

Abstract
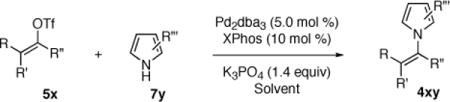
A stereospecific palladium catalyzed N-vinylation of azaheterocycles with vinyl triflates is described. Cyclic and acyclic vinyl triflates along with non-nucleophilic azaheterocycles were found to be substrates for this palladium catalyzed synthesis of N-vinyl pyrrole and indole derivatives.
Graphical abstract
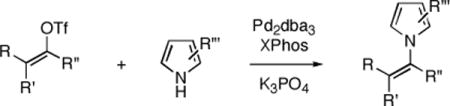
Transition–metal catalyzed carbon–nitrogen bond formation has become a powerful methodology in organic synthesis.1 A variety of highly active palladium and copper catalyst systems have been reported for the N-arylation of amines, amides, azoles, and carbamates.2,3 However, there exist fewer examples of C–N bond formation as a method of N-vinylation.1,4,5 Reports concerning the N-vinylation of dialkyl amines and azoles are even more rare as compared to N-vinylation of amides and carbamates.6,7 A single study on the palladium catalyzed coupling of lithiated azoles with vinyl bromides has been reported.6c Herein we describe a palladium catalyzed stereospecific coupling of vinyl triflates with pyrroles and indoles.
Recently, Buchwald reported the efficient copper catalyzed asymmetric conjugate reduction of a variety of 3-aza-2-enoates (2) to give the corresponding 3-aza-alkanoates (3, eq 1).7 The necessary 3-aza-2-enoate (2) substrates were prepared via a copper catalyzed N-vinylation of azaheterocycles and lactams with Z-3-iodoenoates (1).
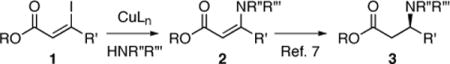 |
(1) |
We were particularly interested in the use of this chemistry for the synthesis of optically active β-azolyl carboxylic acid building blocks for complex alkaloid synthesis. However, the reaction conditions for generating the necessary Z-3-iodoenoates (1), namely treatment of the corresponding ynoates with sodium iodide in acetic acid at elevated temperatures (60→150 °C),8 were not compatible with several substrates of interest. Furthermore, a similar direct synthesis of the isomeric E-3-iodoenoates9 is not available. We envisioned the use of readily available β-ketoesters as precursors for the stereospecific synthesis of both Z- and E-β-pyrrolyl enoates 4 (Scheme 1). Specifically, we sought a catalytic method for the stereospecific N-vinylation of pyrroles with configurationally defined vinyl triflates.10
SCHEME 1.
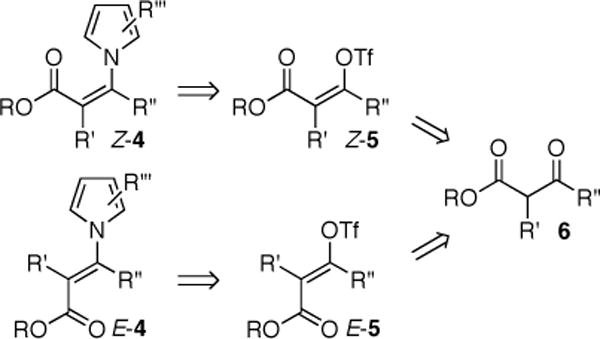
Stereospecific synthesis of β-pyrrolyl enoates.
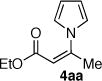
Initial experiments on the coupling of pyrrole (7a) and vinyl triflates 5a–b (eq 2) revealed that copper-based catalyst systems were not effective, typically returning the unreacted triflates. However, the combination of palladium dibenzylideneacetone (Pd2(dba)3) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl (XPhos)11,12 provided a highly active catalyst system for the efficient and selective synthesis of the desired N-vinyl pyrroles. Under optimal conditions, the coupling of the vinyl triflate 5a (1 equiv) with pyrrole (7a, 1.5 equiv) using Pd2dba3 (5 mol%), XPhos (10 mol%) as ligand in the presence of anhydrous potassium phosphate (1.40 equiv) in toluene at 60 °C proceeded to give the desired Z-β-pyrrolyl enoate 4aa with complete stereospecificity in 84% yield after 3.5 h (Table 1, entry 1). Under identical conditions, the coupling of the more sensitive dimethoxy–acetal containing vinyl triflate 5b afforded the corresponding N-vinyl pyrrole 4ba (eq 2) in 79% isolated yield.13
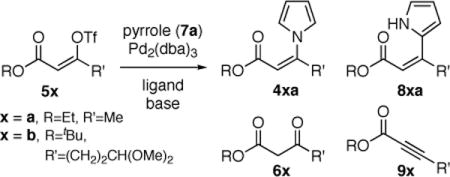 |
(2) |
TABLE 1.
Palladium catalyzed N-vinylation of azaheterocycles with vinyl triflates.
| ||||
|---|---|---|---|---|
| entry | product | temperature (time)a | solventb | yieldc |
| 1 |
|
60 (3.5) | A | 84 |
| 2 3 4 |
|
60 (7) 60 (1) 80 (1.5) |
A B A |
70 91 81 |
| 5 6 7 |
|
60 (25) 60 (1); 80 (6) 60 (2.5); 80 (7) |
A B A |
29d 44 50d |
| 8 9 10 |
|
60 (48) 60 (1); 80 (6) 60 (2.5); 80 (7) |
A B A |
24d 9e 17 |
| 11 12 |

|
60 (2.5); 80 (3) 60 (0.5); 80 (1.5) |
A B |
52 60f |
| 13 14 |
|
60 (2.5); 80 (3) 60 (0.5); 80 (1.5) |
A B |
29 73 |
| 15 |
|
60 (5) | A | 74 |
|
16 17 |
|
60 (2.5); 80 (3) 60 (0.5); 80 (2.5) |
A B |
74 71 |
| 18 |
|
60 (0.5); 80 (2) | B | 70 |
| 19 20 |
|
60 (0.5); 80 (3); 95 (3.5) 60 (0.5); 95 (3.5) |
B B |
53 62 |
| 21 22 |
|
60 (0.5); 80 (3); 95 (3.5) 60 (0.5); 95 (2.5) |
B B |
18g 17g |
| 23 24 |
|
60 (6); 80 (8) 60 (2.5); 110 (2.5) |
A A |
55 91 |
| 25 26 |
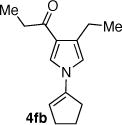
|
60 (5.5); 80 (18.5) 60 (1); 110 (16) |
A A |
43d 72 |
Reaction temperature in °C; reaction time in h.
Solvent: A = toluene, B = toluene–1,4-dioxane (3:1).
Isolated yield after purification.
Remaining vinyl triflate: entry 5, 38%; entry 7, 13%; entry 8, 65%; entry 25, 25%.
Complete consumption of 5a; the ynoate 9a constitutes the mass balance.
Complete conversion to product 4cb (>90% yield) prior to chromatographic purification.
Ethyl 2-oxo-cyclopentanecarboxylate was formed (~50%).
Two commonly observed side products in these coupling reactions were the β-ketoesters 6a–b and the ynoates 9a–b (eq 2). Potassium phosphate tribasic was most effective as the base–additive compared to potassium phosphate dibasic, cesium carbonate, triethylamine and 1,8-diazabicyclo[5.4.0]undec-7-ene; the latter giving predominantly the undesired ynoate byproduct. The use of rigorously dried anhydrous potassium phosphate was found to minimize the formation of both the β-ketoester and the ynoate byproducts.14,15 Attempted palladium catalyzed coupling of preformed pyrrolyllithium with vinyl triflate 5b gave predominantly (~80%) the undesired elimination product 9b (eq 2).6c Furthermore, when N-tributylstannylpyrrole16 was used in place of pyrrole (7a) for the synthesis of N-vinyl pyrrole 4ba, in the absence of anhydrous potassium phosphate, the product mixture contained as much as 50% C-coupled product 8ba (eq 2).17 Interestingly, this undesired C-coupled byproduct was not observed using pyrrole (7a) with the optimal conditions described above.
Considering the synthetic utility of N-vinyl pyrroles and indoles and encouraged by these early results, we sought to explore the generality of this catalytic N-vinylation reaction (Table 1). Representative vinyl triflates18 and azaheterocycles used as substrates in these studies are shown in Chart 1.
CHART 1.
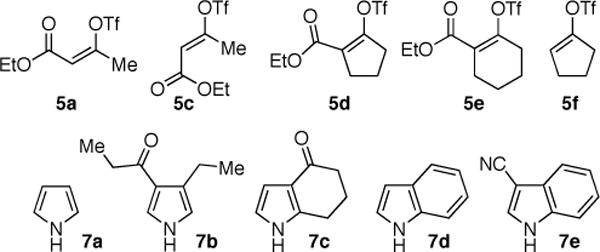
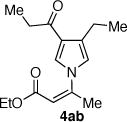
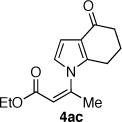
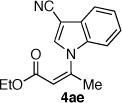
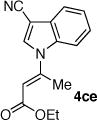
The coupling of Z-vinyl triflate 5a with the less nucleophilic acyl pyrrole 7b at 60 °C affords the desired product 4ab in 70% yield after 7 h (Table 1, entry 2). An enhancement in the rate of this coupling reaction was noted using dioxane as a co-solvent, affording N-vinyl pyrrole derivative 4ab in 91% isolated yield after 1 h at 60 °C (Table 1, entry 3). Alternatively, increasing the reaction temperature to 80 °C gave 4ab in 81% yield after complete consumption of starting material (Table 1, entry 4). The coupling of Z-vinyl triflate 5a with the more sterically hindered tetrahydroindolone 7c was found to be more difficult as compared to the acyl pyrrole 7b (Table 1, compare entries 2 and 5). The use of toluene–dioxane solvent mixture and higher reaction temperatures slightly increased the yield of this coupling reaction to afford 4ac in 50% isolated yield (Table 1, entry 7). The coupling of the cyanoindole 7e, the least reactive azole examined in this study, with Z-vinyl triflate 5a gave only 24% yield of the N-vinyl azaheterocycle 4ae after 48h (Table 1, entry 8). Both the use of toluene–dioxane solvent mixture and higher reaction temperatures were found to increase the formation of the ynoate 9a (eq 2) without a net increase in the yield of the coupled product 4ae (Table 1, entries 9 and 10).15,19
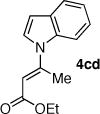
To highlight the stereospecificity of this transformation, we examined the coupling between E-vinyl triflate 5c and various azaheterocycles (Table 1, entries 11–17). In every case, the palladium catalyzed stereospecific C–N bond formation proceeded to afford the desired E-enoate. Acyl pyrrole 7b, tetrahydroindolone (7c), indole (7d), and 3-cyanoindole (7e) were successfully coupled with E-vinyl triflate 5c to give the corresponding N-vinyl azaheterocycle in 60, 73, 74, and 74% yield, respectively (Table 1, entries 12, 14, 15, and 16). Interestingly, the E-vinyl triflate 5c demonstrated an enhanced rate of coupling relative to the Z-vinyl triflate 5a in all cases examined. Additionally, the coupling reactions employing E-vinyl triflate 5c exhibited a much slower rate of ynoate formation as compared to those using Z-vinyl triflate 5a. Where possible, the use of 1,4-dioxane as a co-solvent and higher reaction temperatures for a shorter reaction time afforded more efficient palladium catalyzed N-vinylation.20 An initial short incubation of the reaction components at 60 °C followed by warming to higher temperatures provided the best results.21
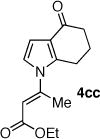
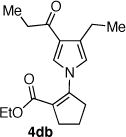
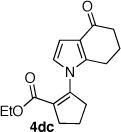
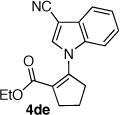
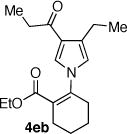
The palladium catalyzed N-vinylation reactions with cyclic vinyl triflates 5d and 5e were found to be slower than the vinyl triflates 5a and 5c. However, the greater thermal stability and the absence of the competing base-induced triflate elimination allowed the use of higher reaction temperatures with these substrates. The coupling of azaheterocycles 7b, 7c and 7e with triflate 5d gave the corresponding N-vinyl products in 70, 62 and 17% yield, respectively (Table 1, entries 18, 20, and 22). In the latter case a significant quantity (~50%) of ethyl 2-oxo-cyclopentanecarboxylate was isolated, suggesting a competitive sulfonyl transfer reaction.19 The coupling of the vinyl triflate 5e with the acyl pyrrole 7b proceeded efficiently at 110 °C to give the N-vinyl pyrrole 4eb in 91% yield (Table 1, entry 24). Neither of the less reactive azaheterocycles 7c nor 7e gave appreciable amounts of the corresponding coupling products with vinyl triflate 5e.
The coupling of vinyl triflate 5f with acyl pyrrole 7b afforded the desired N-vinyl azole 4fb in 72% yield (Table 1, entry 26), demonstrating the possible extension of this chemistry to unactivated vinyl triflates. However, the longer reaction time and the higher reaction temperature needed for the synthesis of product 4fb should be noted. The reduction of the steric bulk of the vinyl triflate and the presence of the electron withdrawing ester, each have a beneficial effect on the rate of the coupling reaction. The relative rate of coupling for the vinyl triflates discussed here was found to be: 5c > 5a > 5d > 5e > 5f (Chart 1). Similarly, the azaheterocycles used in this study may be ranked from most reactive to the least reactive: 7a > 7b > 7d > 7c > 7e (Chart 1). While the deprotonation of the heterocycle is facilitated by the presence of an electron-withdrawing substituent, these less nucleophilic heterocycles exhibit a slower overall rate of coupling. Hence, undesired reactions including triflate elimination and sulfonyl transfer reactions may compete when very non-nucleophilic azaheterocycles are used as substrates.22
In conclusion, a catalytic method for the stereospecific N-vinylation of azaheterocycles using vinyl triflates was described. Both cyclic and acyclic vinyl triflates were found to be substrates for this palladium catalyzed synthesis of N-vinyl pyrrole and indole derivatives. Successful use of non-nucleophilic azaheterocycles in this coupling reaction is noteworthy. The ready availability of both E- and Z-vinyl triflates23 in conjunction with the herein described chemistry offers an attractive method for the synthesis of N-vinyl azaheterocycles.
Experimental Section
Representative experimental procedure: Z-3-Pyrrol-1-yl-but-2-enoic acid ethyl ester (4aa, Table 1, entry 1)
Toluene (1.90 mL) was added to an argon–purged sample of Pd2(dba)3 (17.5 mg, 19.1 μmol, 0.05 equiv), XPhos (18.2 mg, 38.1 μmol, 0.10 equiv), and rigorously anhydrous K3PO4 (113 mg, 534 μmol, 1.40 equiv) in a flame-dried flask. Pyrrole (7a, 40.0 μL, 572 μmol, 1.50 equiv) was then added and the deep red mixture was heated to 60 °C. After 30 min, triflate 5a24 (100 mg, 381 μmol, 1 equiv) was added via syringe, producing a color change to forest green within approximately 10 min then to brown within an additional 1h. After 3.5 h TLC analysis indicated that the reaction was complete, whereupon the mixture was allowed to cool to 23 °C, was diluted with EtOAc (10 mL), and was vacuum filtered through a plug of celite (diam. 2.5 cm, ht. 2.5 cm). The celite-plug and flask were rinsed with an additional 15-mL portion of EtOAc, and the combined organic filtrates were washed with water (7.5 mL) and brine (7.5 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure to give a deep brown residue. Purification of the crude material by flash column chromatography (silica gel: diam. 3.0 cm, ht. 22 cm; 60% EtOAc–hexanes) afforded the vinyl pyrrole 4aa (57.1 mg, 84%) as a yellow oil. 1H NMR (500 MHz, CDCl3, 20°C): 6.90 (app t, J = 2.2 Hz, 2H), 6.23 (app-t, J = 2.2 Hz, 2H), 5.52 (q, J = 1.2 Hz, 1H), 4.10 (q, J = 7.1 Hz, 2H), 2.26 (d, J = 1.3 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (125.7 MHz, CDCl3, 20°C): 165.2, 147.7, 121.4, 110.1, 107.6, 60.5, 24.5, 14.4. FTIR (neat): 2982 (w, C–H), 1718 (s, C=O), 1641 (s, C=C), 1481, 1182, 1051. HRMS–ESI (m/z): calcd for C10H13NO2Na [M + Na]+: 202.0838, found: 202.0839.
Supplementary Material
Acknowledgments
M.M. is a Dale F. and Betty Ann Frey Damon Runyon Scholar supported by the Damon Runyon Cancer Research Foundation (DRS-39-04). M.M. is a Firmenich Assistant Professor of Chemistry. A.E.O. acknowledges a Robert T. Haslam Presidential Graduate Fellowship. We thank Mr. Matthew P. Rainka and Professor Stephen L. Buchwald for helpful discussions. We thank Mr. Robert W. Sindelar for early contributions to this study. We acknowledge generous financial support by the donors of the American Chemical Society Petroleum Research Fund, MIT and Amgen Inc.
Footnotes
Supporting Information Available: Complete experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805–818. [Google Scholar]; (b) Hartwig JF. Acc Chem Res. 1998;31:852–860. [Google Scholar]; (c) Hartwig JF. Angew Chem, Int Ed. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (d) Yang BH, Buchwald SL. J Organomet Chem. 1999;576:125–146. [Google Scholar]; (e) Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131–209. [Google Scholar]; (f) Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Wiley-Interscience; New York: 2002. p. 1051. [Google Scholar]
- 2.(a) Wolfe JP, Wagaw S, Buchwald SL. J Am Chem Soc. 1996;118:7215–7216. [Google Scholar]; (b) Driver MS, Hartwig JF. J Am Chem Soc. 1996;118:7217–7218. [Google Scholar]; For examples of palladium catalyzed N-arylation of azoles, see:; (c) Mann G, Hartwig JF, Driver MS, Fernández-Rivas C. J Am Chem Soc. 1998;120:827–828. [Google Scholar]; (d) Old DW, Harris MC, Buchwald SL. Org Lett. 2000;2:1403–1406. doi: 10.1021/ol005728z. [DOI] [PubMed] [Google Scholar]; (e) Watanabe M, Nishiyama M, Yamamoto T, Koie Y. Tetrahedron Lett. 2000;41:481–483. [Google Scholar]
- 3.For examples of copper-catalyzed N-arylation, see:; (a) Kiyomori A, Marcoux JF, Buchwald SL. Tetrahedron Lett. 1999;40:2657–2660. [Google Scholar]; (b) Collman JP, Zhong M. Org Lett. 2000;2:1233–1236. doi: 10.1021/ol000033j. [DOI] [PubMed] [Google Scholar]; (c) Gujadhur RK, Bates CG, Venkataraman D. Org Lett. 2001;3:4315–4317. doi: 10.1021/ol0170105. [DOI] [PubMed] [Google Scholar]; (d) Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727–7729. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]; (e) Klapars A, Huang X, Buchwald SL. J Am Chem Soc. 2002;124:7421–7428. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]; (f) Kwong FY, Buchwald SL. Org Lett. 2003;5:793–796. doi: 10.1021/ol0273396. [DOI] [PubMed] [Google Scholar]; (g) Ma D, Cai Q, Zhang H. Org Lett. 2003;5:2453–2455. doi: 10.1021/ol0346584. [DOI] [PubMed] [Google Scholar]; (h) Padwa A, Crawford KR, Rashatasakhon P, Rose M. J Org Chem. 2003;68:2609–2617. doi: 10.1021/jo026757l. [DOI] [PubMed] [Google Scholar]; (i) Antilla JC, Baskin JM, Barder TE, Buchwald SL. J Org Chem. 2004;69:5578–5587. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- 4.For intramolecular palladium catalyzed N-vinylation of amides, see:; (a) Palomo C, Aizpurua JM, Legido M, Picard JP, Dunogues J, Constantieux T. Tetrahedron Lett. 1992;33:3903–3906. [Google Scholar]; (b) Cuevas J-c, Patil P, Snieckus V. Tetrahedron Lett. 1989;30:5841–5844. [Google Scholar]; (c) Kozawa Y, Mori M. Tetrahedron Lett. 2002;43:111–114. [Google Scholar]; For recent reports on palladium catalyzed N-vinylation, see:; (d) Wallace DJ, Klauber DJ, Chen C-y, Volante RP. Org Lett. 2003;5:4749–4752. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]; (e) Klapars A, Campos KR, Chen C-y, Volante RP. Org Lett. 2005;7:1185–1188. doi: 10.1021/ol050117y. [DOI] [PubMed] [Google Scholar]
- 5.For a recent review on the use of copper in cross-coupling reaction, see:; (a) Beletskaya IP, Cheprakov AV. Coordin Chem Rev. 2004;248:2337–2364. [Google Scholar]; For amidation of vinyl halides with copper as the catalyst or promoter, see:; (b) Ogawa T, Kiji T, Hayami K, Suzuki H. Chem Lett. 1991:1443–1446. [Google Scholar]; (c) Shen R, Porco JA., Jr Org Lett. 2000;2:1333–1336. doi: 10.1021/ol005800t. [DOI] [PubMed] [Google Scholar]; (d) Shen R, Lin CT, Porco JA., Jr J Am Chem Soc. 2002;124:5650–5651. doi: 10.1021/ja026025a. [DOI] [PubMed] [Google Scholar]; (e) Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667–3669. doi: 10.1021/ol035355c. [DOI] [PubMed] [Google Scholar]; (f) Han C, Shen R, Su S, Porco JA., Jr Org Lett. 2004;6:27–30. doi: 10.1021/ol0360041. [DOI] [PubMed] [Google Scholar]; (g) Pan X, Cai Q, Ma D. Org Lett. 2004;6:1809–1812. doi: 10.1021/ol049464i. [DOI] [PubMed] [Google Scholar]; (h) Hu T, Li C. Org Lett. 2005;7:2035–2038. doi: 10.1021/ol0505555. [DOI] [PubMed] [Google Scholar]; For copper catalyzed and promoted N-alkynylation, respectively, see:; (i) Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J Am Chem Soc. 2003;125:2368–2369. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]; (j) Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011–4014. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a copper catalyzed C–N bond formation using allenyl halides, see:; (k) Trost BM, Stiles DT. Org Lett. 2005;7:2117–2120. doi: 10.1021/ol050395x. [DOI] [PubMed] [Google Scholar]
- 6.For palladium catalyzed N-vinylation of amines, see:; (a) Barluenga J, Fernández MA, Aznar F, Valdés C. Chem Commun. 2002:2362–2363. doi: 10.1039/b207524e. [DOI] [PubMed] [Google Scholar]; (b) Willis MC, Brace GN. Tetrahedron Lett. 2002;43:9085–9088. [Google Scholar]; For palladium-catalyzed N-vinylation of lithiated azoles, see:; (c) Lebedev AY, Izmer VV, Kazyul’kin DN, Beletskaya IP, Voskoboynikov AZ. Org Lett. 2002;4:623–626. doi: 10.1021/ol0172370. [DOI] [PubMed] [Google Scholar]
- 7.Rainka MP, Aye Y, Buchwald SL. Proc Nat Acad Sci, USA. 2004;101:5821–5823. doi: 10.1073/pnas.0307764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For the synthesis of Z-β-iodoenoates, see:; Piers E, Wong T, Coish PD, Rogers C. Can J Chem. 1994;72:1816–1819. [Google Scholar]
- 9.For synthesis of E-β-iodoenoates via an iododestannylation step, see:; (a) Thibonnet J, Launay V, Abarbri M, Duchêne A, Parrain J-L. Tetrahedron Lett. 1998;39:4277–4280. [Google Scholar]; (b) Maguire RJ, Munt SP, Thomas EJ. J Chem Soc, Perkin Trans. 1998;1:2853–2863. [Google Scholar]; (c) Dieter RK, Lu K. J Org Chem. 2002;67:847–855. doi: 10.1021/jo016053w. [DOI] [PubMed] [Google Scholar]; For isomerization of Z-β-iodoenoates, see:; (d) Ohba M, Kawase N, Fujii T. J Am Chem Soc. 1996;118:8250–8257. [Google Scholar]; (e) Dudley GB, Takaki KS, Cha DD, Danheiser RL. Org Lett. 2000;2:3407–3410. doi: 10.1021/ol006561c. [DOI] [PubMed] [Google Scholar]
- 10.For reviews on the preparation and use of vinyl triflates, see:; (a) Ritter K. Synthesis. 1993:735–762. [Google Scholar]; (b) Baraznenok IL, Nenajdenko VG, Balenkova ES. Tetrahedron. 2000;56:3077–3119. [Google Scholar]; (c) Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299–6302. [Google Scholar]
- 11.Use of XPhos gave superior results as compared to tri-(o-tolyl)phosphine, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), and 2-dicyclohexylphosphino-2′,6′-dimethoxy-1,1′-biphenyl (SPhos).
- 12.Tomori H, Fox JM, Buchwald SL. J Org Chem. 2000;65:5334–5341. doi: 10.1021/jo000691h. [DOI] [PubMed] [Google Scholar]; (b) XPhos is commercially available.
- 13.In the absence of palladium catalyst, none of the coupling products were observed.
- 14.Best results were obtained by further drying commercially available anhydrous potassium phosphate (160 °C, 0.1 Torr, 12 h) and its handling under inert atmosphere.
- 15.For a discussion on the competitive elimination reaction to give ynoates in studies concerning the palladium catalyzed C–O and C–S bond formation, see:; Rossi R, Bellina F, Mannina L. Tetrahedron. 1997;53:1025–1044. [Google Scholar]
- 16.For the synthesis of N-(tributylstannyl)-pyrrole, see:; Williamson BL, Stang PJ. Synlett. 1992:199–200. [Google Scholar]
- 17.For use of dialkylaminotributystannanes in N-arylation reactions, see:; Kosugi M, Mameyama M, Migita T. Chem Lett. 1983:927–928. [Google Scholar]; Paul F, Patt J, Hartwig JF. J Am Chem Soc. 1994;116:5969–5970. [Google Scholar]; Guram AS, Buchwald SL. J Am Chem Soc. 1994;116:7901–7902. [Google Scholar]
- 18.See Supporting Information for details.
- 19.for a discussion on the competitive loss of triflates via nucleophilic attack at the sulfur center during N-arylation of amines with electron deficient aryl triflates, see:; (a) Wolfe JP, Buchwald SL. J Org Chem. 1997;62:1264–1267. [Google Scholar]; (b) Louie J, Driver MS, Hamann BC, Hartwig JF. J Org Chem. 1997;62:1268–1273. [Google Scholar]
- 20.For a discussion on the rate of reductive elimination and C–N bond formation in N-arylation of azaheterocycles, see reference 2c.
- 21.The immediate heating of the starting reaction components to temperatures above 60 °C is not recommended. In the case of triflate 5a, omission of this incubation time resulted in a noticeable decrease in the efficiency of the coupling reaction.
- 22.Consistent with this observation, no coupling product was observed using 2-formylpyrrole in conjunction with vinyl triflates 5a, 5c and 5e. Formation of unreactive (azaheterocyclic)n–palladium complexes due to higher concentration of the aza-anion may also be in part responsible for the observed lower reactivity of acidic azaheterocycles; see reference 2c.
- 23.The ease of stereospecific synthesis of fully substituted vinyl triflates as compared to the corresponding vinyl iodides is noteworthy; see references 8–10.
- 24.For the general procedure used to prepare the vinyl triflate 5a, see:; Kim HO, Ogbu CO, Nelson S, Kahn M. Synlett. 1998:1059–1060. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.