

Abstract
Selenophosphate is the active selenium-donor compound required by
bacteria and mammals for the specific synthesis of Secys-tRNA, the
precursor of selenocysteine in selenoenzymes. Although free selenide
can be used in vitro for the synthesis of
selenophosphate, the actual physiological selenium substrate has not
been identified. Rhodanese (EC 2.3.1.1) normally occurs as a persulfide
of a critical cysteine residue and is believed to function as a
sulfur-delivery protein. Also, it has been demonstrated that a
selenium-substituted rhodanese (E-Se form) can exist in
vitro. In this study, we have prepared and characterized an
E-Se rhodanese. Persulfide-free bovine-liver rhodanese (E form) did not
react with SeO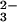 directly, but in the presence of
reduced glutathione (GSH) and SeO
directly, but in the presence of
reduced glutathione (GSH) and SeO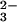 E-Se rhodanese
was generated. These results indicate that the intermediates produced
from the reaction of GSH with SeO
E-Se rhodanese
was generated. These results indicate that the intermediates produced
from the reaction of GSH with SeO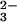 are required for
the formation of a selenium-substituted rhodanese. E-Se rhodanese was
stable in the presence of excess GSH at neutral pH at 37°C. E-Se
rhodanese could effectively replace the high concentrations of selenide
normally used in the selenophosphate synthetase in vitro
assay in which the selenium-dependent hydrolysis of ATP is measured.
These results show that a selenium-bound rhodanese could be used as the
selenium donor in the in vitro selenophosphate synthetase
assay.
are required for
the formation of a selenium-substituted rhodanese. E-Se rhodanese was
stable in the presence of excess GSH at neutral pH at 37°C. E-Se
rhodanese could effectively replace the high concentrations of selenide
normally used in the selenophosphate synthetase in vitro
assay in which the selenium-dependent hydrolysis of ATP is measured.
These results show that a selenium-bound rhodanese could be used as the
selenium donor in the in vitro selenophosphate synthetase
assay.
Selenium is present in several enzymes as a highly specific component that is essential for catalytic activity. In most of these enzymes, selenium is present in the form of selenocysteine residues that are inserted cotranslationally as directed by the UGA codon (1, 2). A few bacterial selenium-dependent enzymes lack selenocysteine and instead contain selenium in a dissociable form that has not been identified (3). The metabolic pathways for the specific insertion of selenocysteine and selenium into proteins are not completely understood.
Although the selenium concentration in the cell is very low,
SeO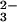 can be incorporated into the amino acid
selenocysteine through specific pathways (4, 5). The selenium oxyanions
must be reduced to selenide in vivo, and it has been
suggested that this reduction is distinct from the sulfate-reduction
pathway (6). In view of the large differences in the relative
concentrations of sulfur and selenium compounds (6) in the environment,
it is apparent that there is a need for specific enzymes and pathways
to ensure that an adequate supply of selenium metabolites exists within
the cell.
can be incorporated into the amino acid
selenocysteine through specific pathways (4, 5). The selenium oxyanions
must be reduced to selenide in vivo, and it has been
suggested that this reduction is distinct from the sulfate-reduction
pathway (6). In view of the large differences in the relative
concentrations of sulfur and selenium compounds (6) in the environment,
it is apparent that there is a need for specific enzymes and pathways
to ensure that an adequate supply of selenium metabolites exists within
the cell.
The metabolic fate of selenium in organisms is still unclear;
however, it is known that the reaction of SeO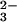 with
several thiols produces selenotrisulfides, according to a reaction
first described by Painter (7). A selenotrisulfide derivative has been
postulated as a possible intermediate in the bioconversion of dietary
inorganic selenium into bioactive selenocompounds (8). In many bacteria
and eukaryotes, glutathione (GSH) is a prime candidate for the thiol
(RSH) in this reaction in vivo, because GSH is the most
abundant low molecular weight thiol in the cell. The reaction of
SeO
with
several thiols produces selenotrisulfides, according to a reaction
first described by Painter (7). A selenotrisulfide derivative has been
postulated as a possible intermediate in the bioconversion of dietary
inorganic selenium into bioactive selenocompounds (8). In many bacteria
and eukaryotes, glutathione (GSH) is a prime candidate for the thiol
(RSH) in this reaction in vivo, because GSH is the most
abundant low molecular weight thiol in the cell. The reaction of
SeO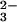 with GSH has been studied extensively in
vitro (9, 10). Some investigators have suggested (6, 11) that
selenodiglutathione (GSSeSG) and its subsequent reduction to
glutathionyl selenide anion (GSSe−) are key
intermediates in the selenium metabolic pathway.
with GSH has been studied extensively in
vitro (9, 10). Some investigators have suggested (6, 11) that
selenodiglutathione (GSSeSG) and its subsequent reduction to
glutathionyl selenide anion (GSSe−) are key
intermediates in the selenium metabolic pathway.
However, at neutral pH GSSeSG is relatively unstable and GSSe− is generated. GSSe− can decompose to produce elemental selenium as the terminal product, or it can be further reduced to hydrogen selenide (HSe−). Although it is thought that HSe− and GSSe− are highly reactive intermediates, there are questions regarding their stability at neutral pH as well as their specific incorporation into selenocysteine. Studies of GSSeSG indicate that it is too labile to be used directly under physiological conditions, especially in the presence of excess GSH.
Alternatively, it is important to consider that cysteine residues of proteins are also reactive toward selenium because of their thiol groups. Rhodanese is well known as a sulfurtransferase that acts as a detoxification enzyme for cyanide (12). It has been reported that this ubiquitous enzyme also has a potential to transfer a selenium atom to cyanide as an acceptor in vitro (13). However, the formation of selenium-substituted rhodanese (E-Se form) has not been investigated fully, and reports on the biological utility of rhodanese in the area of selenium metabolism are not available.
Based on some observations that GSSeSG and rhodanese could serve as
intermediates in the pathway of selenium incorporation into
selenocysteine, we designed experiments to generate E-Se rhodanese in
the presence of SeO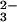 and GSH.
and GSH.
The specific insertion of selenium into selenocysteine (14) and seleno-tRNAs (15) requires the highly reactive reduced-selenium compound monoselenophosphate. The selD gene product selenophosphate synthetase (SPS) catalyzes the synthesis of monoselenophosphate, AMP, and orthophosphate in a 1:1:1 ratio from ATP and selenide in vitro. SPS from Escherichia coli (16) and the closely related enzyme from Haemophilus influenzae (17) have been characterized. In the presence of high levels of free selenide and DTT that are included in the in vitro assay system, the apparent Km values for ATP and selenide are 1 mM and 20 μM, respectively. A Km value of 20 μM for selenide is far above the optimal concentration of selenium needed for the growth of various bacterial species and cultured mammalian cells. In fact, levels above 10 μM are toxic to many bacterial species. Recently, NifS-like proteins and selenocysteine lyase enzymes, which decompose selenocysteine to elemental selenium and alanine, have been considered as candidates for the control of free selenium levels in vivo (18, 19). Although selenocysteine lyase can mobilize a transfer form of selenium (Se*) from l-selenocysteine for selenophosphate biosynthesis, in order for the selenocysteine lyase to be effective as a selenium delivery protein, additional cellular components and proteins may be involved to assist in the specific delivery of the active form of selenium to SPS.
In this article, we report the formation of stable selenium bound to
protein in a reaction with rhodanese and SeO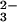 in the
presence of GSH. We also estimated the utility of selenium bound to
rhodanese as a selenium donor in the SPS assay.
in the
presence of GSH. We also estimated the utility of selenium bound to
rhodanese as a selenium donor in the SPS assay.
Materials and Methods
Materials
Bovine-liver rhodanese and sodium SeO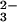 were
obtained from Sigma. [8-14C]ATP was purchased
from ICN. SPS was purified by the procedure of Veres et al.
(16). All buffers and reagents were prepared from the highest
grade chemicals available.
were
obtained from Sigma. [8-14C]ATP was purchased
from ICN. SPS was purified by the procedure of Veres et al.
(16). All buffers and reagents were prepared from the highest
grade chemicals available.
Methods
Preparation of E-Se rhodanese.
Persulfide-free rhodanese (E form) was prepared by adding DTT to an
enzyme solution in 0.1 M Tris⋅HCl buffer at pH 8.0. After a 10-min
incubation, the solution was filtered through a microspin column (G-10,
Amicon) equilibrated with 0.1 M Tris⋅HCl, pH 8.0/1 mM EDTA. E-Se
rhodanese was prepared from the persulfide-free enzyme by reaction with
SeO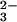 and GSH in PBS at pH 7.4 containing 1 mM EDTA.
After incubation at 37°C for the indicated times, reaction mixtures
were applied to an FPLC fast desalting column (10× 100 mm, Amersham
Pharmacia) to remove the excess SeO
and GSH in PBS at pH 7.4 containing 1 mM EDTA.
After incubation at 37°C for the indicated times, reaction mixtures
were applied to an FPLC fast desalting column (10× 100 mm, Amersham
Pharmacia) to remove the excess SeO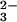 , GSH, and other
small molecules. Rhodanese was eluted in the flow-through fractions as
monitored by protein assay and determination of the selenium content of
each fraction. Selenium content was monitored in all fractions to
demonstrate the elution of low molecular weight compounds including
selenium.
, GSH, and other
small molecules. Rhodanese was eluted in the flow-through fractions as
monitored by protein assay and determination of the selenium content of
each fraction. Selenium content was monitored in all fractions to
demonstrate the elution of low molecular weight compounds including
selenium.
Iodoacetamide treatment for rhodanese.
Iodoacetamide was added to E-form rhodanese after DTT reduction and
incubation in 0.1 M Tris⋅HCl buffer at pH 8.0. Excess
iodoacetamide and alkylated DTT were removed on a Hi-Trap desalting
column (5× 2 ml, Amersham Pharmacia). The selenium-binding procedure
for iodoacetamide-treated enzyme and nontreated enzyme (as a reference)
was performed as described above for the reaction with
SeO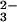 and GSH.
and GSH.
SPS assay with E-Se rhodanese.
The SPS assay was performed anaerobically at 37°C in a reaction mixture containing 100 mM N-tris(hydroxymethyl)glycine (Tricine)/KOH at pH 8.0, 25 mM DTT, 8 mM MgC12, 20 mM KCl, 2 mM ATP, 0.2 μCi (1 Ci = 37 GBq) [8-14C]ATP, 10 μM SPS and E-Se rhodanese. After 30 min, the reactions were terminated by the addition of 1.2 N HClO4 followed by neutralization with 1.4 M KOH. The nucleotides in the supernatant solutions were separated chromatographically on cellulose-polyethyleneimine thin-layer sheets developed in 1.0 M formic acid and 0.5 M LiCl. Nucleotide spots identified by UV-quenching were cut out, and radioactivity was measured by liquid scintillation spectroscopy as described (20).
Analysis of selenium and protein.
Protein concentration was determined by a Bradford procedure (21). Selenium was analyzed by atomic absorption spectroscopy (AZ4100, Perkin–Elmer) with a graphite furnace atomizer (Z-8000, Hitachi, Tokyo).
Results
Reaction of Rhodanese and SeO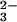 with GSH.
with GSH.
After incubation of E form rhodanese with SeO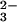 and
GSH for the indicated time, the reaction mixture was applied to an FPLC
fast desalting column. Analysis of the fractions revealed selenium
coeluted with rhodanese in the flow-through volume (Fig.
1A). In contrast, the elution
of selenium was not observed with rhodanese when GSH was omitted from
reaction mixtures (Fig. 1B). Taken together, these results
indicate that an intermediate produced from the reaction of
SeO
and
GSH for the indicated time, the reaction mixture was applied to an FPLC
fast desalting column. Analysis of the fractions revealed selenium
coeluted with rhodanese in the flow-through volume (Fig.
1A). In contrast, the elution
of selenium was not observed with rhodanese when GSH was omitted from
reaction mixtures (Fig. 1B). Taken together, these results
indicate that an intermediate produced from the reaction of
SeO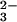 with GSH is required for the generation of a
E-Se rhodanese.
with GSH is required for the generation of a
E-Se rhodanese.
Figure 1.

Profile of the products from the reaction of the E form of rhodanese with selenium after elution on a desalting column. A reaction mixture (100 μl) containing PBS (pH 7.4), 20 μM E form rhodanese and 100 μM selenite was incubated with (A) or without (B) 400 μM GSH. After a 10-min incubation at 25°C, the reaction mixture was applied to a gel filtration column (1.0× 10 cm) and eluted with PBS at pH 7.4. Aliquots of each fraction were assayed to determine selenium (○) and protein (▴) as described in Materials and Methods.
Stability of E-Se Rhodanese.
As shown in Fig. 2, a 10-min reaction of
GSH plus SeO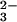 and the subsequent separation by gel
filtration resulted in the elution of low molecular weight
selenium-containing compounds such as GSSeSG and
GSSe−. Increasing the reaction time to 120 min
resulted in a decrease in the amounts of low molecular weight selenium
compounds separated by gel filtration. In contrast, selenium
transferred to rhodanese was recovered completely in high molecular
weight fractions even when the reaction mixture was incubated in PBS at
pH 7.4 for 2 hr at 37°C in the presence of excess GSH. A reaction
mixture including SeO
and the subsequent separation by gel
filtration resulted in the elution of low molecular weight
selenium-containing compounds such as GSSeSG and
GSSe−. Increasing the reaction time to 120 min
resulted in a decrease in the amounts of low molecular weight selenium
compounds separated by gel filtration. In contrast, selenium
transferred to rhodanese was recovered completely in high molecular
weight fractions even when the reaction mixture was incubated in PBS at
pH 7.4 for 2 hr at 37°C in the presence of excess GSH. A reaction
mixture including SeO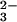 and GSH incubated for more
than 10 min at pH 7.4 deposited sediment that was identified as
elemental selenium. As described before, excess GSH decreases the
stability of GSSeSG by converting it to GSSe−,
which readily decomposes to oxidized GSH and elemental selenium.
However, in the presence of the E form of rhodanese, soluble selenium
remains in solution by forming stable E-Se rhodanese.
and GSH incubated for more
than 10 min at pH 7.4 deposited sediment that was identified as
elemental selenium. As described before, excess GSH decreases the
stability of GSSeSG by converting it to GSSe−,
which readily decomposes to oxidized GSH and elemental selenium.
However, in the presence of the E form of rhodanese, soluble selenium
remains in solution by forming stable E-Se rhodanese.
Figure 2.

Stability of selenium bound to rhodanese and GSSeSG. Reaction mixtures (100 μl) containing PBS at pH 7.4, 100 μM selenite, and 400 μM GSH were incubated with 20 μM E form of rhodanese at 37°C for 120 min (○) and without rhodanese at 25°C for 10 min (□) and for 120 min (▴). Gel filtration profile of a desalting column (1.0× 10 cm) eluted with PBS at pH 7.4. Aliquots of each fraction were assayed to determine the concentration of selenium.
Effect of Iodoacetamide Treatment for Rhodanese.
The active-site cysteine of bovine rhodanese is modified by reaction
with iodoacetate to form an S-carboxymethyl cysteine residue (22).
Substitution of selenium on the carboxymethylated rhodanese was not
observed in the reaction with SeO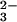 and GSH (Fig.
3). Sulfur-loaded rhodanese also could
not bind selenium produced by the reaction with SeO
and GSH (Fig.
3). Sulfur-loaded rhodanese also could
not bind selenium produced by the reaction with SeO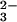 and GSH (data not shown).
and GSH (data not shown).
Figure 3.

Effect of modification of thiol residues by iodoacetamide on the reaction of E form of rhodanese plus selenite in the presence of GSH. E form of rhodanese was treated with iodoacetamide as described in Materials and Methods. Reaction mixtures (100 μl) containing iodoacetamide-treated 20 μM E form rhodanese, 100 μM selenite, and 400 μM GSH were incubated at 25°C. After a 10-min incubation, the reaction mixture was applied to a gel filtration column (1.0× 10 cm) and eluted with PBS at pH 7.4. Aliquots of each fraction were assayed to determine selenium (○) and protein (▴) as described in Materials and Methods.
Optimization to Form E-Se Rhodanese.
Fig. 4 shows that a E-Se rhodanese is
produced most effectively when the ratio of
GSH:SeO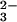 is 4:1. There also is significant
binding of selenium to the E form of rhodanese in the presence of
excess GSH. As shown in Fig. 5, the
amount of selenium bound to rhodanese increased with increasing
concentrations of GSSeSG formed by the reaction of GSH with
SeO
is 4:1. There also is significant
binding of selenium to the E form of rhodanese in the presence of
excess GSH. As shown in Fig. 5, the
amount of selenium bound to rhodanese increased with increasing
concentrations of GSSeSG formed by the reaction of GSH with
SeO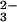 in a molar ratio of 4:1. These results clearly
indicate that 1 mol of the E form of rhodanese can bind 1 molar
equivalent of selenium.
in a molar ratio of 4:1. These results clearly
indicate that 1 mol of the E form of rhodanese can bind 1 molar
equivalent of selenium.
Figure 4.

Effect of the ratio of GSH and selenite on the formation of E-Se rhodanese. Reaction mixtures (100 μl) containing PBS at pH 7.4 and 20 μM E form rhodanese were incubated in the presence of the indicated ratio of GSH and selenite. After a 10-min incubation at 25°C, the reaction mixture was applied to a gel filtration column. Quantitation of bound selenium to rhodanese was determined.
Figure 5.

Effect of the GSSeSG concentration on the formation of E-Se rhodanese. Reaction mixtures (100 μl) containing PBS at pH 7.4 and 20 μM E form rhodanese were incubated in the presence of the indicated concentrations of GSSeSG, theoretically produced by the reaction of GSH with selenite in a molar ratio of 4:1. After a 10-min incubation at 25°C, the reaction mixture was applied to the gel filtration column. The amount of selenium bound per mole of rhodanese was determined by the measurement of selenium and protein contents of peak enzyme fractions from a desalting column.
SPS Assays with E-Se Rhodanese.
To test the ability of a E-Se rhodanese to serve as a selenium donor for selenophosphate biosynthesis, assays were performed in the presence of E-Se rhodanese instead of the high levels of free selenide used routinely. In the in vitro SPS assay, the selenium-dependent hydrolysis of ATP resulted in the formation of AMP and monoselenophosphate in equal amounts (16, 17). However, when selenide was replaced with E form rhodanese, selenophosphate was not formed, as determined by the lack of ATP hydrolysis. In contrast, replacement of selenide in the assay with E-Se rhodanese resulted in the formation of AMP (Fig. 6). Selenium bound to rhodanese was very stable, and the addition of DTT was necessary to observe a selenium-dependent hydrolysis of ATP. In the coupled assay, the observed AMP production was lower with rhodanese-Se as compared with the control assay in which a high concentration of selenide was used. Detailed studies of the reaction conditions have not been performed.
Figure 6.

SPS assay with E-Se rhodanese. Assays were performed anaerobically at 37°C in 50 mM Tricine/KOH, pH 8.0/25 mM DTT/8 mM MgCl2/50 mM KCl/0.1 mM Mg triplex/2 mM ATP/0.2 μCi [14C]ATP/10 μM SPS. The standard assay, performed in the absence of rhodanese, contained 1.5 mM selenide. E-Se rhodanese at the indicated concentrations was added in the absence of selenide. After a 30-min incubation, reactions were terminated, and the reaction products were separated chromatographically as described in Materials and Methods. AMP was quantitated by liquid scintillation spectroscopy.
Discussion
Little is known about the transport and metabolism of
SeO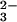 in vivo; however, in many organisms,
GSH is considered to be a major component in selenium metabolism. GSH
nonenzymatically reduces SeO
in vivo; however, in many organisms,
GSH is considered to be a major component in selenium metabolism. GSH
nonenzymatically reduces SeO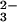 to both GSSeSG and
GSSe− (6, 23). GSSeSG is relatively unstable and
generates a perselenide derivative, glutathioselenolate, which
decomposes rapidly to produce elemental selenium as a terminal product.
GSSe− could be further reduced by GSH to
hydrogen selenide. We considered the possibility that a
selenotrisulfide or perselenide intermediate could donate selenium to a
protein. This protein could bind selenium stably and function as a
selenium transferase to SPS. In this work, we adapted bovine-liver
rhodanese, a member of the sulfurtransferases family as a candidate for
a selenium binding and delivery protein to SPS.
to both GSSeSG and
GSSe− (6, 23). GSSeSG is relatively unstable and
generates a perselenide derivative, glutathioselenolate, which
decomposes rapidly to produce elemental selenium as a terminal product.
GSSe− could be further reduced by GSH to
hydrogen selenide. We considered the possibility that a
selenotrisulfide or perselenide intermediate could donate selenium to a
protein. This protein could bind selenium stably and function as a
selenium transferase to SPS. In this work, we adapted bovine-liver
rhodanese, a member of the sulfurtransferases family as a candidate for
a selenium binding and delivery protein to SPS.
Rhodanese is a widely distributed enzyme in animals, plants, and microorganisms. It catalyzes the transfer of a sulfane sulfur atom from an anionic donor substrate to a thiophilic acceptor by means of a sulfur-substituted enzyme, covalent intermediate. This intermediate enzyme–sulfur complex was demonstrated to be a persulfide group (Cys-SSH) that can stably exist in neutral solution (22, 24).
Canella et al. (13) prepared a selenium derivative of rhodanese by using the synthetic substrate selenosulfate and examined its spectrometric properties. Although the work evaluated the chemical properties of the selenium-derivative enzyme, a physiological significance for a E-Se rhodanese has not been suggested.
To elucidate the potential for rhodanese to exist as a stable
perselenide form (E-Se) and to evaluate whether the selenium bound to
rhodanese can be used by SPS, an E-Se rhodanese was prepared. In this
study, an E-Se enzyme was prepared in a reaction with
SeO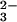 and GSH, both of which are known to be
physiologically available compared with selenosulfate.
and GSH, both of which are known to be
physiologically available compared with selenosulfate.
The stability of GSSeSG was found to depend on the molar ratio of
reduced GSH to SeO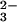 as well as on the pH (9, 25).
Further reduction of GSSeSG to GSSe− was found
to occur when GSH was present in a molar excess of greater than 4:1.
Selenols have a low pKa in the 5–6 range. It has been shown that
GSSeSG is unstable in solution and decomposes at pH values above 2–3.
In the presence of rhodanese, GSH first reacts with
SeO
as well as on the pH (9, 25).
Further reduction of GSSeSG to GSSe− was found
to occur when GSH was present in a molar excess of greater than 4:1.
Selenols have a low pKa in the 5–6 range. It has been shown that
GSSeSG is unstable in solution and decomposes at pH values above 2–3.
In the presence of rhodanese, GSH first reacts with
SeO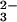 to form GSSeSG. The labile GSSeSG then reacts
with rhodanese at neutral pH to generate a E-Se rhodanese.
to form GSSeSG. The labile GSSeSG then reacts
with rhodanese at neutral pH to generate a E-Se rhodanese.
The results presented here clearly suggest that the E form of rhodanese
can bind selenium at a 1:1 molar ratio. Once reactive selenium binds to
rhodanese, it is more stable than a small molecular perselenide under
physiological conditions. It is likely that selenium is covalently
bound to a cysteine residue—most likely Cys-247—as a mixed
perselenide. GSSeSG, which was formed by reaction of GSH with
SeO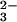 in a molar ratio of 4:1, could provide selenium
directly to the E form of rhodanese. However, it has been proposed that
GSSe− also can react with the E form of
rhodanese, because the E-Se rhodanese can be formed in a reaction when
the GSH/Se ratio is more than 4:1.
in a molar ratio of 4:1, could provide selenium
directly to the E form of rhodanese. However, it has been proposed that
GSSe− also can react with the E form of
rhodanese, because the E-Se rhodanese can be formed in a reaction when
the GSH/Se ratio is more than 4:1.
In the SPS-coupled assay using E-Se rhodanese instead of selenide, AMP formation was not observed in the absence of DTT (Fig. 6), indicating that further reduction of a E-Se rhodanese is necessary to generate the required substrate form of selenium for selenophosphate synthesis. One reason for the low direct availability to SPS of selenium in rhodanese could be the inability of a nucleophilic acceptor to access the active site of bovine-liver rhodanese, thus preventing the efficient transfer of selenium to SPS. It also has been demonstrated that bovine-liver rhodanese binds selenium tightly. The remarkable stability of selenium bound to rhodanese would be useful to pool excess free selenium and mask its toxicity, as in the case of sulfane sulfur (12); however, this property seems to be ineffective for the effective delivery of selenium in vivo. Therefore, for rhodanese-like proteins to be an essential member of a selenium delivery system, additional components may be involved to assist the release of selenium from the enzyme.
Berni and coworkers (26) recently showed that the active site of the
sulfur-substituted form of rhodanese has a specific interaction with
lipoate. It also has been reported that dihydrolipoic acid and
dihydrolipoate amide react with SeO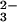 to form
relatively stable selenotrisulfides (27). If dihydrolipoate could
reduce a perselenide in rhodanese instead of DTT, it could serve also
as a cofactor of a selenium transferase in the generation of the
reactive intermediate hydrogen selenide. Although it has not been
confirmed that the E-Se form of rhodanese interacts with
dihydrolipoate, the combination of a rhodanese-like enzyme and
selenotrisulfide derivative of lipoic acid may be involved in a
selenium-delivery system.
to form
relatively stable selenotrisulfides (27). If dihydrolipoate could
reduce a perselenide in rhodanese instead of DTT, it could serve also
as a cofactor of a selenium transferase in the generation of the
reactive intermediate hydrogen selenide. Although it has not been
confirmed that the E-Se form of rhodanese interacts with
dihydrolipoate, the combination of a rhodanese-like enzyme and
selenotrisulfide derivative of lipoic acid may be involved in a
selenium-delivery system.
At present, there are at least two possible pathways for low molecular selenotrisulfides and a protein perselenide to serve as intermediates for selenophosphate formation (Fig. 7). Although these reactions are likely to occur, they have yet to be demonstrated in vivo. It seems reasonable to suppose that a rhodanese-type of enzyme may function as a transferase for the regulation of selenium concentrations in vivo. Future work must focus on the participation of rhodanese-like specific enzymes (selenium transferases) as components of a delivery system for reactive selenium to SPS.
Figure 7.

Proposed reactions for the formation of protein perselenide and pathways for selenophosphate formation. Cysteine-free rhodanese-type enzyme is shown as a protein (S−). Se*, an unidentified transfer form of selenium.
Abbreviations
- GSH
glutathione
- SPS
selenophosphate synthetase
- GSSeSG
selenodiglutathione
- GSSe−
glutathionyl selenide anion
- E-Se
selenium-substituted
- E form
persulfide-free
References
- 1.Stadtman T C. Annu Rev Biochem. 1990;59:111–127. doi: 10.1146/annurev.bi.59.070190.000551. [DOI] [PubMed] [Google Scholar]
- 2.Stadtman T C. J Biol Chem. 1991;266:16257–16260. [PubMed] [Google Scholar]
- 3.Gladyshev V N, Khangulov S V, Stadtman T C. Proc Natl Acad Sci USA. 1994;91:232–236. doi: 10.1073/pnas.91.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock A, Stadtman T C. Biofactors. 1998;1:245–250. [PubMed] [Google Scholar]
- 5.Heider J, Bock A. Adv Microb Physiol. 1993;35:71–109. doi: 10.1016/s0065-2911(08)60097-1. [DOI] [PubMed] [Google Scholar]
- 6.Turner R J, Weiner J H, Taylor D E. Biometals. 1998;11:223–227. doi: 10.1023/a:1009290213301. [DOI] [PubMed] [Google Scholar]
- 7.Painter H E. Chem Rev (Washington, DC) 1941;28:179–213. [Google Scholar]
- 8.Lacourciere G M. Biofactors. 1999;10:237–244. doi: 10.1002/biof.5520100222. [DOI] [PubMed] [Google Scholar]
- 9.Ganther H E. Biochemistry. 1968;7:2898–2905. doi: 10.1021/bi00848a029. [DOI] [PubMed] [Google Scholar]
- 10.Sandholm M, Sipponen P. Arch Biochem Biophys. 1973;99:363–368. doi: 10.1016/s0003-9861(73)80014-1. [DOI] [PubMed] [Google Scholar]
- 11.Ganther H E. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- 12.Westley J. In: Enzymatic Basis of Detoxication. Jacoby W B, editor. Vol. 2. New York: Academic; 1980. pp. 245–262. [Google Scholar]
- 13.Cannela C, Pecci L, Finazzi-Agro A, Federici G, Pensa B, Cavallini D. Eur J Biochem. 1975;55:285–289. doi: 10.1111/j.1432-1033.1975.tb02161.x. [DOI] [PubMed] [Google Scholar]
- 14.Leinfelder W, Forchhammer K, Veprek B, Zehelem E, Bock A. Proc Natl Acad Sci USA. 1990;87:543–547. doi: 10.1073/pnas.87.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veres Z, Tsai L, Scholz T D, Politino M, Balaban R S, Stadtman T C. Proc Natl Acad Sci USA. 1990;89:2975–2979. doi: 10.1073/pnas.89.7.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veres Z, Kim I Y, Scholz T D, Stadtman T C. J Biol Chem. 1994;269:10597–10603. [PubMed] [Google Scholar]
- 17.Lacourciere G M, Stadtman T C. Proc Natl Acad Sci USA. 1999;96:44–48. doi: 10.1073/pnas.96.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esaki N, Nakamura T, Tanaka H, Soda K. J Biol Chem. 1982;257:4386–4391. [PubMed] [Google Scholar]
- 19.Lacourciere G M, Mihara H, Kurihara T, Esaki N, Stadtman T C. J Biol Chem. 2000;275:23769–23773. doi: 10.1074/jbc.M000926200. [DOI] [PubMed] [Google Scholar]
- 20.Lacourciere G M, Stadtman T C. J Biol Chem. 1998;273:30921–30926. doi: 10.1074/jbc.273.47.30921. [DOI] [PubMed] [Google Scholar]
- 21.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Cannella C, Berni R, Rosato N, Finazzi-Agro A. Biochemistry. 1986;25:7319–7323. doi: 10.1021/bi00371a012. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh H S, Ganther H E. Biochemistry. 1975;14:1632–1636. doi: 10.1021/bi00679a014. [DOI] [PubMed] [Google Scholar]
- 24.Aird B A, Horowitz P M. Biochim Biophys Acta. 1988;956:30–38. doi: 10.1016/0167-4838(88)90294-4. [DOI] [PubMed] [Google Scholar]
- 25.Ganther H E. Biochemistry. 1971;10:4089–4098. doi: 10.1021/bi00798a013. [DOI] [PubMed] [Google Scholar]
- 26.Cianci M, Gliubich F, Zanotti G, Berni R. Biochim Biophys Acta. 2001;1481:103–108. doi: 10.1016/s0167-4838(00)00114-x. [DOI] [PubMed] [Google Scholar]
- 27.Self W T, Tsai L, Stadtman T C. Proc Natl Acad Sci USA. 2000;97:12481–12486. doi: 10.1073/pnas.220426897. . (First Published October 24, 2000; 10.1073/pnas.220426897) [DOI] [PMC free article] [PubMed] [Google Scholar]