

Abstract
Overexpression of the zinc enzyme carbonic anhydrase (CA; EC 4.2.1.1) XII is observed in certain human cancers. This bitopic membrane protein contains an N-terminal extracellular catalytic domain, a membrane-spanning α-helix, and a small intracellular C-terminal domain. We have determined the three-dimensional structure of the extracellular catalytic domain of human CA XII by x-ray crystallographic methods at 1.55-Å resolution. The structure reveals a prototypical CA fold; however, two CA XII domains associate to form an isologous dimer, an observation that is confirmed by studies of the enzyme in solution. The identification of signature GXXXG and GXXXS motifs in the transmembrane sequence that facilitate helix–helix association is additionally consistent with dimeric architecture. The dimer interface is situated so that the active site clefts of each monomer are clearly exposed on one face of the dimer, and the C termini are located together on the opposite face of the dimer to facilitate membrane interaction. The amino acid composition of the active-site cleft closely resembles that of the other CA isozymes in the immediate vicinity of the catalytic zinc ion, but differs in the region of the nearby α-helical “130's segment.” The structure of the CA XII–acetazolamide complex is also reported at 1.50-Å resolution, and prospects for the design of CA XII-specific inhibitors of possible chemotherapeutic value are discussed.
The carbonic anhydrases (CAs;
EC 4.2.1.1) constitute a ubiquitous class of enzymes that has evolved
to catalyze the reversible hydration of carbon dioxide to form
bicarbonate ion and a proton, CO2 +
H2O ⇌ HCO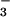 +
H+ (1). In the mechanism of catalysis,
nucleophilic attack of CO2 by a zinc-bound
hydroxide ion is followed by displacement of the resulting zinc-bound
bicarbonate ion by water; subsequent deprotonation regenerates the
nucleophilic zinc-bound hydroxide ion (2–6). To date, 11 enzymatically
active mammalian CAs have been identified, including cytosolic CAs I,
II, III, and VII, mitochondrial CAs VA and VB, secretory CA VI, and
membrane-associated CAs IV, IX, XII, and XIV (1, 7). These isozymes
play roles in physiological processes as diverse as pH regulation;
HCO
+
H+ (1). In the mechanism of catalysis,
nucleophilic attack of CO2 by a zinc-bound
hydroxide ion is followed by displacement of the resulting zinc-bound
bicarbonate ion by water; subsequent deprotonation regenerates the
nucleophilic zinc-bound hydroxide ion (2–6). To date, 11 enzymatically
active mammalian CAs have been identified, including cytosolic CAs I,
II, III, and VII, mitochondrial CAs VA and VB, secretory CA VI, and
membrane-associated CAs IV, IX, XII, and XIV (1, 7). These isozymes
play roles in physiological processes as diverse as pH regulation;
HCO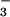 , CO2, and ion transport;
bone resorption; and secretion of gastric and pancreatic juices,
aqueous humor, and cerebrospinal fluid (1, 7). Enzymatically inactive
CA-related proteins are presumed to be structural homologues based on
amino acid sequence identity, but lack an intact zinc binding site
(8–11); these proteins serve as receptors for ligands involved in
signal transduction (11).
, CO2, and ion transport;
bone resorption; and secretion of gastric and pancreatic juices,
aqueous humor, and cerebrospinal fluid (1, 7). Enzymatically inactive
CA-related proteins are presumed to be structural homologues based on
amino acid sequence identity, but lack an intact zinc binding site
(8–11); these proteins serve as receptors for ligands involved in
signal transduction (11).
Recently, CAs IX and XII have been discovered in association with certain human cancers (12–15). CA IX, initially termed MN protein, is overexpressed in tumors of the ovary, endometrium, and uterine cervix, where it is otherwise not normally found (14). CA XII was originally identified by the overexpression of its mRNA in human renal cell cancer (15) and in a lung cancer cell lines (12). Additionally, transcription of both CA IX and CA XII mRNAs is regulated by the von Hippel–Lindau (VHL) tumor suppressor (16). Accordingly, loss of expression of the VHL tumor suppressor in renal cell carcinoma (17) likely explains the up-regulation of CA IX and CA XII in cancer tumor cells (18).
Both CA IX and CA XII are bitopic (single-pass) transmembrane proteins that contain an N-terminal extracellular CA domain, a putative transmembrane α-helix, and a small, ≈30-residue, intracellular C-terminal domain containing potential phosphorylation sites (15, 19, 20). In CA IV, another well-characterized membrane-associated isozyme, the C-terminal polypeptide segment is cleaved and replaced by a glycosylphosphatidylinositol membrane anchor during posttranslational modification (21). Ivanov and colleagues (16) hypothesize that CA IX and CA XII are responsible for acidification of the extracellular milieu surrounding cancer cells, resulting in an environment conducive to tumor growth. Based on the observation that human melanoma cells cultured at acidic pH are more aggressive, Martínez-Zaguilán and colleagues (22) suggest that extracellular acidification triggers an intracellular pH change that activates gelatinase B (matrix metalloproteinase 9), which in turn enhances invasiveness by proteolytic cleavage and remodeling of the extracellular matrix. Consistent with this proposal, treatment of four renal carcinoma cell lines (Caki-1, Caki-2, ACHN, and A-498) with the potent CA-inhibitor acetazolamide results in a substantial dose-dependent attenuation of invasiveness (23). Importantly, the isozymes most prevalent in these cell lines are CA II and CA XII.
Here we report the x-ray crystal structure of the extracellular catalytic domain of CA XII at 1.55-Å resolution, and the structure of its acetazolamide complex at 1.50-Å resolution. The structure of CA XII is compared with the cytosolic isozyme CA II and the membrane-anchored isozyme CA IV; for reference, the catalytic parameters of human CAs II, IV, and XII are summarized in Table 1. The molecular basis for dimer formation in the membrane, catalysis at the membrane surface, and the potential for CA XII isozyme-specific inhibitors are discussed in view of the CA XII crystal structure.
Table 1.
Catalytic constants for CO2 hydration by wild-type human CA isozymes II, IV, and XII
Materials and Methods
Construction of a Bacterial Expression Vector Containing hCA XII cDNA.
The BamHI–NcoI DNA fragment was isolated from the Q291X secretory form of CA XII in pBluescript II KS+ (Stratagene) (15, 18), encoding residues 30–291 from the full-length native sequence. The DNA fragment was cloned into the bacterial expression vector pET-11d (Novagen) by using BamHI and NcoI cloning sites to construct CA XII Q291X pET-11d, and Escherichia coli host cells BL21(DE3) or AD494(DE3) were transformed with the vector.
Expression and Purification of Recombinant Human CA XII.
E. coli AD494(DE3) containing CA XII Q291X pET-11d were grown at 37°C in LB medium containing 100 μg/ml ampicillin to OD600 ≈1.0. The expression of CA XII was induced by adding isopropyl β-D-thiogalactoside (IPTG) and ZnSO4 to 1 mM and 0.6 mM, respectively. The culture was grown for another 3 h and the cells were harvested by centrifugation at 6000 × g for 15 min at 4°C. The bacterial cell pellet was suspended in lysis buffer (20 mM Tris-sulfate, pH 7.5/0.1% Triton X-100/1 mM PMSF/1 mM o-phenanthroline) containing 100 μg/ml lysozyme. The cell homogenate was incubated at room temperature for 15 min and homogenized with a Polytron (Brinkmann) twice for 30 sec each at 4°C. The supernatant, containing soluble proteins, was obtained after centrifugation at 30,000 × g for 30 min. The CA activity in the supernatant and in insoluble membranes was determined.
The clear supernatant was applied to a CA-affinity column (21) that was equilibrated with 10 mM Hepes (pH 7.5). The unbound proteins were removed by washing the column with equilibration buffer followed with 10 mM Hepes (pH 7.5) containing 150 mM NaCl. The human CA XII bound to the affinity column was eluted with 0.1 M sodium acetate (pH 5.5) and 0.5 M sodium perchlorate buffer. The fractions containing CA activity were pooled and concentrated by using a YM-10 membrane (Amicon). The concentrated enzyme preparation was dialyzed against 10 mM Tris sulfate (pH 7.5). The homogeneity of the enzyme was assessed by enzyme activity measurements (24), size exclusion chromatography using Sephacryl S-300, and SDS/PAGE (25).
Crystallization and Structure Determination.
CA XII was crystallized at 4°C by the hanging drop vapor diffusion method. Drops containing 2 μl of 7 mg/ml CA XII in 10 mM Tris sulfate (pH 7.5) were mixed with 2 μl of precipitant buffer [3–5% (wt/vol) polyethylene glycol (PEG) 8000/0.1 M sodium acetate, pH 4.8] and equilibrated over a reservoir of 1 ml of precipitant buffer. Crystals appeared within 1 week and grew to maximum dimensions of 0.3 mm × 0.2 mm × 0.04 mm within 2–3 weeks. Crystals were harvested into a stabilization buffer [10% (wt/vol) PEG 8000/0.1 M sodium acetate, pH 4.8] and then transferred into a cryoprotectant buffer [10% (wt/vol) PEG 8000/20% (vol/vol) glycerol/0.1 M sodium acetate, pH 4.8). For the structure determination of the CA XII–acetazolamide complex, crystals were soaked in a cryoprotectant buffer containing 5 mM acetazolamide for ≈3 days at 4°C.
Diffraction data (1.55-Å resolution) from native CA XII crystals were recorded on a Mar 345 imaging plate detector at the Stanford Synchrotron Radiation Laboratories, beamline 7–1, by using x-radiation with λ = 1.08 Å at T = 97 K. Diffraction data (1.50-Å resolution) from crystals of the CA XII–acetazolamide complex were collected at beamline 19-ID at the Structural Biology Center of the Advanced Photon Source at Argonne National Laboratories (Argonne, IL). Data were recorded on an Electronic Computing Technologies 3 × 3 charge-coupled device detector from crystals frozen at 100 K by using x-radiation with λ = 1.0332 Å. Diffraction data from both crystals were processed with the HKL suite of programs (26). Crystals belonged to space group C2 with unit cell dimensions a = 146.7 Å, b = 44.6 Å, c = 85.2 Å, and β = 94.1°. One homodimer occupied the asymmetric unit.
Initial phases for the CA XII electron density map were determined by molecular replacement using the atomic coordinates of CA II (RCSB accession code 1G0E) (27) as a search model in which all residues except glycine were converted to alanine. Using AMoRe from the CCP4 suite of programs (28, 29), intensity data in the 15.0- to 3.5-Å resolution range were used in a cross-rotation search that yielded two peaks with correlation coefficients of 11.4. Subsequent translation searches using these two highest peaks produced two solutions with correlation coefficients of 7.8. Rigid-body refinement with these solutions yielded an R factor of 0.485. Iterative rounds of model building with O (30) and simulated annealing (5,000 K) and B value refinement by using Crystallography & NMR System (31) converged to a final R factor of 0.194 (Rfree = 0.221).
The final model of the native CA XII dimer contains two zinc ions, two zinc-bound acetate ions, and 537 water molecules in addition to the two polypeptide chains, each of which is complete except for 2 amino acids at the N terminus and 1 amino acid at the C terminus. A total of 88% and 12% of nonproline and nonglycine backbone φ–ψ conformations adopt most favored and additionally allowed conformations, respectively (data not shown). The structure of the CA XII–acetazolamide complex was solved by the difference Fourier technique using the native CA XII dimer structure minus water molecules and zinc-bound acetate ions as a starting model for rigid-body refinement in Crystallography & NMR System. Refinement converged smoothly to a final R factor of 0.190 (Rfree = 0.207). All data collection and refinement statistics are recorded in Table 2. Figs. 3–5 were prepared with MOLSCRIPT or BOBSCRIPT and RASTER3D (32–34).
Table 2.
X-ray data collection and refinement statistics
| CA XII | CA XII–acetazolamide complex | |
|---|---|---|
| Total no. of reflections measured | 402,312 | 636,087 |
| No. of unique reflections | 80,168 | 89,743 |
| Resolution range, Å | 30–1.55 | 50–1.50 |
| Rsym* (last shell), % | 4.6 (28.7) | 5.3 (31.8) |
| Completeness (last shell), % | 96.5 (79.3) | 99.3 (95.4) |
| No. of reflections used for refinement | 74,133 | 84,952 |
| R factor† | 0.194 | 0.190 |
| Rfree‡ | 0.221 | 0.207 |
| No. of nonhydrogen atoms§ | 4178 | 4169 |
| No. of water molecules | 537 | 504 |
| rms deviations from ideality | ||
| Bond lengths, Å | 0.005 | 0.005 |
| Bond angles, ° | 1.3 | 1.4 |
| Dihedral angles, ° | 24.6 | 24.4 |
| Improper dihedral angles, ° | 0.76 | 0.75 |
Rsym = ∑ |I − 〈I〉|/∑ I, where I is the observed intensity and 〈I〉 is the average intensity over all observations of symmetry-related reflections.
R factor = ∑ ∥Fobs| − |Fcalc∥/∑ |Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factor amplitudes, respectively.
Rfree was calculated from a randomly chosen subset of 8% of the reflections excluded from refinement.
For two molecules in the asymmetric unit.
Figure 3.

Schematic drawing showing the CA XII dimer in the membrane; orientation is the same as that in Fig. 2. The extracellular CA domains (molecules A and B) are colored blue and green, respectively. Zinc ions appear as white spheres, disulfide linkages are yellow, and acetazolamide molecules appear as balls-and-sticks. The yellow transmembrane helices are modeled after the structure of the glycophorin A dimer reported by MacKenzie and colleagues (42): both CA XII and glycophorin A contain transmembrane GXXXG dimerization motifs (40, 41). The intracellular C-terminal domains appear as orange spheres. These domains contain potential phosphorylation sites but are currently of unknown structure. Note that membrane association orients the enzyme active sites toward the extracellular milieu, poised for catalysis.
Figure 5.

Electron density map of the CA XII-acetazolamide complex generated with Fourier coefficients |Fo| − |Fc| and phases calculated from the final model less the atoms of acetazolamide (3σ contour). Atoms are colored as in Fig. 4. The 130's segment is in the α-helix on the left.
Results and Discussion
Apparent Molecular Weight in Solution.
The affinity-purified, secretory form of CA XII produced in E. coli is a nonglycoslyated protein that migrates at 30–31 kDa on SDS/PAGE, as predicted from its deduced amino acid sequence (15). However, its elution profile on Sephacryl S-300 in 0.1 M NaCl/0.01 M Tris sulfate (pH 7.5) indicates a molecular mass of 60 kDa, suggesting that the mature form of secretory CA XII in solution is dimeric.
Overall Structure.
The polypeptide fold of CA XII is topologically similar to the β-fold observed in other isozymes, as illustrated by the structure-based sequence alignment of mammalian CAs presented in Fig. 1. In comparison with CA II, the best-studied isozyme, CA XII contains larger loops in the regions of Ala-54A–Asn-54B, Pro-102A, and Arg-240A–Glu-240B.§ One of the two glycosylation sites in CA XII is located at Asn-52, which is on the first of these larger loops. The second glycosylation site, Asn-136, is found in a loop region that superimposes well with the other isozyme structures. A single disulfide linkage is present in CA XII between Cys-23 and Cys-203. This same disulfide linkage is also found in CA IV, where it helps to stabilize the Pro-201–Thr-202 cis-peptide linkage and anchors the loop containing Thr-199 (35). Thr-199 hydrogen bonds to zinc-bound hydroxide and orients the nucleophile for catalysis (36).
Figure 1.

Sequence alignment of human CAs I (RCSB accession code 1CAB; ref. 61), II (1CA2; ref. 47), IV (1ZNC; ref. 35), and XII, mouse CAs IV (2ZNC; ref. 54) and V (1DMY; ref. 58), and rat CA III (1FLJ; ref. 62) based on the alignment of secondary structural elements as observed in the individual crystal structures. Blue and green shading denote β-strands and α-helices, respectively. Residues conserved throughout all of these sequences are colored red and residues at the CA XII dimer interface are underlined. Triangles (▴) mark histidine–zinc ligands and asterisks (*) denote asparagine glycosylation sites. The alignment is numbered according to the sequence of human CA II. The entire sequence of CA XII is shown, and the GXXXG and GXXXS motifs that signal transmembrane helix dimerization are underlined in red; potential phosphorylation sites are double underlined. For all other isozymes, the N and C termini are truncated according to what is visible in each crystal structure.
The most striking difference between CA XII and all other soluble or membrane-associated isozymes is its isolation as a protein with higher order quaternary structure: the soluble CA XII domain is an isologous dimer in solution and in the crystal. Two CA XII domains interact to bury ≈2,200 Å2 of total surface area (Fig. 2). The extent of buried surface area is consistent with surface area/molecular mass values tabulated for biological oligomers (37, 38), and the ≈1,100-Å2 surface area buried per monomer exceeds the cutoff value of ≈860 Å2 per monomer used to discriminate between monomers and homodimers in protein crystals (39). The dimer interface includes both highly conserved residues and residues unique to CA XII among the isozymes of known structure (Fig. 1). However, 19 hydrogen-bond interactions stabilize the dimer interface (Table 3), and only two of these interactions involve a highly conserved residue (Gln-249).
Figure 2.

Electrostatic surface potential of the CA XII dimer. The dimer twofold axis lies vertically in the center of the figure, perpendicular to the membrane surface. Arrows indicate the active site clefts, which are hidden in this view. Surface calculated and color-coded from −15 kT (red) to + 15 kT (blue) with GRASP (63).
Table 3.
Hydrogen bond interactions at the CA XII dimer interface
| Monomer A atom | Monomer B atom | Distance, Å |
|---|---|---|
| Glu-13 Oɛ2 | Lys-250 Nζ | 2.8 |
| Asn-14 O | Ser-17 Oγ | 2.6 |
| Asn-14 Nδ2 | Gln-249 Oɛ1 | 2.8 |
| Asn-14 Nδ2 | Cys-23 O | 3.0 |
| His-34 Nɛ2 | Asp-102 Oδ1 | 2.8 |
| Asp-36 Oδ2 | His-103 Nɛ2 | 2.9 |
| Gln-112 Nɛ2 | Ser-110 O | 2.9 |
| Gln-112 Nɛ2 | Gln-112 Nɛ2 | 2.6 |
| Phe-245 O | Lys-250 Nζ | 2.8 |
| Gln-247 Oɛ1 | Val-248 O | 3.0 |
The dimer interface exhibits noncrystallographic symmetry around a 2-fold axis. Therefore, for each interaction listed above, there is a corresponding symmetry-related interaction that is not listed, except for the self-complementary interaction between Gln-112.
The mode of dimer assembly is consistent with biological function, i.e., the active site clefts and C termini are clearly exposed on opposite faces of the dimer. With the C termini anchored in the cell membrane, the active sites are oriented toward the extracellular milieu of the cell and poised for catalysis (Fig. 3). The two glycosylation sites, Asn-52 and Asn-136, are also exposed on the surface. Interestingly, the glycosylated extracellular domain of CA XII produced in Chinese hamster ovary cells, which has been characterized kinetically (18), crystallizes isomorphously with the nonglycosylated enzyme produced in E. coli (unpublished results). This observation indicates that glycosylation affects neither dimerization nor crystal packing.
The transmembrane segment of CA XII contains signature GXXXG and GXXXS motifs that serve as the framework for the dimerization of transmembrane α-helices (40, 41). The NMR structures of the glycophorin A dimer illustrate the geometry of helix-helix association mediated by a GXXXG motif (42, 43). Accordingly, the occurrence of GXXXG and GXXXS motifs in the transmembrane segment of CA XII strongly suggests that isologous dimer architecture, similar to that of glycophorin A, persists within the membrane in the full-length protein (Fig. 3).
It is reasonable to expect that the ≈30-residue intracellular domain of CA XII also forms an isologous dimer if molecular symmetry is maintained for the entire bitopic membrane protein. Preliminary circular dichroism experiments indicate α-helical content for the isolated domain prepared by solid-phase peptide synthesis (unpublished results). Additionally, this domain contains potential phosphorylation sites characterized by appropriately located acidic or basic residues flanking Ser and Thr positions in the sequence (44). Potentially, intracellular phosphorylation could affect the tertiary and/or quaternary structure of this domain, which in turn could affect other functions. For example, the formation of CA XII complexes with other proteins found in renal carcinoma cell lines is dependent on the presence of an intact, full-length CA XII sequence (unpublished observations). These results may indicate a role for the intracellular domain in the regulation of oligomeric structure, extracellular enzyme activity, and/or signaling pathways. However, additional experiments are needed to probe these possible functions.
Active Site Structure and Catalysis.
The structure in the immediate vicinity of the active site of CA XII is nearly identical to that of CA II and CA IV. The zinc ion is located at the bottom of the conical active site cleft, where it is ligated with distorted trigonal bipyramidal coordination geometry by His-94, His-96, His-119, a water molecule, and an acetate anion. At high pH values, zinc-bound water ionizes to yield nucleophilic zinc-bound hydroxide ion; however, given that the pH of the crystal structure determination is 4.8 and the pKa of this zinc-bound water molecule is 7.1 (18), we conclude that the zinc-bound water species predominates (average O--Zn2+ distance, 2.1 Å). One oxygen atom of the acetate anion present in the crystallization buffer coordinates to zinc (average O–Zn2+ distance, 2.3 Å) and the second oxygen atom accepts hydrogen bonds from the zinc-bound water molecule and the backbone NH of Thr-199 (Fig. 4). This orientation of acetate mimics the position of bicarbonate in that both protrude into the hydrophobic substrate binding pocket, lined by residues Val-121, Leu-141, Val-143, Leu-198, Val-207, and Trp-209 (45, 46). Formate and acetate bind in an analogous fashion to CA II (47, 48).
Figure 4.
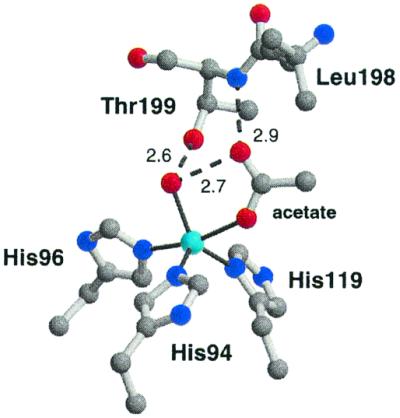
The active site of the native CA XII structure showing the five-coordinate zinc ion. The zinc ion is colored cyan. Oxygen, nitrogen, and carbon atoms are red, blue, and gray, respectively. Hydrogen bonds are shown as dashed lines with distances labeled (Å). Average zinc–ligand distances are as follows: Zn2+–His-94, 2.0 Å; Zn2+–His-96, 2.1 Å; Zn2+–His-119, 2.1 Å; Zn2+–OH2, 2.1 Å; Zn2+–acetate, 2.3 Å.
The catalytic proton shuttle, His-64, is conserved in CA XII and occupies the “out” conformation, as observed in other CA structures solved at low pH (35, 49). Mutagenesis of this residue in CA XII confirms that it plays a role in proton shuttling (18). Interestingly, experiments with His-64 → Ala CA II demonstrate that the proton transfer function is rescued by the addition of imidazole or methylimidazole (27, 50); 4-methylimidazole binds in a location similar to that of the His-64 “out” conformer (27). Although ordered solvent molecules are observed in the active site of CA XII, there is no complete hydrogen-bonded solvent network between zinc-bound solvent and His-64.
Variability in the CA XII active site contour is observed in the vicinity of Thr-91, Gln-92, and the “130's segment” (Ala-131–Ala-134). Previous structural investigations of human CA II implicate Phe-131 as one of the residues important in stabilizing inhibitor binding (51–53). Comparisons of the binding of brinzolamide to human CA II and murine CA IV (which has Val-131) demonstrate that variations in the 130's segment correlate with differences in binding affinities (54). In CA XII, Ala-131 is found, which results in an even wider canyon in the active site cleft of CA XII compared with CA II or CA IV. Consequently, the side chain of Ser-135 is exposed. The particular active-site contour of CA XII in the vicinity of the 130's segment is not observed in any other isozyme and may be exploited in the design of CA XII-specific inhibitors.
Inhibition by Acetazolamide.
Recently it has been demonstrated that 10 μM concentrations of the well-known CA inhibitor acetazolamide inhibit the invasion rate of renal carcinoma tumor cells by 18–74%; immunocytochemical and Western blot analysis confirmed the presence of CA II and CA XII in these cells (23). Acetazolamide binds to human CA II with Kd = 90 nM (55) and the structure of the CA II–acetazolamide complex was reported by Vidgren and colleagues (56). We determined the structure of the CA XII–acetazolamide complex to confirm inhibitor binding to CA XII and to compare CA XII and CA II binding modes. Each active site of the CA XII dimer shows clear electron density for acetazolamide (Fig. 5). There are no gross structural changes in CA XII on acetazolamide binding, in accord with the binding of acetazolamide to CAs I, II, and V (56–58). The native and inhibited forms of CA XII superimpose with rms deviations (all atoms) of 0.12 Å.
In the structure of each CA isozyme complexed with acetazolamide, an ionized sulfonamide NH group coordinates to the zinc ion, displacing zinc-bound solvent. This NH group also donates a hydrogen bond to the hydroxyl group of Thr-199. The hydroxyl group of Thr-200 makes additional hydrogen bond contacts with the two nitrogen atoms of the 1,3,4-thiadiazole ring. The orientation of His-200 in CA I is such that hydrogen bond contacts with the acetazolamide are not possible (57). Finally, the terminal acetamido moiety has the potential to make additional hydrogen bond contacts with the CA active site cleft, but does so in only one of two molecules in the CA XII dimer. In molecule A, there is a hydrogen bond between acetazolamide and Gln-90. An inhibitor molecule larger than acetazolamide could potentially interact within the wide canyon adjacent to the 130's segment, thereby providing CA XII-specific interactions. Future structure-based inhibitor design experiments will reveal whether CA XII-specific inhibitors can be designed with anti-tumorigenic properties.
Acknowledgments
We thank Drs. J. David Cox and Chu-Young Kim for assistance with data collection. This work was supported by National Institutes of Health Grants GM45614 (to D.W.C.) and DK40163 and GM34182 (to W.S.S.). This work is based in part on research conducted at the Stanford Synchrotron Radiation Laboratory (Menlo Park, CA), which is funded by the Department of Energy and the National Institutes of Health. Use of the Argonne National Laboratory Structural Biology Center beamline at the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Biological and Environmental Research.
Abbreviation
- CA
carbonic anhydrase
Footnotes
Data deposition: The atomic coordinates for the native and acetazolamide-inhibited CA XII have been deposited in the Protein Data Bank, www.rscb.org (PDB ID codes 1JCZ and 1JD0, respectively).
The CA XII numbering scheme used in this work is based on that of CA II; additional inserted residues are denoted with the suffix A, B, C, etc.
References
- 1.Chegwidden W R, Carter N D, Edwards Y H. The Carbonic Anhydrases. Boston: Birkhäuser; 2000. [Google Scholar]
- 2.Coleman J E. J Biol Chem. 1967;242:5212–5219. [PubMed] [Google Scholar]
- 3.Lindskog S, Coleman J E. Proc Natl Acad Sci USA. 1973;70:2505–2508. doi: 10.1073/pnas.70.9.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman D N, Lindskog S. Acc Chem Res. 1988;21:30–36. [Google Scholar]
- 5.Lindskog S, Liljas A. Curr Opin Struct Biol. 1993;3:915–920. [Google Scholar]
- 6.Christianson D W, Fierke C A. Acc Chem Res. 1996;29:331–339. [Google Scholar]
- 7.Sly W S, Hu P Y. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- 8.Krueger N X, Saito H. Proc Natl Acad Sci USA. 1992;89:7417–7421. doi: 10.1073/pnas.89.16.7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnea G, Silvennoinen O, Shaanan B, Honegger A M, Canoll P D, D'Eustachio P, Morse B, Levy J B, LaForgia S, Huebner K, et al. Mol Cell Biol. 1993;13:1497–1506. doi: 10.1128/mcb.13.3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy J B, Canoll P D, Silvennoinen O, Barnea G, Morse B, Honegger A M, Huang J-T, Cannizzaro L A, Park S-H, Druck T, et al. J Biol Chem. 1993;268:10573–10581. [PubMed] [Google Scholar]
- 11.Peles E, Nativ M, Campbell P L, Sakurai T, Martinez R, Lev S, Clary D O, Schilling J, Barnea G, Plowman G D, et al. Cell. 1995;82:251–260. doi: 10.1016/0092-8674(95)90312-7. [DOI] [PubMed] [Google Scholar]
- 12.Torczynski, R. M. & Bollon, A. P. (1996) U.S. Patent 5,589,579.
- 13.McKiernan J M, Buttyan R, Bander N H, Stifelman M D, Katz A E, Chen M-W, Olsson C A, Sawczuk I S. Cancer Res. 1997;57:2362–2365. [PubMed] [Google Scholar]
- 14.Zavadá J, Závadová Z, Pastoreková S, Ciampor F, Pastorek J, Zelník V. Int J Cancer. 1993;54:268–274. doi: 10.1002/ijc.2910540218. [DOI] [PubMed] [Google Scholar]
- 15.Türeci O, Sahin U, Vollmar E, Siemer S, Göttert E, Seitz G, Parkkila A-K, Shah G N, Grubb J H, Pfreundschuh M, et al. Proc Natl Acad Sci USA. 1998;95:7608–7613. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov S V, Kuzmin I, Wei M-H, Pack S, Geil L, Johnson B E, Stanbridge E J, Lerman M I. Proc Natl Acad Sci USA. 1998;95:12596–12601. doi: 10.1073/pnas.95.21.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Orcutt M L, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 18.Ulmasov B, Waheed A, Shah G N, Grubb J H, Sly W S, Tu C, Silverman D N. Proc Natl Acad Sci USA. 2000;97:14212–14217. doi: 10.1073/pnas.97.26.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pastorek J, Pastoreková S, Callebaut I, Mornon J P, Zelnik V, Opavsky R, Zat'ovicová M, Liao S, Portetelle D, Stanbridge E J, et al. Oncogene. 1994;9:2877–2888. [PubMed] [Google Scholar]
- 20.Opavský R, Pastoreková S, Zelník V, Gibadulinová A, Stanbridge E J, Závada J, Kettmann R, Pastorek J. Genomics. 1996;33:480–487. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 21.Zhu X L, Sly W S. J Biol Chem. 1990;265:8795–8801. [PubMed] [Google Scholar]
- 22.Martínez-Zaguilán R, Seftor E A, Seftor R E B, Chu Y-W, Gillies R J, Hendrix M J C. Clin Exp Metastasis. 1996;14:176–186. doi: 10.1007/BF00121214. [DOI] [PubMed] [Google Scholar]
- 23.Parkkila S, Rajaniemi H, Parkkila A-K, Kivelä J, Waheed A, Pastoreková S, Pastorek J, Sly W S. Proc Natl Acad Sci USA. 2000;97:2220–2224. doi: 10.1073/pnas.040554897. . (First Published February 25, 2000; 10.1073/pnas.040554897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundaram V, Rumbolo P, Grubb J H, Strisciuglio P, Sly W S. Am J Hum Genet. 1986;38:125–136. [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Duda D, Tu C, Qian M, Laipis P, Agbandje-McKenna M, Silverman D N, McKenna R. Biochemistry. 2001;40:1741–1748. doi: 10.1021/bi002295z. [DOI] [PubMed] [Google Scholar]
- 28.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 29.Collaborative Computational Project No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 30.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 32.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 33.Esnouf R M. J Mol Graphics. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 34.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 35.Stams T, Nair S K, Okuyama T, Waheed A, Sly W S, Christianson D W. Proc Natl Acad Sci USA. 1996;93:13589–13594. doi: 10.1073/pnas.93.24.13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merz K M., Jr J Mol Biol. 1990;214:799–802. doi: 10.1016/0022-2836(90)90333-H. [DOI] [PubMed] [Google Scholar]
- 37.Miller S, Lesk A M, Janin J, Chothia C. Nature (London) 1987;328:834–836. doi: 10.1038/328834a0. [DOI] [PubMed] [Google Scholar]
- 38.Jones S, Marin A, Thornton J M. Protein Eng. 2000;13:77–82. doi: 10.1093/protein/13.2.77. [DOI] [PubMed] [Google Scholar]
- 39.Ponstingl H, Henrick K, Thornton J M. Proteins Struct Funct Genet. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 40.Russ W P, Engelman D M. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 41.Senes A, Gerstein M, Engelman D M. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 42.MacKenzie K R, Prestegard J H, Engelman D M. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 43.Smith S O, Song D, Shekar S, Groesbeek M, Ziliox M, Aimoto S. Biochemistry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 44.Blom N, Gammeltoft S, Brunak S. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 45.Xue Y, Vidgren J, Svensson L A, Liljas A, Jonsson B-H, Lindskog S. Proteins Struct Funct Genet. 1993;15:80–87. doi: 10.1002/prot.340150110. [DOI] [PubMed] [Google Scholar]
- 46.Xue Y, Liljas A, Jonsson B-H, Lindskog S. Proteins Struct Funct Genet. 1993;17:93–106. doi: 10.1002/prot.340170112. [DOI] [PubMed] [Google Scholar]
- 47.Håkansson K, Carlsson M, Svensson L A, Liljas A. J Mol Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 48.Håkansson K, Briand C, Zaitsev V, Xue Y, Liljas A. Acta Crystallogr D. 1994;50:101–104. doi: 10.1107/S0907444993009667. [DOI] [PubMed] [Google Scholar]
- 49.Nair S K, Christianson D W. J Am Chem Soc. 1991;113:9455–9458. [Google Scholar]
- 50.Tu C, Silverman D N, Forsman C, Jonsson B-H, Lindskog S. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 51.Cappalonga Bunn A M, Alexander R S, Christianson D W. J Am Chem Soc. 1994;116:5063–5068. [Google Scholar]
- 52.Boriack-Sjodin P A, Zeitlin S, Chen H-H, Crenshaw L, Gross S, Dantanarayana A, Delgado P, May J A, Dean T, Christianson D W. Protein Sci. 1998;7:2483–2489. doi: 10.1002/pro.5560071201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim C-Y, Chang J S, Doyon J B, Baird T T, Jr, Fierke C A, Jain A, Christianson D W. J Am Chem Soc. 2000;122:12125–12134. [Google Scholar]
- 54.Stams T, Chen Y, Boriack-Sjodin P A, Hurt J D, Liao J, May J A, Dean T, Laipis P, Silverman D N, Christianson D W. Protein Sci. 1998;7:556–563. doi: 10.1002/pro.5560070303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nair S K, Krebs J F, Christianson D W, Fierke C A. Biochemistry. 1995;34:3981–3989. doi: 10.1021/bi00012a016. [DOI] [PubMed] [Google Scholar]
- 56.Vidgren J, Liljas A, Walker N P C. Intl J Biol Macromol. 1990;12:342–344. doi: 10.1016/0141-8130(90)90040-h. [DOI] [PubMed] [Google Scholar]
- 57.Chakravarty S, Yadava V S, Kumar V, Kannan K K. J Biosci. 1985;8:491–498. [Google Scholar]
- 58.Boriack-Sjodin P A, Heck R W, Laipis P J, Silverman D N, Christianson D W. Proc Natl Acad Sci USA. 1995;92:10949–10953. doi: 10.1073/pnas.92.24.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khalifah R G. J Biol Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 60.Baird T T, Jr, Waheed A, Okuyama T, Sly W S, Fierke C A. Biochemistry. 1997;36:2669–2678. doi: 10.1021/bi962663s. [DOI] [PubMed] [Google Scholar]
- 61.Kannan K K, Ramanadham M, Jones T A. Ann NY Acad Sci. 1984;429:49–60. doi: 10.1111/j.1749-6632.1984.tb12314.x. [DOI] [PubMed] [Google Scholar]
- 62.Mallis R J, Poland B W, Chatterjee T K, Fisher R A, Darmawan S, Honzatko R B, Thomas J A. FEBS Lett. 2000;482:237–241. doi: 10.1016/s0014-5793(00)02022-6. [DOI] [PubMed] [Google Scholar]
- 63.Nicholls A, Sharp K A, Honig B. Proteins Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]