

Abstract
Ribosomally synthesized and posttranslationally modified peptide (RiPP) pathways produce a diverse array of natural products. A subset of these pathways depends on radical S-adenosylmethionine proteins to modify the RiPP-produced peptide. Mycofactocin biosynthesis is one example of an S-adenosylmethionine protein-dependent RiPP pathway. Recently, it has been shown that MftC catalyzes the oxidative decarboxylation of the C-terminal tyrosine (Tyr-30) on the mycofactocin precursor peptide MftA; however, this product has not been verified by techniques other than MS. Herein, we provide a more detailed study of MftC catalysis and report a revised mechanism for MftC chemistry. We show that MftC catalyzes the formation of two isomeric products. Using a combination of MS, isotope labeling, and 1H and 13C NMR techniques, we established that the major product, MftA*, is a tyramine-valine-cross-linked peptide formed by MftC through two S-adenosylmethionine-dependent turnovers. In addition, we show that the hydroxyl group on MftA Tyr-30 is required for MftC catalysis. Furthermore, we show that a substitution in the penultimate MftA Val-29 position causes the accumulation of an MftA** minor product. The 1H NMR spectrum indicates that this minor product contains an αβ-unsaturated bond that likely arises from an aborted intermediate of MftA* synthesis. The finding that MftA* is the major product formed during MftC catalysis could have implications for the further elucidation of mycofactocin biosynthesis.
Keywords: enzyme mechanism, iron-sulfur protein, peptide biosynthesis, radical, S-adenosylmethionine (SAM), MftC, mycofactocin
Introduction
Mycofactocin has been proposed to be a new member of the peptide-derived redox cofactor family that consists of members such as pyrroloquinoline quinone and topaquinone (1, 2). The mycofactocin biosynthetic pathway is composed of six genes, mftABCDEF, beginning with the peptide MftA. MftA is ∼30–60 amino acids in length and contains a conserved C-terminal sequence (-IDGXCGVY). The C terminus is putatively modified by the remaining gene products to form mycofactocin, which is anticipated to serve as a redox cofactor for other nicotinamide-dependent proteins (2, 3). However, the biosynthesis, the structure, and the bona fide physiological function(s) of mycofactocin remains unknown. To unravel the biosynthesis of mycofactocin and thereby solve its structure, our laboratory and others have approached the pathway through an in vitro analysis of pathway proteins (4–6). The discovery of the first step in mycofactocin biosynthesis, the MftC-dependent oxidative decarboxylation of the C-terminal tyrosine of MftA, was an important start to understanding mycofactocin biosynthesis (4, 6). However, basic mechanistic questions regarding MftC as well as structural characterization of the MftC-catalyzed product remain unresolved.
MftC is a radical S-adenosylmethionine ((SAM)2 herein referred to as RS) protein responsible for the first step of the biosynthesis of mycofactocin (2, 4, 6). MftC belongs to a subfamily of RS proteins that contain an ∼100-amino acid C-terminal domain annotated as a SPASM domain (subtilosin A, pyrroloquinoline quinone, anaerobic sulfatase maturating enzyme, and mycofactocin) (7). Currently, >16,000 RS-SPASM protein sequences can be found in the Interpro database (IRP023885); however, <0.1% of the proteins have been characterized to date. RS-SPASM proteins are primarily associated with RiPP (ribosomally synthesized and posttranslationally modified peptide) biosynthetic pathways (7, 8). Indeed, all nine RS-SPASM proteins that have been characterized to date have been shown to modify a peptide or protein (4, 6, 9–16). Furthermore, these RS-SPASM proteins have been shown to contribute vital biochemical transformations for a variety of peptide products that include bacteriocins, quorum sensing molecules, and redox cofactors (10–13, 15, 16).
To modify their peptide substrates, RS-SPASM proteins reductively cleave SAM to generate a 5′-deoxyadenosine radical (dAdo·). The dAdo· is directed by the RS-SPASM protein to abstract a hydrogen from the peptide substrate, subsequently resulting in the formation of dAdo and an alkyl radical. The alkyl radical further reacts to form intramolecular thioether or carbon—carbon bonds (e.g. Refs. 10 and 16), decarboxylation of the C terminus (e.g. Refs. 4 and 6), or the transformation of cysteine to formyl glycine (e.g. Ref. 9). The enzyme's ability to control and direct the highly oxidative dAdo· is paramount to preventing inactivation of the enzyme or the formation of unwanted byproducts. However, by loosening the restrictions of the enzyme on its control of the dAdo·, new and intriguing derivatives of natural products can be formed (17, 18).
Recently, published LC-MS/MS and HPLC data suggested that MftC catalyzes the oxidative decarboxylation of the C-terminal tyrosine on the peptide MftA, putatively forming an αβ-unsaturated bond (Fig. 1) (4, 6). Moreover, this reaction only occurs with the peptide chaperone MftB present (6). A mechanism was proposed that suggests the dAdo· generated by the reductive cleavage of SAM by MftC abstracts the Cβ hydrogen from the C-terminal tyrosine on MftA, leading to the formation of a Cβ radical (Fig. 1) (6). Upon the loss of an electron and the deprotonation of the phenolic proton, a benzenone is formed. Through the loss of the CO2 and acidification of benzenone, the phenol is restored, and the αβ-unsaturated bond is formed. Currently, the proposed mechanism and the product of MftC catalysis remain unconfirmed. Herein, we report on the products of the MftC-catalyzed reaction, the mechanism of the reaction, and we describe how amino acid substitutions on MftA can lead to new enzymatic activity and chemically diverse products.
Figure 1.
Previously proposed reaction mechanism for MftC.
Results
MftC catalyzed the formation of two products
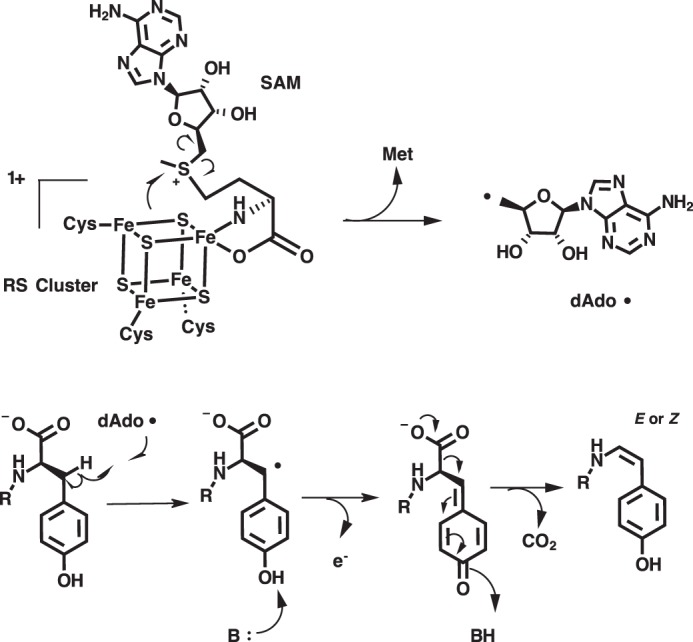
The catalytic activity of MftC was verified by carrying out reactions containing MftA, MftB, MftC, DTH, and DTT and analyzed by HPLC monitoring the absorbance at 280 nm. This procedure was performed similar to previous reports with the exception that the running buffer was replaced with sodium phosphate and a C4 HPLC column was used instead of a C18 HPLC column (6). The MftA M1W variant was used to increase the spectroscopic handle at 280 nm, thus enabling a facile means for identifying starting material and products. Under these new analytical conditions, the chromatogram of the starting material showed a major species eluting at 13.1 min (Fig. 2A, black). The absorbance spectrum at the elution peak maximum indicated that the absorbance maximum is 278 nm (Fig. 2B, black). The MftC reaction resulted in two new peaks that elute at 14.3 and 14.6 min (Fig. 2A, red). The resolution of the 14.6-min peak is an improvement over our previous separation where it was found as a shoulder of the 14.3-min peak (6). By normalizing the peak integrations at 280 nm, this 14.3-min species, herein referred to as MftA*, accounted for ∼90% of the product formed, whereas the minor peak, herein referred to as MftA**, accounted for ∼10% of product formed. The absorbance spectra for both MftA* and MftA** at peak maxima were also compared. The absorbance spectrum for MftA* indicated that the peak absorbance remained at 278 nm (Fig. 2B, red). However, the absorbance spectrum for MftA** was clearly different, with a peak absorbance at 282 nm and a shoulder feature extending out to ∼325 nm (Fig. 2B, blue).
Figure 2.
MftC catalyzed the formation of two MftA products annotated as MftA* and MftA**. A, HPLC analysis of the reactions shows the conversion of MftA M1W (black) to MftA* (14.3 min) and MftA** (14.5 min). Analyses were carried out on a C4 HPLC column. B, the absorbance spectra of MftA M1W (black) and MftA* (red) are similar, whereas the absorbance spectra for MftA** (blue) indicates the appearance of a new spectral feature from 300–325 nm and a red-shift in peak maximum. mAu, milliabsorbance units. *, MftA*; **, MftA**.
Further analysis of the products was carried out by mass spectrometry. Both MftA* and MftA** were isolated by HPLC and analyzed by HRMS and compared with the starting material. HRMS analysis of the starting material found the mass to be m/z = 1185.54 (MH3+, theoretical m/z = 1185.54, supplemental Fig. S1A). The same analysis was carried out on MftA* and MftA**, and both were found to have the same mass of m/z = 1170.20 (MH3+), a difference of m/z = 15.33 from MftA. It should be noted that all masses reported herein were in the +3 charge state (MH3+, z = 3) unless otherwise stated. Therefore, a difference of m/z = 15.33 is equivalent to a difference of 46 Da. This 46-Da change in mass corresponds to the loss of 1 carbon, 2 oxygens, and 2 hydrogens from MftA (MH3+, theoretical m/z = 1170.20, supplemental Fig. S1, B and C) and is in good agreement with previous accounts (4, 6). Taken together with the differences in the absorbance spectra, we propose that MftC catalysis results in the formation of two isomeric products, MftA* and MftA**.
13C NMR and 1H NMR of MftA* suggested that an unsaturated carbon-carbon bond is not present
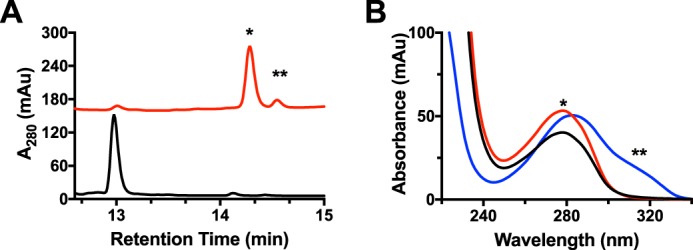
Although the mechanism proposed is reasonable under the assumption that the αβ-unsaturated bond is formed, the only evidence that an αβ-unsaturated is present in MftA* comes indirectly from HRMS analysis and more recently through the incorporation of Marfey's reagent to a proposed downstream product (4–6). Therefore, to conclusively determine if an αβ-unsaturated bond is formed by MftC, the sole tyrosine (Tyr-30) on MftA was uniformly labeled with 13C for use in 13C NMR analysis. The rationale behind this experiment is 2-fold; first, the 13C NMR spectrum would indicate the loss of the carboxylate carbon, confirming HRMS experiments (4, 6). Second, upon formation of the unsaturated bond, the signals from the Cα and Cβ carbons would undergo a substantial chemical shift (δ ∼50–80 ppm) downfield. Both results would unambiguously confirm that the product of the MftC-catalyzed reaction contains an unsaturated bond.
Because MftA contains only a single tyrosine, 13C labeling of the peptide tyrosine was carried out by growing Escherichia coli RF4 (DE3) cells (19) containing the mftA M1W/pET28HST plasmid in M9 minimal media supplemented with individual amino acids and 50 mg/liter 13C9,15N tyrosine. The peptide was expressed and purified to homogeny following standard protocols. The peptide was analyzed by HRMS and found to be fully labeled, having a m/z = 1188.88 (MH3+, theoretical m/z = 1188.54, supplemental Fig. S2A). Accordingly, the 13C NMR spectrum for unreacted peptide contained the expected nine carbons with notable saturated carbon signals at δ ∼35 and ∼55 ppm and the carboxylate carbon at δ ∼178 ppm (Fig. 3A, black). After an overnight reaction with MftC, MftB, SAM, DTH, and DTT, the product MftA* was isolated by HPLC and subjected to mass analysis and 13C NMR experiments. The mass spectrum of MftA* indicated a loss of 47 Da (MH3+, found m/z = 1173.21; theoretical m/z = 1173.21, supplemental Fig. S2B), in good agreement with the loss of one 13C, two oxygens, and two hydrogens (4, 6). Analysis of the reacted peptide by 13C NMR indicated that the signal for the carboxylate carbon is absent, strongly supporting that MftA* is decarboxylated. However, the NMR spectrum also indicated that the chemical shift for the β carbon remains at δ ∼35 ppm, whereas the α carbon underwent a modest downfield shift to δ ∼61 ppm (Fig. 3A, red). The chemical shifts for both α and β remain in the saturated regime of 13C NMR and suggest that an αβ-unsaturated bond is not formed by MftC under these conditions (for full NMR spectra, see supplemental Fig. S3, A and B). Together with the mass spectrum data, this indicates that a new intramolecular bond is formed at the Cα or Cβ position.
Figure 3.
13C (A) and 1H (B) NMR analyses of MftA* did not indicate that an αβ-unsaturated bond is present. MftA is represented by black spectra, and MftA* is represented by red spectra.
To confirm this finding, 1H NMR was conducted on the MftA* as it only contains three aromatic residues, histidine, tryptophan, and tyrosine, and the non-aromatic unsaturated regime (δ 4.5–6.5 ppm) should not contain any signal. As expected, the 1H NMR spectrum for the HRMS-verified unmodified MftA indicates that 11 hydrogens are in the aromatic region. The tyrosine hydrogens show as two doublets at δ ∼6.7 and ∼7.0 ppm, the tryptophan hydrogens show as two doublets between δ 7.4 and 7.5 ppm, two triplets at δ ∼7.1 and 7.2, and a singlet at δ ∼6.7 ppm, and the histidine hydrogens show as two singlets at δ ∼7.1 and ∼7.8 ppm (Fig. 3B, black). After an overnight reaction with MftC, MftB, SAM, DTH, and DTT, the major product was isolated by HPLC, verified by HRMS, and used in 1H NMR experiments. Interestingly, the 1H NMR spectrum of MftA* did not contain any new signal in the unsaturated regime. However, the C2 and C6 hydrogens on tyrosine underwent a modest change to δ ∼7.1 ppm (Fig. 3B, red). This result corroborates the 13C NMR findings (for full NMR spectra, see supplemental Fig. S3, C and D). Two new peaks did become apparent at δ ∼7.4 and 8.4 ppm; however, these features are likely attributed to protected amide hydrogens. Direct 1H NMR experiments on MftA** could not be conducted due to its limited yield; however, an alternative strategy was used to deduce if a double bond is present, and this strategy will be discussed later.
To validate the expected chemical shifts from an αβ-unsaturated MftA*, the model compound l-Val-p-(2-aminoethenyl)phenol was synthesized following modifications of previously reported synthetic routes (supplemental Schemes S1 and S2) (20, 21). We synthesized both cis and trans model compounds because the isomeric form of the peptide product had yet to be identified. The 13C NMR of the cis isomer clearly indicates that the chemical shift of Cα and Cβ are δ ∼109 and 121 ppm, whereas in the trans isomer the Cα and Cβ are δ ∼112 and 123 ppm (supplemental Fig. S4, A and B). Likewise, the 1H NMR spectra for the model compounds clearly show the presence of a pair of doublets at δ ∼6.7 and 5.7 ppm and δ ∼6.3 and 7.5 ppm for the cis and trans isomers, respectively (supplemental Fig. S4, C and D). The NMR data for both model compounds contrast with NMR data for MftA*. Whereas the model compounds have key 13C and 1H NMR spectral features that indicate the presence of a Cα/Cβ-unsaturated bond, the NMR spectra of MftA* are missing these features. Therefore, 13C and 1H NMR data are inconsistent with MftA* containing an αβ-unsaturated bond.
The major product of the MftC reaction was a cross-linked Val-Tyr*
Previous MS/MS experiments have demonstrated that b fragments can be found up to the penultimate residue (Val-29) on modified MftA (4, 6). Collectively, with the NMR results from the previous section, it was hypothesized that MftA* could consist of a carbon-carbon bond cross-link between the tyrosine Cα or Cβ and the valine side chain. To test this hypothesis, a peptide with deuterium-labeled valine, Val-29 (D8), could be used in HRMS experiments to probe if a new bond was being formed between the penultimate valine and the tyrosine Cα or Cβ. If a new C-C bond was being formed between Val-29 (D8) and the tyrosine Cα or Cβ, a loss of ∼47 Da would be expected. Therefore, Val-29 of MftA was labeled with valine (D8, 98%) using growth conditions that suppressed scrambling (e.g. addition of excess leucine and isoleucine). It should be noted that the triple mutant MftA construct containing V13I, V18I, and V21I was used so that only the valine at position 29 was labeled (Val-29). The labeled peptide was purified using standard protocol. Analysis of Val-29 (D8) MftA by mass spectrometry showed 50% incorporation (MH3+, found m/z = 1183.89, theoretical m/z = 1183.89, Fig. 4A). The peptide was subsequently reacted with MftB, MftC, SAM, DTT, and DTH to form MftA*. HRMS analysis of MftA* indicated the product was 47-Da lighter (MH3+, found m/z = 1168.22; Fig. 4B), in good agreement with the loss of 1 carbon, 2 oxygens, 1 hydrogen, 1 deuterium (MH3+, theoretical m/z = 1168.22). This finding supports the formation of a carbon-carbon bond between the tyrosine Cα or Cβ and the valine side chain.
Figure 4.

Analysis of the mass spectrum of Val-29 (D8)-labeled MftA M1W-purified peptide (A) and Val-29 (D8)-labeled MftA M1W* (B) indicated that Val-29 forms a new intramolecular bond. Mass spectra of Val-29 (3-D)-labeled MftA M1W (C) and Val-29 (3-D) labeled MftA M1W* (D) indicates that the new intramolecular bond is formed on the Cβ of Val-29. All collected spectral data are represented in black, and the predicted spectra for the deuterium-labeled MftA MH3+ ions are shown in red. The predicted spectra for unlabeled MftA MH3+ is shown in blue for panels A and B.
We expected that a new bond would be formed between tyrosine Cα/Cβ and the Cβ of valine. To validate this hypothesis, we incorporated 3-D-labeled valine at Val-29 on MftA. Similar to before, if the new bond is formed on the Cβ of Val-29 as expected, mass analysis of Val-29 (3-D)-labeled MftA* would show a loss of 47 Da. Analysis of unreacted Val-29 (3-D) MftA by HRMS showed complete incorporation (MH3+, found m/z = 1181.55 theoretical m/z = 1181.55; Fig. 4C). After an overnight reaction with MftC, HRMS analysis of the MftA* found the mass to be m/z = 1165.88 (MH3+, Fig. 4D). This difference of 47 Da corresponding to the loss of 1 carbon, 2 oxygens, 1 hydrogen, and 1 deuterium (MH3+, theoreticalm/z = 1165.88) and is consistent with the expectation of an intramolecular bond between the Cβ position of Val-29 and the Cα/Cβ of tyrosine.
The MftA V29A mutant accumulated a reaction intermediate
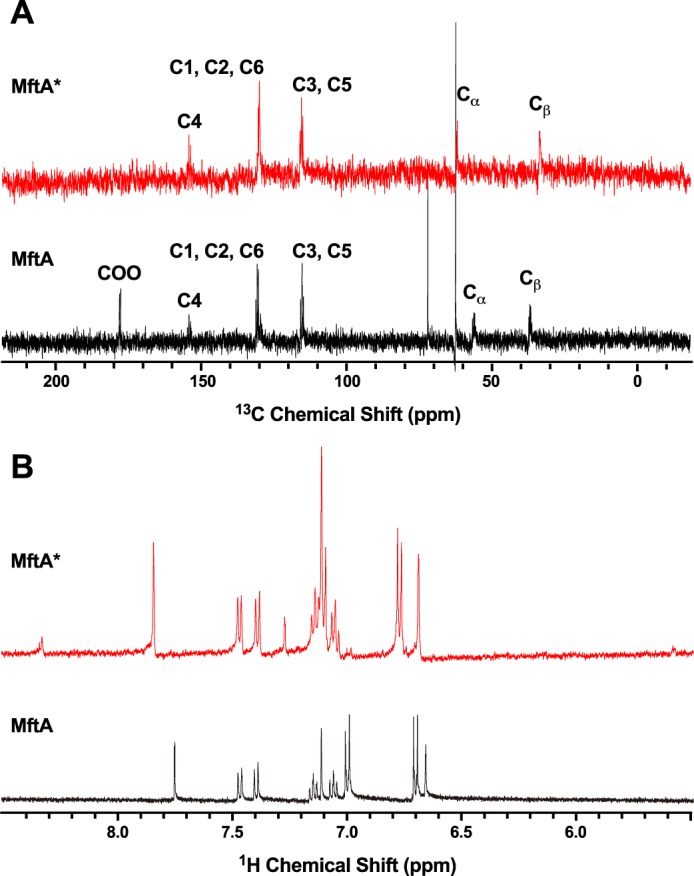
After the demonstration that Val-29 is an important component in MftA*, it was predicted that a mutation at the Val-29 position would be detrimental to the formation of MftA*. Therefore, the MftA M1W/V29A mutant was generated. As mentioned previously, when MftC reactions were carried out with wild-type MftA, two products were found to elute at 14.3 min (90%) and 14.6 min (10%) when analyzed by HPLC. The absorbance maximum of the major peak is clearly defined at 278 nm, whereas the minor peak had an absorbance maximum at 282 nm with a shoulder that extended to ∼325 nm. However, in the case of the reaction containing MftA M1W/V29A, HPLC analysis showed a reverse trend with a minor peak for MftA M1W/V29A* eluting at 13.8 min (10%) and a major peak for MftA M1W/V29A** eluting at 14.2 min (90%) (Fig. 5A). The relative amount of MftA M1W/V29A** varied between 50 and 90% when compared with MftA M1W/V29A* (data not shown) depending on reaction conditions. Moreover, the absorbance maximum of MftA M1W/V29A* was 278 nm, whereas the absorbance maximum of MftA M1W/V29A** was 282 nm, with a shoulder extending to ∼325 nm (Fig. 5B, red and blue, respectively). Interestingly, the absorbance spectrum is similar to the synthesized model compound trans-l-Val-p-(2-aminoethenyl)phenol but not of the cis-l-Val-p-(2-aminoethenyl)phenol isomer (supplemental Fig. S5). Further analysis of the MftA M1W/V29A** by HRMS indicated a mass of m/z = 1160.85 (MH3+, supplemental Fig. 6A), a change of −46 Da from the starting material (MH3+: 1176.18 m/z, supplemental Fig. 6B). This mass is identical to the −46 Da that is expected in the oxidative decarboxylated product that forms the αβ bond unsaturated product.
Figure 5.
The MftA M1W/V29A mutant led to the accumulation of MftA M1W/V29A** product. HPLC analysis of the reactions shows the conversion of MftA M1W/V29A (black) to MftA* (13.7 min) and MftA** (14.1 min). Analyses were carried out on a C4 HPLC column. B, the absorbance spectra of MftA M1W/V29A (black), MftA M1W/V29A* (red), and MftA M1W/V29A** (blue) were similar to what was found for reactions with MftA M1W. C, 1H NMR analysis of MftA M1W/V29A** demonstrated the formation of an αβ-unsaturated indicated by appearance of doublets at ∼6.3 and ∼7.2 ppm (indicated by **). mAu, milliabsorbance units.
To verify that the MftA V29A mutant did in fact contain a double bond, 1H NMR was conducted on the reaction product MftA M1W/V29A**. The 1H NMR spectrum for the unreacted MftA M1W/V29A showed the same predicted number of protons and chemical shifts as described above (Fig. 5C, black). After an overnight reaction, MftA M1W/V29A** was isolated and analyzed by 1H NMR. Interestingly, two new doublets appeared at δ ∼6.3 and 7.3 ppm (Fig. 5C, blue). By measuring the J-coupling constant for the well resolved doublet (δ ∼6.3 ppm, J = 14.7 Hz), we determined that MftA M1W/V29A** is in the trans configuration. The chemical shifts of the new doublets and the coupling constant is in good agreement with the synthesized trans-l-Val-p-(2-aminoethenyl)phenol model compound (δ ∼6.3 and 7.5 ppm, J = 14.7 Hz).
MftC utilized 2 eq of SAM to cross-link Tyr-30 and Val-29
To test which hydrogen on tyrosine is abstracted by the dAdo radical, deuterium was specifically labeled in the Cα and Cβ positions on the C-terminal tyrosine of MftA, and the dAdo formed from the reaction was subjected to HRMS analysis. The rationale for this experiment is that if the Cβ position is labeled with deuterium, dAdo resulting from dAdo· abstraction of a deuterium would have a mass of ∼1-Da greater. These types of experiments have been used to successfully determine which hydrogen is abstracted by the RS-SPASM proteins AlbA and SkfB (22, 23). Labeling of the peptide was conducted similarly to the 13C-labeling experiments except that the M9 minimal media was supplemented with 100 mg/liter Ring-2,6-D2, 2-D (Cα-labeled), or 3,3-D2-tyrosine (Cβ-labeled). The Cα and Cβ deuterium-labeled peptides that were expressed and purified resulted in 40 and 100% incorporation, respectively, calculated from simulations of the HRMS data (supplemental Fig. S7, A and B).
Optimized conditions for the deuterium incorporation experiments contained 200 μm Cα-labeled tyrosine or Cβ-labeled tyrosine-labeled MftA, 200 μm MftB, 100 μm MftC, 400 μm SAM, 10 mm DTT, and 2 mm DTH. Under these conditions approximately half of MftA and SAM was consumed, and a maximum of 20 and 50% incorporation of deuterium in dAdo was expected for the Cα- and Cβ-labeled peptides, respectively. Two major factors led to the expected incorporation values; first, the amount of deuterium-labeled tyrosine incorporated into the peptide would directly affect the incorporation value (e.g. for a single turnover, 50% labeling would result in a 50% incorporation). The second factor is the observation that MftC catalyzes the uncoupled reductive cleavage of SAM (i.e. MftA is not present), which results in the formation of unlabeled dAdo (e.g. for a reaction with 2 eq of SAM and 1 eq of 100% labeled peptide, the result would be 50% deuterium labeled dAdo) (6). The resulting dAdo mixture from the reaction was purified by reverse phase HPLC and analyzed by HRMS. In the controls, mass spectrometry results for commercial dAdo showed a mass of m/z = 252.1098 (MH+, theoretical m/z = 252.1091; Fig. 6A), and the simulated spectrum for deuterium-labeled dAdo predicts a mass at m/z = 253.1154 (MH+, Fig. 6B). HRMS analysis of the dAdo isolated from the reaction with Cα-labeled MftA provided a mass of m/z = 252.1095 (MH+, Fig. 6C) similar to the dAdo control. Conversely, the mass spectrum of the dAdo isolated from the reaction with Cβ-labeled MftA showed two prominent masses at m/z = 252.1095 and m/z = 253.1158 (Fig. 6D). The species at m/z = 252.1095 corresponded to unlabeled dAdo, whereas the species at m/z = 253.1158 corresponded to the deuterium-enriched dAdo species. By comparing the ion abundances for the dAdo, it was estimated that deuterium was incorporated into ∼25% of the dAdo, half of the predicted maximum. These results strongly indicate that the dAdo· abstracts the Cβ hydrogen in the MftC-catalyzed decarboxylation of MftA.
Figure 6.

Shown are HRMS analyses of dAdo control (A), simulated incorporation of one deuterium in dAdo (B), dAdo isolated from a reaction with Tyr-30 (ring-2,6-D2, 2-D) MftA (Cα-labeled) (C), dAdo isolated from a reaction with Tyr-30 (3,3-D2) MftA (Cβ-labeled) (D), and dAdo isolated from a reaction with Val-29 (3-D) MftA (Cβ-labeled) (E).
After determining that Val-29 played an important role in MftC catalysis, it was hypothesized that MftC required 2 eq of SAM, one for the oxidative decarboxylation of tyrosine and the other for the generation of a cross-link between Tyr-30 to Val-29. Moreover, because the MftC reactions with the MftA M1W/V29A mutant accumulated trace amounts of MftA* product, we anticipated that the second dAdo· was responsible for abstraction of a hydrogen atom from the Cβ of Val-29. To test this hypothesis, site-specific Val-29 (3-D)-labeled MftA (described above) was used in optimal assays containing 200 μm MftA, 200 μm MftB, 100 μm MftC, 1 mm SAM, 10 mm DTT, and 2 mm DTH. Under these conditions, we expected a maximum of 20% deuterium incorporation into dAdo. The resulting dAdo mixture was purified by HPLC and subjected to HRMS analysis. The mass spectrum of the dAdo isolated from the reaction with Val-29 (3-D) MftA shows two prominent masses at m/z = 252.1100 and m/z = 253.1155 (Fig. 6E), similar to previous results when the MftA substrate was labeled with deuterated tyrosine. Comparison of the ion abundances for dAdo indicated that deuterium was incorporated into ∼15% of the dAdo. The observation of reaction-derived deuterium enrichment in dAdo supports the conclusion that the hydrogen atom from Cβ of valine is abstracted by dAdo· and indicates that 2 eq of SAM are likely being consumed to form MftA*.
MftC catalyzed the conversion of MftA** to MftA*
Current data suggest that MftA** is an intermediate in the formation of MftA*. If this is correct, MftC should catalyze the conversion of MftA** to MftA*. To confirm this hypothesis, we isolated a sufficient quantity MftA** by HPLC (Fig. 7, blue) and tested it as a substrate for MftC. An overnight reaction containing MftA**, MftB, MftC, SAM, DTT, and DTH was subjected to HPLC analysis. The chromatogram of the MftC reaction with MftA** shows the disappearance of the MftA** peak and the appearance of the MftA* peak (Fig. 7, red). The absorbance spectra of the starting material and product (Fig. 7, inset) also suggests that MftC converted MftA** to MftA*. This result clearly indicates that MftC catalyzes the conversion of MftA** to MftA*. Moreover, this result indicates that MftA** is an intermediate in the formation of MftA*.
Figure 7.
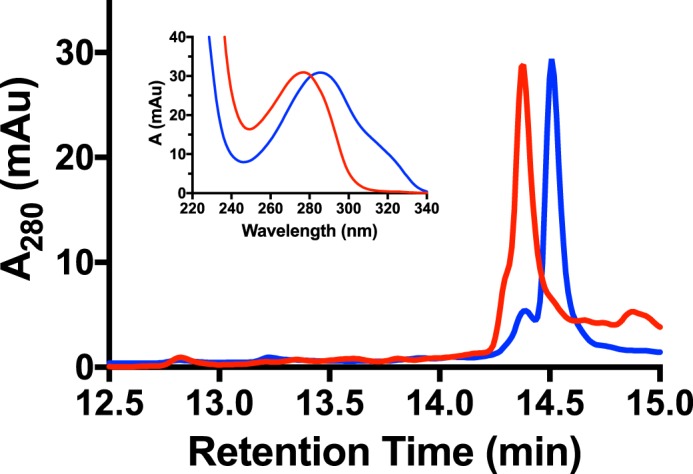
MftC catalyzed the conversion of MftA** to MftA*. HPLC analysis of MftA** starting material (blue) and of a reaction with MftC (red) indicates a change in retention time and absorbance spectrum (inset). Analyses were carried out on a C4 HPLC column. mAu, milliabsorbance units.
Site-directed mutagenesis of Tyr-30 indicated that a labile proton was required for catalysis
The original reaction mechanism for MftC (Fig. 1) was proposed to be dependent upon the loss of the phenolic proton from the MftA tyrosine to form a benzenone. To test if the phenol on MftA Tyr-30 participates in the chemical mechanism, single amino acid replacements of the residue were prepared by site-directed mutagenesis, and the resulting peptides were tested as substrates for MftC. Again, the M1W MftA variant was used as the template for site-directed mutagenesis to increase the spectroscopic handle at 280 nm, as previously discussed (6). All starting materials and reactions were analyzed by HPLC using a C4 column and phosphate buffer, monitoring the absorbance at 280 nm and the absorbance spectrum between 220 and 400 nm. Lastly, samples were analyzed by HRMS.
The reference dataset for MftA Tyr-30 peptide is shown in Fig. 8A. For clarity, all peptide substrates are shown in black, and all reaction products are shown in red. Analysis by HPLC shows that the retention time for the decarboxylated product is ∼1-min longer than starting material, in agreement to with the previous report (6). Moreover, the UV-visible absorbance spectrum of the peptide product shows a peak absorbance of 278 nm. Mass spectrum analysis of the MftA Tyr-30 peptide showed a parent peak of m/z = 1185.54 (MH3+, theoretical m/z = 1185.54). In addition, peaks for the single- and double-oxidized MftA Tyr-30 are present with an increase of m/z = 5.33 and 10.66, respectively. The oxidized forms of the peptide are found in most of the samples that were analyzed and are likely associated with oxidation of a methionine residue. Lastly, the mass spectrum for the reaction shows decarboxylated MftA Tyr-30 peptide at m/z = 1170.20 (MH3+, theoretical m/z = 1170.20) with various oxidation states. These results are identical to those previously discussed.
Figure 8.

HPLC, UV-visible, and HRMS analysis of MftA M1W (A), MftA M1W/Y30F (B), MftA M1W/Y30S (C), and MftA M1W/Y30W (D). Each variant has a corresponding HPLC chromatogram (middle column), a UV-visible spectrum (middle column, inset), and an ion chromatogram (right column). HPLC analyses were carried out on a C4 HPLC column. For all HPLC and HRMS chromatograms, black is the unreacted MftA variant, and red represents the reaction with the MftA variant. For the UV-visible spectra, black is the unreacted MftA variant, red is the reacted MftA variant, and cyan is dAdo adduct to the Y30W variant. mAu, milliabsorbance units.
The mutant MftA Y30F was tested as a substrate for MftC using the same conditions as the MftA Tyr-30 peptide. As shown by Fig. 8B, HPLC analysis of the reaction mixture did not yield evidence of catalytic turnover. Moreover, HRMS analysis of the reaction shows a single species with unreacted MftA Y30F peptide (m/z = 1180.20, MH3+). This suggests that the tyrosine hydroxyl group is necessary for the decarboxylation of MftA. To validate this finding, the MftA mutants Y30S and Y30W were prepared and tested as substrates. These mutants were chosen based upon their size, aromaticity, and/or ability to donate a proton. Evidence for decarboxylated products (−46 Da) with reactions that included MftA Y30S (Fig. 8C, unreacted MH3+: 1160.19 m/z; modified: 1144.86 m/z) and MftA Y30W (Fig. 8D, unreacted MH3+: 1193.21 m/z; modified: 1177.87 m/z) could be found by HRMS analysis. In addition, new species were also observed in the HPLC analysis of the reactions for both MftA Y30S (retention time: unreacted 9.6 min; modified 10.1 min) and MftA Y30W (retention time: unreacted 10.3 min; modified 11.8 min). The nature of these products is discussed below.
MftC catalyzed a diverse array of chemical transformations
Upon further scrutiny of the HPLC and HRMS data, potentially new enzymatic activities were discovered for MftC. In reactions containing MftA Y30S, a new, more abundant species with an observed m/z = 1150.19 (MH3+, Fig. 8C), became apparent. The difference in mass (−30 Da) corresponds to the loss of one carbon, one oxygen, and two hydrogens. This loss matches the loss of formaldehyde, likely removed from the serine methanol, resulting in the formation of glycine. In reactions containing MftA Y30W, two new observations were made. First, in the HPLC trace, a new species eluted ∼30 s before the starting material, in direct contrast to the previous observations that demonstrated the decarboxylated product elutes ∼30–60 s later than starting material. As shown in Fig. 8D, this new species contained a maximum absorbance at 260 nm and a shoulder at 280 nm. The second observation was made in the mass spectrum dataset where a new peptide species at m/z = 1276.24 (MH3+), corresponding to a mass increase of 249.1 Da, was found. This mass increase is likely the addition of a dAdo group (249.1 Da) directly to the tryptophan. It should be noted that dAdo elutes at an earlier time than the MftA Y30W starting material and has an absorbance maximum of 260 nm. Taken together, it is likely that the dAdo· adds directly to the tryptophan residue.
Discussion
A more in-depth characterization of MftC catalysis led us to discover that two isomeric MftA products, MftA* and MftA**, were formed by MftC in our laboratory. Characterization of the major product, MftA* by 13C and 1H NMR, indicates that it does not contain an unsaturated bond; these data, therefore, are inconsistent with the product that we, and others, previously proposed (4, 6). Indeed, by using deuterium-labeling experiments, we demonstrated that the MftA* product was a tyramine-valine cross-link. Although our analyses did not provide us the exact location of the new carbon-carbon bond, we propose that the new bond bridges the tyramine Cα and the valine Cβ. We base this proposal on two factors as follows. First, our 13C NMR data is most consistent with simulated 13C NMR spectra for models with a cross-link between tyramine Cα and the valine Cβ (Scheme 2) (24–26). Second, in reactions with site-specific Val-29 (3-D)-labeled MftA, we observed deuterium enrichment of dAdo, indicating that the deuterium on Cβ of Val-29 is being abstracted. In addition, we saw various amounts (10–50%, data not shown) of the MftA M1W/V29A* product. This indicates that Cβ of the alanine mutant, which is the equivalent to the Cβ of valine, is capable of being cross-linked to tyramine. Lastly, we provided UV-visible spectral and 1H NMR data as evidence that product MftA** has an α/β-unsaturated bond. Although this is consistent to what has been previously proposed as the product for MftC, in our hands MftA** is the minor product and is likely the result of an incomplete reaction.
Interestingly, we found that MftC uses 2 eq of SAM to catalyze the formation of MftA*. The first turnover of SAM results in the oxidative decarboxylated MftA** product through the abstraction of a hydrogen on the Cβ of tyrosine (Fig. 8). Recent mechanistic studies have demonstrated that the RS-SPASM proteins SkfB and AlbA form intramolecular thioether bonds by directing the dAdo· to abstract the Cα hydrogen from the peptide backbone, which in turn forms the thioether bond with cysteine (22, 23). In contrast, RS-SPASM proteins such as QhpD and StrB abstract the Cβ and Cγ hydrogens from their respective substrates (14, 16). In the second turnover of MftC with SAM, we propose here that the dAdo· abstracts the Cβ hydrogen from the adjacent valine side chain. This newly formed alkyl radical could subsequently attack the neighboring Cα/Cβ unsaturated bond at the Cα position resulting in the formation of a bridging carbon-carbon bond. The attack conformation by the alkyl radical could be achieved by free bond rotation along the amide C-N bond, the Val-29 O=C-Cα bond, and the Val-29 Cα—Cβ bond. The remaining electron from the double bond could form a radical on Tyr-30 Cβ or the phenol ring (Fig. 9). Upon injection of another proton and electron the radical is quenched, resulting in the tyramine-valine cross-linked peptide. Although this is not the first time a RS-SPASM protein has been attributed to forming carbon—carbon bonds (e.g. PqqE and StrB), this is the first reported incidence in which a single RS-SPASM protein catalyzes two distinct chemistries on the same substrate through two turnovers of SAM.
Figure 9.

Revised mechanism for MftC catalysis.
Mutational analysis established that the phenol of tyrosine is critical for MftC catalysis. Importantly, the Y30F mutant, where the hydroxyl group is absent from the phenyl ring, was not reactive with MftC. However, when the tyrosine is replaced with other proton-donating residues (e.g. Y30S or Y30W), MftC was still capable of catalysis. Although the evidence we provide does suggest that the phenol is important in the MftC catalysis, it is unclear if the mechanism transitions through a benzenone intermediate or a tyrosine radical intermediate. In our effort to verify that the phenol on MftA was required for our proposed catalytic mechanism, we also discovered that MftC catalyzes a range or transformations. Depending on the size of the side chain, MftC can catalyze other, non-native chemistries such as the removal of the methanol group of serine or the addition of dAdo directly to tryptophan. Although these side activities have not been previously reported for other RS-SPASM proteins, they are not uncommon among RS proteins in general. For instance, a recent report on NosL, a tryptophan lyase, demonstrated the incorporation of dAdo into the double bond of a non-native indole methyl acrylate substrate (17).
Although we provide an in-depth mechanistic study of MftC, many questions remain. Mainly, in terms of mycofactocin biosynthesis, is MftA* or MftA** the physiologically relevant product of MftC catalysis? Recently, Bruender and Bandarian (5) demonstrated that MftE, a peptidase in mycofactocin biosynthesis, cleaves the modified peptide on the N terminus of Val-29. In addition, by using Marfey's reagent and tandem mass spectrometry, they determined that their product was derived from MftA**, resulting in l-Val-p-(2-aminoethenyl)phenol with unknown cis or trans conformation. Although there is no doubt that MftE cleaved MftA**, it is uncertain if the MftA** is the native substrate for MftE due to the very low conversion to the product, as was evident by the HPLC trace provided, even in the presence of 50 μm MftE (5). We propose that the MftA* may be the relevant product of MftC catalysis. We base this proposal on two factors; first, the product distribution of MftA* and MftA** does not change significantly throughout a time-course reaction (data not shown), indicating that MftA is not fully converted to MftA** and then to MftA*. Second, it is unlikely that MftC would catalyze a non-native reaction to completion. This idea is reinforced by our mutational studies where non-native product formation never went to completion.
In summary, we have presented evidence that MftC catalyzes the formation of a cross-linked valine-tyramine product (MftA*) and evidence for its chemical mechanism. These results provide the mycofactocin community direction for future characterization of the pathway. For instance, if MftA* is the physiologically relevant product, then it could be used in experiments with MftE or other mycofactocin biosynthetic proteins. However, if MftA** is the proper product, then we have identified the isomeric form and have provided a synthetic route to trans-l-Val-p-(2-aminoethenyl)phenol for future studies with other proteins in the pathway. Because either case is possible, we are pursuing both avenues simultaneously.
Experimental procedures
Expression and purification of MftC
The mftC gene (Uniprot: A0PM49) from Mycobacterium ulcerans Agy99 was cloned into the pET-28a vector (Novagen) using NdeI and XhoI restriction sites. The sequence verified mftC/pET28 plasmid was co-transformed into E. coli BL21 (DE3) competent cell line with the plasmid pPH151, which contains the isc operon. An overnight culture was used to inoculate 6 liters of terrific broth (TB) medium. The cells were grown at 37 °C and 200 rpm until A600 ∼0.8–1.0 at which point His6-MftC production was induced with sodium fumarate (0.75 g/liter), FeCl3 (50 μm), and 1 mm isopropyl-β-thiogalactopyranoside (IPTG). The cells were grown anaerobically overnight at 21 °C. After the overnight growth, the cells were pelleted by centrifugation at 5500 × g for 10 min. The resulting cell pellet was transferred into the anaerobic chamber and suspended in lysis buffer (50 mm Tris, 200 mm NaCl, 25 mm imidazole, pH 7.6). CHAPS (1%), lysozyme (0.1%) and DNase (0.05 mg/g cell paste) were added to the suspension and stirred for 30 min at room temperature. The lysate was poured into sealable tubes, removed from the chamber, and centrifuged at 30,000 × g for 15 min. The clarified lysate was transferred back into the anaerobic chamber, and the supernatant was loaded onto a 5-ml His-Trap column (GE Healthcare) using an ÄKTA Start FPLC. The bound protein was washed with lysis buffer, and MftC was eluted by the addition of elution buffer (50 mm Tris, 200 mm NaCl, 300 mm imidazole, pH 7.6). MftC-containing fractions were pooled and buffer-exchanged to the storage buffer (50 mm HEPES, 100 mm NaCl, pH 7.5) using a PD-10 column (GE Healthcare). The solution was concentrated using Pall 30-kDa spin columns and reconstituted using the procedures below.
Reconstitution of MftC
All reagents discussed below were transferred as powder into the anaerobic chamber and dissolved in anaerobic water. All procedures discussed below were carried out anaerobically. DTT (10 mm) was added to freshly purified MftC, and the solution was stirred for 30 min at 4 °C. Next, 12 eq (compared with MftC) of FeCl3 was added to the protein and incubated for 30 min at 4 °C with stirring. After the incubation with FeCl3, 12 eq (compared with MftC) of Na2S was added to the protein and incubated for 1 h at 4 °C with stirring. The solution was then centrifuged at 16,000 × g for 2 min, and the soluble fraction was buffer-exchanged into storage buffer (50 mm HEPES, 100 mm NaCl, pH 7.5) using a PD-10 column. The final solution was concentrated using Pall 30 kDa spin columns, aliquoted, and stored at −80 °C.
Expression and purification of GST-MftB
The mftB gene (Uniprot: A0PM48) from M. ulcerans Agy99 was cloned into pGEX6p-1 vector (GE Healthcare) using BamHI and XhoI restriction sites to obtain the GST-MftB-fused protein. The sequence-verified mftB/pGEX6p-1 plasmid was transformed into E. coli T7 Express Iq (DE3) (New England BioLabs) and grown overnight. The overnight culture was used to inoculate 1 liter of LB medium. The cultures were grown at 37 °C at 200 rpm to an A600 ∼0.6. The expression of GST-MftB was then induced with 1 mm IPTG, and the temperature reduced to 21 °C. After an overnight incubation, the cells were pelleted by centrifugation at 5500 × g for 10 min. The resulting cell pellet was suspended in lysis buffer (50 mm HEPES, 100 mm NaCl, 10% glycerol, pH 7.5). Lysozyme (0.1%) and DNase (0.05 mg/g of cell paste) were added to the suspension and stirred for 10 min at room temperature. The cell suspension was sonicated, and the lysate was clarified by centrifugation at 30,000 × g for 15 min. The clarified lysate was then transferred into the anaerobic chamber, and the supernatant was loaded onto a 5-ml GSTrap column (GE Healthcare) using an ÄKTA Start FPLC system. The bound protein was washed with lysis buffer and eluted by elution buffer (50 mm HEPES, 100 mm NaCl, 10% glycerol, 30 mm glutathione, pH 7.5). Protein fractions were pooled, concentrated using a Pall 10-kDa spin column, and buffer-exchanged into lysis buffer using a PD-10 column. The final solution was concentrated to ∼1 mm, aliquoted, and stored at −80 °C.
Expression and purification of MftA
The undesignated mftA gene sequence from M. ulcerans Agy99 was cloned into pET28HST vector using NdeI and XhoI restriction sites as previously described (6). Sequence-verified mftA/pET28HST plasmid DNA was transformed into E. coli BL21 Star (DE3) (Invitrogen). An overnight culture was used to inoculate 2 liters of LB medium. The cells were grown at 37 °C and 200 rpm until an A600 of ∼0.6 and then were induced with 1 mm IPTG and incubated at 21 °C overnight. The cells were pelleted by centrifugation at 5500 × g for 10 min. The resulting pellet was suspended in lysis buffer (50 mm Tris, 200 mm NaCl, 25 mm imidazole, pH 7.6), sonicated, and clarified by centrifugation at 30,000 × g for 15 min. The supernatant was loaded onto a 5-ml HisTrap column using an ÄKTA FPLC system. The bound protein was washed with lysis buffer and eluted with elution buffer (50 mm Tris, 200 mm NaCl, 300 mm imidazole, pH 7.6). The His6-SUMO-MftA-containing fractions were pooled and buffer-exchanged into phosphate buffer (5 mm Na2HPO4, 1 mm tris(2-carboxyethyl)phosphine, pH 7.5) over a HiPrep 26/10 Desalting column (GE Life Sciences) and concentrated using a Pall 10-kDa spin column. After concentrating, tobacco etch virus protease was added, and the solution was incubated at 4 °C overnight. The peptide/SUMO/tobacco etch virus protease solution was loaded onto semi-prep 10 × 250-mm C4 5-μm reverse-phase column (Phenomenex) using a Shimadzu Prominence-i LC-2030C HPLC. The mobile phase was 5 mm sodium phosphate (pH 7.5, buffer A) and 5 mm sodium phosphate in 70% acetonitrile (pH 7.5, buffer B). The MftA peptide fraction was collected and lyophilized overnight. The dried peptide was transferred into the anaerobic chamber, dissolved in anaerobic deionized water, aliquoted, and stored at −80 °C.
Labeling MftA with deuterium-labeled amino acid
To obtain tyrosine deuterium-labeled variants of MftA, an overnight E. coli BL21 Star (DE3) culture containing the mftA/pET28HST plasmid was used to inoculate supplemented M9 minimal medium (glycerol (4 ml/liter), Na2HPO4 (25 mm), KH2PO4 (25 mm), NH4Cl 50 mm), Na2SO4 (5 mm), MgSO4 (1 mm), CaCl2 (100 μm), FeCl3 (50 μm), pH 6.7. In addition, the media contained 19 unlabeled amino acids (100 mg/liter) and the corresponding deuterium-labeled amino acid (100 mg/liter, Cambridge Isotope Laboratory). The growth and purification of the labeled peptide were performed as described above.
Labeling MftA with 13C-labeled tyrosine
To obtain 13C-labeled variants of MftA, an overnight E. coli RF4 (DE3) culture transformed with the mftA/pET28HST plasmid was used to inoculate M9 minimal medium described above except with 13C-labeled tyrosine (Cambridge Isotope Laboratory, 100 mg/liter). The growth and purification of the labeled peptide were performed as described above.
Site-directed mutagenesis
All the mutants were prepared using QuikChange protocol (Stratagene) with Phusion polymerase (New England BioLabs). The mftA/pET28HST plasmid with the N-terminal methionine codon replaced with tryptophan (M1W) was used as template for PCR. The mutants were sequence-verified and transformed into E. coli BL21 Star (DE3) competent cells (Invitrogen).
MftA reactions
Reactions were performed in an anaerobic chamber. Where possible, reagents were transferred to the chamber as powder and dissolved in anaerobic deionized water. Reactions were prepared using the following order of addition: reaction buffer (50 mm HEPES, 100 mm NaCl, pH 7.5), 2 mm sodium dithionite, 8 mm DTT, 100 μm MftA, 100 μm GST-MftB, 1 mm SAM, 50 μm MftC. Reactions were carried out at room temperature for 18 h. The reactions were then removed from the chamber and centrifuged to remove any precipitate, and the supernatant was analyzed by reverse-phase chromatography. HPLC analysis was performed on a Shimadzu Prominence-i LC-2030C HPLC using either a 4.6 × 250-mm C4 5-μm column (Phenomenex) or a 4.6 × 250-mm C18 5-μm column (Phenomenex) and 5 mm sodium phosphate (pH 7.5, buffer A) and 5 mm sodium phosphate in 70% acetonitrile (pH 7.5, buffer B) as the mobile phase. Wavelengths between 200 and 400 nm were monitored, and the chromatogram was reported at 280 nm. MftA* and MftA** products were isolated and analyzed by HRMS.
High-resolution mass spectrometry
All samples were analyzed by the Biological Mass Spectrometry Facility at the University of Colorado Anschutz Medical Campus. Samples were desalted using a C18 ZipTip (EMD Millipore) and subject to LC-ESI-MS using a Thermo Scientific LTQ Orbitrap Velos Pro and a nanoflow liquid chromatography system. Data were analyzed using XCalibur Qual Browser v. 3.0.63 (Thermo Scientific).
Deuterium-labeled MftA assay
All reactions were carried out in an anaerobic chamber, and the reagents were prepared as described earlier. Reactions with ring-2,6-D2, 2-D, and 3,3-D2-tyrosine were prepared in the following order of addition: reaction buffer (50 mm HEPES, 100 mm NaCl, pH 7.5), 2 mm sodium dithionite, 8 mm DTT, 200 μm labeled-MftA, 200 μm GST-MftB, 400 μm SAM, 100 μm MftC. For reactions containing Val(3-D)-labeled MftA, the conditions were the same except 1 mm SAM was used. Analysis of the reactions were carried out as described above. The products of the reactions were tracked on the chromatogram monitoring absorbance at 260 nm. The corresponding fraction of dAdo was collected, lyophilized, and dissolved in deionized water. The samples were analyzed by HRMS.
MftA product purification assay
All reactions were carried out in an anaerobic chamber, and the reagents were prepared as described earlier. Individual 100-μl reactions were prepared with the following order of addition: buffer (50 mm HEPES, 100 mm NaCl, pH 7.5), 2 mm sodium dithionite, 8 mm DTT, 300 μm MftA, 300 μm GST-MftB, 1 mm SAM, 300 μm MftC. Reactions were carried out at room temperature for 4 h. MftA* and MftA** were separated using the previously discussed HPLC conditions. The corresponding fractions of modified MftA were collected, lyophilized, and dissolved in H2O (for MS analysis) or D2O (for NMR analysis).
Nuclear magnetic resonance assay
All NMR spectra were recorded on a Bruker UltraShield 500/54 Plus spectrometer. Spectra were processed and analyzed in TopSpin v. 2.1 program (Bruker). All peptide NMR samples were prepared in D2O. Suppression of H2O signal was applied at a frequency of 2353.37 Hz. Signals were integrated, and the coupling constants were calculated in MestReNova v. 10.0.1 program (Mestrelab Research).
Synthesis of E-l-Val-p-(2-aminoethenyl)phenol
To a mixture of N-butoxycarbonyl-valine (5 mmol, 1.08 × g) in DCM (4 ml) was added octopamine hydrochloride (5 mmol, 0.94 g), hydroxybenzotriazole (7.5 mmol, 1.14 g), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide; 6 mmol, 1.15 g) and N,N-diisopropylethylamine (1.1 ml). The reaction mixture was stirred at 25 °C overnight, diluted with DCM, then washed with a saturated aqueous solution of NaHCO3. The organic layer was collected, and the aqueous layer was washed again with DCM. The combined organic extracts were washed with aqueous NH4Cl, dried over Na2SO4, and concentrated to afford a gummy oil. Acetic anhydride (10 ml) and pyridine (3 ml) were added, and the mixture was heated at 100 °C for 1 h. The mixture was cooled and poured on ice, and the resulting solution was extracted with three portions of ethyl acetate. The combined organic extracts were washed with water and brine and dried over Na2SO4. The solvent was removed under reduced pressure and purified using flash column chromatography to afford 89% (1.79 g) of 1. A mixture of 1 (1.79 g, 4.25 mmol) and K2CO3 (1.68 g, 1.22 mmol) in 13 ml of DMSO was heated at 95 °C under N2 for 2 h. The mixture was cooled to 25 °C and poured onto ice. The resulting solution was extracted with six portions of ethyl acetate. The combined organic extracts were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure and purified by flash column chromatography. To the resulting product (1.45 g, 3.4 mmol) in a round-bottom flask was added (0.23 g, 4.12 mmol) of sodium methoxide in methanol (10 ml) and stirred at room temperature for 1 h. The resulting mixture was then extracted with three portions of ethyl acetate dried over Na2SO4. The solvent was removed under reduced pressure and purified by flash column chromatography to afford 1.02 g, 69% of the trans product (2). Data were 1H NMR (DMSO) 7.47 (d, 1H, J = 14.73), 7.38 (d, 2H, J = 8.47), 7.04 (d, 2H, J = 8.68), 6.30 (d, 1H, J = 14. 73), 3.18 (d, 1H, J = 5.73), 2.00 (m, 1H), 1.01 (d, 3H, J = 6.84), 0.97 (d, 3H, J = 6.85); 13C NMR (DMSO) 169.4, 149.3, 134.1, 127.4 (2C), 123.3 (2C), 122.2, 112.4, 60.0, 31.3, 20.1, 16.2.
Synthesis of Z-l-Val-p-(2-aminoethenyl)phenol
An oven-dried flask was charged with the N-butoxycarbonyl-valine-amide (220 mg, 1.00 mmol), bis(2-methallyl)-cycloocta-1,5-diene-ruthenium(II) (16.0 mg, 0.05 mmol), 1,4-bis(dicyclohexylphosphino) butane (27.0 mg, 0.06 mmol), and ytterbium triflate (24.8 mg, 0.04 mmol) and flushed with nitrogen. Subsequently, dry N,N-dimethylformamide (3.0 ml), alkyne (264 mg, 2.00 mmol), and water (108 μl, 6.00 mmol) were added via syringe. The resulting solution was stirred for 6 h at 60 °C and then poured into an aqueous sodium bicarbonate solution (30 ml). The resulting mixture was extracted 3 times with 20-ml portions of ethyl acetate, the combined organic layers were washed with water and brine, dried with Na2SO4, and filtered, and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography to afford 197 mg of 41% of the Z-methoxy-product (3). A 50-ml oven-dried two-neck round-bottom flask equipped with a stir bar and a rubber septum and thermometer was cooled under nitrogen and charged with product 3 (197 mg, 0.57 mmol) dissolved in 10 ml of anhydrous DCM. A 25-ml oven-dried addition funnel was attached and filled with BBr3 (1.70 mmol, 0.161 ml), and the set up was cooled to −78 °C using dry ice and acetone. The BBr3 solution was then added dropwise at that temperature, a CaCl2 filled drying tube was placed on top of the flask, and the reaction mixture was left to stir overnight. The mixture was then cooled to −78 °C and quenched with saturated aqueous NaHCO3. The mixture was allowed to slowly warm up to room temperature and transferred into a separatory funnel. The aqueous layer was extracted with three portions of ethyl acetate, and the combined organic extracts were washed with brine and dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was then purified by flash column chromatography to afford 160 mg, 81% of the Z-product (4). Data were 1H NMR (DMSO) 7.31 (d, 2H, J = 8.70), 6.96 (d, 2H, J = 8.78), 6.70 (d, 1H, J = 9.68), 5.65 (d, 1H, J = 9.69), 3.21 (d, 1H, J = 4.06), 2.09 (m, 1H), 0.93 (d, 3H, J = 6.89), 0.78 (d, 3H, J = 6.89), 13C NMR (DMSO) 169.4, 149.3, 134.1, 127.4 (2C), 123.3 (2C), 122.2, 112.4, 60.0, 31.3, 20.1, 16.2.
Author contributions
B. K., R. A., M. T., and J. A. L. designed, performed, and analyzed all the biochemical experiments. J. A. L. wrote the paper.
Supplementary Material
Acknowledgment
We thank Dr. Monika Dzieciatkowska (University of Colorado Anschutz Medical Campus) for performing the high-resolution LC/MS measurements.
This work was supported by the University of Denver. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S7 and Schemes S1 and S2.
- SAM
- S-adenosylmethionine
- RS
- radical S-adenosylmethionine
- dAdo
- 5′-deoxyadenosine
- DTH
- dithionite
- HRMS
- high-resolution mass spectrometry
- SPASM
- subtilosin, pyrroloquinoline quinone, anaerobic sulfatase maturating enzyme, mycofactocin
- SUMO
- small ubiquitin-like modifier
- IPTG
- isopropyl-β-thiogalactopyranoside
- DCM
- dichloromethane.
References
- 1. Klinman J. P., and Bonnot F. (2014) The intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev. 114, 4343–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haft D. H. (2011) Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics. 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haft D. H., Pierce P. G., Mayclin S. J., Sullivan A., Gardberg A. S., Abendroth J., Begley D. W., Phan I. Q., Staker B. L., Myler P. J., Marathias V. M., Lorimer D. D., and Edwards T. E. (2017) Mycofactocin-associated mycobacterial dehydrogenases with non-exchangeable NAD cofactors. Sci. Rep. 7, 41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruender N. A., and Bandarian V. (2016) The radical S-adenosyl-l-methionine enzyme MftC catalyzes an oxidative decarboxylation of the C terminus of the MftA peptide. Biochemistry 55, 2813–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruender N. A., and Bandarian V. (2017) The creatininase homolog MftE from Mycobacterium smegmatis catalyzes a peptide cleavage reaction in the biosynthesis of a novel RiPP. J. Biol. Chem. 292, 4371–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khaliullin B., Aggarwal P., Bubas M., Eaton G. R., Eaton S. S., and Latham J. A. (2016) Mycofactocin biosynthesis: modification of the peptide MftA by the radical S-adenosylmethionine protein MftC. FEBS Lett. 590, 2538–2548 [DOI] [PubMed] [Google Scholar]
- 7. Haft D. H., and Basu M. K. (2011) Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 193, 2745–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Latham J. A., Iavarone A. T., Barr I., Juthani P. V., and Klinman J. P. (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 290, 12908–12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benjdia A., Leprince J., Guillot A., Vaudry H., Rabot S., and Berteau O. (2007) Anaerobic sulfatase-maturating enzymes: Radical SAM enzymes able to catalyze in vitro sulfatase post-translational modification. J. Am. Chem. Soc. 129, 3462–3463 [DOI] [PubMed] [Google Scholar]
- 10. Flühe L., Knappe T. A., Gattner M. J., Schäfer A., Burghaus O., Linne U., and Marahiel M. A. (2012) The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 8, 350–357 [DOI] [PubMed] [Google Scholar]
- 11. Bruender N. A., Wilcoxen J., Britt R. D., and Bandarian V. (2016) Biochemical and spectroscopic characterization of a radical SAM enzyme involved in the formation of a peptide thioether cross-link. Biochemistry 55, 2122–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flühe L., Burghaus O., Wieckowski B. M., Giessen T. W., Linne U., and Marahiel M. A. (2013) Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J. Am. Chem. Soc. 135, 959–962 [DOI] [PubMed] [Google Scholar]
- 13. Barr I., Latham J. A., Iavarone A. T., Chantarojsiri T., Hwang J. D., and Klinman J. P. (2016) The pyrroloquinoline quinone (PQQ) biosynthetic pathway: Demonstration of de novo carbon-carbon cross-linking within the peptide substrate (PqqA) in the presence of the Radical SAM enzyme (PqqE) and its peptide chaperone (PqqD). J. Biol. Chem. 291, 8877–8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakai T., Ito H., Kobayashi K., Takahashi Y., Hori H., Tsubaki M., Tanizawa K., and Okajima T. (2015) The radical S-adenosyl-l-methionine enzyme QhpD catalyzes sequential formation of intra-protein sulfur-to-methylene carbon thioether bonds. J. Biol. Chem. 290, 11144–11166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wieckowski B. M., Hegemann J. D., Mielcarek A., Boss L., Burghaus O., and Marahiel M. A. (2015) The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 589, 1802–1806 [DOI] [PubMed] [Google Scholar]
- 16. Schramma K. R., Bushin L. B., and Seyedsayamdost M. R. (2015) Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan cross-link. Nat. Chem. 7, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhandari D. M., Fedoseyenko D., and Begley T. P. (2016) Tryptophan lyase (NosL): a cornucopia of 5′-deoxyadenosyl radical mediated transformations. J. Am. Chem. Soc. 138, 16184–16187 [DOI] [PubMed] [Google Scholar]
- 18. Wang Y., Schnell B., Baumann S., Müller R., and Begley T. P. (2017) Biosynthesis of branched alkoxy groups: iterative methyl group alkylation by a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc. 139, 1742–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iwasaki T., Fukazawa R., Miyajima-Nakano Y., Baldansuren A., Matsushita S., Lin M. T., Gennis R. B., Hasegawa K., Kumasaka T., and Dikanov S. A. (2012) Dissection of hydrogen bond interaction network around an iron-sulfur cluster by site-specific isotope labeling of hyperthermophilic archaeal rieske-type ferredoxin. J. Am. Chem. Soc. 134, 19731–19738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goossen L. J., Salih K. S., and Blanchot M. (2008) Synthesis of secondary enamides by ruthenium-catalyzed selective addition of amides to terminal alkynes. Angew. Chem. Int. Ed. Engl. 47, 8492–8495 [DOI] [PubMed] [Google Scholar]
- 21. Snider B. B., Song F., and Foxman B. M. (2000) Total syntheses of (+/−)-anchinopeptolide D and (+/−)-cycloanchinopeptolide D. J. Org. Chem. 65, 793–800 [DOI] [PubMed] [Google Scholar]
- 22. Bruender N. A., and Bandarian V. (2016) SkfB abstracts a hydrogen atom from Cα on SkfA to initiate thioether cross-link formation. Biochemistry 55, 4131–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benjdia A., Guillot A., Lefranc B., Vaudry H., Leprince J., and Berteau O. (2016) Thioether bond formation by SPASM domain radical SAM enzymes: Cα H-atom abstraction in subtilosin A biosynthesis. Chem. Commun. (Camb.) 52, 6249–6252 [DOI] [PubMed] [Google Scholar]
- 24. Castillo A. M., Patiny L., and Wist J. (2011) Fast and accurate algorithm for the simulation of NMR spectra of large spin systems. J. Magn. Reson. 209, 123–130 [DOI] [PubMed] [Google Scholar]
- 25. Banfi D., and Patiny L. (2008) Resurrecting and processing NMR spectra on-line. Chim. Int. J. Chem. 62, 280–281 [Google Scholar]
- 26. Aires-de-Sousa J., Hemmer M. C., and Gasteiger J. (2002) Prediction of 1H NMR chemical shifts using neural networks. Anal. Chem. 74, 80–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.