Introduction
Hepatitis C virus (HCV)4 infects over 170 million individuals worldwide. Although newly developed treatment combinations based on direct-acting antiviral drugs cure a high proportion of infections (1, 2), the vast majority of HCV-infected patients do not have access to these therapies because they are unaware of their infection, do not have access to a proper healthcare system, or cannot afford the high cost of treatment.
Chronic HCV infection is not only a leading cause of chronic liver disease, including cirrhosis and hepatocellular carcinoma, but also induces systemic disorders. In particular, HCV infection was reported to be an independent risk factor for type 2 diabetes (T2D), regardless of the presence of cirrhosis (3–5). Indeed, a meta-analysis showed a 1.8-fold excess risk of T2D among HCV-positive patients compared with HBV-positive/HCV-negative ones (6). In a longitudinal study, the risk of developing diabetes was up to 12 times higher in HCV-infected patients than in the general population (7). In patients with chronic hepatitis C, T2D is an independent predictor of a more rapid progression of liver fibrosis (8) and impairs the response to antiviral treatments based on PEGylated interferon α and ribavirin (9, 10). In addition, patients with cirrhosis and T2D have an increased risk of developing hepatocellular carcinoma (11, 12). In patients with compensated HCV-related cirrhosis, diabetes and marked insulin resistance are independently associated with higher liver morbidity and mortality (13).
T2D results from a combination of mechanisms, including insulin resistance (IR), resulting in increased hepatic glucose production and altered insulin secretion. Several arguments suggest that HCV is pro-diabetogenic per se (for a review, see Ref. 14, 15). Indeed, IR persists in patients who do not respond to antiviral treatment despite a decrease in body mass index (16). Although still controversial, a correlation between IR and the HCV RNA level, a surrogate marker of viral replication, has been reported (17), and the incidence of IR is higher in patients infected with genotypes 1 and 4 than in those infected with genotypes 2 or 3 (18, 19). This genotype specificity of HCV-induced IR could result from amino acid sequence differences across genotypes in the core sequence (20, 21) and HCV core-induced hypoadiponectinemia (22–24). The cure of infection is often associated with IR reduction, in particular in patients infected with genotype 1 (25–28).
The direct involvement of HCV in T2D has also been suggested in experimental models. HCV core-mediated down-regulation of insulin receptor substrates 1 and 2 (IRS-1/2) and protein phosphatase 2A (PP2A)-dependent Akt dephosphorylation mediated by the nonstructural NS5A protein have been suggested to play a role in HCV-induced IR (29–33). Other studies showed that HCV modulates the activity of transcription factors such as peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α) and FoxO1 and FoxA2, both implicated in metabolic enzyme expression (34–37).
Thus, clinical observations and experimental data strongly suggest that HCV directly induces T2D in infected patients. However, the molecular mechanisms underlying HCV-induced T2D, the associated IR, and glucose metabolism abnormalities remain unknown. In this study, we show that FL-N/35 HCV-transgenic mice that express the full-length HCV genotype 1 open reading frame (38) are at a prediabetic stage, exhibiting glucose intolerance and IR, mirroring the situation in HCV-infected patients. Mechanistically, we demonstrate that the HCV proteins directly impair Glut2-mediated hepatic glucose intake and insulin-driven shutdown of gluconeogenesis by down-regulating IRS-2 and altering FoxO1 phosphorylation and nuclear exclusion.
Results
Mice expressing the full-length HCV open reading frame are insulin-resistant and glucose-intolerant
We studied the impact of the HCV proteins on the prevalence of insulin resistance in our murine model. Transgenic and WT mice did not display different fasting and fed glycemias (Fig. 1A). In contrast, fasting and fed insulinemias were significantly higher in HCV-transgenic mice than in their WT littermates (Fig. 1B). As a result, the homeostatic model assessment of insulin resistance index was significantly higher in HCV-transgenic than in WT mice (13.0 ± 3.4 versus 7.9 ± 0.7 microunits/mg, p < 0.05), suggesting that HCV mice are resistant to insulin.
Figure 1.

A and B, glycemia and insulinemia, respectively, in 14- to 18-month-old FL-N/35 transgenic mice expressing the full-length HCV open reading frame (HCV-tg) and in control WT littermates in fasting or fed states. The results are presented as box plots (median and 95th percentiles). n, number of animals tested; NS, not significant. C and D, IPGTT in 14- to 18-month-old FL-N/35 transgenic mice expressing the full-length HCV open reading frame and in control WT littermates. IPGTT results are shown as plasma glucose concentrations using the glycemia level at injection as the 100% reference (C) and areas under the curve of plasma glucose concentrations (D). E and F, glucose turnover rate under basal conditions and during hyperinsulinemic–euglycemic clamps in 14- to 18-month-old FL-N/35 transgenic mice expressing the full-length HCV open reading frame and in control WT littermates. E, glucose turnover rate under basal conditions (BASAL), GIR, GUR, and hepatic endogenous glucose production (EGP). F, post-clamp glucose uptake in subcutaneous white adipose tissue (WATs) and visceral white adipose tissue (WATv) and in skeletal muscles (S, soleus; TA, tibialis anterior).
Because orally administered glucose can induce secretion of glucagon-like peptide 1 (GLP-1) by the gut, which increases insulin secretion by pancreatic β cells, mice were subjected to intraperitoneal glucose tolerance tests (IPGTT), which bypass the effect of GLP-1 and are associated with lower glucose-stimulated insulin secretion than oral glucose tolerance tests. As shown in Fig. 1, C and D, IPGTT analyses confirmed that HCV-transgenic mice are glucose-intolerant.
Body weight and fat mass did not differ between HCV-transgenic mice and their WT littermates. Weight gain was similar in both groups until 13 months of age, followed by a steady state until 20 months of age (supplemental Fig. 1A). Moreover, body fat assessments by tomography scanner (39) showed similar body mass distributions in both groups, with no evidence of excessive fat storage in HCV-transgenic mice, ruling out an effect of accumulated lipids in the IR of HCV-transgenic mice (supplemental Fig. 1B), i.e. excluding a possible role of a metabolic syndrome in our findings. Together, these results indicate that the sole expression of HCV proteins in the livers of transgenic mice is associated with insulin resistance and glucose intolerance, i.e. a prediabetic stage unrelated to a metabolic syndrome.
Insulin resistance in HCV-transgenic mice is of both hepatic and muscular origin
To complement these data, glucose turnover rate and metabolic flux were measured in HCV-transgenic and non-transgenic animals by means of hyperinsulinemic–euglycemic clamps. The basal glucose turnover rate was similar in WT and HCV-transgenic mice (Fig. 1E). The glucose infusion rate (GIR), i.e. the amount of glucose needed to maintain euglycemia, and the whole-body glucose utilization rate (GUR) were significantly lower in HCV-transgenic than in WT mice, indicating a defect in glucose uptake from the bloodstream despite hyperinsulinemia (Fig. 1E). Furthermore, endogenous glucose production was significantly higher in HCV-transgenic animals (Fig. 1E), indicating a defect in insulin-driven shutdown of gluconeogenesis and/or glycogenolysis in the liver. As shown in Fig. 1F, glucose uptake in the soleus oxidative skeletal muscle (S, soleus) and, to a lesser extent, the visceral white adipose tissue (WATv) were significantly lower in HCV-transgenic mice than in WT animals, whereas no difference was observed in glucose uptake in subcutaneous white adipose tissue (WATs). Together with our previous analyses, these results confirm that mice expressing the full-length HCV open reading frame are insulin-resistant and indicate that insulin resistance is of both hepatic and muscular origin.
Impaired liver glucose intake in HCV-transgenic mice is at least partly due to Glut2 down-regulation by HCV proteins
To examine the underlying mechanisms surrounding the IR observed in HCV-transgenic mice, we next examined the membrane expression of glucose and hexose transporter 2 (Glut2), which has been implicated as an important regulator of hepatic glucose intake (40, 41). Western blot experiments showed significantly lower amounts of Glut2 in membrane fractions from HCV-transgenic mouse livers compared with WT controls (Figs. 2, A and B), without significant differences in its transcript levels (0.85 ± 0.33-fold-change in HCV-transgenic compared with wild-type animals). Correspondingly, glucose uptake in cultured mouse primary hepatocytes was significantly lower in HCV-transgenic hepatocytes than in non-transgenic ones (Fig. 2C). These observations may, at least in part, account for the insulin resistance observed in the presence of the HCV proteins.
Figure 2.
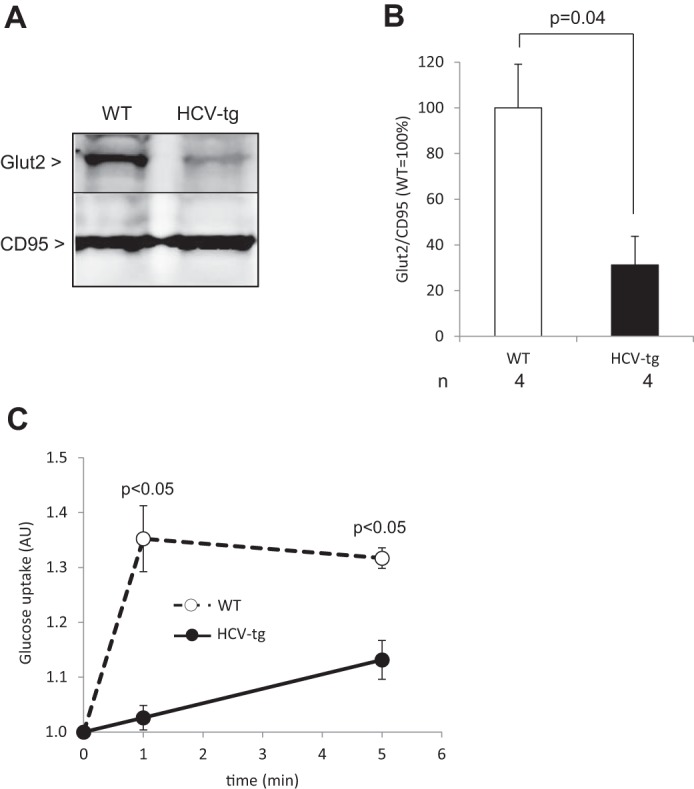
A, representative examples of Glut2 protein levels in liver plasma membrane extracts from HCV-transgenic (HCV-tg) and control WT mice by Western blot analysis, normalized to CD95 expression (representative animals). B, average Glut2 protein levels in liver plasma membrane extracts from HCV-tg and control WT mice by Western blot analysis, normalized to CD95 expression. C, dynamics of glucose uptake in mouse primary hepatocytes isolated from HCV-tg and WT animals (AU: arbitrary units).
Glycogen storage is not altered in HCV-transgenic mice
Next we examined whether the HCV proteins affected glycogen synthesis by examining the expression and activation of glycogen synthase kinase 3, the enzyme that inhibits glycogen synthase. Western blotting showed no difference in the expression or phosphorylation of GSK3 on Ser21 between transgenic and non-transgenic animals (Figs. 3, A and B). In addition, the amounts of liver-stored glycogen did not differ between fasting HCV-transgenic and WT mice, respectively (Fig. 3C). Therefore, HCV does not impact hepatic glycogen storage.
Figure 3.
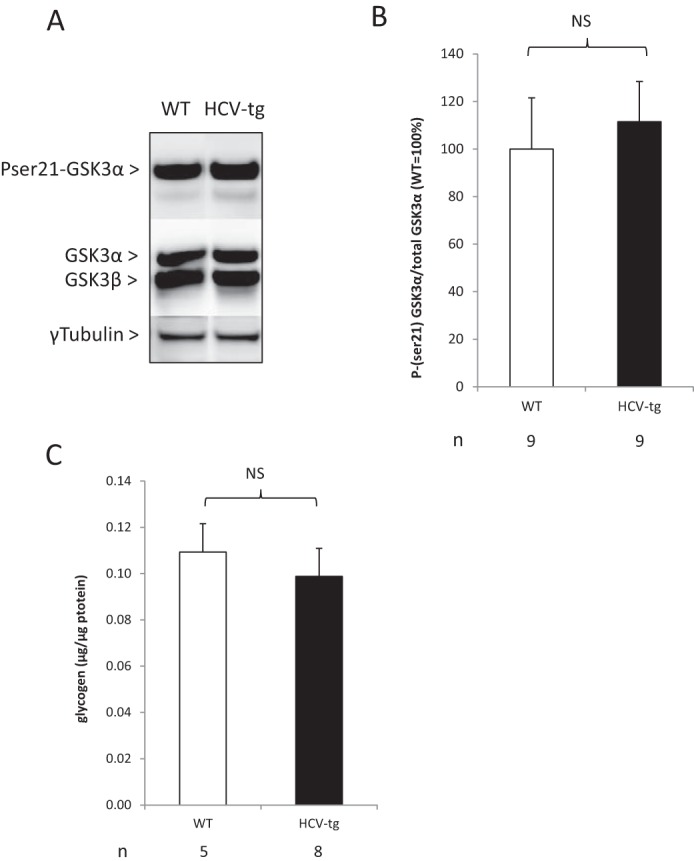
A, representative examples of Ser(P)21 GSK3α phosphorylation status in liver extracts from HCV-transgenic (HCV-tg) and control WT mice by Western blot analysis, normalized to total GSK3 expression (representative animals). B, average Ser(P)21 GSK3α protein levels in liver extracts from HCV-tg and control WT mice by Western blot analysis, normalized to total GSK3 expression. NS, not significant. C, total liver glycogen content in HCV-tg and control WT mice, normalized to total liver protein weight.
Insulin-driven shutdown of hepatic gluconeogenesis is impaired in HCV-transgenic mice
Given our previous data, we postulated that the regulators of glucose production, which operate in the switch from gluconeogenesis to glycolysis in response to insulin, may be affected by the HCV proteins. Therefore, we examined the transcription levels of key regulators of hepatic gluconeogenesis by means of quantitative RT-PCR in liver RNA extracts from fasting HCV-transgenic and non-transgenic mice. They included glucokinase (GK), fructose 1,6-bisphosphatase (PFKFB3), phosphoenolpyruvate carboxykinase (PEPCK, PCK1 gene), and glucose-6-phosphatase (Glc-6-PC). As shown in Fig. 4A, the expressions of GK and PFKFB3 were reduced by ∼5-fold and 1.5-fold, respectively, in the livers of HCV-transgenic mice compared with WT animals. The amount of PCK1 transcripts was 2-fold higher in transgenic than in WT animals, whereas Glc-6-PC mRNA levels were identical in both groups. Correspondingly, Western blotting of crude liver extracts revealed that GK and PEPCK protein expression was lower and higher, respectively, in HCV-transgenic mice than in non-transgenic animals, as shown in Fig. 4B. Furthermore, although insulin injection increased GK mRNA expression in WT mice, GK mRNA levels were reduced in HCV-transgenic mice in response to insulin (Fig. 4C), demonstrating that regulation of gluconeogenesis is perturbed by the HCV proteins. Together, these data indicate that HCV transgenic mice develop hepatic IR leading to higher gluconeogenesis and lower glycolysis.
Figure 4.
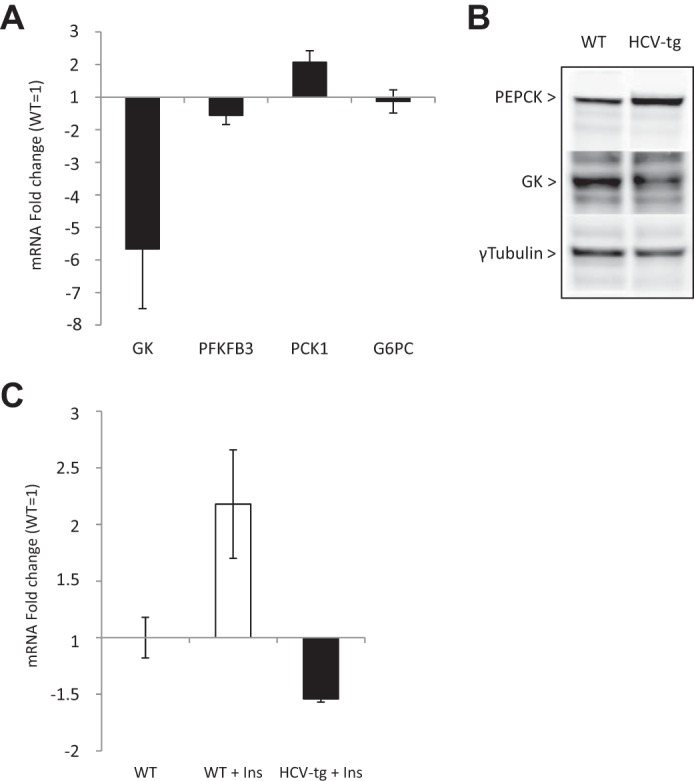
A, mRNA quantification of glycolysis and gluconeogenesis enzymes in total liver RNA extracts from fasting HCV-tg and control WT mice. Gene transcript levels in HCV-tg animals are expressed as –fold changes and were normalized using WT mRNA expression levels as the reference. B, PEPCK and GK protein levels assessed by Western blotting in crude liver protein extracts from fasting HCV-tg or WT mice. C, mRNA quantification of GK in total liver RNA extracts from WT mice injected with PBS (WT) or WT (WT Ins) and HCV-transgenic (HCV-tg Ins) mice injected with insulin. Gene transcript levels are expressed as -fold changes and were normalized using PBS-injected WT mRNA expression levels as the reference.
IRS2 expression is down-regulated in HCV-transgenic mice
To further understand the mechanisms for the IR observed in HCV-transgenic mice, we next examined the hepatic insulin receptor pathway. Similar basal amounts of insulin receptor were found in liver extracts from HCV-transgenic and non-transgenic mice by Western blot analysis (data not shown). In contrast, indirect immunofluorescence revealed lower basal levels of IRS2 in primary hepatocytes from HCV-transgenic mice than in those from WT animals (Fig. 5A). Moreover, both basal levels of IRS2 and those induced following intraperitoneal insulin injection were decreased in liver extracts from HCV-transgenic mice compared with the WT (Fig. 5, B and C). However, IRS2 mRNA levels were identical in transgenic and non-transgenic mouse livers by quantitative RT-PCR assay (Fig. 5D), ruling out modulation of IRS2 expression by HCV proteins at the transcriptional level. These results suggest that IRS2 expression is down-regulated at the posttranscriptional level in HCV-transgenic mice and demonstrates that the HCV proteins perturb the hepatic insulin receptor pathway.
Figure 5.

A–C, IRS2 protein levels studied by immunofluorescence (A) and Western blotting (B, representative animals; C, quantification) in primary mouse hepatocytes from HCV-tg and control WT mice. D, IRS2 mRNA expression by RT-qPCR in total RNA extracts from HCV-Tg and WT mice. E, Ser(P)728-PKCϵ, Thr(P)389-p70S6K, and SOCS3 protein levels assessed by Western blotting in liver extracts from HCV-tg or WT mice injected with insulin or vehicle (representative animals). F–H, quantitative results for Ser(P)728-PKCϵ (F), Thr(P)389-p70S6K (G), and SOCS3 (H) protein level expression. I, SOCS3 mRNA expression assessed by RT-qPCR in total RNA extracts from WT and HCV-tg mice. J, IRS2 protein levels assessed by Western blotting in primary mouse hepatocytes from WT and HCV-tg mice treated with MG132 or PBS. NS, not significant.
Down-regulation of IRS2 expression in HCV-transgenic mice occurs via a SOCS3-dependent mechanism
To elucidate the underlying mechanisms for these observations, we examined whether the degradation of IRS2 was affected by the viral proteins. In mouse primary hepatocytes isolated from WT and HCV-transgenic mice, treatment with the proteasome inhibitor MG132 restored IRS2 levels in HCV-transgenic animals to those observed in WT animals (Fig. 5E), demonstrating that HCV down-regulated IRS2 levels by inducing proteasomal degradation.
Several kinases have been implicated in regulating the phospho-dependent degradation of IRS2, including PKCϵ, which phosphorylates IRS2 at serine 612 and induces its degradation by the proteasome (42); p70 S6 kinase (p70S6K), activated through a negative feedback loop resulting from the prolonged action of insulin, which phosphorylates IRS2 at multiple serine residues and induces its degradation (43); and suppressor of cytokine signaling 3 (SOCS3), which induces IRS1/2 degradation and inhibits IRS2 tyrosine phosphorylation, therefore blocking insulin signal transduction (44, 45). We therefore examined the levels and activation of these kinases in our murine model of HCV-induced IR.
Western blot analyses of liver extracts showed similar levels of PKCϵ phosphorylation at serine 728 in HCV-transgenic and WT mice (Fig. 5, F and G). In agreement, fasted HCV-transgenic mice did not show significantly lower levels of p70S6K phosphorylation at serine 389 than WT animals (Fig. 5, F and H). In contrast, the expression of SOCS3 protein (Fig. 5, F and I) and mRNA (Fig. 5J) was significantly higher in the livers of fasted HCV-transgenic mice than in those of their WT littermates. In addition, TNFα transcript levels were 2.5 ± 0.5 times higher in HCV-transgenic than in WT mice. Because TNFα directly regulates SOCS3 expression, these data provide a potential explanation for the higher levels of SOCS3 expression in HCV-transgenic mice (data not shown). Together, these results indicate that the hepatic insulin signaling is impaired in mice expressing the full-length HCV open reading frame, through the proteasomal degradation of IRS2, potentially via a SOCS3-dependent mechanism.
Down-regulation of IRS2 expression down-regulates the hepatic insulin signaling pathway
Because our data strongly suggest that HCV proteins induce the degradation of hepatic IRS2, we examined the downstream effects on hepatic insulin receptor signaling. In normal hepatic tissues, phosphoinositide-dependent protein kinase 1 (PDK1) is activated downstream of IRS2 activation through phosphorylation at serine 241. In keeping with our previous data, Western blot analysis of liver extracts showed significantly lower amounts of Ser(P)241 PDK1 in HCV-transgenic animals subjected to vehicle injections than in their WT littermates. Moreover, insulin treatment induced PDK1 hyperphosphorylation in the livers of WT mice but not in HCV-transgenic animals (Fig. 6, A and B). Furthermore, phosphorylation of the downstream target of PDK1, Akt, at threonine 308 was also significantly lower in HCV-transgenic than in WT animals, both in the presence or absence of insulin treatment (Fig. 6, C and D). However, despite this hypophosphorylation at threonine 308, phosphorylation of AKT at serine 473 and Akt kinase activity, assessed using recombinant GSK3 protein, were constitutively enhanced in the livers of older (14–18 months old) HCV-transgenic mice (Fig. 6, E and F), confirming our previous observation in younger HCV-transgenic mice (46). These data suggest that, in our murine model, although decreased IRS2 expression has a knock-on effect on PDK1 activation, global Akt activity is elevated, probably because of an effect of the viral proteins on kinases regulating Akt Ser473 phosphorylation.
Figure 6.

A, Ser(P)241-PDK1 assessed by Western blotting in liver extracts from WT and HCV-tg mice injected with insulin or vehicle. The results were normalized using total PDK1 expression (representative animals). B, quantitative results for Ser(P)241-PDK1 protein levels. C, Thr(P)308-Akt assessed by Western blotting in liver extracts from WT and HCV-tg mice injected with insulin or vehicle. The results were normalized using total Akt expression (representative animals). D, quantitative results for Thr(P)308-Akt protein levels. E, quantitative results for Ser(P)473-Akt protein levels. F, analysis of Akt activity by kinase assay in hepatic lysates from WT and HCV-tg animals. Akt activity was normalized to the relative expression of Akt in the lysates and is expressed as the percentage of WT Akt activity.
FoxO1 phosphorylation at serine 256 by Akt is altered in HCV-transgenic mice
To examine whether the increased Akt activity observed in the presence of the HCV proteins elevated insulin signaling, we next examined the expression and phosphorylation of the forkhead in rhabdomyosarcoma transcription factor (FKHR or FoxO1) (47, 48). Akt regulates glucose homeostasis by phosphorylating FoxO1 at serine 256. In keeping with a role for HCV in perturbing hepatic insulin signaling, significantly lower amounts of Ser(P)256 FoxO1 were observed in liver extracts from vehicle-injected and insulin-treated HCV-transgenic mice compared with their WT littermates (Fig. 7, A and B). A similar result was found in cultured mouse primary hepatocytes using indirect immunofluorescence to detect Ser(P)256 FoxO1 (Fig. 7, C, left panels, no insulin, and D). These findings were in keeping with the higher PCK1 and the lower GK and PFKFB3 transcript levels reflecting higher endogenous glucose production in the livers of HCV-transgenic mice. However, the reduced phosphorylation of FoxO1 in the presence of enhanced Akt kinase activity in the livers of HCV-transgenic mice suggests that HCV protein expression uncouples the activities of Akt/FoxO1 and that another IRS2-independent mechanism is involved.
Figure 7.

A, Ser(P)256-FoxO1 assessed by Western blotting and quantified in liver extracts from WT and HCV-tg mice injected with insulin or vehicle and normalized using γ-tubulin expression (representative animals). B, quantitative results for Ser(P)256-FoxO1 levels. C, indirect immunofluorescence on cultured primary mouse hepatocytes from WT and HCV-tg mice. Cells were treated with insulin or PBS (no insulin) for 20 min prior to analysis. Ser(P)256-FoxO1 antibody was detected using a secondary antibody coupled to Alexa 498. Nuclei were stained using DAPI. D, quantification of fluorescence intensity within the cytoplasm and the nucleus of primary hepatocytes using ImageJ software.
Impairment of FoxO1 phosphorylation at serine 256 alters its nuclear exclusion in HCV-transgenic mice
The activity of FoxO1 is regulated by nuclear/cytoplasmic shuttling. Indeed, serine 256 phosphorylation of FoxO1 triggers its nuclear exclusion, thus removing it from gene promoters (49). We demonstrated that HCV proteins inhibit FoxO1-P. We therefore examined the nuclear/cytoplasmic localization of FoxO1 in hepatocytes isolated from both HCV-transgenic and WT mice. In the absence of insulin, Ser(P)256-FoxO1 was mainly located in the nuclei of primary hepatocytes from both HCV-transgenic and WT animals. As expected, after insulin injection, Ser(P)256-FoxO1 relocated in the cytoplasm of WT hepatocytes. However, it remained in the nuclei of hepatocytes from HCV-transgenic mice (Fig. 7, C and D). These results show that, despite elevated Akt activity, HCV inhibits the phosphorylation of FoxO1 at serine 256, thereby inducing its nuclear retention and decreasing its FoxO1 transcriptional activities, leading to impaired insulin signaling.
Discussion
In patients with HCV, chronic infection has been shown to be frequently associated with insulin resistance and type 2 diabetes, accounting for significant virus-related morbidity and mortality, independent of the severity of liver disease (Refs. 18, 50, 51; for a review, see Refs. 4, 52–54). In this study, we showed that, like infected patients, transgenic mice expressing the full-length HCV open reading frame are glucose-intolerant and insulin-resistant and that this is due to the sole expression of HCV proteins in the liver without the need for viral replication, local inflammation, or advanced liver disease such as fibrosis or cirrhosis. HCV core protein-expressing mice were reported previously to be insulin-resistant, but, in contrast with our HCV-transgenic mice, they did not display glucose intolerance or altered endogenous glucose production (29). These differences could be due to the very young age of the HCV core transgenic mice (2 months) compared with the animals used in our study and due to the expression of only one HCV protein in the model, whereas ours express the full spectrum of HCV proteins at nearly physiological levels, better mimicking HCV protein expression in infected humans (29). Our results also showed that IR is of both peripheral (striated muscle) and hepatic origin in HCV-transgenic mice, a result in keeping with clinical data showing little or no involvement of adipose tissue in this process (55). Although HCV protein expression in our model is hepatocyte-restricted, this peripheral effect may result from the endocrine effects of inflammatory cytokines (56), such as IL-8, whose overexpression is induced by the HCV NS5A protein, as we showed recently (57).
Our results suggest that glucose intake is perturbed in HCV-transgenic mice, probably as a result of reduced amounts of Glut2 in the plasma membrane. Such a defect in Glut2 expression at the plasma membrane was also reported in Huh7.5 cells replicating the genotype 2 culture-adapted JFH1 HCV clone (58, 59) as well as in the livers of HCV-infected patients (59). Our results demonstrate that this effect is solely related to the expression of HCV proteins in vivo. In addition, we found that insulin-driven shutdown of gluconeogenesis was defective in the livers of HCV-transgenic mice, which explains the higher endogenous glucose production observed in these mice during the clamp experiments. Previously, an increase in PCK1 and Glc-6-PC mRNAs and glucose production in Huh-7.5 cells harboring HCV genotype 1b RNA replicons or infected with the JFH1 strain has been reported (37). Other groups observed an increase in gluconeogenic activities in Huh-7 cells or in murine hepatocytes expressing the HCV NS5A protein as well as in livers from HCV-infected patients and engineered mice infected with HCV (60–62). Nevertheless, our results extend these observations in an in vivo model and demonstrate that insulin resistance is directly due to the expression of the HCV proteins.
In HCV-infected patients, hepatic IRS1 and IRS2 levels are diminished compared with non-infected patients (25, 31). In addition, HCV clearance is accompanied by normalization of IRS1 and IRS2 levels, proving the direct role of HCV in this perturbation (25). In our experiments, levels of IRS2 protein were reduced in HCV-transgenic mice, and this reduction was reversed by proteasome inhibitors. This in vivo result confirms previous findings in HCV core transgenic mouse livers and HCV core-transfected human hepatoma cells, showing proteasomal degradation of IRS1 and IRS2, possibly resulting from an increased production of proinflammatory cytokines, such as TNFα, involving the activation of the JNK pathway by the HCV core protein (29, 31, 63). In keeping with these data, our HCV-transgenic mice displayed higher levels of TNFα in their livers, suggesting a role for pro-inflammatory cytokines in this process and providing a potential mechanistic explanation for our observations.
Serine phosphorylation of IRS-1 was also described in HCV NS5A-expressing hepatoma cells (60). Furthermore, it was suggested that IRS1 down-regulation, rescued by MG132 treatment, is dependent upon mTORC1 activation in JFH1-infected hepatocytes (64). Accordingly, we observed that HCV protein expression activated Akt, an upstream activator of mTORC1, in our HCV-transgenic mice. It has been shown that SREBP1 suppresses IRS2-mediated insulin signaling in hepatocytes (65, 66) via a negative feedback loop involving transcriptional regulation of the IRS2 gene. We reported previously that HCV triggers the activation of SREBP1 in the livers of HCV-transgenic mice (67). However, IRS2 mRNA levels were unchanged in our study, suggesting that this mechanism is not involved in our observations.
Our results suggest that HCV-mediated SOCS3 overexpression is responsible for IRS2 degradation in HCV-transgenic mice. Accordingly, hepatic SOCS3 levels were reported to be higher in HCV-infected patients than in healthy individuals (68, 69). Furthermore, HCV core protein expression was reported to induce SOCS3 overexpression and IRS degradation, leading to IFN-α signaling impairment (31, 70). Interestingly, SOCS3 expression was shown to be an independent predictor of IFN-α treatment response, especially in genotype 1–infected patients (71, 72). It was also demonstrated, in a non-infected mouse model, that overexpression of SOCS induces IRS1 and IRS2 degradation, subsequently inducing insulin resistance (44). HCV core substitutions at positions Arg70 and Leu91 have been suggested to enhance SOCS3 expression in an IL-6/UPR-dependent mechanism (73). Interestingly, the HCV clone used to generate the FL-N/35 mouse model used in this study contains the core L91M substitution. However, we observed no UPR stimulation in the livers of these mice (67). Whether SOCS3 overexpression and the resulting degradation of IRS2 is sufficient to trigger insulin resistance and type 2 diabetes is questionable. A recent study suggested that HBV can also induce SOCS3 expression and IRS1 degradation in the liver, thereby inhibiting insulin signaling (74). Nevertheless, no association between HBV infection and type 2 diabetes or insulin resistance has been reported (75–77). Thus, other perturbations are probably necessary, justifying our assessment of the integrity of the IR pathway downstream of IRS2 in the livers of HCV-transgenic mice.
Impaired gluconeogenesis inhibition by insulin generally reflects reduced Akt activation. However, we found that Akt kinase activity was constitutively enhanced in the livers of HCV-transgenic mice, in agreement with our previous findings in younger animals (46). In addition, Akt activity was significantly augmented in HCV-transgenic mice injected with insulin. This result might appear as paradoxical in the context of impaired insulin-driven gluconeogenesis shutdown and IRS2/PDK1 down-regulation described above. Akt regulates glucose homeostasis gene expression through the phosphorylation of FoxO1 at serine 256, which, in turn, triggers its nuclear exclusion, thus removing it from gene promoters (for a review, see Ref. 78). Thus, the lower amounts of Ser(P)256-FoxO1 associated with the impairment of its insulin-driven nuclear exclusion in the hepatocytes of HCV-transgenic mice were also unexpected in the context of increased Akt activity in the same hepatocytes but in keeping with the elevated levels of expression of gluconeogenic genes. Akt phosphorylation at threonine 308 has been reported to be essential for glucose uptake whereas phosphorylation at serine 473 is not (79). The hypophosphorylation of Akt at threonine 308 we observed in the livers of HCV-transgenic mice could therefore explain the impairment of glucose uptake. However, others reported that the modulation of Akt phosphorylations had no effect on FoxO1 phosphorylation (35). Another study, reporting similar observations in Huh7.5 cell lines harboring an HCV genotype 1b replicon or JFH1 infection, suggested an Akt-independent mechanism involving JNK pathway activation through mitochondrial reactive oxygen species production (37). A correlation was indeed reported between oxidative stress and insulin resistance, through the analyses of hepatic thioredoxin levels, in HCV-infected patients (80). In addition, we and others have shown an accumulation of reactive oxygen species in the liver of HCV-transgenic mice (Refs. 46, 81–83; for a review, see Ref. 84), which could be at least partly responsible for the uncoupling of the Akt/FoxO1 signaling we observed.
In summary, we used the FL-N/35 transgenic mouse model, which expresses the full-length HCV open reading frame at nearly physiological levels in the liver of these animals, to demonstrate that, like in HCV-infected patients, HCV-transgenic mice are insulin-resistant and glucose-intolerant and that these effects are solely due to the expression of the viral proteins in hepatocytes. This model also allowed us to unravel the molecular mechanisms by which HCV protein expression alters glucose metabolism. Our data suggest that the impaired glucose intake in the liver of transgenic animals is partly due to a lower expression of Glut2 in the presence of HCV proteins. We also observed that the insulin-driven hepatic neoglucogenesis switch to glycolysis was impaired in HCV-transgenic mice. This resulted, at least in part, from an alteration of the hepatic insulin receptor cascade because of the down-regulation of IRS2 expression, potentially via a SOCS3-dependent mechanism, as a result of increased TNFα expression. Finally, the Akt/FoxO1 uncoupling we observed in HCV-transgenic animals coincided with an alteration of FoxO1 nuclear exclusion induced by HCV protein expression, which subsequently generated alterations in glucose homeostasis despite enhanced Akt activation in HCV-transgenic animals. Our results suggest that HCV-infected patients should be assessed for insulin resistance and prediabetes in their routine evaluation to avoid the double burden of diabetes mellitus and chronic liver disease. They also suggest that insulin resistance, glucose intolerance, and diabetes mellitus should be considered extrahepatic manifestations of chronic HCV infection and imply that patients presenting signs of severity should be prioritized for treatment with the new safe and efficacious IFN-free, all-oral combination regimens.
Experimental procedures
Animals
Animals were housed under specific pathogen–free conditions. 2- to 20-month-old C57BL6 male mice transgenic for the full-length HCV open reading frame (FL-N/35 lineage) (38) were used for body weight monitoring. 14- to 18-month-old HCV-transgenic mice were used for all of the other experiments in this study. Age-matched WT male littermates were used as controls. Animals harboring liver tumors at necropsy were removed from the study. The animals were housed in a temperature-controlled environment with a 12-h light/dark cycle and ad libitum access to water and diet (D04 from SAFE, Augy, France; 6.1% carbohydrate, 3.1% fat, and 15.8% protein).
When indicated, mice were injected with 0.25 units/g insulin lispro (Humalog® KwikPen, 100 IU/ml, Eli Lilly, Indianapolis, IN) diluted in PBS, either directly in the portal vein in anesthetized animals sacrificed 2–5 min after injection or intraperitoneally in animals sacrificed 1 h after injection. After sacrifice by means of CO2 intoxication, liver tissue fragments were immediately snap-frozen in liquid nitrogen and stored at −80 °C before analysis. In the “fasting animal” group, animals were sacrificed after a 24-h fasting period. In the “fed animal” group, 24 h-fasting animals were fed using regular chow and glucose-supplemented water and sacrificed 3 h later.
Cell cultures
Hepatoma-derived Huh7 cells, purchased from the ATCC (Manassas, VA), were maintained in DMEM supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Murine hepatocytes from both HCV-transgenic and non-transgenic animals were isolated by portal vein perfusion of collagenase. Freshly isolated hepatocytes were cultured in DMEM supplemented with 10% fetal calf serum, 10 units/ml penicillin, 10 μg/ml streptomycin, 10 μg/ml insulin, 5.5 μg/ml transferrin, and 5 ng/ml sodium selenite. Four hours after perfusion, the medium was removed, and fresh medium supplemented with 0.1 μmol/liter dexamethasone (Sigma-Aldrich, St. Louis, MO) and 50 ng/ml epidermal growth factor was added. When indicated, cells were starved overnight by withdrawal of fetal calf serum and cultured in low-glucose (1g/liter) DMEM and then incubated with 5 μg/ml insulin lispro for 20 min.
Total RNA isolation and RT-qPCR
Total RNA was isolated from frozen mouse livers using the Ambion PARIS RNA and protein isolation kit (Thermo Fisher Scientific, Waltham, MA), according to the instructions of the manufacturer. Then RNA quality and quantity were assessed by means of a 2100 Bioanalyzer and RNA Nano Chips (Agilent Technologies, Santa Clara, CA). RNA integrity number was calculated using Agilent software, and samples displaying an RNA integrity number below 6 were discarded. 1 μg of RNA was reverse-transcribed using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). Real-time quantitative PCR was performed using TaqMan reagents and an Applied Biosystems 7300 thermal cycler (Applied Biosystems, Carlsbad, CA). Specific primers and probes were purchased from Applied Biosystems (supplemental Table 1). The expression level of each gene studied was normalized to that of 18S ribosomal RNA by using the comparative Ct method and expressed as -fold-change using the wild-type mRNA expression level for standardization.
Preparation of protein extracts and immunoblot analysis
Crude protein extracts were prepared by homogenization of frozen mouse livers (50–100 μg) in tissue lysis buffer from the Ambion PARIS RNA and protein isolation kit, supplemented with protease inhibitors (cOmpleteTM EDTA-free protease inhibitor mixture, Sigma-Aldrich) and phosphatase inhibitors (PhosSTOPTM, Sigma-Aldrich), using a tissue homogenizer (MP Fast Prep24, MP Biomedicals, Santa Ana, CA) and MP Lysing Matrix A tubes. Proteins were quantified using the BCA assay (Thermo Fisher Scientific). For protein analysis, polypeptides from crude extracts were separated on precast 4–15% SDS-polyacrylamide gels (Criterion XT precast gels, Bio-Rad) and transferred onto polyvinylidene fluoride membranes (GE Healthcare), and proteins were detected by immunoblotting. The primary antibodies used are listed in supplemental Table 2. The membranes were incubated with the corresponding secondary antibodies coupled to HRP-conjugated donkey anti-rabbit IgG or sheep anti-mouse IgG (GE Healthcare). Labeled antibodies were detected with ECL Prime detection kits (GE Healthcare). Chemiluminescent signals were detected and quantified using an Image Quant Las 4000 minicamera and Image Quant software (GE Healthcare). A polyclonal mouse γ-tubulin antibody (supplemental Table 2) was used as a loading control for total lysates.
Akt kinase assay was performed as described previously (46) using an Akt non-radioactive kinase kit (Cell Signaling Technology, Danvers, MA). Briefly, hepatic samples (0.5 mg) were lysed and active Akt was immunoprecipitated, and its ability to phosphorylate GSK-3 in vitro was analyzed by Western blotting. Relative Akt activities are expressed as the levels of phospho-GSK3 normalized to Akt expression in the unprecipitated input.
Body mass composition
Body mass composition (lean tissue mass, fat mass) was analyzed prior to the clamp studies using a whole-body composition analyzer (EchoMRI 100, EchoMRI, Houston, TX) according to the instructions of the manufacturer. Briefly, awake animals were weighed before they were placed in a mouse holder and inserted into the MRI analyzer. Readings of body composition were obtained within 1 min. Body composition was expressed as a percentage of body weight.
Glucose tolerance tests and glucose and insulin assays
IPGTT was performed in overnight fasted mice. Briefly, glucose (2 mg/g) was injected intraperitoneally. Glycemia was assessed by means of a glucometer (Accu-Check Go and the corresponding test strips, Roche) from 2 μl of blood collected from the tip of the tail vein 0, 10, 20, 40, 60, 90 and 120 min after glucose injection. Insulinemia was measured before glucose injection on blood samples collected from the mouse tail vein using heparinized capillaries. Plasma was separated by centrifugation, and the insulin concentration was assessed using an ultrasensitive mouse insulin ELISA kit (Mercodia, Uppsala, Sweden), according to the instructions of the manufacturer.
Glucose turnover rate at basal state and during hyperinsulinemic–euglycemic clamps
One week before the experiment, an indwelling catheter (BD Biosciences) was inserted into the right jugular veins of mice anesthetized with isoflurane. To measure the glucose turnover rate under basal conditions, a 5-μCi bolus of [3-3H]glucose was injected through the jugular vein, followed by a continuous infusion of [3-3H]glucose (15 μCi) at a constant rate of 1 μl/min for 90 min. Blood samples were collected at the end of the experiment for radioactivity measurement (see below).
The experiment aimed at measuring the glucose turnover rate during hyperinsulinemic–euglycemic clamping was conducted in conscious mice fasting for 5 h as described previously (85). Briefly, a 5-μCi bolus of [3-3H]glucose and a priming dose of insulin (83 milliunits/kg; Actrapid, Novo Nordisk, Bagsvaerd, Denmark) dissolved in isotonic saline was injected through the jugular vein, followed by a continuous infusion of [3-3H]glucose (15 μCi) and insulin (2 milliunits/kg/min) at a constant rate of 1 μl/min to maintain blood glucose levels at 100 mg/dl. During the clamping, blood was sampled from the cut tail every 10 min to determine glucose levels and to adjust the rate of unlabeled glucose infusion to maintain euglycemia. The euglycemic conditions were reached within 30–40 min and subsequently maintained for 60 min. Steady-state specific glucose radioactivity and plasma glucose were determined during the last 20 min of the clamping. During the glucose clamping (50–70 min after the onset of insulin infusion), the glucose disposal rate, which reflects the GUR, is equal to the rate of glucose appearance, which results from endogenous glucose production, added to the amount of infused glucose necessary to maintain euglycemia (GIR, expressed in milligrams/minute/kilogram). To measure [3-3H]glucose radioactivity, blood samples were deproteinized with Ba(OH)2 and ZnSO4, and the supernatant was evaporated to dryness at 50 °C to remove tritiated water. The dry residue was dissolved in 0.5 ml of water, to which 10 ml of scintillation solution was added (Aqualuma Plus, Lumac), and radioactivity was determined in a Packard Tri-Carb 460C liquid scintillation system.
Glucose uptake
Primary mouse hepatocytes were isolated and plated in 6-well plates as described previously (86). Twenty-four hours later, cells were washed with Hanks' buffered salt solution (HBSS, Thermo Fisher Scientific), incubated for 30 min with HBSS supplemented with 100 nm insulin, and washed with HBSS. Then cells were incubated for 1–5 min with HBSS supplemented with 100 μm 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-d-glucose (Sigma-Aldrich). Fluorescence was measured using a microplate fluorimeter (Mithras LB940, Berthold Technologies GmbH & Co., Bad Wildbad, Germany) set with FITC filters.
Indirect immunofluorescence
For indirect immunofluorescence studies and proximity-dependent DNA ligation assays, Huh7.5 cells transiently expressing HCV core protein (genotype 1b (87)) or primary mouse hepatocytes were fixed with 2% paraformaldehyde in PBS for 10 min at room temperature and permeabilized with ice-cold methanol at −20 °C for 10 min, washed with PBS, and incubated for 1 h at room temperature with blocking buffer (Duolink assay, Sigma-Aldrich). The primary antibody was diluted in antibody diluent reagent (Duolink assay, Sigma-Aldrich) and incubated for 1 h at room temperature. For indirect immunofluorescence, bound antibodies were detected with an Alexa Fluor–conjugated secondary antibody (Life Technologies), whereas cellular DNA was stained with DAPI (Life Technologies). Slides were mounted using Prolong Gold antifading solution (Life Technologies), and images were captured using a Zeiss Axioskop 40 microscope in conjunction with the Zeiss MRm Axiocam and Axiovision V40 4.7 software.
Statistical analysis
Results are expressed as means ± S.E. Fisher—Yates–Terry or Mann–Whitney non-parametric rank tests were used when appropriate. p values of less than 0.05 were considered statistically significant.
Ethics statement
Animal housing and procedures were conducted in accordance with the recommendations from the Direction des Services Vétérinaires, Ministry of Agriculture of France according to European Communities Council Directive 2010/63/EU and according to recommendations for health monitoring from the Federation of European Laboratory Animal Science Associations. Protocols involving animals were approved by the local ethics committee (protocol approval DU Exp 38): Comité d'Ethique pour l'Expérimentation Animale no. 51 du Centre d'Expérimentation et de Recherche Fonctionnelle Expérimentale de la Genopole d'Evry (Housing Agreement B21-228-107).
Author contributions
Study conception and design: H. L., J. M. P., F. F., and C. M.; Acquisition of data: H. L., M. R. I., M. R. H., J. P., A. G., M. M., F. D., C. B., and A. P.; Analysis and interpretation of data: H. L., M. R. H., J. M. P., C. M., and F. F.; Drafting of the manuscript: H. L.; Critical revision: J. M. P., C. M., F. F., and M. R. H.
Supplementary Material
Acknowledgments
We thank Philippe Delis, head of the Center for Exploration and Experimental Functional Research (Centre d'Expérimentation et de Recherche Fonctionnelle Expérimentale, Genopole, Evry, France) and Hélène Gautier for animal housing and care.
This work was supported by the National Agency for Research on AIDS and Viral Hepatitis. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables 1 and 2.
- HCV
- hepatitis C virus
- T2D
- type 2 diabetes
- HBV
- hepatitis B virus
- IPGTT
- intraperitoneal glucose tolerance test(s)
- GIR
- glucose infusion rate
- GUR
- glucose utilization rate
- GK
- glucokinase
- PFKFB3
- fructose 1,6-bisphosphatase
- PEPCK
- phosphoenolpyruvate carboxykinase
- Glc-6-PC
- glucose-6-phosphatase
- RT-qPCR
- quantitative real-time PCR
- HBSS
- Hanks' buffered salt solution
- tg
- transgenic
- UPR
- unfolded protein response.
References
- 1. Pawlotsky J. M. (2015) Hepatitis C treatment: the data flood goes on: an update from the liver meeting 2014. Gastroenterology 148, 468–479 [DOI] [PubMed] [Google Scholar]
- 2. Pawlotsky J. M. (2014) New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146, 1176–1192 [DOI] [PubMed] [Google Scholar]
- 3. Allison M. E., Wreghitt T., Palmer C. R., and Alexander G. J. (1994) Evidence for a link between hepatitis C virus infection and diabetes mellitus in a cirrhotic population. J. Hepatol. 21, 1135–1139 [DOI] [PubMed] [Google Scholar]
- 4. Lecube A., Hernández C., Genescà J., Esteban J. I., Jardí R., and Simó R. (2004) High prevalence of glucose abnormalities in patients with hepatitis C virus infection: a multivariate analysis considering the liver injury. Diabetes Care 27, 1171–1175 [DOI] [PubMed] [Google Scholar]
- 5. Alexander G. J. (2000) An association between hepatitis C virus infection and type 2 diabetes mellitus: what is the connection? Ann. Int. Med. 133, 650–652 [DOI] [PubMed] [Google Scholar]
- 6. White D. L., Ratziu V., and El-Serag H. B. (2008) Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J. Hepatol. 49, 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mehta S. H., Brancati F. L., Strathdee S. A., Pankow J. S., Netski D., Coresh J., Szklo M., and Thomas D. L. (2003) Hepatitis C virus infection and incident type 2 diabetes. Hepatology 38, 50–56 [DOI] [PubMed] [Google Scholar]
- 8. Foxton M. R., Quaglia A., Muiesan P., Heneghan M. A., Portmann B., Norris S., Heaton N. D., and O'Grady J. G. (2006) The impact of diabetes mellitus on fibrosis progression in patients transplanted for hepatitis C. Am. J. Transplant. 6, 1922–1929 [DOI] [PubMed] [Google Scholar]
- 9. Eslam M., Aparcero R., Mousa Y. I., Grande L., Shaker Y., Ali A., Del Campo J. A., Khattab M. A., and Romero-Gomez M. (2012) Insulin resistance impairs viral dynamics independently of ethnicity or genotypes. J. Clin. Gastroenterol. 46, 228–234 [DOI] [PubMed] [Google Scholar]
- 10. Serfaty L., Forns X., Goeser T., Ferenci P., Nevens F., Carosi G., Drenth J. P., Lonjon-Domanec I., DeMasi R., Picchio G., Beumont M., and Marcellin P. (2012) Insulin resistance and response to telaprevir plus peginterferon α and ribavirin in treatment-naive patients infected with HCV genotype 1. Gut 61, 1473–1480 [DOI] [PubMed] [Google Scholar]
- 11. Kawamura Y., Arase Y., Ikeda K., Hirakawa M., Hosaka T., Kobayashi M., Saitoh S., Yatsuji H., Sezaki H., Akuta N., Suzuki F., Suzuki Y., and Kumada H. (2010) Diabetes enhances hepatocarcinogenesis in noncirrhotic, interferon-treated hepatitis C patients. Am. J. Med. 123, 951–956.e1 [DOI] [PubMed] [Google Scholar]
- 12. Arase Y., Kobayashi M., Suzuki F., Suzuki Y., Kawamura Y., Akuta N., Kobayashi M., Sezaki H., Saito S., Hosaka T., Ikeda K., Kumada H., and Kobayashi T. (2013) Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology 57, 964–973 [DOI] [PubMed] [Google Scholar]
- 13. Calzadilla-Bertot L., Vilar-Gomez E., Torres-Gonzalez A., Socias-Lopez M., Diago M., Adams L. A., and Romero-Gomez M. (2016) Impaired glucose metabolism increases risk of hepatic decompensation and death in patients with compensated hepatitis C virus-related cirrhosis. Dig. Liver Dis. 48, 283–290 [DOI] [PubMed] [Google Scholar]
- 14. Negro F. (2011) Mechanisms of hepatitis C virus-related insulin resistance. Clin. Res. Hepatol. Gastroenterol. 35, 358–363 [DOI] [PubMed] [Google Scholar]
- 15. Kaddai V., and Negro F. (2011) Current understanding of insulin resistance in hepatitis C. Expert Rev. Gastroenterol. Hepatol. 5, 503–516 [DOI] [PubMed] [Google Scholar]
- 16. Romero-Gómez M., Del Mar Viloria M., Andrade R. J., Salmerón J., Diago M., Fernández-Rodríguez C. M., Corpas R., Cruz M., Grande L., Vázquez L., Muñoz-De-Rueda P., López-Serrano P., Gila A., Gutiérrez M. L., Pérez C., et al. (2005) Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 128, 636–641 [DOI] [PubMed] [Google Scholar]
- 17. Hsu C. S., Liu C. J., Liu C. H., Wang C. C., Chen C. L., Lai M. Y., Chen P. J., Kao J. H., and Chen D. S. (2008) High hepatitis C viral load is associated with insulin resistance in patients with chronic hepatitis C. Liver Int. 28, 271–277 [DOI] [PubMed] [Google Scholar]
- 18. Moucari R., Asselah T., Cazals-Hatem D., Voitot H., Boyer N., Ripault M. P., Sobesky R., Martinot-Peignoux M., Maylin S., Nicolas-Chanoine M. H., Paradis V., Vidaud M., Valla D., Bedossa P., and Marcellin P. (2008) Insulin resistance in chronic hepatitis C: association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology 134, 416–423 [DOI] [PubMed] [Google Scholar]
- 19. Tsochatzis E., Manolakopoulos S., Papatheodoridis G. V., Hadziyannis E., Triantos C., Zisimopoulos K., Goulis I., Tzourmakliotis D., Akriviadis E., Manesis E. K., and Archimandritis A. J. (2009) Serum HCV RNA levels and HCV genotype do not affect insulin resistance in nondiabetic patients with chronic hepatitis C: a multicentre study. Aliment. Pharmacol. Ther. 30, 947–954 [DOI] [PubMed] [Google Scholar]
- 20. Sumida Y., Kanemasa K., Hara T., Inada Y., Sakai K., Imai S., Yoshida N., Yasui K., Itoh Y., Okanoue T., and Yoshikawa T. (2011) Impact of amino acid substitutions in hepatitis C virus genotype 1b core region on liver steatosis and glucose tolerance in non-cirrhotic patients without overt diabetes. J. Gastroenterol. Hepatol. 26, 836–842 [DOI] [PubMed] [Google Scholar]
- 21. Hung C. H., Hu T. H., Lee C. M., Huang C. M., Chen C. H., Wang J. H., and Lu S. N. (2013) Amino acid substitutions in the core region associate with insulin resistance in chronic hepatitis C. Intervirology 56, 166–171 [DOI] [PubMed] [Google Scholar]
- 22. Uraki S., Tameda M., Sugimoto K., Shiraki K., Takei Y., Nobori T., and Ito M. (2015) Substitution in amino acid 70 of hepatitis C virus core protein changes the adipokine profile via Toll-like receptor 2/4 signaling. PLoS ONE 10, e0131346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen L., Liu W., Lai S., Li Y., Wang X., and Zhang H. (2013) Insulin resistance, serum visfatin, and adiponectin levels are associated with metabolic disorders in chronic hepatitis C virus-infected patients. Eur. J. Gastroenterol. Hepatol. 25, 935–941 [DOI] [PubMed] [Google Scholar]
- 24. Anty R., Gelsi E., Giudicelli J., Mariné-Barjoan E., Gual P., Benzaken S., Saint-Paul M. C., Sadoul J. L., Huet P. M., and Tran A. (2007) Glucose intolerance and hypoadiponectinemia are already present in lean patients with chronic hepatitis C infected with genotype non-3 viruses. Eur. J. Gastroenterol. Hepatol. 19, 671–677 [DOI] [PubMed] [Google Scholar]
- 25. Kawaguchi T., Ide T., Taniguchi E., Hirano E., Itou M., Sumie S., Nagao Y., Yanagimoto C., Hanada S., Koga H., and Sata M. (2007) Clearance of HCV improves insulin resistance, β-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am. J. Gastroenterol. 102, 570–576 [DOI] [PubMed] [Google Scholar]
- 26. Romero-Gómez M., Fernández-Rodríguez C. M., Andrade R. J., Diago M., Alonso S., Planas R., Solá R., Pons J. A., Salmerón J., Barcena R., Perez R., Carmona I., and Durán S. (2008) Effect of sustained virological response to treatment on the incidence of abnormal glucose values in chronic hepatitis C. J. Hepatol. 48, 721–727 [DOI] [PubMed] [Google Scholar]
- 27. Arase Y., Suzuki F., Suzuki Y., Akuta N., Kobayashi M., Kawamura Y., Yatsuji H., Sezaki H., Hosaka T., Hirakawa M., Ikeda K., and Kumada H. (2009) Sustained virological response reduces incidence of onset of type 2 diabetes in chronic hepatitis C. Hepatology 49, 739–744 [DOI] [PubMed] [Google Scholar]
- 28. Thompson A. J., Patel K., Chuang W. L., Lawitz E. J., Rodriguez-Torres M., Rustgi V. K., Flisiak R., Pianko S., Diago M., Arora S., Foster G. R., Torbenson M., Benhamou Y., Nelson D. R., Sulkowski M. S., et al. (2012) Viral clearance is associated with improved insulin resistance in genotype 1 chronic hepatitis C but not genotype 2/3. Gut 61, 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shintani Y., Fujie H., Miyoshi H., Tsutsumi T., Tsukamoto K., Kimura S., Moriya K., and Koike K. (2004) Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 126, 840–848 [DOI] [PubMed] [Google Scholar]
- 30. Miyamoto H., Moriishi K., Moriya K., Murata S., Tanaka K., Suzuki T., Miyamura T., Koike K., and Matsuura Y. (2007) Involvement of the PA28γ-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J. Virol. 81, 1727–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawaguchi T., Yoshida T., Harada M., Hisamoto T., Nagao Y., Ide T., Taniguchi E., Kumemura H., Hanada S., Maeyama M., Baba S., Koga H., Kumashiro R., Ueno T., Ogata H., et al. (2004) Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 165, 1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aytug S., Reich D., Sapiro L. E., Bernstein D., and Begum N. (2003) Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology 38, 1384–1392 [DOI] [PubMed] [Google Scholar]
- 33. Bernsmeier C., Duong F. H., Christen V., Pugnale P., Negro F., Terracciano L., and Heim M. H. (2008) Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J. Hepatol. 49, 429–440 [DOI] [PubMed] [Google Scholar]
- 34. Bernsmeier C., Calabrese D., Heim M. H., and Duong H. T. (2014) Hepatitis C virus dysregulates glucose homeostasis by a dual mechanism involving induction of PGC1α and dephosphorylation of FoxO1. J. Viral Hepat. 21, 9–18 [DOI] [PubMed] [Google Scholar]
- 35. Banerjee A., Meyer K., Mazumdar B., Ray R. B., and Ray R. (2010) Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 84, 5936–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., and Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 37. Deng L., Shoji I., Ogawa W., Kaneda S., Soga T., Jiang D. P., Ide Y. H., and Hotta H. (2011) Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 85, 8556–8568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lerat H., Honda M., Beard M. R., Loesch K., Sun J., Yang Y., Okuda M., Gosert R., Xiao S. Y., Weinman S. A., and Lemon S. M. (2002) Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122, 352–365 [DOI] [PubMed] [Google Scholar]
- 39. Lubura M., Hesse D., Neumann N., Scherneck S., Wiedmer P., and Schürmann A. (2012) Non-invasive quantification of white and brown adipose tissues and liver fat content by computed tomography in mice. PLoS ONE 7, e37026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thorens B. (1996) Glucose transporters in the regulation of intestinal, renal, and liver glucose fluxes. Am. J. Physiol. 270, G541–G553 [DOI] [PubMed] [Google Scholar]
- 41. Leturque A., Brot-Laroche E., and Le Gall M. (2009) GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 296, E985–E992 [DOI] [PubMed] [Google Scholar]
- 42. De Fea K., and Roth R. A. (1997) Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry 36, 12939–12947 [DOI] [PubMed] [Google Scholar]
- 43. Zhang J., Gao Z., Yin J., Quon M. J., and Ye J. (2008) S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rui L., Yuan M., Frantz D., Shoelson S., and White M. F. (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398 [DOI] [PubMed] [Google Scholar]
- 45. Ueki K., Kondo T., and Kahn C. R. (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 24, 5434–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Higgs M. R., Lerat H., and Pawlotsky J. M. (2013) Hepatitis C virus-induced activation of β-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 32, 4683–4693 [DOI] [PubMed] [Google Scholar]
- 47. Nakae J., Park B. C., and Accili D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a wortmannin-sensitive pathway. J. Biol. Chem. 274, 15982–15985 [DOI] [PubMed] [Google Scholar]
- 48. Zhang X., Gan L., Pan H., Guo S., He X., Olson S. T., Mesecar A., Adam S., and Unterman T. G. (2002) Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms: direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 277, 45276–45284 [DOI] [PubMed] [Google Scholar]
- 49. Rena G., Prescott A. R., Guo S., Cohen P., and Unterman T. G. (2001) Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem. J. 354, 605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cavaghan M. K., Ehrmann D. A., and Polonsky K. S. (2000) Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J. Clin. Invest. 106, 329–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kahn B. B. (1998) Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell 92, 593–596 [DOI] [PubMed] [Google Scholar]
- 52. Bugianesi E., Salamone F., and Negro F. (2012) The interaction of metabolic factors with HCV infection: does it matter? J. Hepatol. 56, S56–65 [DOI] [PubMed] [Google Scholar]
- 53. Naing C., Mak J. W., Wai N., and Maung M. (2013) Diabetes and infections-hepatitis C: is there type 2 diabetes excess in hepatitis C infection? Curr. Diab. Rep. 13, 428–434 [DOI] [PubMed] [Google Scholar]
- 54. Negro F., Forton D., Craxì A., Sulkowski M. S., Feld J. J., and Manns M. P. (2015) Extrahepatic morbidity and mortality of chronic hepatitis C. Gastroenterology 149, 1345–1360 [DOI] [PubMed] [Google Scholar]
- 55. Vanni E., Abate M. L., Gentilcore E., Hickman I., Gambino R., Cassader M., Smedile A., Ferrannini E., Rizzetto M., Marchesini G., Gastaldelli A., and Bugianesi E. (2009) Sites and mechanisms of insulin resistance in nonobese, nondiabetic patients with chronic hepatitis C. Hepatology 50, 697–706 [DOI] [PubMed] [Google Scholar]
- 56. Shoelson S. E., Lee J., and Goldfine A. B. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maqbool M. A., Imache M. R., Higgs M. R., Carmouse S., Pawlotsky J. M., and Lerat H. (2013) Regulation of hepatitis C virus replication by nuclear translocation of nonstructural 5A protein and transcriptional activation of host genes. J. Virol. 87, 5523–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsui C., Shoji I., Kaneda S., Sianipar I. R., Deng L., and Hotta H. (2012) Hepatitis C virus infection suppresses GLUT2 gene expression via downregulation of hepatocyte nuclear factor 1α. J. Virol. 86, 12903–12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kasai D., Adachi T., Deng L., Nagano-Fujii M., Sada K., Ikeda M., Kato N., Ide Y. H., Shoji I., and Hotta H. (2009) HCV replication suppresses cellular glucose uptake through down-regulation of cell surface expression of glucose transporters. J. Hepatol. 50, 883–894 [DOI] [PubMed] [Google Scholar]
- 60. Parvaiz F., Manzoor S., Iqbal J., McRae S., Javed F., Ahmed Q. L., and Waris G. (2014) Hepatitis C virus nonstructural protein 5A favors upregulation of gluconeogenic and lipogenic gene expression leading towards insulin resistance: a metabolic syndrome. Arch. Virol. 159, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 61. Kuo Y. C., Chen I. Y., Chang S. C., Wu S. C., Hung T. M., Lee P. H., Shimotohno K., and Chang M. F. (2014) Hepatitis C virus NS5A protein enhances gluconeogenesis through upregulation of Akt-/JNK-PEPCK signalling pathways. Liver Int. 34, 1358–1368 [DOI] [PubMed] [Google Scholar]
- 62. Chen J., Wang N., Dong M., Guo M., Zhao Y., Zhuo Z., Zhang C., Chi X., Pan Y., Jiang J., Tang H., Niu J., Yang D., Li Z., Han X., et al. (2015) the metabolic regulator histone deacetylase 9 contributes to glucose homeostasis abnormality induced by hepatitis C virus infection. Diabetes 64, 4088–4098 [DOI] [PubMed] [Google Scholar]
- 63. Banerjee S., Saito K., Ait-Goughoulte M., Meyer K., Ray R. B., and Ray R. (2008) Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 82, 2606–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bose S. K., Shrivastava S., Meyer K., Ray R. B., and Ray R. (2012) Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 86, 6315–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsunekawa S., Demozay D., Briaud I., McCuaig J., Accili D., Stein R., and Rhodes C. J. (2011) FoxO feedback control of basal IRS-2 expression in pancreatic β-cells is distinct from that in hepatocytes. Diabetes 60, 2883–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ide T., Shimano H., Yahagi N., Matsuzaka T., Nakakuki M., Yamamoto T., Nakagawa Y., Takahashi A., Suzuki H., Sone H., Toyoshima H., Fukamizu A., and Yamada N. (2004) SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 6, 351–357 [DOI] [PubMed] [Google Scholar]
- 67. Lerat H., Kammoun H. L., Hainault I., Mérour E., Higgs M. R., Callens C., Lemon S. M., Foufelle F., and Pawlotsky J. M. (2009) Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. J. Biol. Chem. 284, 33466–33474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Huang Y., Feld J. J., Sapp R. K., Nanda S., Lin J. H., Blatt L. M., Fried M. W., Murthy K., and Liang T. J. (2007) Defective hepatic response to interferon and activation of suppressor of cytokine signaling 3 in chronic hepatitis C. Gastroenterology 132, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Walsh M. J., Jonsson J. R., Richardson M. M., Lipka G. M., Purdie D. M., Clouston A. D., and Powell E. E. (2006) Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut 55, 529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bode J. G., Ludwig S., Ehrhardt C., Albrecht U., Erhardt A., Schaper F., Heinrich P. C., and Häussinger D. (2003) IFN-α antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 17, 488–490 [DOI] [PubMed] [Google Scholar]
- 71. Kim K. A., Lin W., Tai A. W., Shao R. X., Weinberg E., De Sa Borges C. B., Bhan A. K., Zheng H., Kamegaya Y., and Chung R. T. (2009) Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J. Hepatol. 50, 705–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. El-Saadany S., Ziada D. H., El Bassat H., Farrag W., El-Serogy H., Eid M., Abdallah M., Ghazy M., and Salem H. A. (2013) The role of hepatic expression of STAT1, SOCS3 and PIAS1 in the response of chronic hepatitis C patients to therapy. Can. J. Gastroenterol. 27, e13–e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Funaoka Y., Sakamoto N., Suda G., Itsui Y., Nakagawa M., Kakinuma S., Watanabe T., Mishima K., Ueyama M., Onozuka I., Nitta S., Kitazume A., Kiyohashi K., Murakawa M., Azuma S., et al. (2011) Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J. Virol. 85, 5986–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim K., Kim K. H., and Cheong J. (2010) Hepatitis B virus X protein impairs hepatic insulin signaling through degradation of IRS1 and induction of SOCS3. PLoS ONE 5, e8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang C. C., Tseng T. C., and Kao J. H. (2015) Hepatitis B virus infection and metabolic syndrome: fact or fiction? J. Gastroenterol. Hepatol. 30, 14–20 [DOI] [PubMed] [Google Scholar]
- 76. Park S. H., Kim D. J., and Lee H. Y. (2009) Insulin resistance is not associated with histologic severity in nondiabetic, noncirrhotic patients with chronic hepatitis B virus infection. Am. J. Gastroenterol. 104, 1135–1139 [DOI] [PubMed] [Google Scholar]
- 77. Imazeki F., Yokosuka O., Fukai K., Kanda T., Kojima H., and Saisho H. (2008) Prevalence of diabetes mellitus and insulin resistance in patients with chronic hepatitis C: comparison with hepatitis B virus-infected and hepatitis C virus-cleared patients. Liver Int. 28, 355–362 [DOI] [PubMed] [Google Scholar]
- 78. Gross D. N., van den Heuvel A. P., and Birnbaum M. J. (2008) The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 79. Kondapaka S. B., Zarnowski M., Yver D. R., Sausville E. A., and Cushman S. W. (2004) 7-Hydroxystaurosporine (UCN-01) inhibition of Akt Thr308 but not Ser473 phosphorylation: a basis for decreased insulin-stimulated glucose transport. Clin. Cancer Res. 10, 7192–7198 [DOI] [PubMed] [Google Scholar]
- 80. Mitsuyoshi H., Itoh Y., Sumida Y., Minami M., Yasui K., Nakashima T., and Okanoue T. (2008) Evidence of oxidative stress as a cofactor in the development of insulin resistance in patients with chronic hepatitis C. Hepatol. Res. 38, 348–353 [DOI] [PubMed] [Google Scholar]
- 81. Chouteau P., Defer N., Florimond A., Caldéraro J., Higgs M., Gaudin A., Mérour E., Dhumeaux D., Lerat H., and Pawlotsky J. M. (2012) Hepatitis C virus (HCV) protein expression enhances hepatic fibrosis in HCV transgenic mice exposed to a fibrogenic agent. J. Hepatol. 57, 499–507 [DOI] [PubMed] [Google Scholar]
- 82. Higgs M. R., Lerat H., and Pawlotsky J. M. (2010) Downregulation of Gadd45β expression by hepatitis C virus leads to defective cell cycle arrest. Cancer Res. 70, 4901–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Furutani T., Hino K., Okuda M., Gondo T., Nishina S., Kitase A., Korenaga M., Xiao S. Y., Weinman S. A., Lemon S. M., Sakaida I., and Okita K. (2006) Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 130, 2087–2098 [DOI] [PubMed] [Google Scholar]
- 84. Higgs M. R., Chouteau P., and Lerat H. (2014) “Liver let die”: oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 95, 991–1004 [DOI] [PubMed] [Google Scholar]
- 85. Troy S., Soty M., Ribeiro L., Laval L., Migrenne S., Fioramonti X., Pillot B., Fauveau V., Aubert R., Viollet B., Foretz M., Leclerc J., Duchampt A., Zitoun C., Thorens B., et al. (2008) Intestinal gluconeogenesis is a key factor for early metabolic changes after gastric bypass but not after gastric lap-band in mice. Cell Metab. 8, 201–211 [DOI] [PubMed] [Google Scholar]
- 86. Disson O., Haouzi D., Desagher S., Loesch K., Hahne M., Kremer E. J., Jacquet C., Lemon S. M., Hibner U., and Lerat H. (2004) Impaired clearance of virus-infected hepatocytes in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 126, 859–872 [DOI] [PubMed] [Google Scholar]
- 87. Piodi A., Chouteau P., Lerat H., Hézode C., and Pawlotsky J. M. (2008) Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology 48, 16–27 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.