

Abstract
MyD88 and FcR common gamma chain (Fcer1g, FcRγ) elicit proinflammatory responses to exogenous antigens. Deletion of these receptors in autoimmune models has generally led to reduced overall disease. In B cells, Myd88 is required for anti-DNA and anti-RNA autoAb responses, while Fcer1g is not expressed in these cells. The roles of these receptors in myeloid cells during B cell autoimmune activation remain less clear. To investigate the roles of Myd88 and Fcer1g on non-B cells, we transferred anti-self IgG (rheumatoid factor) B cells and their physiologic target antigen, anti-chromatin Ab, into mice lacking Fcer1g, Myd88 or both and studied the extrafollicular plasmablast response. Surprisingly, we found a markedly higher and more prolonged response in the absence of either molecule, an effect that was accentuated in doubly-deficient recipients, with a 40-fold increase compared to WT recipients at d10. This enhancement was dependent on CD40L, indicating that Myd88 and FcRγ, presumably on myeloid APC, was required to downregulate T cell help for the EF response. To extend the generality we then investigated a classic T cell dependent response to NP-CGG and found a similar effect. These results thus reveal novel regulatory roles in the B cell response for receptors that are typically proinflammatory.
Introduction
Autoantibody responses are a hallmark of systemic and organ-specific autoimmune syndromes (1). While the specificities of serum antibodies (Abs) in many such diseases are relatively well-understood, the cellular pathways that generate these Abs remain more obscure. Paradigms for understanding these self-reactive B cell responses derive chiefly from parallels with induced immune responses to foreign antigens. Such responses classically proceed via germinal center (GC) formation, sometimes after a brief period of extrafollicular (EF) plasmablast (PB) generation. GC responses are the classical sources of long-lived plasma cells and memory B cells, though the latter may also form outside of the GC (2).
Despite this classical view, it has become apparent that B cell autoimmune responses can include a prominent EF PB response, often without a significant GC response (3). Similar responses have recently been observed after challenge with certain bacterial pathogens as well (4-6). This dynamic EF response can proceed continually throughout the disease process or infection, presumably recruiting new cohorts of B cells to differentiate into PBs, which in turn have a half-life of just a few days. This has been observed in multiple animal models and can be inferred in humans and mice by the loss of some, though not all, autoAbs after B cell depletion therapy (7-10). Most prominently, anti-DNA and anti-self-IgG (rheumatoid factor, RF) responses use the EF pathway (11, 12). In contrast to the GC, how these EF responses are promoted and regulated is poorly defined, both in immunity and autoimmunity.
Though EF responses are in some cases T cell-independent or partially so (13-16), T cells can participate in EF responses as well. Recently, a population of T cells that resemble TFH, that are nonetheless localized extrafollicularly and hence have been called TEFH, has been identified (17, 18). These play a role in both autoreactive and immunization-induced EF PB responses. TEFH both amplify and alter the quality of EF responses, but they are relatively little studied compared to TFH. In particular it is not clear how they are initially activated and also how or even if they are regulated. In this respect, their follicular counterpart, the TFH can be antagonized at a later stage of the response by a form of regulatory TFH (TFR) (19); T cells or other factors that might regulate the EF response have not yet been identified.
In addition to T cell help for B cells, it is generally thought that all immune responses require some form of innate immune activation in order to initiate and possibly also in order to be sustained (20). Classically, for responses to microbial and vaccine Ags, the DC is the initial APC for CD4 T cells and hence it must be activated by one or more innate immune receptors (21). However, in autoimmunity a growing body of evidence implicates the B cell as a predominant and even initiating APC for CD4 T cells (22, 23). If the B cell were an initiating APC, this would require the B cell to receive an innate immune signal. Indeed, for autoimmune responses that focus on typical lupus autoantigens, engagement of TLR7 and/or TLR9 along with the BCR is strictly required for initial B cell activation, and for subsequent differentiation and autoantibody production (24). Moreover, in lupus-prone mice deletion of Myd88 in B cells, but not in DCs, abrogated autoantibody production and greatly ameliorated target organ disease that was T cell-mediated (25). Commensurately, deletion of B cells but not DCs, markedly reduced the peripheral accumulation of activated/memory phenotype T cells (26, 27).
On the other hand, the potential functions of innate immune signaling in myeloid cells—such as DCs, neutrophils and macrophages—in activating and/or regulating autoreactive EF B cell responses remain obscure. Myeloid cells express multiple TLRs, most of which signal via MyD88 (28, 29). In addition, they express a variety of “activating” FcR that recognize immune complexes (ICs), which are prevalent in diseases like lupus and Rheumatoid Arthritis (30). In addition to providing activating signals transduced via FcRγ and Syk, FcR also can direct TLR ligands contained in ICs to intracellular compartments where they can engage certain TLRs (31), potentially leading to crosstalk between these two receptors.
The consequences of such potential ligand interactions for the B cells response are not well studied. Myeloid cells are sources of BAFF and APRIL, cytokines known to promote and possibly regulate PB differentiation (3). It is not fully understood what signals are naturally generated to elicit these cytokines during the EF response, though TLR or FcR signals may be upstream in some cases (32-34). Myeloid cells, particularly DCs and macrophages, also express MHCII and could be important APCs for T cells that promote EF responses; alternatively, or in addition, they could promote the development of various types of regulatory T cell that could function to rein in the response, by analogy to TFR.
Here we have taken a genetic and cell transfer approach to evaluate functions of innate immune signaling via both MyD88 and FcRγ in EF responses. We began with the goal of understanding autoreactive anti-IgG (also known as rheumatoid factor or RF) B cell activation after exposure to ICs that contain the endogenous DNA or RNA (35, 36). Such ICs are natural ligands of receptors that are upstream of both FcRγ and MyD88. Previously we showed that responses to such ICs depend on the nucleic acid-specific TLRs 7 and 9 expressed in B cells and hence the DNA and RNA in this context are endogenous TLR ligands (37). T cells also contribute to the response, in particular to amplify the IgG AFC response, though they are not required for it (16, 37). This response is an ideal model for the types of TLR-dependent B cell activation that is prominent in lupus and other systemic autoimmune diseases, particularly when ICs are abundant. Since the roles of MyD88 in B cells are relatively well-known (25, 37, 38), we have used cell transfer to restrict its deficiency to the non-B cell component, which in this context we expect to be mainly comprised of myeloid cells. Though FcRγ is a downstream signaling effector of a variety of receptors, in the context of an acute immune response driven by ICs we expect the major role for this molecule will be in FcR-mediated signaling in myeloid cells.
We transferred small numbers of B cells expressing an anti-IgG2a specificity from V genes “knocked in” to the native Ig loci to genetically deficient or WT control recipients and then challenged with IgG2a anti-nucleosome protein (35, 36). IgG anti-nucleosome binds to ubiquitous nuclear material in vivo to generate RF B cell stimulatory complexes that in turn lead to EF PB development that closely mimics the spontaneous response. We then assessed the B cell response under these conditions.
Though we expected to find stimulatory roles for both MyD88 and FcRγ we instead found rather striking paradoxical enhancement of the response in the absence of each of the receptors, whch was further accentuated in doubly-deficient animals. These studies thus reveal an unexpected role for these receptors when expressed in non-B cells during an EF PB response that is typical of TLR-driven autoimmunity. Moreover, we also found a parallel effect on a hapten driven T-dependent response, suggesting that these paradoxical inhibitory roles of non-B cell expressed MyD88 and FcRγ may be even more general in regulating the humoral immune response.
Materials and Methods
Mice
AM14 site directed transgenic (sd-Tg) mice (36) and Vh186.2 KI (B1.8) mice (39) were backcrossed to the BALB/c strain for at least 8 generations. Myd88 deficient mice (40) were obtained from R. Medzhitov (Yale University, New Haven, CT, USA) and backcrossed to the BALB/c strain for at least 8 generations. Fcer1g deficient mice (41) on the BALB/c strain were purchased from Taconic (Germantown, NY). Ccr2 deficient mice on the BALB/c background (42) were obtained from B. Rollins (DFCI, Boston, MA, USA). Tcra-deficient mice were derived from mice obtained from K. Bottomly (Yale University) (43). All mice were housed under specific pathogen free conditions and all experiments were performed with the approval of the Yale University and University of Pittsburgh IACUC.
B cell isolation and transfer
Transfer of AM14 (16) and B1-8 B cells (44) has been described previously. Briefly, B cells were isolated from single cell suspensions of splenocytes using the EasySep Mouse B Cell Enrichment kit (StemCell Technologies, Vancouver, Canada) per manufacturer's instructions. Purity was verified to be 90-95% by flow cytometry. 3 million AM14 B cells or B1-8 B cells containing 200,000 NP-specific cells in sterile PBS were injected i.v. into recipient mice.
In vivo B cell activation
Growth and injection of PL2-3 (45) has been described previously (16). 500 μg PL2-3 was injected i.p. at days 0, 2, and 4 of the experiment. Alternatively, mice were injected i.p. with 50 μg of NP-CGG precipitated in alum at day 0. Day 0 was defined as being within 24 hours after B cell transfer.
In vivo Ab administration
We used MR-1, which blocks CD40L (46); hamster isotype control UC8-1B9 (anti-DNP-KLH) (ATCC); and 1A8, which mediates cell depletion by binding Ly6G (47). Abs were grown and purified as previously described (48). 200 ug MR-1 or hamster IgG isotype control, was injected i.p. on days -1 and 3 of the experiment. 1 mg of 1A8 was injected between days 3 and 5 of the experiment.
Flow Cytometry and cell sorting
Preparation of splenocytes has been described (48). The following Ab were produced in lab or purchased as indicated: 4-44 (anti-AM14 idiotype) biotin, alexa 647, or alexa 488; anti-mouse Ly6G alexa 488 (1A8); anti-mouse CD11b alexa 647 (M1/70) or APC-Cy7 (M1/70, Biolegend); anti-mouse CD11c biotin (N418, eBioscience) or BUV737 (N418, Becton-Dickinson); anti-mouse TCRβ biotin or PerCP-Cy5.5 (H57-597, Biolegend); anti-mouse CD19 BUV395 (1D3, Becton-Dickinson); anti-mouse CD4 Pacific Blue (GK1.5); anti-mouse CD62L alexa 647 (MEL-14); anti-mouse CD44 alexa 488 (1M7) or APC-Cy7 (1M7, Biolegend); anti-mouse CD40 PE (3/23, Becton-Dickinson); anti-mouse PSGL1 phycoerythrin (2PH1, Becton-Dickinson); anti-mouse FoxP3 allophycocyanin (FJK-16s, eBioscience); anti-mouse kappa (187.1); anti-mouse CD86 Pacific Blue (GL1) or PE (GL1, Becton-Dickinson); anti-mouse CD80 BV605 (16-10A1, Becton-Dickinson) or PE-Cy7 (16-10A1, Biolegend); anti-mouse I-A/I-E biotin or APC-Cy7 (M5/114.15.2, Biolegend); anti-mouse CD95 PE-Cy7 (Jo2, Becton-Dickinson); and anti-mouse CD138 allophycocyanin (281-2, Biolegend). Streptavidin PE-Cy7 (BD) or Streptavidin PE-Cy5.5 (Invitrogen) were used as secondary reagents. Nitro-iodo-phenyl hapten coupled to either allophycocyanin or PE, NIP-APC and NIP-PE, respectively, have been described (49). Flow cytometry data were collected on a Becton-Dickinson LSRII or Fortessa and analyzed in FlowJo (Tree Star). Cell sorting was performed on a Becton-Dickinson FACS Aria II.
Quantitative RT-PCR
RNA was prepared from sorted CD11b+ Ly6G- cells using the QIAgen RNeasy Plus Mini kit according to manufacturer's instructions. cDNA was prepared using iScript Reverse Transcription Supermix (Bio-Rad). qPCR was performed on a Roche LightCycler 96 using KAPA SYBR Fast Master Mix and the following primers: Il6 TTCCCTACTTCACAAGTCCG and CAAGTGCATCATCGTTGTTC; Gapdh TCCCACTCTTCCACCTTCGA and AGTTGGGATAGGGCCTCTCTT.
ELISpot Assay
Detection of 4-44+ AFCs (35) and NP-specific AFCs using ELISpot has been described (50). Briefly, Immulon 4 plates were coated with either anti-mouse IgM (B7-6, produced in lab) or polyclonal anti-mouse IgG2a (Southern Biotech) for 4-44 AFCs or with NP2-BSA or NP16-BSA and blocked with PBS and 1% BSA. NP-BSA conjugates were produced and characterized as described (49). Splenocytes were incubated at 37 degrees with 5% CO2 for 4.5-6 hours. 4-44+ AFCs were enumerated by detection with 4-44 biotin and Streptavidin-alkaline phosphatase while NP AFCs were detected using anti-IgM or anti-IgG1 alkaline phosphatase (Southern Biotech). Color was developed using 5-Bromo-4-chloro-3-indolyl phosphate (BCIP) in agarose. AFCs were counted using a dissecting microscope.
Detection of Serum Cytokines
MCP-1 was detected from serum using the Bio-Plex Mouse Cytokine MCP-1 set per manufacturer's instructions (Bio-Rad, Hercules, CA, USA).
Results
B cell extrinsic Myd88 and Fcer1g regulate RF B cell contraction
To test whether MyD88 and FcRγ expressed on non-B cells modulated the TLR-dependent EF RF B cell response to anti-chromatin Ab, we utilized our well-characterized model in which WT AM14 B cells are transferred to BALB/c recipients and activated by the IgG2aa anti-chromatin Ab, PL2-3 (16, 35). We then compared the response in WT recipients with recipients that lacked either Myd88 or Fcer1g, or with recipients that lacked both, tracking cells using the 4-44 anti-idiotype Ab that detects the transferred RF B cells (Fig. 1A) (48). We found that at day 5, the response in terms of total RF cells or AFCs was similar in all groups analyzed (Fig. 1B-D). Thus, while we previously showed that Myd88 is required in AM14 B cells in this response, these data demonstrate that Myd88 and Fcer1g are not required in any other cell type to initiate the AM14 B cell response to anti-chromatin Ab.
Figure 1.
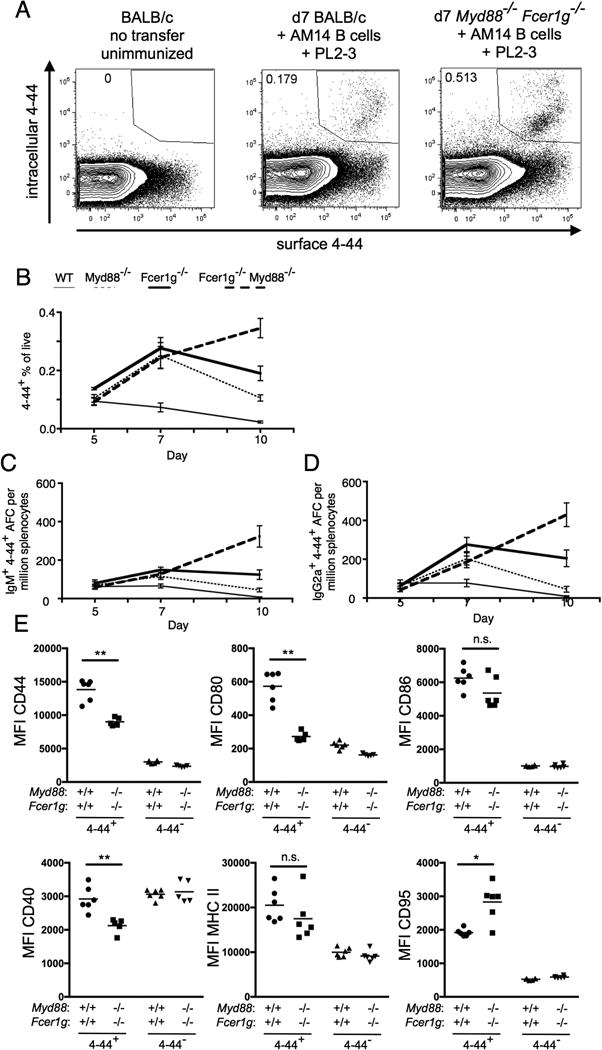
FcRγ and MyD88 control contraction of the EF RF response. BALB/c or Myd88-deficient BALB/c or Fcer1g-deficient BALB/c or MyD88-deficient Fcer1g-deficient BALB/c mice were sacrificed on day 5, 7, or 10 following transfer of purified AM14 sd-Tg B cells and administration of PL2-3 as indicated in Materials and Methods. (A) Representative staining of surface and intracellular 4-44 double positive cells in indicated recipients at d7 following transfer and immunization. (B) Surface and intracellular 4-44 double positive cells as gated in (A) were quantitated by flow cytometry. (C-D) Splenic 4-44+ AFC of IgM (C) or IgG2a (D) isotype were measured by ELISpot. At least 4 mice per group and 2 independent experiments per time point were compiled. Data are represented as means +/- SEM. (E) Expression of indicated activation markers on 4-44+ or 4-44- B cells at d7 in the indicated recipients was determined by flow cytometry. Statistical comparisons by Mann-Whitney two-tailed test, * p<0.05; ** p<0.01
The AM14 response in WT recipients remained relatively stable from day 5 to day 7 and contracted by day 10 (Fig. 1B-D, thin solid line). Unexpectedly, the response in mice deficient in either Myd88 or Fcer1g continued to expand from day 5 to day 7 (Fig. 1B-D, thin dashed line and thick solid line). Indeed, the frequency of 4-44+ cells at day 7 was more than 3-fold increased in deficient recipients compared to that found in WT recipients (Fig. 1B). Furthermore, 4-44+ IgM (Fig. 1C) and 4-44+ IgG2a AFCs (Fig. 1D) were increased roughly 2-fold in deficient recipients compared to that found in WT recipients. Following this, in recipients deficient in either Myd88 or Fcer1g, there was partial contraction by day 10, but this contraction was much less pronounced than in WT mice such that at day 10 in Myd88 deficient recipients, the total 4-44+ population and both IgM and IgG2a 4-44+ AFCs were increased 5-fold compared to that found in the WT recipients (Fig. 1B-D). At day 10 in Fcer1g deficient recipients, the 4-44+ response was markedly increased (10-fold) compared to WT (Fig. 1B-D). Dramatically, in recipients lacking both Myd88 and Fcer1g, contraction was not observed by day 10. Instead, the response continued such that in these recipients, the total 4-44+ population was increased 15-fold compared with WT (Fig. 1B, thick dashed line), while the AFC response was 40-fold increased compared with WT (Fig 1C and 1D, thick dashed line). Thus, our results indicate synergy between the two pathways in a profound regulation of the late phase of the autoreactive B cell response.
To better understand how host deficiency in Myd88 and Fcer1g affected the AM14 response to anti-chromatin PL2-3 immunization, we examined the expression of B cell activation markers on transferred idiotype-positive B cells and idiotype-negative host B cells. At d7 following transfer and immunization, WT AM14 B cells transferred into the WT recipient, despite being fewer in number (Fig 1B), expressed more CD44, CD80 and CD40 than cells transferred into Myd88-/-Fcer1g-/- recipients, somewhat less CD95, and similar levels of MHC II and CD86 (Fig 1E). These differences suggest that the activation signals received by WT AM14 B cells in the presence of anti-chromatin Ab is of equal or better quality in the WT versus deficient recipients, and thus does not explain the dysregulation observed in the deficient recipient mice.
The effect of myeloid Myd88 and Fcer1g on RF B cell activation
To understand how MyD88 and FcRγ regulate the WT AM14 B cell response to anti-chromatin Ab, we profiled myeloid cell subsets following transfer of AM14 B cells to WT or deficient mice and activation with anti-chromatin Ab in vivo at day 7. We first analyzed CD11bhi cells, characterizing Ly6G+ cells as neutrophils (51) and Ly6Glo cells likely as a combination of monocytes and macrophages (referred to hereafter as macrophages). CD11bhi Ly6G+ neutrophils were increased in WT recipients compared to those lacking Myd88, regardless of Fcer1g presence or absence (Fig. 2A). There was also an increased frequency of macrophages in the spleens of WT recipients compared with all deficient recipients (Fig. 2B). DCs (ascertained by gating out neutrophils and gating on CD11bint CD11chi cells) were decreased in mice lacking only Fcer1g, but not in other deficient recipients (Fig. 2C).
Figure 2.
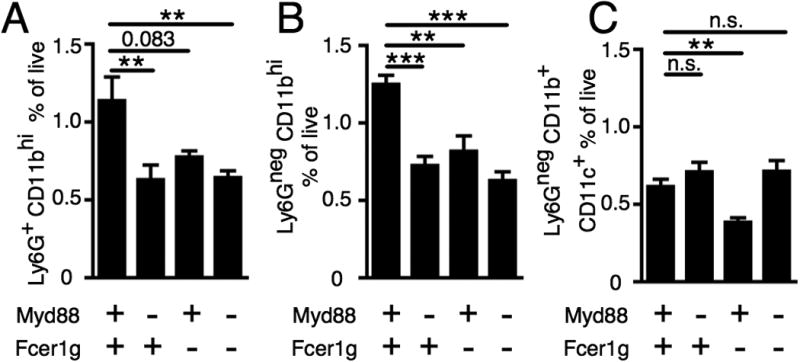
FcRγ and MyD88 are required for increased neutrophils and macrophages. BALB/c or Myd88-deficient BALB/c or Fcer1g-deficient BALB/c or MyD88-deficient Fcer1g-deficient BALB/c mice were sacrificed on day 7 following transfer of purified AM14 sd-Tg B cells and administration of PL2-3 as indicated in Materials and Methods. (A) CD11bhi Ly6G+ cells were quantitated. (B) CD11bhi Ly6Gneg cells were quantitated. (C) Ly6Gneg CD11b+ CD11c+ cells were quantitated. At least 6 mice per group and 3 independent experiments were compiled. Data are represented as means +/- SEM. **p<0.01 and ***p<0.001 by Mann-Whitney two-tailed test.
In addition to differences in macrophage frequency, we observed differences in macrophage activation state following adoptive transfer of AM14 B cells and immunization with anti-chromatin PL2-3. At d7, we saw an increase in expression of CD86 and CD40 in WT but not Myd88-/-Fcer1g-/- recipients (Fig S1A and S1B) with no difference in MHCII or CD80 expression (Fig S1C and S1D). We also sorted CD11b+ Ly6G- cells and performed qRT-PCR and observed an increase in expression of IL-6 in WT but not Myd88-/-Fcer1g-/- recipients at d7 (Fig S1E). We did not observe differences in the expression of several other genes, including IFNγ, IL10, BAFF, Arg1 or Nos2; nor did we see differences in BAFF, APRIL or IL1b mRNA expression in sorted CD11b+ Ly6G+ neutrophils (not shown). Thus, at this time point, increased activation of macrophages does not readily explain the increase in the AM14 B cell response in Myd88-/-Fcer1g-/- hosts.
Given differences in macrophage population recruitment and that MCP-1 is a chemokine that attracts macrophages, we tested serum for MCP-1 following activation of transferred AM14 B cells with anti-chromatin Ab in vivo. Interestingly, at day 5 following anti-chromatin Ab activation, MCP-1 was increased in WT recipients but not recipients lacking Myd88 and Fcer1g (Fig. S2A).
Since CCR2 is a receptor that mediates macrophage migration in response to MCP-1 (52, 53) we hypothesized that CCR2+ macrophages might be an important cell type for controlling contraction of AM14 B cells after activation by anti-chromatin Ab. To test this, we compared WT to Ccr2-deficient recipients (42) following AM14 B cell transfer and activation with anti-chromatin Ab (Fig. S2B and 3). As expected, we found the percentage of CD11bhi Ly6Gneg cells to be greatly reduced in Ccr2 deficient recipients compared with WT recipients (Fig. S2B). However, CCR2 was not required for contraction of transferred WT AM14 B cells following activation with PL2-3 (Fig. 3). Although in one experiment there was an increase in the 4-44+ response in Ccr2 deficient recipients compared with WT recipients at day 7, this result was not reproducible. Unlike in FcRγ and MyD88 deficient recipients at day 10 (Fig. 1 B-D), contraction of the 4-44+ response did not significantly differ between WT and Ccr2 deficient recipients. This pattern was consistent in total 4-44+ cells (Fig. 3A), 4-44+ IgM AFCs (Fig. 3B), and 4-44+ IgG2a AFCs (Fig. 3C). Thus, we conclude that recruitment of CCR2+ macrophages is not the mechanism underlying FcRγ and MyD88 dependent regulation of the AM14 B cell response to anti-chromatin Ab.
Figure 3.
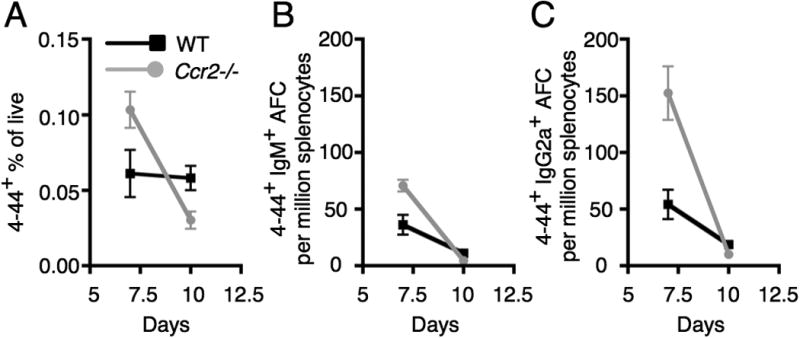
Contraction of the EF RF response does not require Ccr2. BALB/c or Ccr2-deficient BALB/c mice were sacrificed on day 7 or 10 following transfer of purified AM14 sd-Tg B cells and administration of PL2-3 as indicated in Materials and Methods. (A) Surface and intracellular 4-44 double positive cells were quantitated by flow cytometry. (B-C) Splenic 4-44+ AFC of IgM (B) or IgG2a (C) isotype. Data are represented as means +/- SEM. At least 5 mice per group and 2 independent experiments per time point were compiled.
In our initial flow cytometric analysis of Myd88 and Fcer1g deficient mice following AM14 B cell transfer and in vivo activation with anti-chromatin Ab, we also identified a trend in which decreased neutrophils correlated with dysregulated AM14 B cell responses. To test whether neutrophils were functionally contributing to the control of the AM14 B cell response, we depleted neutrophils using the anti-Ly6G depleting Ab clone 1A8 (47) (Fig. S3A-C). We then examined whether the AM14 B cell response was dysregulated at day 7. In our analysis of 4-44+ cells (Fig. S3D), 4-44+ IgM AFCs (Fig. S3E), and 4-44+ IgG2a AFCs (Fig. S3F), the AM14 B cell response was indistinguishable in the presence and absence of neutrophils.
B cell extrinsic Myd88 and Fcer1g control RF B cells via CD40L
Given that the AM14 response to anti-chromatin Ab is enhanced by Ag specific T cell help (16), it was important to analyze the T cell subsets in Myd88 and Fcer1g deficient mice compared with wild type mice during AM14 B cell activation with anti-chromatin Ab. We first examined CD4+ CD62Llo CD44hi PSGL1lo T cells, which identifies T cells that help B cells (18). There was an increased frequency of these cells in mice that lacked Myd88, both in the presence or absence of Fcer1g (Fig. 4A). We also examined FoxP3+ regulatory CD4 T cells (54). There was a lower frequency of FoxP3+ CD4 T cells in mice deficient in Fcer1g, both in the presence and absence of Myd88 (Fig. 4B). Interestingly, the smallest d7 AM14 B cell response, that seen in WT recipients, (Fig 1B-D) correlated with the presence of the fewest B helper T cells and greatest frequency of regulatory T cells. The strongest AM14 B cell response, that seen in Myd88-/-Fcer1g-/- recipients, correlated with a higher frequency of B helper T cells and lower frequency of regulatory T cells. Deficiency in Myd88 alone affected T extrafollicular helper cell frequency but not regulatory T cell frequency, while deficiency in Fcer1g affected regulatory T cell frequency with only a modest effect on T extrafollicular helper cell frequency (Fig 4A-B) indicating that the synergistic effects of Myd88 and Fcer1g may act via distinct pathways.
Figure 4.
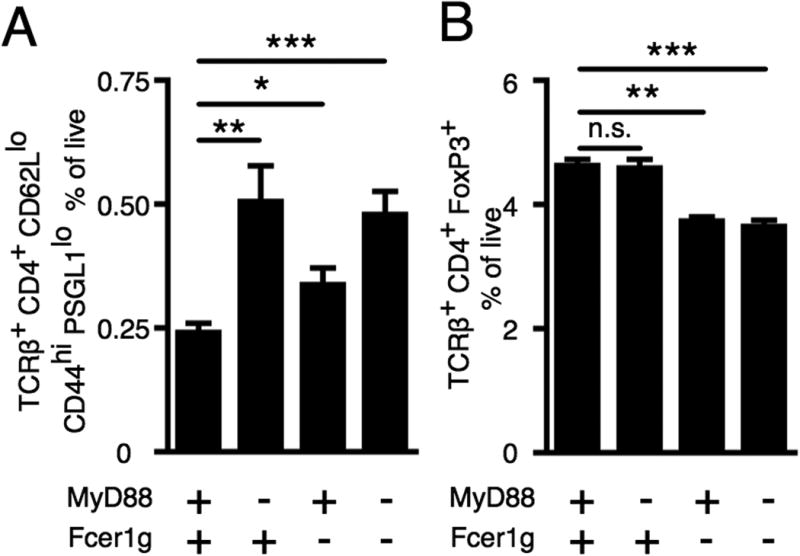
FcRγ and MyD88 influence T cell subsets. BALB/c or Myd88-deficient BALB/c or Fcer1g-deficient BALB/c or MyD88-deficient Fcer1g-deficient BALB/c mice were sacrificed on day 7 following transfer of purified AM14 sd-Tg B cells and administration of PL2-3 as indicated in Materials and Methods. (A) TCR beta+ CD4+ CD62Lneg CD44+ PSGL1lo cells were quantitated. (B) TCR beta+ CD4+ FoxP3+ cells were quantitated. At least 6 mice per group and 2 independent experiments were compiled. Data are represented as means +/- SEM. *p<0.05, **p<0.01, and ***p<0.001 by Mann-Whitney two-tailed test.
Since there was variation in B helper T cell and regulatory T cell frequency in Myd88 and Fcer1g deficient mice and its correlation with the RF B cell response, we sought to test the role of T cell help in Myd88 and Fcer1g dependent control of this response. To do this, we used the anti-CD40L blocking Ab, clone MR-1 (46). We previously used this Ab to demonstrate that T cell help amplified the AM14 B cell response at day 5, although there was a substantial T-independent component as well (16). Here we wanted to test whether the dysregulation observed in the absence of Myd88 and Fcer1g was dependent on CD40L; if so this would indicate a mechanistic link between cells expressing Myd88 and Fcer1g and T cells, consistent with the trends observed above. Commensurate with effective blocking of CD40L, there was a reduction in CD44hi CD62Llo CD4 T cells at day 7. This was similar in both WT recipients and recipients lacking both Myd88 and Fcer1g (Fig. S4). Similar to our previously published work at day 5, there was a reduction in the total 4-44+ cells at day 7 in CD40L-blocked WT recipients compared with isotype control treated WT recipients (Fig. 5A). Although not significant, there was a trend towards reduction of 4-44+ IgM AFCs and 4-44+ IgG2a AFCs following CD40L blocking at day 7 in WT recipients (Fig. 5B and 5C). This difference was significant at day 5 in our previous studies (16). As in Fig. 1, the striking increase in total 4-44+ cells in Myd88 and Fcer1g deficient recipients compared with WT recipients was again observed when both groups were given the isotype control Ab (Fig. 5). Dramatically, in the Myd88 and Fcer1g deficient mice treated with CD40L blocking Ab, this excessive response in the gene-deficient recipients was fully reversed (Fig. 5). Thus, the dysregulation of the AM14 B cell response in the absence of Myd88 and Fcer1g is CD40L-dependent.
Figure 5.
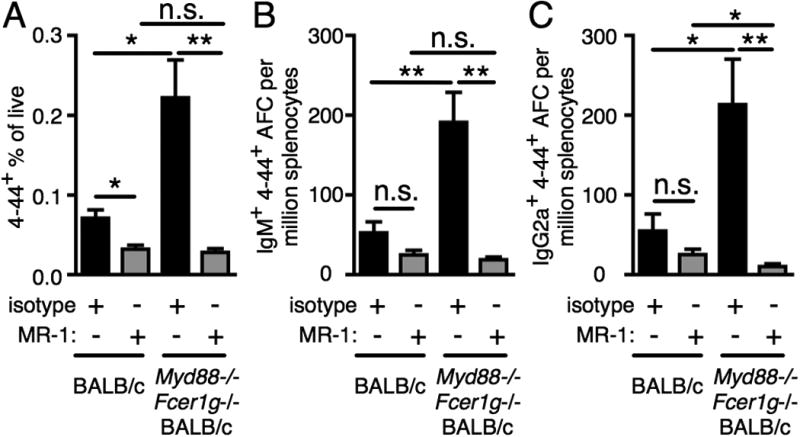
FcRγ and MyD88 regulate CD40L dependent cells. BALB/c or MyD88-deficient Fcer1g-deficient BALB/c mice were sacrificed on day 7 following transfer of purified AM14 sd-Tg B cells, CD40L blocking, and administration of PL2-3 as indicated in Materials and Methods. (A) Surface and intracellular 4-44 double positive cells were quantitated by flow cytometry. (B-C) Splenic 4-44+ AFC of IgM (B) or IgG2a (C) isotype. At least 6 mice per group and 2 independent experiments per time point were compiled. Data are represented as means +/- SEM. *p<0.05 and **p<0.01 by Mann-Whitney two-tailed test.
Fcer1g and MyD88 dependent regulation of Ab is not restricted to autoreactive B cells
As the AM14 B cell response to anti-chromatin antibodies is facilitated by T cells, and as the dysregulation we observed in the absence of Fcer1g and MyD88 is dependent on CD40L, we wondered whether the unsuspected regulatory roles of Fcer1g and MyD88 in the EF response would apply during a classic T cell dependent GC response. To assess this, we used B1-8i mice, which carry a BCR heavy chain that recognizes the hapten NP when paired with the λ1 light chain (39). We transferred these B cells into wild type mice, or mice deficient in both Fcer1g and MyD88 and immunized with NP-CGG in alum. While there was a trend toward expansion of the high-affinity NP2-specific IgM AFCs in the absence of Fcer1g and MyD88 (Fig. 6A), the frequency of high (NP2-) and moderate (NP16-) affinity NP-specific IgG1 AFCs was increased more than 2-fold (Fig. 6B and C). Thus, FcRγ and/or MyD88 regulate both self-reactive and anti-foreign antibody responses.
Figure 6.
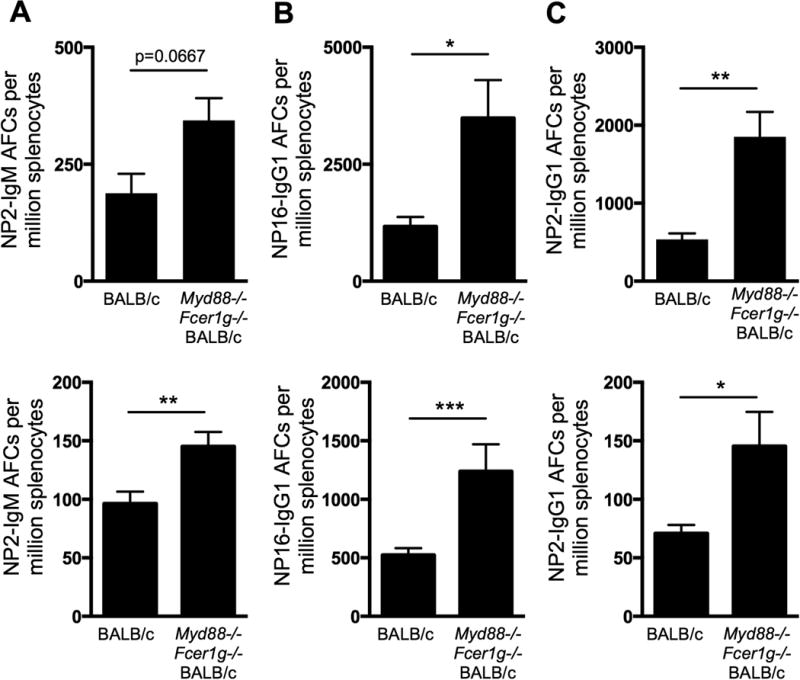
FcRγ and MyD88 regulate T cell dependent antibody production. BALB/c or MyD88-deficient Fcer1g-deficient BALB/c female BALB/c mice were immunized with NP-CGG in alum one day after adoptive transfer of purified female B1.8 B cells and were sacrificed on day 11 post immunization. Splenic NP2-binding IgM+ AFCs (A), NP16-binding IgG1+ AFCs (B) or NP2-binding IgM+ AFCs (C) were identified by ELISpot. Two independent experiments with at least 4 mice per group are shown. Data are represented as means +/- SEM. * p<0.05, ** p<0.01, *** p<0.001 by Mann-Whitney two-tailed test.
Discussion
Though it is common to consider certain molecules and their associated pathways as either positively activating or negatively regulating, in practice such a simplified view is not always accurate. Often, initially “positive” signals induce their own counterregulation; or, such positive signals become reprogrammed to negative in certain contexts. The signaling adapter MyD88 and the ITAM-containing signaling adapter for multiple FcR and C-type lectins, FcRγ, are both typically considered to promote rather than regulate multiple aspects of the immune response (3). For this reason we expected that these two receptors, when expressed on non-B cells, would mediate signals that enhance the response of autoreactive B cells to nucleic acid-containing immune complexes. Indeed, IgG ICs are excellent ligands for multiple “activating” FcR that rely on FcRγ and the contents of the ICs we are using have been shown to contain TLR7 and TLR9 ligands that can activate myeloid cells in a MyD88-dependent manner (32).
Nonetheless, and quite unexpectedly, the absence of either one of these receptors led to an environment that was much more conducive to plasmablast formation by autoreactive B cells. The response was even further enhanced in the absence of both signaling molecules. The most notable effect was the marked prolongation of the plasmablast response, such that by day 10 in the doubly-deficient recipient mice, its magnitude was 15-40 fold higher than in WT recipients. In WT recipients the response had contracted while in the double mutants it had continued to expand. This is indicative of the failure to induce one or more counterregulatory mechanisms. This enhancing effect is not explained by existing paradigms for the function of these adapter molecules or the cells they were likely to be expressed in.
A search for the mechanism underlying this effect began with identifying the cells most likely to be important. In this case, given the effect of FcRγ, we focused on the myeloid compartment, as neither T cells nor B cells express this molecule to a significant degree. The situation is similar for MyD88, albeit host B cells of course do express MyD88 (and would lack its expression in the knockout mice); however, we transferred WT B cells and measured their responses, focusing us on cells of the host. In addition to expression of the key molecules, myeloid cells are found in close proximity to plasmablasts at the T-B border, red pulp and in the marginal zone (12). Myeloid cells could exert direct effects on B lineage cells, such as activated B cells or PBs, and/or could affect the quantity and quality of T help for such B cells. The latter could occur during the cognate MHCII-based presentation of Ag by myeloid cells to such T cells; T cells could also be influenced by cytokines elaborated by myeloid cells.
It is noteworthy that others have implicated myeloid cells in humoral immunity, typically in the context of stimulatory, not regulatory, function. For example, certain types of DCs can trap antigen and present it to B cells to promote Ab responses (55, 56). In addition, myeloid cells—in particular granulocytes—have been identified as major sources of BAFF and APRIL, both of which are important in promoting PB differentiation and survival both in spleen and bone marrow (3, 57). TACI, a B cell-expressed receptor for both BAFF and APRIL has complex effects that can be both positive and negative (58). Regulatory roles for myeloid cells, including macrophages, have also been reported in the context of autoimmunity (59, 60). Recently, Jørgensen and colleagues found that Gr-1high CD11b+ cells were responsible for suppressing autoreactive B cell responses in male mice in the NZB/W mouse model of lupus (61).
In this regard, it was intriguing that we did see alterations in myeloid cells in the mutant mice—in particular less accumulation of CD11bhi cells of both Ly6Ghi and Ly6Glo phenotypes (i.e. macrophages and neutrophils). Nonetheless, we were unable to confirm a non-redundant role for neutrophils. Since we did not deplete multiple cell types at the same time, however, we cannot rule out that macrophages and neutrophils play redundant roles. We hope to evaluate this in future work as it will be ultimately of interest to understand whether in fact myeloid cell types have critical roles in mediating the effect and if so, which ones. As discussed below, DCs seem likely as critical actors given the lack of effect of macrophage or neutrophil deletion as well as the effects on T cells.
Fortunately, we did gain insight into the cellular mechanisms underlying the regulation by Fcerg1 and Myd88: we were able to demonstrate a role for these two molecules in regulating T cell help for augmenting the B cell response. The first clue came from a correlation in that regulatory T cells were reduced in Fcerg1-deficient mice while cells with a TFH phenotype were increased in the absence of Myd88. This was intriguing because it suggested two possible mechanisms, each linked to a different receptor. We were then able to specifically test this, hypothesizing that enhancement of the response in doubly-deficient mice would be lost when B-T collaboration was interrupted by blocking CD40L. Indeed, the enhancement was completely reversed. This suggests that FcRγ and MyD88 regulate the T cell dependent component of the response and that in the absence of this regulation T cell help becomes less restricted, thus prolonging and enlarging B cell expansion.
Such a connection is important because it implies that both FcRγ and MyD88 transmit negative regulatory signals to APC during the late part of the B cell immune response. This feedback loop, which is based on two receptors that are generally thought to transmit positive signals, was previously unrecognized. How the meaning of these signals is altered in the context of the late B cell immune response is yet to be determined. Given the capability of DCs to efficiently activate T cells (21), this would further imply that expression of FcRγ and MyD88 in DCs would be critical in regulating T cells as the response progresses. To test this will require an inducible system of cell-type specific deletion for these receptors.
It is important to point out that induction of negative regulators after signaling via both MyD88 and Syk-family non-receptor tyrosine kinases has been described. For example, stimulation via FcRγ can lead to induction of an inhibitory state known as ITAMi (62, 63) and numerous molecules that regulate MyD88 signaling are induced by MyD88 signals themselves (64). However, it is also possible that failure to directly sustain activation of DCs in some fashion restricts the induction or activation of regulatory type T cells, which could include both classical regulatory T cells (65) as well as FasL expressing CD4 T cells that could kill Fas-positive activated B and T cells (66, 67). In this regard, it is interesting to note that Fas expression was actually elevated on AM14 B cells transferred to the Myd88-/-Fcer1g-/- recipients (Fig 1E), suggesting that host cells were not able to eliminate them.
Another important question regards the sources of ligands for the two key regulatory receptors. In autoimmunity the ligands are fairly clear: IgG ICs that contain molecules recognized by TLR7 or 9. These have been broadly implicated in activating autoreactive B cells as well as DCs and such ligands are abundantly present naturally in a constitutive fashion during established lupus-like autoimmunity (3). Here we have supplied them exogenously without adjuvant in order to observe the temporal evolution of the response. It is also possible that during stimulation IL-1 family cytokines are generated, for example via pyroptotic macrophages responding to activation; such cytokines could be additional upstream mediators of MyD88-transduced signals (68). We observed few differences in cytokine production by macrophages at d7, but it remains possible that the macrophage response to anti-chromatin had returned to baseline by this time.
Though most of our work focused on the EF PB response, we did seek to extend it by determining if a similar regulatory response might exist in the alum-induced anti-NP-CGG response. Although this response is fundamentally different from the induced PB response, indeed we did find a similar regulatory circuit. In this case it is likely that Fcer1g is an important regulatory player, as IgG is made early in the response and presumably ICs form as well. Additionally, alum is reported to activate the inflammasome, thus liberating IL-1β and possibly other IL-1 family member cytokines (69). These could cryptically serve as regulatory cytokines signaling via MyD88 at the later stages of an alum-driven immune response. The finding that some form of FcRγ /MyD88-dependent regulation extends to classical responses that generate both GCs and PBs further generalizes the applicability of the mechanism. The relevance of this finding is likely not limited to anti-hapten responses or to autoantibodies such as RF and anti-DNA that are generated via the EF pathway. The EF response to Salmonella typhimurium was recently shown by our lab and others to be dominated by PBs (4, 14, 17) and thus could be subject to similar regulation, a proposition that has yet to be tested.
In conclusion, FcRγ and MyD88 are two critical molecules in signaling pathways that are very widely engaged during a variety of immune responses and in a multiplicity of cell types. Previously most of the roles ascribed to them have been activating and proinflammatory. Our data are significant in uncovering a latent but potent negative regulatory role for them in the late stages of B cell responses, especially those dominated by PB at EF sites. Such roles could limit acute responses such as shown in the anti-NP response, but could be particularly significant in chronic responses such as autoimmunity and chronic infection.
Supplementary Material
Acknowledgments
The authors would like to acknowledge C. Zhang for technical assistance and the staff of the Yale Animal Resources Center, especially J. St Laurent and J. Fonck for excellent animal husbandry. We would also like to acknowledge Dr. Joanne Reed for critical reading of the manuscript.
Sources of support: This work was supported by NIH grant R01-AI073722 to M.J.S. and NIH Kirschstein National Research Service Award Predoctoral Fellowship 1F31-AI071694 to R.A.S.
Abbreviations
- AFC
Antibody Forming Cell
- APRIL
A proliferation-inducing ligand
- BAFF
B-cell activating factor
- EF
Extrafollicular
- DC
Dendritic Cell
- GC
Germinal Center
- IC
Immune Complex
- NP-CGG
(4-hydroxy-3-nitrophenyl)acetyl conjugated to chicken gamma globulin
- PB
Plasmablast
- RF
Rheumatoid Factor
- sd-Tg
site directed Transgenic
- TFH
T follicular helper cell
- WT
Wild Type
References
- 1.Naparstek Y, Plotz PH. The role of autoantibodies in autoimmune disease. Annu Rev Immunol. 1993;11:79–104. doi: 10.1146/annurev.iy.11.040193.000455. [DOI] [PubMed] [Google Scholar]
- 2.Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- 3.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, Gupta NT, Kleinstein SH, Vigneault F, Gilbert TJ, Meffre E, McSorley SJ, Shlomchik MJ. Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity. 2015;43:120–131. doi: 10.1016/j.immuni.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Racine R, Jones DD, Chatterjee M, McLaughlin M, Macnamara KC, Winslow GM. Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J Immunol. 2010;184:5085–5093. doi: 10.4049/jimmunol.0902710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hastey CJ, Elsner RA, Barthold SW, Baumgarth N. Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J Immunol. 2012;188:5612–5622. doi: 10.4049/jimmunol.1103735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. 2006;24:467–496. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 8.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–3361. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 9.Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci U S A. 2010;107:4658–4663. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferraro AJ, Drayson MT, Savage CO, MacLennan IC. Levels of autoantibodies, unlike antibodies to all extrinsic antigen groups, fall following B cell depletion with Rituximab. Eur J Immunol. 2008;38:292–298. doi: 10.1002/eji.200737557. [DOI] [PubMed] [Google Scholar]
- 11.Chang SH, Kim TJ, Kim YJ, Liu Y, Min SY, Park MJ, Park HS, Lee SK, Nam KH, Kim HY, Mohan C, Kim HR. The lupus susceptibility locus Sle1 facilitates the peripheral development and selection of anti-DNA B cells through impaired receptor editing. J Immunol. 2014;192:5579–5585. doi: 10.4049/jimmunol.1201558. [DOI] [PubMed] [Google Scholar]
- 12.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 13.Bitsaktsis C, Nandi B, Racine R, MacNamara KC, Winslow G. T-Cell-independent humoral immunity is sufficient for protection against fatal intracellular ehrlichia infection. Infect Immun. 2007;75:4933–4941. doi: 10.1128/IAI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner KM, Lane PJ, MacLennan IC. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J Immunol. 2007;178:6200–6207. doi: 10.4049/jimmunol.178.10.6200. [DOI] [PubMed] [Google Scholar]
- 15.Gil-Cruz C, Bobat S, Marshall JL, Kingsley RA, Ross EA, Henderson IR, Leyton DL, Coughlan RE, Khan M, Jensen KT, Buckley CD, Dougan G, MacLennan IC, Lopez-Macias C, Cunningham AF. The porin OmpD from nontyphoidal Salmonella is a key target for a protective B1b cell antibody response. Proc Natl Acad Sci U S A. 2009;106:9803–9808. doi: 10.1073/pnas.0812431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweet RA, Ols ML, Cullen JL, Milam AV, Yagita H, Shlomchik MJ. Facultative role for T cells in extrafollicular Toll-like receptor-dependent autoreactive B-cell responses in vivo. Proc Natl Acad Sci U S A. 2011;108:7932–7937. doi: 10.1073/pnas.1018571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, Yu D, Fagarasan S, Tarlinton DM, Cunningham AF, Vinuesa CG. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. 2011;208:1377–1388. doi: 10.1084/jem.20102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wing JB, Sakaguchi S. Foxp3(+) T(reg) cells in humoral immunity. Int Immunol. 2014;26:61–69. doi: 10.1093/intimm/dxt060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 22.Giles JR, Kashgarian M, Koni PA, Shlomchik MJ. B Cell-Specific MHC Class II Deletion Reveals Multiple Nonredundant Roles for B Cell Antigen Presentation in Murine Lupus. J Immunol. 2015;195:2571–2579. doi: 10.4049/jimmunol.1500792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod'homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, Jenne DE, Wekerle H, Sobel RA, Bernard CC, Shlomchik MJ, Zamvil SS. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210:2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol. 2011;23:106–112. doi: 10.1016/j.smim.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 28.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 29.Thomas CJ, Schroder K. Pattern recognition receptor function in neutrophils. Trends Immunol. 2013;34:317–328. doi: 10.1016/j.it.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 31.van Egmond M, Vidarsson G, Bakema JE. Cross-talk between pathogen recognizing Toll-like receptors and immunoglobulin Fc receptors in immunity. Immunol Rev. 2015;268:311–327. doi: 10.1111/imr.12333. [DOI] [PubMed] [Google Scholar]
- 32.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Su K, Ji C, Szalai AJ, Wu J, Zhang Y, Zhou T, Kimberly RP, Edberg JC. Immune opsonins modulate BLyS/BAFF release in a receptor-specific fashion. J Immunol. 2008;181:1012–1018. doi: 10.4049/jimmunol.181.2.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, Hilbert DM. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 35.Herlands RA, William J, Hershberg U, Shlomchik MJ. Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor B cells at extrafollicular sites. Eur J Immunol. 2007;37:3339–3351. doi: 10.1002/eji.200737752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweet RA, Christensen SR, Harris ML, Shupe J, Sutherland JL, Shlomchik MJ. A new site-directed transgenic rheumatoid factor mouse model demonstrates extrafollicular class switch and plasmablast formation. Autoimmunity. 2010;43:607–618. doi: 10.3109/08916930903567500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 39.Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, Rajewsky K. B cell development under the condition of allelic inclusion. Immunity. 1997;6:225–233. doi: 10.1016/s1074-7613(00)80325-8. [DOI] [PubMed] [Google Scholar]
- 40.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 41.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 42.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dittrich AM, Chen HC, Xu L, Ranney P, Connolly S, Yarovinsky TO, Bottomly HK. A new mechanism for inhalational priming: IL-4 bypasses innate immune signals. J Immunol. 2008;181:7307–7315. doi: 10.4049/jimmunol.181.10.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014;15:631–637. doi: 10.1038/ni.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Losman MJ, Fasy TM, Novick KE, Monestier M. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp(-)+/+ mouse. Oligoclonality of the antibody response and recognition of a determinant composed of histones H2A, H2B, and DNA. J Immunol. 1992;148:1561–1569. [PubMed] [Google Scholar]
- 46.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 48.Shlomchik MJ, Zharhary D, Saunders T, Camper SA, Weigert MG. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- 49.Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- 52.Rollins BJ. JE/MCP-1: an early-response gene encodes a monocyte-specific cytokine. Cancer Cells. 1991;3:517–524. [PubMed] [Google Scholar]
- 53.Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A. 1994;91:2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 55.Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- 56.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G, Knowles DM, Tam W, Chiu A, Bussel JB, Serrano S, Lorente JA, Bellosillo B, Lloreta J, Juanpere N, Alameda F, Baro T, de Heredia CD, Toran N, Catala A, Torrebadell M, Fortuny C, Cusi V, Carreras C, Diaz GA, Blander JM, Farber CM, Silvestri G, Cunningham-Rundles C, Calvillo M, Dufour C, Notarangelo LD, Lougaris V, Plebani A, Casanova JL, Ganal SC, Diefenbach A, Arostegui JI, Juan M, Yague J, Mahlaoui N, Donadieu J, Chen K, Cerutti A. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2012;13:170–180. doi: 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Li J, Zhang YM, Zhang XM, Tao J. Effect of TACI signaling on humoral immunity and autoimmune diseases. J Immunol Res. 2015;2015:247426. doi: 10.1155/2015/247426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilmon MA, Wagner NJ, Garland AL, Lin L, Aviszus K, Wysocki LJ, Vilen BJ. Macrophages prevent the differentiation of autoreactive B cells by secreting CD40 ligand and interleukin-6. Blood. 2007;110:1595–1602. doi: 10.1182/blood-2006-12-061648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gilbert MR, Wagner NJ, Jones SZ, Wisz AB, Roques JR, Krum KN, Lee SR, Nickeleit V, Hulbert C, Thomas JW, Gauld SB, Vilen BJ. Autoreactive preplasma cells break tolerance in the absence of regulation by dendritic cells and macrophages. J Immunol. 2012;189:711–720. doi: 10.4049/jimmunol.1102973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trigunaite A, Khan A, Der E, Song A, Varikuti S, Jorgensen TN. Gr-1(high) CD11b+ cells suppress B cell differentiation and lupus-like disease in lupus-prone male mice. Arthritis Rheum. 2013;65:2392–2402. doi: 10.1002/art.38048. [DOI] [PubMed] [Google Scholar]
- 62.Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, Henin D, Benhamou M, Pretolani M, Blank U, Monteiro RC. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 63.Ben Mkaddem S, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, El Benna J, Launay P, Goujon JM, Benhamou M, Bruhns P, Monteiro RC. Shifting FcgammaRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J Clin Invest. 2014;124:3945–3959. doi: 10.1172/JCI74572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 65.Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, Steinman RM. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med. 2003;198:235–247. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hao Z, Duncan GS, Seagal J, Su YW, Hong C, Haight J, Chen NJ, Elia A, Wakeham A, Li WY, Liepa J, Wood GA, Casola S, Rajewsky K, Mak TW. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29:615–627. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takahashi Y, Ohta H, Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity. 2001;14:181–192. doi: 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- 68.Magna M, Pisetsky DS. The Role of Cell Death in the Pathogenesis of SLE: Is Pyroptosis the Missing Link? Scand J Immunol. 2015;82:218–224. doi: 10.1111/sji.12335. [DOI] [PubMed] [Google Scholar]
- 69.Lambrecht BN, Kool M, Willart MA, Hammad H. Mechanism of action of clinically approved adjuvants. Curr Opin Immunol. 2009;21:23–29. doi: 10.1016/j.coi.2009.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.