

Abstract
The tyrosine kinase WEE1 controls the timing of entry into mitosis in eukaryotes and its genetic deletion leads to pre-implantation lethality in mice. Here, we show that besides the premature mitotic entry phenotype, Wee1 mutant murine cells fail to complete mitosis properly and exhibit several additional defects that contribute to the deregulation of mitosis, allowing mutant cells to progress through mitosis at the expense of genomic integrity. WEE1 interacts with the anaphase promoting complex, functioning as a negative regulator, and the deletion of Wee1 results in hyper-activation of this complex. Mammary specific knockout mice overcome the DNA damage response pathway triggered by the mis-coordination of the cell cycle in mammary epithelial cells and heterozygote mice spontaneously develop mammary tumors. Thus, WEE1 functions as a haploinsufficient tumor suppressor that coordinates distinct cell division events to allow correct segregation of genetic information into daughter cells and maintain genome integrity.
Introduction
Early work in fission yeast suggested that the WEE1 kinase has an important role in a checkpoint that coordinates cell growth and cell division at the G2/M transition.1–3 Studies have shown that loss of WEE1 activity causes cells to enter mitosis before sufficient growth has occurred, resulting in the generation of two abnormally small daughter cells,1 known as the wee phenotype. Conversely, a gene dosage increase of wee1 causes delayed entry into mitosis and an increase in cell size.3 WEE1 is an atypical tyrosine kinase4 and adds an inhibitory phosphorylation to a highly conserved tyrosine residue (Tyr-15) of the cyclin dependent kinase 1 (CDK1; also known as cell-division control protein 2).5–10 In contrast, CDC25 is a protein phosphatase that dephosphorylates Tyr-15 of CDK1, thereby promoting entry into mitosis,11–15 and this type of regulation highlights the central role of CDK1 in orchestrating mitotic events. However, it has been suggested previously that there are phenotypes associated with the loss of WEE1 activity that cannot be explained by overall elevation of CDK1 activity,16 suggesting that there are alternative unknown substrates of WEE1.
In mammalian cells, the G2/M checkpoint is part of the cellular machinery that maintains genomic stability, protecting the genome from the deleterious effects of genotoxic agents.17,18 Upon DNA damage, several checkpoints prevent genomic instability by enforcing cell-cycle arrest at the G1/S transition, S-phase or G2/M transition, thus facilitating damage repair, apoptosis or cellular senescence.17,19–23 Consistent with this dogma, mutations or loss of gene expression involved in cell-cycle regulation have been found in both hereditary and sporadic cancers.17,18,20,24 Most commonly, cancer cells are characterized by a deficient G1/S checkpoint, primarily because of mutations in the broadly important tumor suppressor p53.25,26 Mitotic catastrophe, however, can be avoided if these cells retain a functional G2/M checkpoint. In contrast, G2-checkpoint abrogation can result in increased DNA damage and subsequent cell death through the apoptotic program in cancer cells compared with normal cells,27 suggesting that removing cell-cycle checkpoints may be a more effective therapeutic strategy than blocking the replication of cancer cells. This explains why G2-abrogation has been proposed as a therapeutic strategy for cancer, particularly when combined with conventional DNA-damaging agents.28,29
The anaphase-promoting complex/cyclosome (APC/C) is a multi-subunit member of the RING finger family of ubiquitin ligases and has an essential role in the regulation of mitotic progression.30–32 Through its two co-activators, CDH1 and CDC20, APC/C mediates ubiquitination of many substrates that have distinct functions during mitosis, including Aurora-A and -B, cyclins-A and -B, survivin, Plk1, Nek2A and securin.32 In mammalian cells, CDC20 activates APC/C in early mitosis until anaphase, while CDH1 acts in late mitosis and during G1 phase.33 Mis-regulation of APC/C members, its co-activators and downstream proteins could cause genetic instability and consequently tumorigenesis.34–37 Although both WEE1 and APC/C have important functions during mitosis, the relationship between these proteins is unknown.
We have previously showed that deletion of Wee1 in mice results in embryonic lethality at the blastocyst stage,38 which accounts for the lack of an in vivo model to study its role in normal adult tissues. In this study, using conditional and tissue-specific Wee1 mutant mice and cells, we show that, besides the well-established role of WEE1 in preventing premature entry of mitosis, WEE1 modulates activity of APC/C and Wee1 deficiency, affects mitotic progression through perturbing the maintenance of genomic integrity. Furthermore, we create an animal model carrying a conditional deletion of Wee1 in the mammary gland. Our analysis revealed that WEE1 is indispensable for maintaining genomic stability and it functions as a haploinsufficient tumor suppressor. These results provide further insight into the dual function of WEE1 in tumor inhibition and promotion, which is contingent upon the cellular context.
Results
Wee1 deficiency prevents completion of mitosis
Using a Wee1 tamoxifen inducible knockout (Wee1Co/ −;Tm-Cre) mouse embryonic fibroblast (MEF) cell line, we have previously shown that Wee1-deficient cells enter mitosis prematurely.38 Time-lapse live imaging was done in Wee1Co/ −;Tm-Cre cells treated with 4-hydroxytamoxifen (4-HT) for 24 h, which is the earliest time point that achieves significant knockdown of Wee1 (Supplementary Figure 1a). Wee1Co/ −;Tm-Cre cells either untreated or treated with ethanol were used as control. In these experiments, the control cells completed mitosis within 60 min (Figure 1A), whereas impaired mitosis was detected in Wee1-deficient cells, as revealed by either prolongation of mitosis without completion (Figure 1B) or mainly, by mitotic exit without cytokinesis (Figure 1C). As summarized in Figure 1D, 91 and 92% of mitoses were normal in control cells, while the number of normal mitoses was significantly reduced to 58% from a total of 131 Wee1Co/ −;Tm-Cre MEFs treated with 4-HT. Interestingly, the majority of cells with mitotic defects aborted mitosis without formation of daughter cells (76%).
Figure 1.
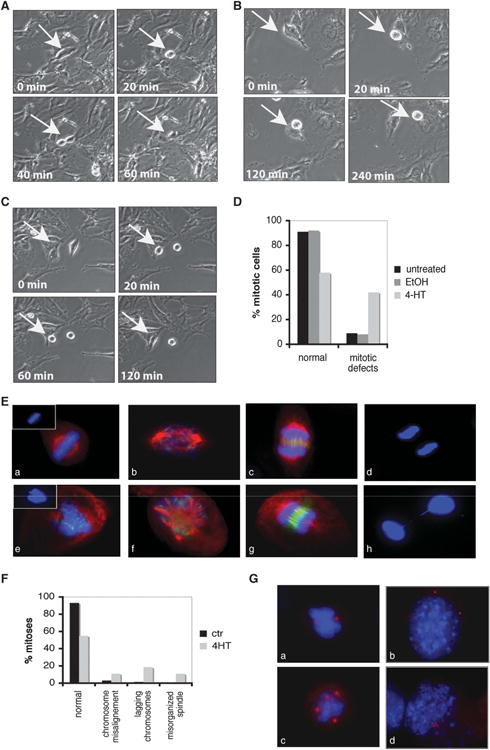
Wee1 mutant cells display mitotic aberrations. (A–C) Time-lapse images of dividing Wee1Co/−;TM-Cre MEFs either treated with ethanol (ctr) (A) or 4-hydroxytamoxifen (4-HT) (B, C). Representative images show either normal mitosis (A) or mitotic cells that cannot complete mitosis (B) or cells that withdraw from mitosis (C). (D) Summary of mitotic abnormalities in Wee1 mutant MEFs during live imaging. (E) Tamoxifen inducible Wee1 knockout MEFs either in the absence (a, b, c, d) or presence (e, f, g, h) of 4-HT were stained with antibodies against α-tubulin (red), Aurora-B (green) and 4′,6-diamidino-2-phenylindole (DAPI; blue). (F) Quantification of the mitotic defects observed in Wee1-deficient cells after treatment with 4-HT. (G) Wee1 knockout MEFs either in the absence (a, b) or presence (c, d) of 4-HT were stained with pericentrin (red) and DAPI (blue).
To further investigate the mitotic abnormalities in Wee1-deficient cells, we examined mitotic spindle organization and chromosome alignment and segregation in these cells. Normal mitotic spindles and chromosome alignment were detected by staining for α-tubulin (red), Aurora-B (green) and DNA (blue) in control cells, as shown in Figure 1E (a–d). In contrast, the analysis revealed various defects including disorganized and multipolar spindles, loosely congregated chromosomes and lagging chromosomes during anaphase in cells upon 4-HT treatment (Figure 1E (e–h), and Figure 1F for quantification). Because centrosomes are the primary microtubule organizing centers and have an essential role in mitotic spindle organization and chromosome segregation, we evaluated possible defects in centrosome organization by immunostaining with two known centrosome markers, γ-tubulin or pericentrin. In addition to premature entry into the cell cycle detected previously,38 deletion of Wee1 caused various centrosomal defects in the number and position of centrosomes (Figure 1G (c,d)), which were rarely observed in control cells (Figure 1G (a,b)). Thus, Wee1-deficient cells exhibit additional mitotic defects and fail to complete mitosis properly, suggesting that WEE1 has a fundamental role in orchestrating the progression of mitosis.
Wee1 deletion results in degradation of mitotic regulators
To investigate a mechanism associated with this phenotype, the levels of proteins involved in the mitotic process were determined. The levels of Aurora-A, Aurora-B, Cyclin B1, Plk1 and securin were decreased in asynchronous Wee1Co/ −;Tm-Cre MEFs upon 4-HT treatment (Figure 2a). Similar results were obtained in cells synchronized in mitosis as shown in Figure 2b. Moreover, reduced levels of both Cyclin B1 and Aurora-A were found in 293T cells after WEE1 knockdown with short hairpin RNA (shRNA; Figure 2c) and in HeLa cells after WEE1 knockdown (Supplementary Figure 1b) or when treated with a WEE1 inhibitor (6-Butyl-4-(2-chlorophenyl)-9-hydroxypyrrolo[3,4-c]carbazole-1,3-(2H,6H)-dione) (Supplementary Figure 1c). Conversely, WEE1 overexpression led to increased levels of these proteins (Figure 2d) and the kinase activity of the protein is required for regulating the levels of mitotic proteins (Figure 2e) since no change was observed after overexpressing the kinase-dead mutant protein.
Figure 2.
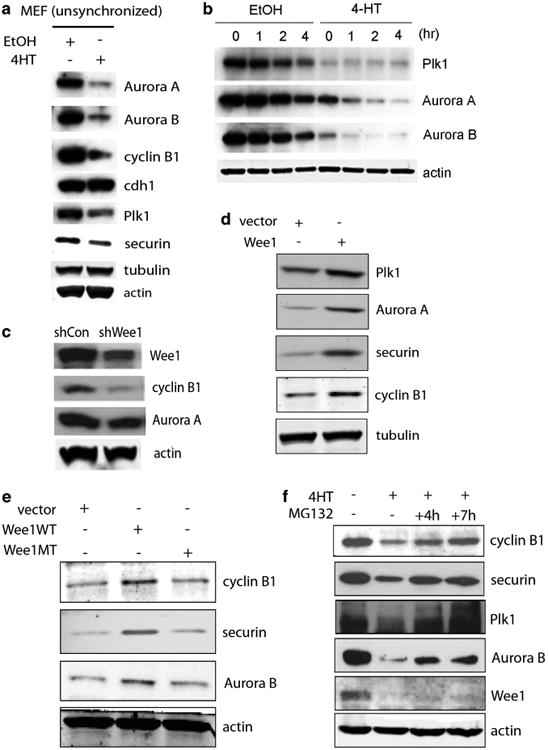
WEE1 controls levels of cell-cycle regulators. (a) Western blots showing levels of Aurora-A, Aurora-B, Cyclin B1, Cdh1, securin and Plk1 in unsynchronized tamoxifen inducible Wee1 knockout MEFs. Cells were treated either with ethanol (ctr) or 4-hydroxytamoxifen (4-HT). (b) Western blot analysis in synchronized tamoxifen inducible Wee1 knockout MEFs after mitotic release. Cells were synchronized by serum starvation for 72 h, replaced with normal media containing nocodazole for 18 h, and then mitotic cells collected by shake off were replated and harvested at indicated time points for western blot analysis. (c) 293T cell were transfected with WEE1 shRNA or non-targeting control shRNA for 24 h to collect whole-cell lysates for immunoblot analysis. (d) 293T cells were transfected with human WEE1 complementary DNA and levels of different cell-cycle proteins were checked by western blotting. (e) Wild-type (WT) and kinase-dead mutant (MT) WEE1 constructs were expressed in 293T cells and levels of cell-cycle proteins were examined by immunoblot. (f) Tamoxifen inducible Wee1 knockout MEFs were cultured in the presence of MG132 for the indicated time points and western blot was performed for Cyclin B1, securin, Plk1 and Aurora-B.
Next, we made a first attempt to investigate whether WEE1 affects these proteins by inhibiting CDK1 activity, which is a well-established mechanism for WEE1 to regulate the G2/M transition.39 First, the levels of these mitotic proteins were affected by WEE1 in unsynchronized cells, where most cells are in G1 phase and the CDK1 activity is low (Figures 2a, c, d and e, and Supplementary Figures 1b and c).40,41 Moreover, levels of mitotic regulators were changed both under conditions that CDK1 phosphorylation was reduced after Wee1 deletion (Figure 2a and Supplementary Figure 1a) and under conditions where CDK1 was fully phosphorylated on Tyr-15 and no difference in phosphorylation was observed after over-expressing WEE1 (Figures 2d and e and Supplementary Figures 1d and e). More importantly, treatment with a CDK1 inhibitor, R0-3306, failed to rescue the defect in the levels of APC/C targets after deletion of Wee1 (Supplementary Figures 1f and h), suggesting that other WEE1 targets may mediate, at least in some part, this response.
In this regard, all the cell-cycle-related proteins tested share a common characteristic in that they are all substrates of the APC/C complex. Thus, it is reasonable to test whether lower protein levels might be associated with mis-regulation of APC/C activity. To test this hypothesis, cells were treated with MG132 followed by examination of levels of these proteins. Interestingly, we found that Cyclin B1, securin, Plk1 and Aurora-B returned to normal levels in Wee1-deficient cells when MG132 was added (Figure 2f), suggesting that WEE1 affects mitotic regulators at the post-translational level by affecting protein degradation. Of note, when we checked the cell-cycle profile of these cells after deleting Wee1, we didn't detect a significant difference within the first 24 h indicating that the observed reduced protein levels couldn't be attributed to the fact that the cells cannot cycle (Supplementary Figures 2a and b). Finally, MG132 had no effect on control cells (Supplementary Figure 2c), and there was no difference in the levels of cell-cycle regulators at early time points after treating cells with 4-HT (8 h) where there is no difference in WEE1 levels (Supplementary Figure 2d), implying that proteasome-mediated protein degradation is involved in the observed phenotype after deleting Wee1.
WEE1 forms a complex with APC/C and affects its activity
APC/C is an E3 ubiquitin ligase that targets cell-cycle proteins for degradation by the 26S proteasome. In our effort to investigate the underlying mechanism through which WEE1 affects the protein stability of mitotic regulators, we identified interactions between WEE1 and components of the APC/C (Figure 3a). Given our earlier observation that MG132 reverses the reduced levels of APC/C substrates in the absence of WEE1, we hypothesized that it might interact with APC/C when the complex is activated after binding of the co-activators, Cdh1 and Cdc20. Indeed, we confirmed an interaction between WEE1 with Cdh1 and Cdc20 in cells using reciprocal immunoprecipitation (Figures 3b and c). Moreover, we could detect interaction of endogenous WEE1 with Cdc27, another major component of the APC/C (Figure 3d), as well as Cdh1 and Cdc20 (Figure 3e). Since it has been shown that Cdh1 and Cdc20 activate APC/C by binding to Cdc27 (or APC3),30,31 it remained to be determined if WEE1 affects the interaction between these proteins. Under our experimental conditions, no difference in the interaction between Cdc27 and mitotic regulators (Cdh1 and Cdc20) or the substrates (Aurora-A, Plk1 and securin), was detected, suggesting that the formation of the complex is not affected by WEE1 (Supplementary Figure 3).
Figure 3.
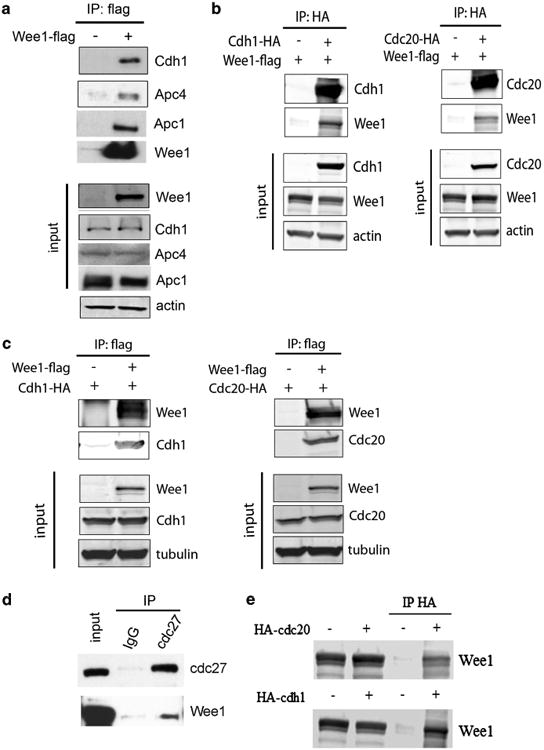
WEE1 interacts with APC/C. (a) Flag-WEE1 was immunoprecipitated and immunoblots of interacting proteins of the APC/C complex are shown. (b, c) Reciprocal immunoprecipitation of ectopically expressed Cdh1 (left) and Cdc20 (right) with WEE1 by using antibodies either against HA-tagged Cdh1 and Cdc20 (b) or flag-tagged WEE1 (c). (d, e) Co-immunoprecipitation of endogenous WEE1 with APC/C after immunoprecipitating endogenous Cdc27 in HeLa cells (d) as well as overexpressed Cdc20 and Cdh1 in 293T cells (e).
Since Wee1 deletion decreases the protein levels of mitotic regulators and WEE1 interacts with APC/C it seemed reasonable to propose that APC/C is activated in the absence of WEE1. To explore this possibility the activity of APC/C was directly measured after deleting Wee1. APC/C was immunopurified from mitotic Wee1Co/ −;Tm-Cre MEFs treated either with 4-HT or ethanol and an in vitro activity assay was performed using ubiquitination of Cyclin B1 as a readout. In accordance with the lower levels of cell-cycle proteins, the activity of APC/C was enhanced in the absence of WEE1 (Figure 4a). To further confirm that it inhibits the APC/C, we examined activity by checking in vivo ubiquitination in asynchronous cells after blocking proteasome degradation. Control and WEE1 knock-down (shWee1) HeLa cells were transfected with a myc-tagged Aurora-A construct and a haemagglutinin (HA)-ubiquitin construct. After treatment with MG132, immunoprecipitated Aurora-A showed enhanced ubiquitination in the shWee1 cells (Figure 4b), an effect that was stronger when Cdh1 was overexpressed to induce APC/Cdh1 activity (lanes 4 and 5). Same results were obtained when Wee1Co/ −;Tm-Cre MEFs were treated with 4-HT to delete Wee1 (Figure 4c and Supplementary Figure 4a). In addition, increased ubiquitination of Cyclin B1 was observed, as revealed by western blotting, when HeLa cells were treated with a specific WEE1 inhibitor (Supplementary Figure 4b). As a positive control for the ubiquitination assay, enhanced APC/C activity was found in cells over-expressing Cdh1, which positively regulates the complex (Supplementary Figure 4b). However, addition of recombinant WEE1 had no effect on in vitro APC activity in APC purified either from Wee1+/+ or Wee1-/- cells (Supplementary Figure 4c).
Figure 4.
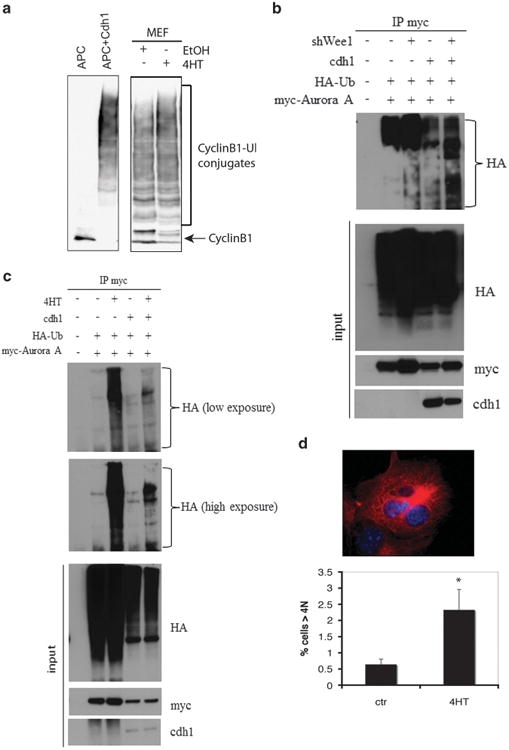
WEE1 negatively regulates APC/C activity. (a) APC/C was isolated from interphase Xenopus egg extracts and incubated in the absence (lane 1) or presence of recombinant human Cdh1 protein (lane 2), or APC/C was isolated from cell lysates from Wee1Co/−;TM-Cre MEFs treated either with ethanol (ctr) (lane 3) or 4-HT (lane 4). The ubiquitination activity of APC/C was assayed with a Myc-tagged N-terminal fragment of human cyclin B1. The reaction mixtures were separated on SDS–PAGE and blotted with the anti-Myc antibody. The positions of the cyclin B1 substrate and the cyclin B1-ubiquitin conjugates are labeled. (b) Control and WEE1 knocked-down (shWee1) HeLa cells were transfected with a myc-tagged Aurora-A construct as well as HA-ubiquitin. After treatment with MG132, Aurora-A was immunoprecipitated using myc-agarose beads and samples were blotted with an anti-HA antibody. Same experiment was performed in cells overexpressing Cdh1 to induce APC/Cdh1 activity (lanes 4 and 5). (c) After transfecting Myc-tagged Aurora-A and HA-ubiquitin, Aurora-A was immunoprecipitated from Wee1Co/−;TM-Cre MEFs either untreated or treated with 4-HT in the presence of MG132. Samples were blotted with an anti-HA antibody. Same experiment was performed in cells overexpressing Cdh1 to induce APC/Cdh1 activity (lanes 4 and 5). (d) DNA content of Wee1Co/−;TM-Cre MEFs treated either with ethanol or 4-HT for 48h. Percentage of cells with >4N DNA content is shown (P < 0.05). Characteristic image of a multi-nucleated cell is shown in upper panel.
Deregulation of the APC/C is associated with mitotic defects, including the development of spontaneous genomic instability.34,42 Wee1Co/ −;Tm-Cre MEFs treated with 4-HT showed an increase of >4N cells relative to control cells measured by flow cytometry. In particular, a fourfold increase of >4N cells was observed following treatment with 4-HT for 48 h as well as presence of bi-or multi-nucleated cells (Figure 4d and Supplementary Figure 4d). Thus, these results indicate that Wee1 deficiency affects levels of APC/C substrates by deregulating APC/C, leading to impaired mitosis and maintenance of genome integrity.
Targeted deletion of Wee1 in mammary epithelium caused increased cellularity
Genomic instability is a well-established early event in mammary gland transformation,43 as such, it seemed reasonable to delete Wee1 in the mammary gland to study its role in adult tissues. For this purpose, we generated a conditional knockout allele of Wee1 (Wee1Co) that carried loxP sites flanking exons 9 and 10 (Supplementary Figures 5a and b) that was crossed with a MMTV-Cre mouse generating a Wee1Co/Co;MMTV-Cre mouse strain (Supplementary Figures 5c and e). The mammary glands of Wee1Co/Co;MMTV-Cre mice appeared denser, which was most obvious in the alveoli visualized in high power images (Figure 5a), otherwise no other defects were observed. Histological analysis revealed increased cellularity in the mammary duct (Figure 5b), increased branching morphogenesis, and alveolar formation in heterozygous glands (Figures 5a and b). We also observed 14.2 and 4.6% BrdU-positive cells in homozygotes and heterozygotes mice, respectively, while no proliferation was detected in wild-type mouse epithelial cells (Figure 5c), implying that mutant cells might experience replication stress. No signs of apoptosis were detected in the mutant glands (Figure 5d), suggesting that increased proliferation of mammary cells in the absence of apoptosis may result in the enhanced branching morphogenesis in the Wee1 mutant mice.
Figure 5.
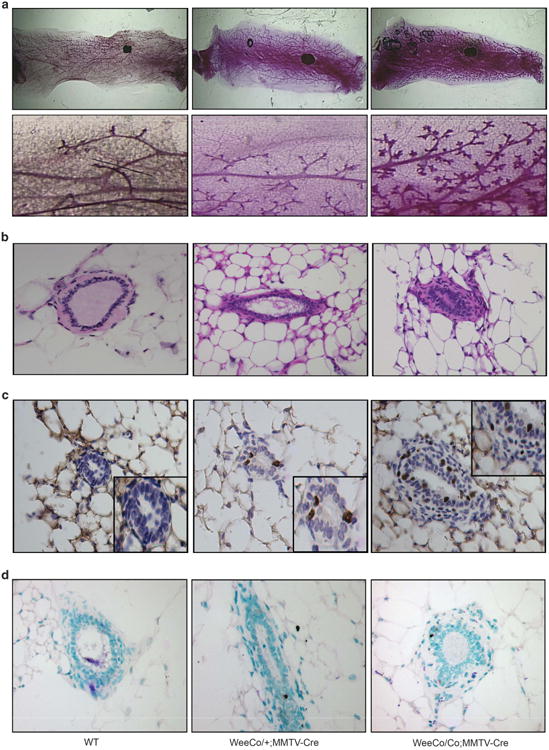
Conditional deletion of Wee1 in mammary gland caused increased cellularity. (a) Whole-mount imaging of mammary glands from 6-month-old virgin WT (left), Wee1Co/+;MMTV-Cre (middle) and Wee1Co/Co;MMTV-Cre mice (right). Boxed areas are enlarged and placed underneath. (b–d) Hematoxylin and eosin stained sections (b), BrdU-positive cells (c) and TUNEL-positive nuclei (d) in 6-month-old virgin WT (left), Wee1Co/+;MMTV-Cre (middle) and Wee1Co/Co;MMTV-Cre mice (right). Boxed areas in c are enlarged areas showing BrdU-positive cells.
Wee1 mammary gland deletion results in mis-coordinated cell cycle and DNA damage
Next, we studied the position of these BrdU+ cells in the cell cycle by using markers for different stages. We found that these cells had indeed finished G1 phase since they were not co-stained with phospho-p27, a protein that marks the G1/S transition in mammalian cells (Supplementary Figure 6a). However, when sections were checked for phosphorylated serine 10 of histone-H3, a commonly used mitotic marker, ∼4 and 12% cells were pH3-positive cells in heterozygotes and homozygotes, respectively (Figures 6a and b). Interestingly, the number of cells containing BrdU or phospho-H3 co-staining revealed ∼ 1:1 ratio. To make sure that these cells are indeed in mitosis, tissues were stained for pH3 and Aurora-B, a marker for mitotic progression. Aurora-B (green) co-localized exclusively with phospho-H3 (red) to the same large condensed nuclear foci in Wee1 mutant mammary cells (Supplementary Figure 6b). It is unlikely that these Wee1 mutant cells could have entered the M phase within the 2h time frame of pulsed BrdU labeling before killing the mice. Instead, this suggests that Wee1 mutant cells progressed into mitosis while still undergoing DNA replication.
Figure 6.
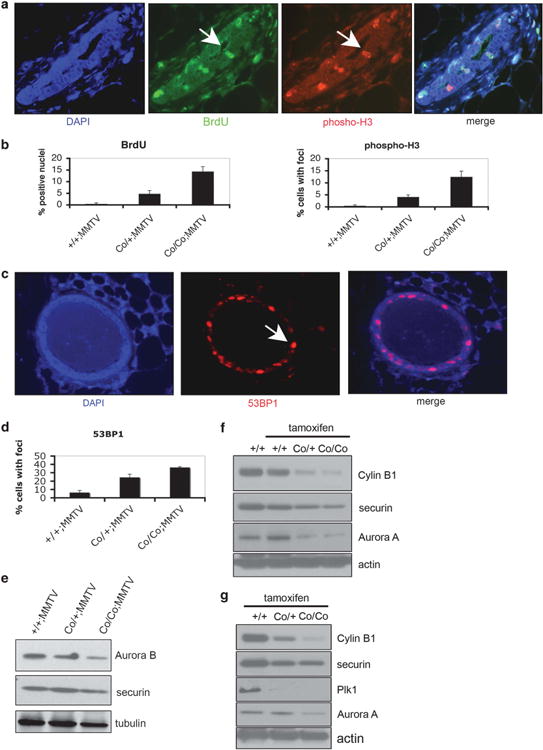
Wee1 deletion in mammary gland results in mis-coordination of the cell cycle. (a) Mammary gland sections of Wee1+/+;MMTV-Cre, Wee1Co/+;MMTV-Cre and Wee1Co/Co;MMTV-Cre mice were stained with anti-phosphorylated histone-H3 Ser10 (pH3; red) and anti-BrdU antibodies (green). Representative images from Wee1 mutant mammary glands are shown and arrows indicate positive stained cells. Nuclei were counterstained with DAPI. (b) Epithelial cells from tissue sections stained as described before were counted and the percentage of either BrdU (left) or phospho-H3 (right) positive cells is shown. (c) Wee1-defficiency in mammary gland causes DNA damage. Mammary glands from WT, Wee1Co/+;MMTV-Cre and Wee1Co/Co;MMTV-Cre mice were stained with an antibody against 53BP1. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and arrows indicate positive stained cells from mutant mammary glands. (d) Quantification of the percentage of mammary cells displaying positive staining for 53BP1. (e) Mammary glands from Wee1+/+;MMTV-Cre, Wee1Co/+;MMTV-Cre and Wee1Co/Co;MMTV-Cre mice were immunoblotted with antibodies against Aurora-B and securin. (f) Mammary glands from Wee1+/+;TM-Cre, Wee1Co/+;TM-Cre and Wee1Co/Co;TM-Cre mice either untreated or treaded with tamoxifen were immunoblotted with antibodies against Aurora-A and securin. (g) Liver (upper) and testis (lower) from same mice as described in (f) were checked for levels of mitotic regulators by immunoblot.
Mutant mammary glands exhibited extensive DNA damage as compared with normal mammary glands after staining tissue sections with an antibody against 53BP1, which is a very sensitive marker of the DNA damage response, by using indirect immunofluorescence (Figure 6c). These results were further confirmed by staining for phosphorylated-H2AX at S139, as a second measurement of DNA damage (Supplementary Figure 6c). Quantitative scoring of punctate nuclear foci found a dose dependent effect of the deletion of Wee1. In particular, Wee1Co/+;MMTV-Cre mice showed 15.3% nuclei positive for DNA damage, whereas the number was increased to 29.4% in Wee1Co/Co;MMTV-Cre mice. In contrast, wild-type mammary glands displayed foci in ∼ 5% of the cells after staining for phospho-H2AX. The DNA damage foci formation suggests that these Wee1 mutant cells undergo an aberrant S-phase resulting in spontaneous DNA damage, which can be carried over to mitosis.
Reduced in vivo levels of cell-cycle regulators in Wee1 mutant mice We showed earlier that Wee1 mutant cells have increased APC/C activity, which might allow cells to progress through mitosis at the expense of genomic stability. In this regard, it was determined that Wee1 deletion results in reduced levels of both Aurora-B and Cyclin B1 in the mammary gland (Figure 6e). The effect of WEE1 in regulating mitotic regulators was further confirmed in 2–3-weeks-old Wee1Co/Co;Tm-Cre mice after acute deletion of Wee1 by tamoxifen injection (Supplementary Figure 5f). Indeed, reduction of levels of mitotic regulators was observed in tissues, such as mammary gland in female mice and liver and testis in male mice, after acute knockdown of Wee1 (Figures 6f and g and Supplementary Figures 7a and b). These results suggest that the impaired APC/C activity in vivo allows cells experiencing extensive DNA damage to escape from surveillance mechanisms, thus contributing to genomic instability.
Wee1 is a mammary haploinsufficient tumor suppressor in vivo
To determine an in vivo genomic instability permissive phenotype in mice lacking Wee1, cohorts of both wild-type and mutant animals were followed. In total, 20 Wee1+/+;MMTV-Cre (control), 20 Wee1Co/+;MMTV-Cre and 27 Wee1Co/Co;MMTV-Cre mice were monitored. Beginning at 16-months-of-age, heterozygous mice developed mammary tumors with an incidence of 25% up to 24 months. In contrast, no tumors were observed in either control or Wee1Co/Co;MMTV-Cre mice (Figure 7a), implying that Wee1 is a haploinsufficient tumor suppressor. Hematoxylin and eosin stain and immunostaining found that these tumors were well-differentiated and positive for ER, HER2, CK18 and CK19 (Figures 7b, c and d), indicating that these are luminal-type cancers, presumably derived from differentiated luminal epithelial cells. Interestingly, when metaphase spreads were prepared from tumor cell lines developed from Wee1 mutant tumors, double minute chromosomes were detected (Figure 7e), which have been associated with premature chromosome condensation. Moreover, chromosome spreads from three different primary tumors, which showed that only 4.2% of metaphases exhibited normal chromosome number, while the majority of cancer cells were polyploid. This observation is in accordance with the cytokinesis defects observed after Wee1 deletion as shown in vitro, suggesting that genome instability induced by Wee1 deficiency results in the tumor permissive phenotype observed in these mice. Last, we detected upregulation of proteins involved in the senescence response in homozygote compared with heterozygote mice, as revealed by western blotting (p16 and p21) and immunohistochemistry (p53-Ser15 phosphorylation and γH2AX) (Supplementary Figures 7c and d). Thus, although loss of one copy of Wee1 has a weaker phenotype, this allows cells to escape from the senescence response, which seems to be a necessary condition for the development of tumors in Wee1 mutant mice. This senescence response might be, in part, mediated by p53, as tumor onset in the mice double heterozygous for Wee1 and p53 (Wee1Co/+;p53+/ −;MMTV-Cre) (n=12) was accelerated in comparison with that of Wee1Co/+;MMTV-Cre mice (from 16-months to 10-months-of-age, Figure 7a).
Figure 7.
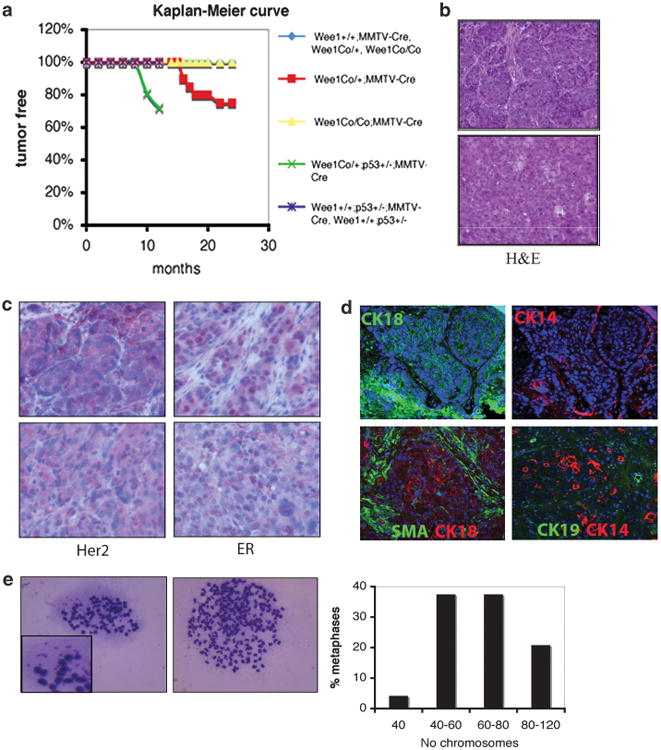
Tumor formation in mice carrying mammary-specific deletion of Wee1. (a) Kaplan–Meier survival curve of mice with different genotypes as indicated. (b) Representative hematoxylin and eosin stained (H&E) slides from mammary tumors from two Wee1Co/+;MMTV-Cre mice. (c) Sections from mammary tumors from Wee1Co/+;MMTV-Cre mice were stained with antibodies against HER2 and ER by immunohistochemistry. (d) Indirect immunofluorescence in mammary tumors from Wee1Co/+;MMTV-Cre mice with antibodies against SMA, CK14, CK18 and CK19. (e) Metaphases of cell lines developed from primary tumors showing double minute chromosomes (left) and polyploidy (middle). Quantification of chromosome numbers in Wee1-deficient tumor cells is shown (right).
Finally, the finding that Wee1 serves as a tumor suppressor in vivo was further supported by analyzing data from human breast cancers deposited in the Oncomine cancer microarray database. In all six studies, WEE1 expression was decreased in tumor samples compared with normal breast tissue and expression levels of WEE1 from three studies are shown in Supplementary Figures 8a and c. Of note, it was observed that even when tumors with different histological grades were included in the analysis (G1, G2 or G-3, Elston system), WEE1 was decreased independently of the tumor grade (Supplementary Figure 8d). Collectively, these data support the idea that WEE1 is indispensable for regulation of the cell cycle and maintenance of genome integrity and may function as a tumor suppressor.
Discussion
In this study we showed that Wee1 mutant cells, in addition to premature mitotic entry, exhibit impaired mitosis characterized by delayed progression and completion without normal cytokinesis, among other defects. Second, we demonstrated that WEE1 negatively regulates APC/C, as Wee1 deficiency causes increased activity of APC/C, which also allows mutant cells to progress through mitosis at the expense of genomic integrity. Finally, we demonstrated that Wee1 acts as a haploid tumor suppressor since mutant mammary gland develop tumors. These data suggest that WEE1 coordinates distinct cell-division events in somatic cells to allow correct segregation of genetic information into daughter cells and maintenance of genomic integrity.
Functions of WEE1 in modulating activity of APC/C
Cell cycle progression from G2 phase into mitosis is tightly regulated by several coordinated cellular events, including activation of CDK1.44 WEE1 kinase is a potent mitotic inhibitor by phosphorylating CDK1 at Tyr-15, which inactivates CDK1 and delays entry into mitosis, while Cdc25 phosphatase promotes entry into mitosis by removing the inhibitory phosphorylation of CDK1.7,8,12,13 In addition to premature mitotic entry, our data indicates that Wee1 deficiency causes increased activity of APC/C. We showed that acute deletion of Wee1 in MEFs and shRNA-mediated knockdown in 293T cells significantly reduces these downstream targets, while ectopic overexpression of WEE1 increases their levels (Figure 2). Using Cyclin B1 and Aurora-A as reporters, we demonstrated that Wee1 deficiency increased ubiquitin levels of both proteins in vitro and in vivo (Figure 4, and Supplementary Figure 4). Of note, a recent study indicated that SWE1, a WEE1 homology in yeast, inhibits APC/C activity through CDK1.45 However we found that pharmacologic Cdk1 inhibition could not rescue the levels of mitotic regulators in the absence of Wee1. Thus, it is possible that WEE1 regulates APC/C activity both in a CDK1-dependent and a CDK1-independent manner, therefore simply inhibiting CDK1 alone could not generate an obvious effect. Further studies will be needed to uncover the mechanistic details regarding the regulatory role of WEE1 on APC/C.
On the other hand, the activity of WEE1 must be precisely regulated to ensure cell-cycle progression under normal growth conditions. It has been shown that WEE1 is subjected to at least two waves of hyper-phosphorylation by Cyclin B1-Cdk1 and Cdc5 in yeast, leading to its ubiquitin-mediated degradation.46,47 Interestingly, both Cyclin B1 and Cdc5 are downstream targets of APC/C. In mammalian cells, WEE1 is also degraded by the APC/C target protein, Polo-like kinase 1, which phosphorylates and degrades WEE1 at the onset of mitosis.48 These studies indicate that WEE1 and APC/C constitute a negative feedback loop to regulate each other during cell-cycle progression, that is, APC/C mediates WEE1 degradation to promote mitotic progression, while WEE1 inhibits APC/C activity to delay this progression to maintain genomic integrity. On the other hand, loss of the inhibitory role of WEE1 on APC/C exerts the opposite effect, as Wee1-deficient cells fail to arrest properly during mitosis in the presence of DNA damage, leading to abnormal mitosis and genomic instability.
WEE1 as potential target for cancer therapy
WEE1 has been proposed as a therapeutic target for the treatment of cancer.49 Consistent with this hypothesis, WEE1 has recently attracted the attention as a promising target in anticancer treatment. Several preclinical studies have shown that WEE1 inhibitors can be very effective when combined with either chemopreventive compounds50–52 or irradiation53,54 in various cancer cell lines. Furthermore, RNA interference screening studies revealed reduced cell viability in the absence of WEE1 confirming its role as a potential therapeutic target.55 Similar results were obtained when WEE1 inhibition was tested in several studies using xenograft animal models49,51 with the most striking effects observed in a glioblastoma model, where tumor growth was significantly reduced after inhibition of WEE1.56 Our data indicates that these results may be related to the function of the protein and can be explained due to enhanced APC/C activity after losing or inhibiting WEE1. In particular, this affects stability of important mitotic regulators resulting in reduced levels of these proteins, which may cause profound growth defects in cancer cells after immediate knocking down or treatment with pharmacological inhibitors of WEE1.
Long-term loss of Wee1 in mammary epithelium induces tumor formation
Our data indicates that 25% of Wee1 heterozygous mice with mammary epithelium disruption of Wee1 developed tumors after a long latency, while mice with homozygous deletion of Wee1 were tumor free. This finding suggests that Wee1 acts as a haploid tumor suppressor although it is not contradictory to the view that WEE1 can serve as a therapeutic target for cancer therapy considering the fact that long-term loss of Wee1 results in profound genetic instability. Strikingly, although both heterozygous and homozygous loss of Wee1 in mammary epithelium causes mis-coordinated cell-cycle progression and genetic instability, the abnormalities in Wee1 homozygous glands are more severe than Wee1 heterozygous glands. Thus, it is conceivable that while the prolonged genetic instability could eventually result in malignancy as demonstrated in numerous cases,18 the homozygous loss of Wee1 is harmful for cell growth due to profound genetic instability, which may induce secondary responses such as cell-cycle arrest, apoptosis or senescence. Consistent with this view, we detected upregulation of proteins involved in the senescence response in homozygotes compared with heterozygote mice. In contrast to what we have seen before in Wee1-/- embryos, where embryonic lethality was mainly due to increased apoptosis,38 we could not detect apoptotic cells in adult mammary epithelial cells, suggesting that cellular senescence is the major antitumor barrier. Heterozygotes exhibit a milder phenotype regarding mis-coordination of cell cycle and DNA damage, thus escaping senescence, whereas deregulation of APC/C allows cells to enter and complete mitosis at the expense of genomic instability leading to mammary tumor formation after a long latency. Interestingly, when we checked Wee1Co/+;MMTV-Cre mice heterozygotes for the p53 gene we could detect earlier onset of tumorigenesis (Figure 7a). This observation illustrates that like many other tumor suppressors that are involved in maintenance of genome integrity, such as Brca1 (ref. 57), p53 mutation/inactivation accelerates tumorigenesis associated with Wee1 deficiency.
In summary, using a Cre/LoxP approach to mutate Wee1 in fibroblast cells and mammary epithelial cells, we demonstrated that Wee1 acts as a haploid tumor suppressor regulating mitosis at different time points through different mechanisms. Besides the well-documented role of WEE1 in controlling entry into mitosis, here we show that WEE1 may regulate APC/C as well. The increased APC/C activity after deleting Wee1 opposes the function of mechanisms that hold cells in mitosis in the presence of stressful conditions such as replication stress and DNA damage. These cells become competent to progress through mitosis, but this phenomenon results in the impairment of genomic integrity. Thus, Wee1 deficiency causes mis-coordination of the cell cycle in mammary epithelial cells, triggers the DNA damage response pathway and supports inappropriate progress through mitosis at the expense of genomic integrity. Collectively, these data suggest that WEE1 coordinates distinct cell-division events in somatic cells to allow the correct segregation of genetic information into daughter cells and maintain genomic stability.
Materials and Methods
Mice
Mice carrying the floxed exon 8 and exon 9 of the Wee1 gene were bred with the D strain of the MMTV-Cre transgenic mice58,59 to obtain Wee1Co/+;MMTV-Cre and Wee1Co/Co;MMTV-Cre mice as well as control mice with various genotypes. In addition, the same mice were crossed with mice expressing a transgene insert, which contains a fusion protein between Cre and a mutated form of the ligand binding domain of the estrogen receptor (Cre-ER),60 to generate Wee1Co/Co;Cre-ER mice. To induce deletion of Wee1, a solution of 0.5 mg of tamoxifen, was injected for 5 consecutive days and animals were killed 5 days after the last injection. (See Supplementary Section for more details). All experiments were approved by the Animal Care and Use Committee of the National Institute of Diabetes, Digestive and Kidney Diseases (ACUC, NIDDK).
Plasmids, small interfering RNA and lentiviral shRNA
The mouse and human pCMV-Wee1 expression vectors were from Open Biosystems (Pittsburgh, PA, USA). The vectors were confirmed by direct sequencing. The human wild-type (WT) and mutant (MT) expression vector tagged with either Flag or HA epitope were kindly provided by Dr. Nagi G Ayad (University of Miami, Coral Gables, FL, USA). pCS2-Cdh1-HA and pCS2-Cdc20-HA plasmids were obtained from MW Kirschner through Addgene (Addgene, Cambridge, MA, USA, plasmid 11596 and 11594). The ubiquitin expression vector, pcDNA3-Flag-Ubiquitin, was from Dr KL Guan (University of California-San Diego, La Jolla, CA, USA). For knockdown of WEE1, lentiviral-based shRNA was obtained from Thermo Scientific (Pittsburgh, PA, USA).
Transfection of cells
For transient transfection, 293T and HeLa cells were transfected using polyethylenimine (PEI) at a ratio 1/3 (μg DNA/μl PEI), while MEFs were transfected using FuGENE HD Transfection Reagent at a ration 1/5 (μg DNA/μl FuGENE) according to the manufacturer's instructions.
Immunofluorescence
Cells were grown, treated, fixed and stained directly either in chamber slides, or on coverslips. Growth medium was removed and cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature. After three washes with PBS for 5 min each, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min followed by two washes with PBS for 5 min each. For blocking, 3% bovine serum albumin (BSA) in PBS was used for 1 h at room temperature followed by overnight incubation with primary antibodies in 3% BSA in PBS at 4 °C. Then cells were rinsed three times with PBS for 5 min each, and fluorochrome-conjugated secondary antibodies in 3% BSA were added for 1 h at room temperature avoiding light exposure. After three washes with PBS for 5 min each, slides were coverslipped with Prolong Gold Antifade Reagents (Life Technologies, Grand Island, NY, USA) and cells were examined immediately using a Leica DMR microscope (Leica, Wetzlar, Germany). For long-term storage, slides were kept at 4 °C protected from light.
For paraffin embedded tissues, sections were first deparaffinized and hydrated and then placed in water bath at 95–100 °C for 15 min for antigen retrieval by using Citrate buffer (Thermo Scientific, Waltham, MA, USA). Slides were allowed to cool for 20 min followed by three washes with PBS for 5 min each. For permeabilization and reduction of unspecific fluorescence, 0.5% Triton X-100 was used for 5 min at 37 °C and 0.5 mg/ml Sodium borohydride was used for 10 min at room temperature, respectively. For blocking as well as primary and secondary antibody incubations, same procedure was followed as described above. Primary antibodies against BrdU, Aurora-B (BD Biosciences, San Jose, CA, USA), phospho-H2AX, phospho-H3 (Upstate, Lake Placid, NY, USA), phospho-27 (Santa Cruz Biotechnology, Dallas, TX, USA), 53BP1 (Novus Biologicals, Littleton, CO, USA), a-tubulin, γ-tubulin (Sigma-Aldrich, St Louis, MO, USA), pericentrin, CK18 (Abcam, Cambridge, MA, USA), SMA (Dako, Carpinteria, CA, USA) and CK14 (Covance, Princeton, NJ, USA) were used.
Time lapse
Cells were plated on a SmartSlide-6 Microincubator multiwell plate (Wafergen BioSystems, Fremont, CA, USA) with heated base and lid for one day in a 37 °C CO2 incubator and 24 h later the plate was placed to an Olympus IX81 Inverted Microscope (Olympus America, Center Valley, PA, USA). Temperature and gas were controlled by the SmartSlide 150 system instrument (WaferGen Biosystems). All parameters were set according to the manufacturer by using the Smartware software (Western Digital, Irvine, CA, USA). Images were captured every 5 min and analyzed with Slidebook 4.2 software (Olympus America).
DNA content and flow cytometry
Cells were collected and fixed with 70% ethanol on ice. After centrifugation, cells were washed with PBS and resuspended in 25 μg/ml propidium iodide solution. Staining was performed at room temperature for 30 min followed by flow cytometer (FACS) analysis using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). When mammary tumors from Wee1 conditional knockout mice were used for FACS analysis the procedure was followed as described earlier. Briefly, tumors were minced, digested and filtered through a 40-mm mesh to obtain single-cell suspension. After blocking and lineage depletion, cells were stained at a concentration of 1×106 cells per 100 μl of buffer (PBS pH 7.2, 0.5% BSA, 2 mM EDTA) with antibodies against CD24 (anti CD24-PE, BD Biosciences) and CD29 (anti CD29-FITC, Chemicon, Temecula, CA, USA).
Western blot
For western blotting, cells were harvested, lysed, and 40 μg of total protein (as determined by Bio-Rad protein assay, Bio-Rad Laboratories, Hercules, CA, USA) was loaded onto Tris-Glycine gels (Invitrogen, Grand Island, NY, USA). After transferring to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), membranes were blocked with either 5% nonfat dried milk in PBS or 5% BSA in Tris buffered saline with tween 20 and then incubated with primary antibodies. Horseradish peroxidase-conjugated secondaries antibodies (GE Healthcare, Pittsburgh, PA, USA) were used and protein was visualized using enhanced Immobilon Western Chemiluminescent HRP Substrate (Millipore). Antibodies against WEE1 (H-300), p53 (Pab240), p21 (F-5), p16 (M-156), Cdc2 (17), Cyclin B1 (H-433, GNS1), Cdc20 (E-7), HA (Y-11), myc (9E-10) (Santa Cruz Biotechnology), phospho-Cdc2 (9111), phospho-p53 (9284) (Cell Signaling, Danvers, MA, USA), Tubulin (B-5-1-2), Actin (AC-15), Flag (M2) (Sigma-Aldrich), Aurora-A (35C1), Aurora-B (ab2254), securin (DCS-280), Cdc27 (AF3.1), Cdh1 (DH01) (Abcam, Cambridge, MA, USA), Plk1 (35-206) (Invitrogen), phospho-H2AX (JBW301) and HP1a (15.19s2) (Upstate), APC1, APC4 and APC6 (Bethyl Laboratories, Montgomery, TX, USA) were used.
In vivo detection of APC/C activity
MEFs were transfected with Myc-Aurora-A in the presence or absence of 4-HT and were treated with MG132 (10 mM) for 4 h. After cell lysis, lysates were immunoprecipitated with anti-Myc antibody. The immunoprecipitates were washed three times with lysis buffer and subjected to immunoblot analysis. Alternatively, HeLa cells were transfected with Flag-ubiquitin followed by immunoprecipitation with anti-Flag antibody and immunoblot analysis using antibody against Cyclin B1.
Immunoprecipitation
Cell or tissue lysates were incubated overnight with appropriate antibodies followed by incubation with protein A or G sepharose beads (Santa Cruz Biotechnology) for 4 h at 4 °C. After washing five times, immunocomplexes were resolved using SDS–PAGE and analyzed by western blot. For immunoprecipitation of ectopically expressed proteins anti-Flag, HA and myc- agarose-conjugated beads (Sigma-Aldrich) were used.
Supplementary Material
Acknowledgments
We are grateful for the critical reading and helpful discussion of members of the Deng laboratory. This work was supported in part by the Intramural Research Program of the NIDDK, NCI and CCR, NIH. DG is supported by 1R01CA152601-01 from the NCI, BC093803 from the DOD, SPORE P50CA98131 and a Hirshberg Foundation for Pancreatic Cancer Research Seed Grant Award. Melissa Stauffer, PhD, of Scientific Editing Solutions, provided editorial assistance.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Nurse P. Genetic control of cell size at cell division in yeast. Nature. 1975;256:547–551. doi: 10.1038/256547a0. [DOI] [PubMed] [Google Scholar]
- 2.Thuriaux P, Nurse P, Carter B. Mutants altered in the control co-ordinating cell division with cell growth in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 1978;161:215–220. doi: 10.1007/BF00274190. [DOI] [PubMed] [Google Scholar]
- 3.Fantes PA, Nurse P. Control of the timing of cell division in fission yeast. Cell size mutants reveal a second control pathway. Exp Cell Res. 1978;115:317–329. doi: 10.1016/0014-4827(78)90286-0. [DOI] [PubMed] [Google Scholar]
- 4.Squire CJ, Dickson JM, Ivanovic I, Baker EN. Structure and inhibition of the human cell cycle checkpoint kinase, Wee1A kinase: an atypical tyrosine kinase with a key role in CDK1 regulation. Structure. 2005;13:541–550. doi: 10.1016/j.str.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Featherstone C, Russell P. Fission yeast p107wee1 mitotic inhibitor is a tyrosine/serine kinase. Nature. 1991;349:808–811. doi: 10.1038/349808a0. [DOI] [PubMed] [Google Scholar]
- 6.Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature. 1989;342:39–45. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- 7.Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell. 1991;64:1111–1122. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- 8.Parker LL, Atherton-Fessler S, Piwnica-Worms H. p107wee1 is a dual-specificity kinase that phosphorylates p34cdc2 on tyrosine 15. Proc Natl Acad Sci USA. 1992;89:2917–2921. doi: 10.1073/pnas.89.7.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 10.Parker LL, Sylvestre PJ, Byrnes MJ, 3rd, Liu F, Piwnica-Worms H. Identification of a 95-kDa WEE1-like tyrosine kinase in HeLa cells. Proc Natl Acad Sci USA. 1995;92:9638–9642. doi: 10.1073/pnas.92.21.9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunphy WG, Kumagai A. The cdc25 protein contains an intrinsic phosphatase activity. Cell. 1991;67:189–196. doi: 10.1016/0092-8674(91)90582-j. [DOI] [PubMed] [Google Scholar]
- 12.Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67:197–211. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- 13.Kumagai A, Dunphy WG. The cdc25 protein controls tyrosine dephosphorylation of the cdc2 protein in a cell-free system. Cell. 1991;64:903–914. doi: 10.1016/0092-8674(91)90315-p. [DOI] [PubMed] [Google Scholar]
- 14.Millar JB, McGowan CH, Lenaers G, Jones R, Russell P. p80cdc25 mitotic inducer is the tyrosine phosphatase that activates p34cdc2 kinase in fission yeast. EMBO J. 1991;10:4301–4309. doi: 10.1002/j.1460-2075.1991.tb05008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strausfeld U, Labbe JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, et al. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242–245. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- 16.Stumpff J, Kellogg DR, Krohne KA, Su TT. Drosophila Wee1 interacts with members of the gammaTURC and is required for proper mitotic-spindle morphogenesis and positioning. Curr Biol. 2005;15:1525–1534. doi: 10.1016/j.cub.2005.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–1426. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability-an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 19.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 20.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 21.Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 2004;3:997–1007. doi: 10.1016/j.dnarep.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Indovina P, Marcelli E, Di Marzo D, Casini N, Forte IM, Giorgi F, et al. Abrogating G/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer Biol Ther. 2013;15:380–388. doi: 10.4161/cbt.27623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simanski S, Madoux F, Rahaim RJ, Chase P, Schurer S, Cameron M, et al. Identification of Small Molecule Inhibitors of Wee1 Degradation and Mitotic Entry. Probe Reports from the NIH Molecular Libraries Program: NIH; Bethesda, MD, USA: 2010. [PubMed] [Google Scholar]
- 24.Towler WI, Zhang J, Ransburgh DJ, Toland AE, Ishioka C, Chiba N, et al. Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum Mutat. 2013;34:439–445. doi: 10.1002/humu.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 26.Michael D, Oren M. The p53 and Mdm2 families in cancer. Curr Opin Genet Dev. 2002;12:53–59. doi: 10.1016/s0959-437x(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 27.Dixon H, Norbury CJ. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle. 2002;1:362–368. doi: 10.4161/cc.1.6.257. [DOI] [PubMed] [Google Scholar]
- 28.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98:523–528. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–519. [PubMed] [Google Scholar]
- 30.Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 31.Pines J. The APC/C: a smorgasbord for proteolysis. Mol Cell. 2009;34:135–136. doi: 10.1016/j.molcel.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009;4:2. doi: 10.1186/1747-1028-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16:55–63. doi: 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. [DOI] [PubMed] [Google Scholar]
- 35.Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–499. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saeki T, Ouchi M, Ouchi T. Physiological and oncogenic Aurora-A pathway. Int J Biol Sci. 2009;5:758–762. doi: 10.7150/ijbs.5.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmit TL, Zhong W, Nihal M, Ahmad N. Polo-like kinase 1 (Plk1) in non-melanoma skin cancers. Cell Cycle. 2009;8:2697–2702. doi: 10.4161/cc.8.17.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tominaga Y, Li C, Wang RH, Deng CX. Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int J Biol Sci. 2006;2:161–170. doi: 10.7150/ijbs.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- 40.Timofeev O, Cizmecioglu O, Settele F, Kempf T, Hoffmann I. Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J Biol Chem. 2010;285:16978–16990. doi: 10.1074/jbc.M109.096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trovesi C, Falcettoni M, Lucchini G, Clerici M, Longhese MP. Distinct Cdk1 requirements during single-strand annealing, noncrossover, and crossover recombination. PLoS Genet. 2011;7:e1002263. doi: 10.1371/journal.pgen.1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engelbert D, Schnerch D, Baumgarten A, Wasch R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene. 2008;27:907–917. doi: 10.1038/sj.onc.1210703. [DOI] [PubMed] [Google Scholar]
- 43.DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 44.Murray AW, Solomon MJ, Kirschner MW. The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature. 1989;339:280–286. doi: 10.1038/339280a0. [DOI] [PubMed] [Google Scholar]
- 45.Lianga N, Williams EC, Kennedy EK, Dore C, Pilon S, Girard SL, et al. A Wee1 checkpoint inhibits anaphase onset. J Cell Biol. 2013;201:843–862. doi: 10.1083/jcb.201212038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harvey SL, Charlet A, Haas W, Gygi SP, Kellogg DR. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell. 2005;122:407–420. doi: 10.1016/j.cell.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 47.Simpson-Lavy KJ, Brandeis M. Clb2 and the APC/C(Cdh1) regulate Swe1 stability. Cell Cycle. 2010;9:3046–3053. doi: 10.4161/cc..9.115.12457. [DOI] [PubMed] [Google Scholar]
- 48.van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 49.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 51.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther. 2004;3:305–313. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 53.Posthumadeboer J, Wurdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, et al. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11:156. doi: 10.1186/1471-2407-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radio-sensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–8217. [PubMed] [Google Scholar]
- 55.Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS ONE. 2009;4:e5120. doi: 10.1371/journal.pone.0005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244–257. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dine J, Deng CX. Mouse models of BRCA1 and their application to breast cancer research. Cancer Metastasis Rev. 2013;32:25–37. doi: 10.1007/s10555-012-9403-7. [DOI] [PubMed] [Google Scholar]
- 58.Robinson GW, Hennighausen L. MMTV-Cre transgenes can adversely affect lactation: considerations for conditional gene deletion in mammary tissue. Anal Biochem. 2011;412:92–95. doi: 10.1016/j.ab.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.