

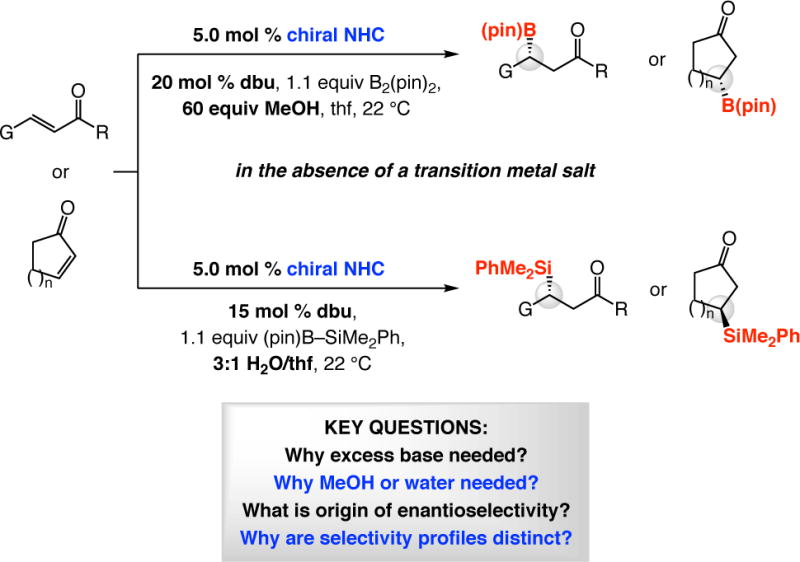
Abstract
Broadly applicable enantioselective C–B and C–Si bond forming processes catalyzed by an N-heterocyclic carbene (NHC) were recently introduced; these boryl and silyl conjugate addition reactions (BCA and SCA, respectively), which proceed without the need for a transition metal complex, represent reaction pathways that are distinct from those facilitated by transition metal-containing species (e.g., Cu-, Ni-, Pt-, Pd- or Rh-based). The Lewis base (NHC) catalyzed transformations are valuable to chemical synthesis, as they can generate high enantioselectivities and possess unique chemoselectivity profiles. Here, the results of investigations that elucidate the principal features of the NHC-catalyzed BCA and SCA processes are detailed. Spectroscopic evidence is provided illustrating why the presence of excess base and MeOH or H2O is required for efficient and enantioselective boryl and silyl addition reactions. It is demonstrated that the proton sources influence the efficiency and/or enantioselectivity of NHC-catalyzed enantioselective transformations in several ways. The positive, and at times adverse, impact of water (biphasic conditions) on catalytic enantioselective silyl addition reactions is analyzed. It is shown that a proton source can facilitate non-enantioselective background reactions and NHC decomposition, requiring the catalyst to surpass such complications. Stereochemical models are presented that account for the identity of the observed major enantiomers, providing a rationale for the differences in selectivity profiles of BCA and SCA processes.
Graphical abstract
Introduction
The landscape of organic chemistry would be considerably more limited were it not for the multifaceted attributes of organoboron compounds. Also important are Si-containing organic molecules, entities that have significantly impacted the art of chemical synthesis. Accordingly, development of reliable catalytic enantioselective processes that generate C–B1,2,3,4,5,6,7 or C–Si8,9 bonds remains a compelling goal of research in modern chemistry. One area of activity relates to boryl and silyl conjugate addition (BCA and SCA, respectively) reactions that are facilitated by different transition metal species,10 and in particular Cu-based chiral complexes.11,12,13 For example, cyclic β-hydroxyl carbonyl products as well as acyclic or cyclic variants that contain a tertiary alcohol may be prepared by catalytic BCA methods (after C–B bond oxidation); these entities, which cannot be accessed through catalytic enantioselective aldol addition protocols or the related approaches,14 facilitate synthesis of a variety of biologically active molecules.
It was reported in 2009 that achiral N-heterocyclic carbenes15 (NHCs) can catalyze the addition of (pinacolato)diboron to α,β-unsaturated carbonyl compounds (Scheme 1).16, 17 Enantioselective variants,18 including those promoted by chiral phosphines,19 and transformations involving a borosilyl reagent (to give a C–Si bond),20 were subsequently introduced. Phosphine-21 and NHC-catalyzed boryl additions to tosylimines and alkynes soon followed.22 Activation of B–B bonds by an external23 and then a neighboring Lewis basic alkoxide24 were later exploited to effect diboron additions to unactivated alkenes, and related transformations involving propargyl alcohols were outlined.25 Metal alkoxides have been shown to catalyze reactions of silylboron reagents with organohalide substrates, affording the corresponding silanes.26 In the significant majority of Lewis base catalyzed boryl and silyl additions, the presence of a proton source (typically MeOH) is necessary;16a in many instances, excess base is needed for optimal efficiency and/or stereoselectivity. Nonetheless, a rigorous mechanistic basis for the latter requirements is lacking.
Scheme 1.
The Catalytic Processes and the Key Mechanistic Questions
Boryl and silyl conjugate additions that are facilitated by transition metal-free Lewis base catalysts mark a new chapter in organoboron and organosilicon chemistries; these are mechanistically distinct reactions that offer complementary functional group compatibility profiles (vs those promoted by organometallic species). However, in addition to the aforementioned issues vis-à-vis the need for a proton source and excess base, a number of questions regarding the details of the catalytic cycles for C–B and C–Si bond formation (Scheme 1), including those that are promoted by NHCs, remain unaddressed.27 An appreciation of such attributes will be crucial to the success of future endeavors regarding catalyst and method development in this emerging area of research. Herein, we detail the results of studies performed aimed at finding a plausible answer for the above issues and various other questions relating to the inner workings of NHC-catalyzed boryl and silyl conjugate additions.
Background
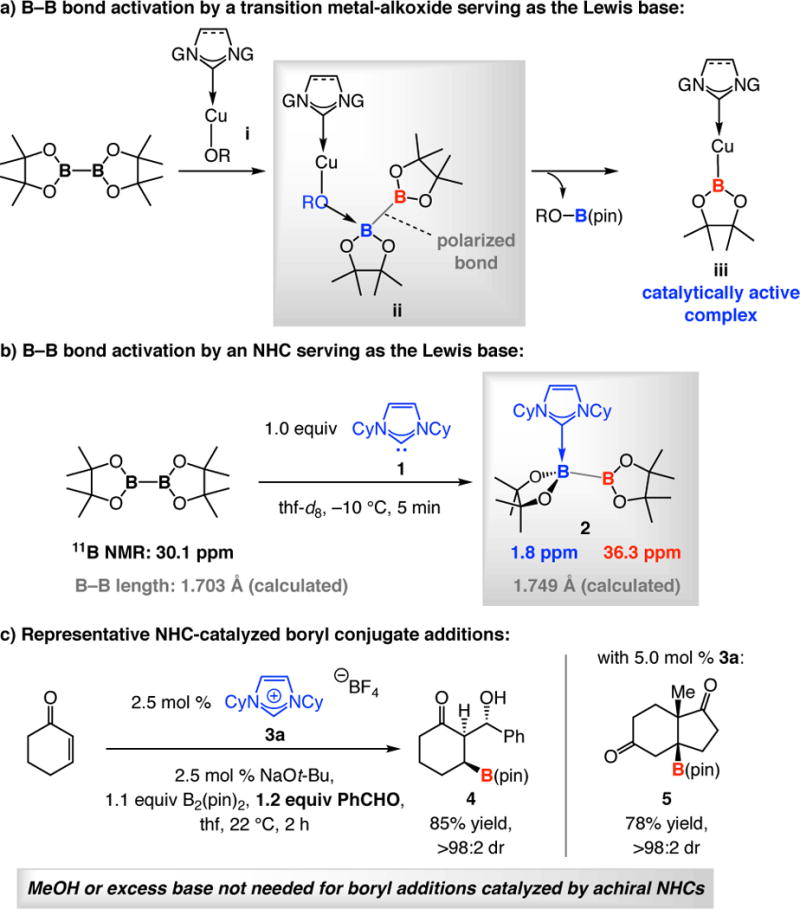
The viability of Lewis base catalyzed BCA is manifested in the manner with which initial activation of a B–B bond might occur by the oxygen atom of an NHC– or phosphine–Cu–OR complex (i→iii via ii, Scheme 2a).28 In addition to increasing Cu Lewis acidity, according to the well-established tenets of Lewis base enhancement of Lewis acidity;29 hence, the Lewis base (e.g., an NHC) elevates the nucleophilicity of the alkoxide ligand, facilitating the formation of ii, which is the principal player in a Cu–boryl addition. The question would then be whether efficient activation can be achieved directly by an organic Lewis basic molecule. This notion finds support in X-ray crystallographic investigations showing that adducts derived from association of bis(catecholato)diboron and bis(thiocatecholato)diboron with 4-methylpyridine and dimethylphenylphosphine, respectively, cause perceptible weakening of B–B bonds.30,31
Scheme 2.
Discovery of NHC-Catalyzed Boryl Conjugate Additiona
aG = alkyl or aryl group; B(pin) = (pinacolato)boron.
We initially established that NHC•B2(pin)2 complexes can be readily generated. Spectroscopic analysis pointed to the formation of a Lewis base bound B atom (cf. 2 derived from NHC 1, Scheme 2b), and calculations indicated B–B bond lengthening. The validity of the originally suggested NHC•diboron complex 2 was afterwards substantiated by the notable X-ray crystallographic studies of Marder and co-workers.32
While formation of a Lewis base•diboron complex does not guarantee a catalytic process, we soon discovered that NHCs do promote BCA reactions. The multicomponent transformation in Scheme 2c, affording 4 as a single stereoisomer, is illustrative;16a this reaction underscores the functional group compatibility of the NHC-catalyzed processes versus those promoted by Cu-based complexes (vs 31% yield with NHC–Cu species due to boryl addition to the aldehyde). We found that phosphines are catalytically less active,18a and alkoxides (e.g., NaOt-Bu) proved to be less capable promoters (e.g., compared to i).16a
Enantioselective boryl additions to α,β-unsaturated carbonyl compounds (acyclic and cyclic enones, esters, Weinreb amides, or enals) can be performed with chiral NHC catalysts and 1,8-diazabicyclo[5.4.0]undec-7-ene (dbu) as the base (Scheme 3a).18a,33 The high er values are mechanistically revealing: they clearly indicate that an NHC-bound chiral complex is responsible for the C–B bond forming process. Further, enantioselective addition to the more challenging trisubstituted enones is feasible; this is exemplified by the transformation promoted by the enantiomerically pure NHC derived from 7b (Scheme 3b).18b The BCA en route to natural product crassinervic acid proceeded readily despite the presence of a phenol and an aryl aldehyde, moieties that greatly diminish the activity of phosphine– or NHC–Cu complexes.34 NHC-Catalyzed enantioselective BCA reactions afford products that cannot be easily prepared otherwise in high enantioslectivity (e.g., acetate aldol product that contain a Weinreb amide33 and ketone aldol products such as 818b in Scheme 3b) and are practical and robust; contrary to reactions with Cu complexes, use of a dry box is not needed.
Scheme 3.
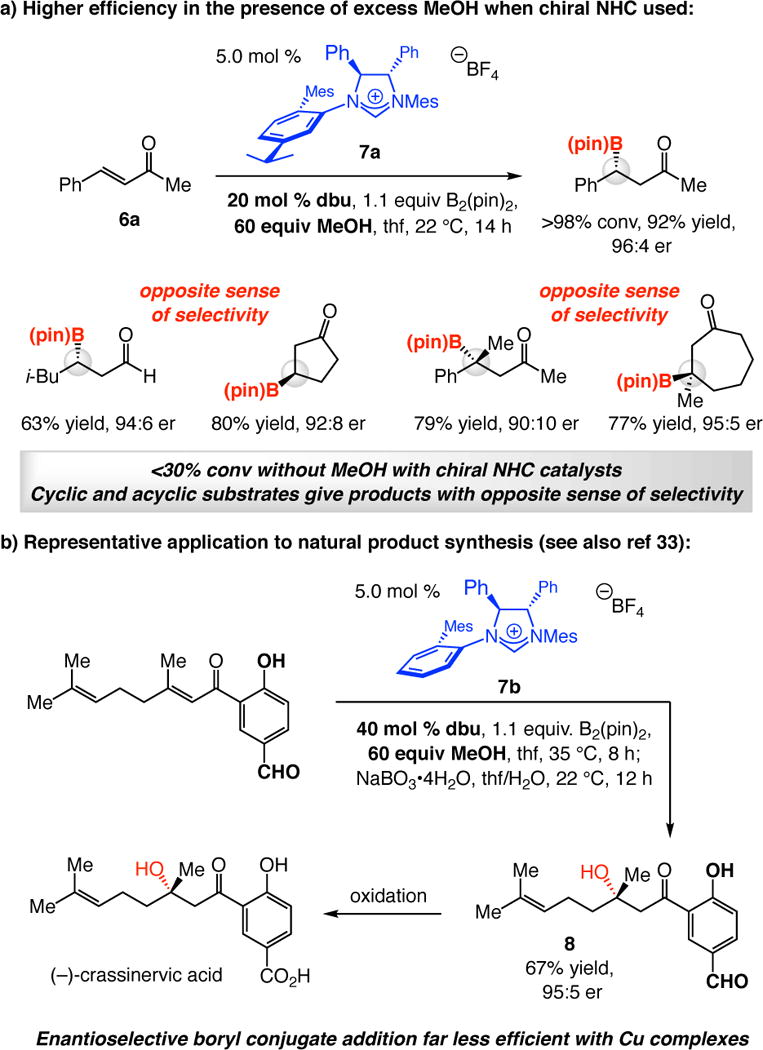
Enantioselective NHC–Catalyzed Boryl Conjugate Additionsa
aMes = 2,4,6-Me3-C6H2. The er values for some of the β-boryl ketones in Scheme 3a correspond to reactions with NHC catalysts that are slight structural variants of 7a; see ref 18 for details. The stereoselectivity profiles, however, are as shown.
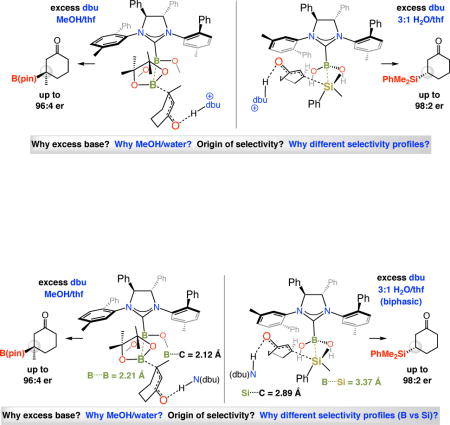
The above progress raises several fundamental questions: (1) Why is it that, unlike additions promoted by achiral NHC catalysts, the presence of MeOH is required for high conversion in enantioselective BCA reactions? (2) Why is excess base (dbu), beyond what is necessary for NHC formation, generally needed for achieving high yields of the desired products? (3) If the transferred boryl group is not the one that is associated with the NHC catalyst, then precisely how is a high degree of stereochemical induction observed with the Lewis base promoter being at a relatively remote site? (4) Why is the sense of addition to the acyclic substrates opposite to that of the cyclic enones (cf. Scheme 3a)?
NHC-Catalyzed silyl conjugate addition (SCA) with dimethylphenylsilylpinacolatoboron [PhMe2Si–B(pin); Scheme 4]35 further broadens the scope of the approach and, on several fronts, is distinct from the related boryl additions. Unlike the BCA processes, regardless of whether an achiral or a chiral NHC is involved, there is minimal conversion without a protic additive.20 Not only is a proton source needed in SCA reactions, water is the superior choice (vs MeOH with BCA; cf. Scheme 4). Similar to the boryl additions, excess dbu enhances efficiency, but in further contrast to BCA processes, the same sense of absolute stereochemistry is generated in the C–Si bond forming reactions regardless of whether cyclic or linear α,β-unsaturated carbonyl compounds are employed.
Scheme 4.
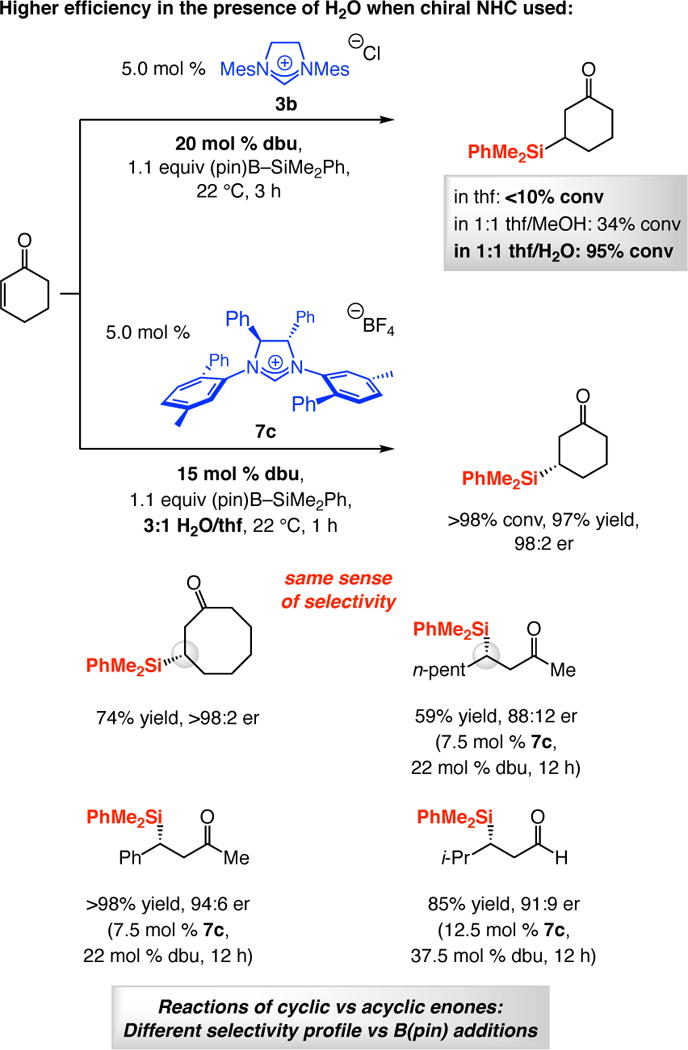
NHC-Catalyzed Silyl Conjugate Addition
Results and Discussion
1. Probing the possibility of a radical-based mechanism
We began by looking into whether the NHC-catalyzed BCA or SCA reactions involve odd-electron intermediates, a scenario that may find support in the pioneering investigations of Curran, Malacria and co-workers. The latter researchers have provided convincing evidence that NHC–borane complexes are capable of facilitating a C–H and C–C bond forming transformations by radical-based routes.36 We judged the possibility of a radical pathway to be especially relevant because the calculated homolytic bond dissociation energy (BDE) for B–B bonds in B2(pin)2 and NHC•B2(pin)2 (NHC = 1,3-dimethylimidazol-2-ylidene) is 104 kcal/mol and 63 kcal/mol, respectively [at unrestricted B97D/6-31+G(d,p) level of theory].37 The aforementioned values are lower than those calculated36a for BDE of B–H bonds in NHC–BH3 complexes (~74–80 kcal/mol), which are within the range measured for established radical precursors (e.g., 74 kcal/mol for n-Bu3Sn–H).38
Subjection of 3-cyclopropyl-cyclohexenone 9 to the BCA conditions afforded β-boryl ketone 10 in 85% yield without any detectable amounts of 11 (Scheme 5a), which would be formed if a carbon radical were generated at the carbon β to the carbonyl group. A similar observation was made with the cyclopropyl moiety positioned at the site adjacent to the C=O bond: 12 was isolated in 67% yield as a single stereoisomer without any cyclopropyl cleavage. To ascertain that a radical mechanism is not in effect, we examined the reactions of the derived diphenylcyclopropane; here, the corresponding odd electron species would undergo cleavage at a rate that is above the rate of diffusion-controlled processes.39 We did not detect any 13, likely because of steric hindrance, but did obtain β-boryl ketone 14 in 45% yield. Again, the product resulting from rupture of the cyclopropyl moiety was not observed. The identity of the BCA product was further substantiated by determination of the crystal structure of 15, generated from reaction of 14 with atmospheric oxygen (Scheme 5a). NHC-catalyzed C–Si bond formation does not seem to involve odd-electron intermediates either, as the conversion of 16 to β-silyl ketone 17 and complete absence of 18 indicates (Scheme 5b).
Scheme 5.
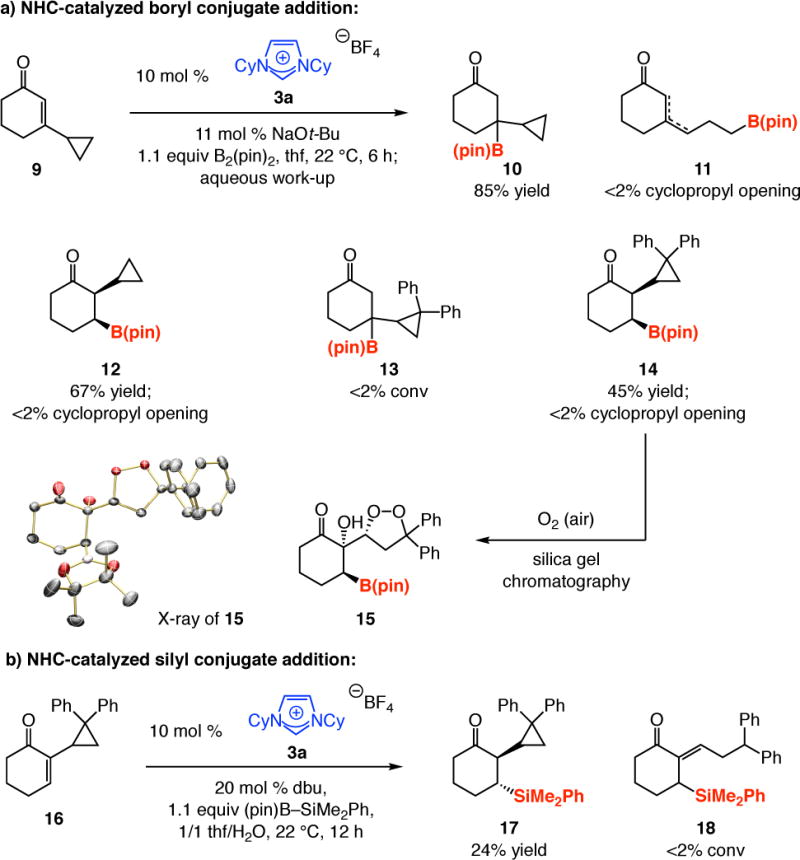
Probing the Possibility of a Radical-Based Mechanism
aCatalytic BCA reactions leading to 12 and 14 were performed with 20 mol % 3a and 22 mol % NaOtBu. See the Supporting Information for details.
2. The influence of excess base and MeOH on boryl conjugate additions
We then turned to addressing the question of why BCA reactions with achiral NHCs can be performed without MeOH (e.g., with 3a in Scheme 2c), whereas when the corresponding chiral catalysts are used, excess amount of a proton source is required (cf. Scheme 3a). When we made inquiry regarding the possible association of the chiral NHC derived from imidazolinium salt 7c with B2(pin)2 in the absence of MeOH, we did not detect any change in the 11B NMR spectrum (6.0 h, 25 °C; Scheme 6); this is in contrast to when achiral NHC 1 was employed, where there was >98% conversion within just five minutes. In the case of the chiral NHC, only after 30 equivalents of MeOH and six equivalents of dbu were introduced were we able to detect 20–30% of a new signal at 5.1 ppm (11B NMR), likely arising from NHC•diboron complex 19a and/or 19b (Scheme 6). (As will be detailed below, the aforementioned signal is not due to the derived methoxy complex.)
Scheme 6.
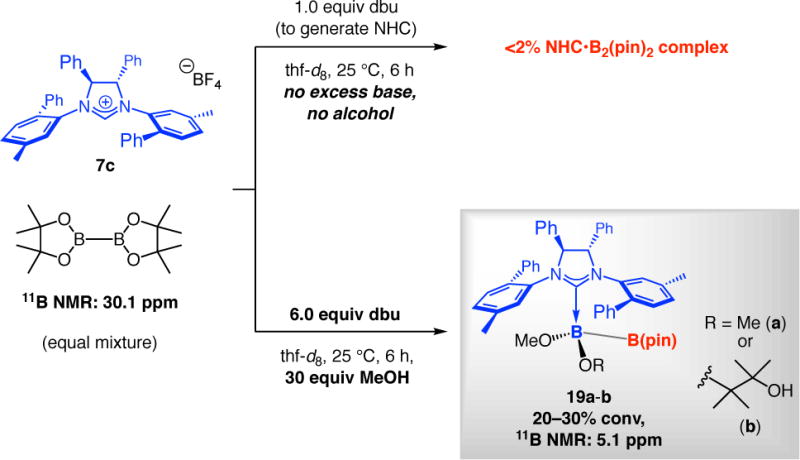
Probing the Effect of Base and MeOH on B–B Bond Activation
Concurrent availability of base and MeOH likely causes one of the pinacolato units of B2(pin)2 to rupture, producing a more accessible boron center (Scheme 7a).42 With MeOH, but in the absence of base, B2(pin)2 remains unaltered (Scheme 7b). The structurally modified and sterically less hindered boron site can readily associate with the more sizeable chiral NHC (e.g., 7a–c). Thus, with the smaller achiral NHC, complex formation and B–B bond activation does not require a priori cleavage of the pinacolato ring. Utilization of less congested diboron derivatives [vs B(pin)] to boost reactivity is an established strategy in organoboron chemistry;40 in situ conversion of a B(pin) unit to more accessible derivative was recently illustrated.41 The B(pin) moiety within the BCA product is more immune to reaction with MeOH probably because of the shorter B–C bond (1.5 Å vs 1.7 Å for B–B bond) as well as the presence of a neighboring sp3-hybridized C (vs an sp2-hybridized B).
Scheme 7.
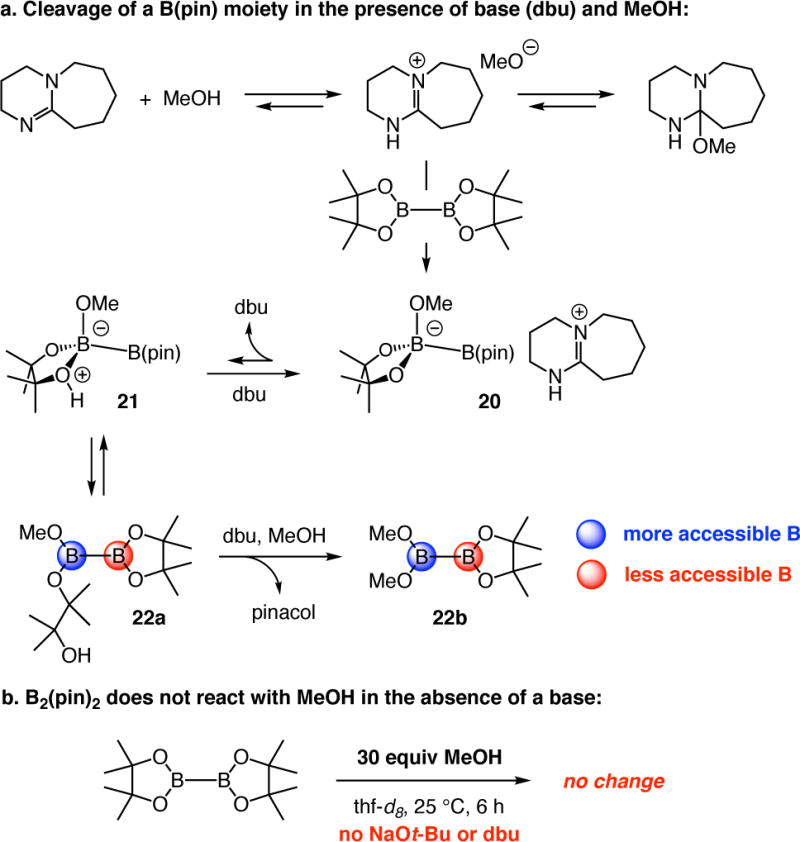
Susceptibility of B2(pin)2 to Alcohol and Base
Spectroscopic analysis provided additional information about the role of excess base and alcohol. Reaction of B2(pin)2 with dbu and MeOH generated free pinacol, as indicated by analysis of 1H NMR spectra of the unpurified mixtures,42 and resulted in a peak at 7.5 ppm in the 11B NMR spectrum, a signal that may be attributed to the methoxy-bound diboron species 23a–b (peak a, Figure 1); the intensity of the signal increased with time. Additionally, two broad peaks appeared between 25–35 ppm (b and c, Figure 1). The more upfield peak b may be attributed to the B atom undergoing hybridization change due to the rapid equilibrium between B2(pin)2 and 20. As the sample was cooled to −10 °C, signal b shifted upfield towards ~10 ppm, indicating an increasing preference for the formation of borate 20 (Figure 1). The large and broad peak c almost certainly corresponds to the sp2-hybridized B atoms of B2(pin)2 and other species such as 20, 22a–b and 23a–b. The most upfield small signal (marked by a blue box), which can only be detected at ≤10 °C, is likely derived from association of the more weakly Lewis basic and larger dbu (vs methoxide) with the diboron species (24a–b); thus, the more weakly held complex is more readily dissociated. As will be described, the amount of this latter entity increases in the presence of a highly Lewis basic NHC. Methoxide activation of a B–B bond via 20 or 23a–b points to the possibility of methoxide-catalyzed boryl addition.23,24 Accordingly, treatment of enone 6a (Scheme 3) with 1.1 equiv B2(pin)2, 20 mol % dbu, and 60 equivalents of MeOH generated ~20% of the corresponding rac-β-boryl ketone after 14 hours (22 °C). The chiral NHC catalyst must be overcoming this latter competitive background process since high enantioselectivity can be attained.
Figure 1.
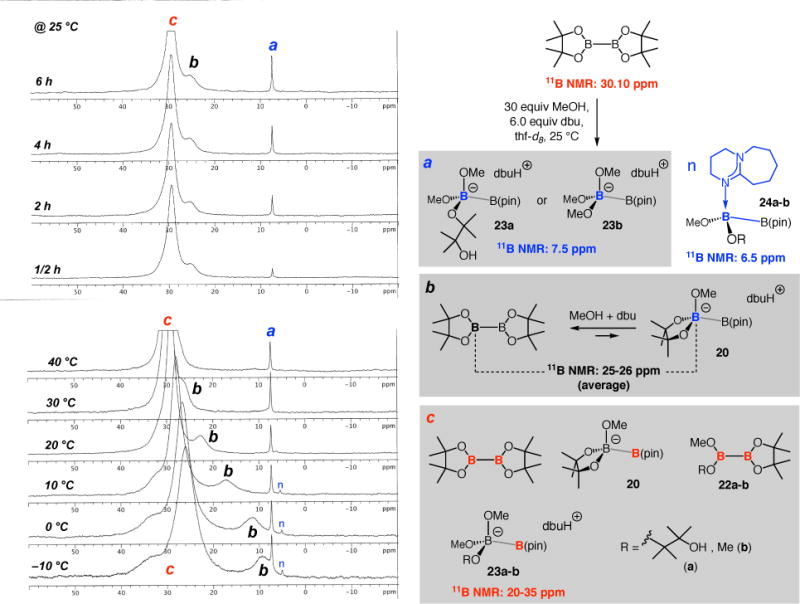
The effect of dbu on loss of pinacol and association of Lewis basic groups with the more exposed B center.
3. Details of the formation of chiral NHC•diboron complex
Spectroscopic studies with samples that contain enantiomerically pure imidazolinium salt 7c shed light on the interaction of various Lewis bases with in situ-generated sterically accessible/more reactive diboron species (Figure 2). Within 30 minutes at 25 °C, a peak at 7.5 ppm (c) and another at 6.5 ppm (b) could be detected. After two hours, the two signals grew in strength and another upfield singlet emerged at 5.0 ppm (a). In six hours, the intensity of the three peaks (a–c) increased extensively. The chemical shift at 7.5 ppm (c) corresponds to the methoxy-bound B atoms of diboron species 23a–b, as established by the experiments depicted in Figure 1, and the signal at 5.0 ppm arises from the NHC•diboron complexes 19a–b, based on the spectroscopic observations illustrated in Scheme 6. The signal at 6.5 ppm (b, Figure 2) is attributable to dbu•diboron complexes 24a–b; as before, though this particular peak could not be detected at 25 °C in the absence of the NHC (cf. Figure 1), it could be seen by cooling the sample to below 10 °C (cf. Figure 1).42 The signal corresponding to 24a–b also appears as a much smaller peak in the spectra in Figure 1 recorded at ≤10 °C; this difference in signal intensity is to be expected because the sterically less demanding methoxide can more effectively compete with dbu (vs chiral NHC). The increased abundance of 24a–b in the presence of NHC suggests that the Lewis base accelerates B(pin) cleavage,43 likely in the same manner as illustrated in Scheme 7a for a methoxide ion, to provide a higher concentration of the dbu•diboron complex. The broad signals d, e and f in Figure 2 have been already analyzed (cf. signals b and c, Figure 1).
Figure 2.
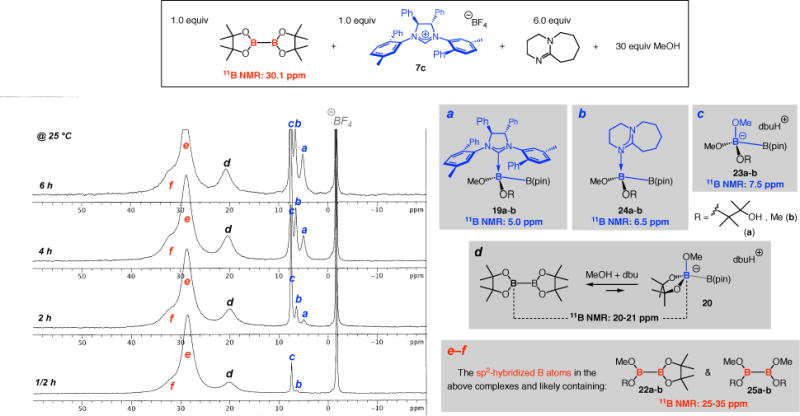
11B NMR spectra for reaction of B2(pin)2 with 100 mol % 7c with 6.0 equiv dbu and 30 equiv MeOH (thf-d8, 25 °C).
Variable temperature NMR experiments showed that if the sample is allowed to warm from −10 to 40 °C (Figure 3), a Lewis base (methoxy, dbu or NHC) may begin to dissociate from its diboron host. Accordingly, signals a and b disappeared and peak d became visible at 40 °C, pointing to a shift in equilibrium in favor of sp2-hybridized B atoms. Methoxide complexes 23a–b (signal c) proved to be more resistant towards dissociation (vs 19a–b or 24a–b), as the signal at 7.5 ppm persisted at elevated temperatures. Such stability profiles may be partly due to the smaller size of a methoxide (less steric relief upon dissociation), which is congruent with the more rapid appearance of the corresponding peak versus those that relate to NHC- (a) or dbu-based diboron complexes (b) (cf. Figure 2 at 0.5 and 2 h). As mentioned vis-à-vis peak b in Figure 1 for MeO•diboron species 20, higher temperatures led to conversion of Lewis base•diboron complexes to the free diboron species that contain only sp2-hybridized B atoms; this is evidenced by the downfield movement of signal e in Figure 3 when the sample was allowed to warm from −10 to 40 °C.
Figure 3.
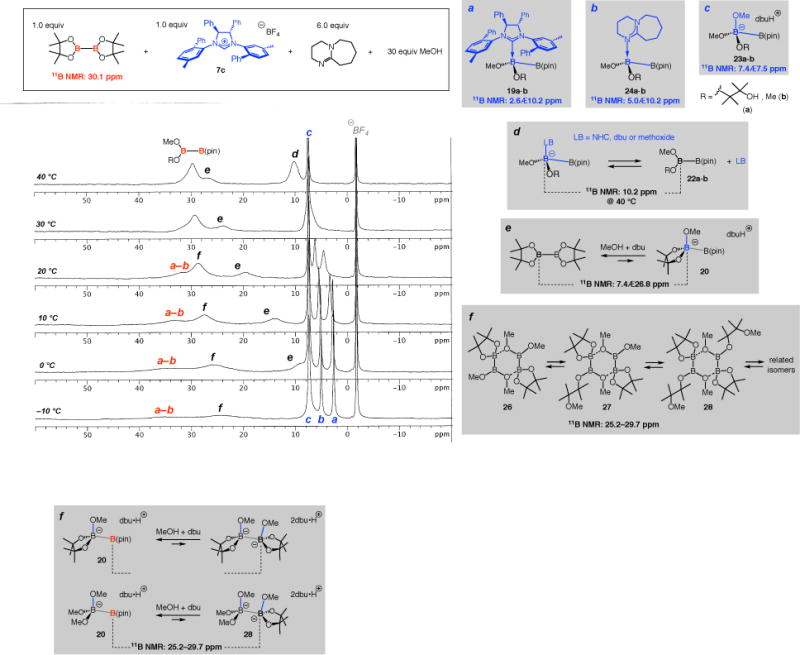
11B NMR spectra at different temperatures for reaction of B2(pin)2, 100 mol % 7c with 6.0 equiv dbu and 30 equiv MeOH after 6 h (thf-d8).
The above spectroscopic studies show that electronic variations extend beyond changes in the chemical shift of the B(pin) group that is directly bound to a Lewis base. Spectral data point to increased Lewis acidity at the unbound boron site (sp2-hybridized). Thus there is a chemical shift change from the B atoms of B2(pin)2 (30.1 ppm in 11B NMR) to the more downfield signal for the uncoordinated sp2-hybridized B atom of the NHC•diboron complex derived from carbene 1a (36.3 ppm; cf. Scheme 2b). The electronic influence of Lewis base activation is further manifested by the most downfield signals in Figure 3 (a–b, red); these peaks may be attributed to the sp2-hybridized B atoms of NHC-, methoxide- and dbu-bound complexes 19, 23 and 24. These findings are again consistent with the principles of Lewis base activation of Lewis acids,29,43 indicating that alteration of electron density extends to the neighboring B(pin) unit as well (net increase at the more electronegative O atoms). The increase in Lewis acidity of the unbound boron atom, caused by association of a Lewis base to the diboron compound, should not be expected to have an adverse effect on the rate of a BCA process, as it is electron density of the more polarized B–B bond that likely kick-starts that C–B bond forming process. It might be suggested that the enhancement in Lewis acidity of the uncoordinated boron might cause to associate with a Lewis base as well. However, DFT calculations clearly indicate37 that a doubly coordinated species would be substantially higher in energy probably as a result of steric congestion (severe eclipsing interactions) caused by vicinal full-substituted B atoms. Furthermore, the resulting B–B bond would be less polarized and the activation barrier towards addition to an enone would be more energetically demanding.
The above studies provide valuable clues regarding optimization of the experimental conditions, namely that increasing the amount of Lewis base (e.g., dbu) should lead to faster cleavage of a B(pin) unit of B2(pin)2. NHC•diboron complex formation would then be facilitated and B–B bond activation and an increase in the efficiency of the BCA process would ensue. Such expectations are supported by the representative data in Scheme 8. Whereas with 20 mol % dbu, there was 27% and 75% conversion to 29a and 29b, respectively, after 14 h (22 °C), with 100 mol % base the BCA proceeded to full conversion. This enhancement in enantioselectivity suggests that, at lower concentration of dbu and with less efficient chiral NHC binding, there is likely residual catalysis by an achiral Lewis base (most likely the methoxide ion).23–24
Scheme 8.
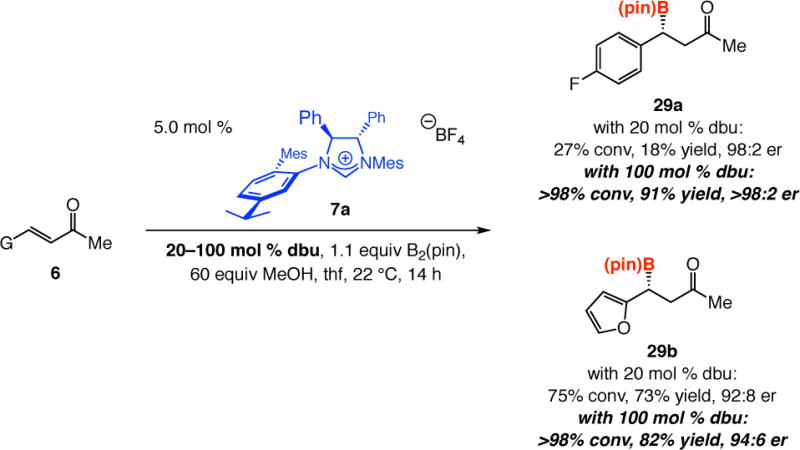
Influence of Amount of Base on NHC-Catalyzed Boryl Additions
4. Influence of excess base and H2O on NHC-catalyzed silyl conjugate additions
There are several key differences between NHC-catalyzed BCA and SCA reactions. Unlike boryl additions, there is no C–Si bond formation without a protic additive despite involvement of a smaller achiral NHC. Moreover, SCA reactions are less efficient when carried out in the presence of MeOH; transformations proceed readily when conducted in a water/thf mixture that is turned emulsive due to the presence of the borosilane reagent. It is therefore possible that activation of a more sterically demanding borosilane [vs B2(pin)2] requires a more accessible B(OH)2 unit and/or a medium that can better accommodate the formation of polar intermediates and transition states.
Inefficient generation of a Lewis base•borosilane complex is not the reason why SCA processes are slow when they are performed in the absence of a proton source. Treatment of imidazolium salt 3c with (pin)B–SiMe2Ph at 25 °C with 1.05 equivalents of dbu (without any H2O) led to complete loss of the signal for the borosilane reagent (33.4 ppm, 11B NMR spectrum) in five minutes (Figure 4); this time, a broad peak appeared at 8.0 ppm that may be attributed to NHC•borosilane complex 30.44 It is therefore more likely that because of the bulkier dimethylphenylsilyl moiety [vs B(pin) of B2(pin)2], efficient approach and subsequent association of a species such as 30 with an enone substrate requires the conversion of the pinacolato group to a B(OH)2 unit.
Figure 4.
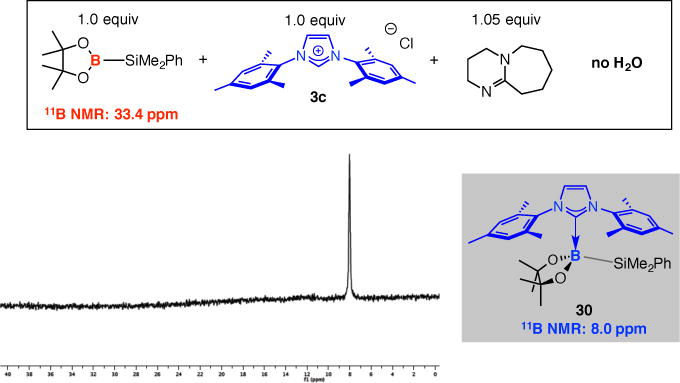
11B NMR spectrum for reaction of (pin)B–SiMe2Ph with an achiral NHC in the presence of only dbu (thf-d8, 25 °C).
Hydrolysis of the B(pin) group is likely to be equally (if not more) critical when the more sizeable chiral NHC catalysts are involved. In the presence of chiral imidazolinium salt 7c, excess amounts of dbu (6.0 equiv) and 6.0 equivalents of H2O, the borosilane reagent was completely consumed in one hour. Still, a signal corresponding to the NHC•borosilane complex could not be detected (Scheme 9).42
Scheme 9.
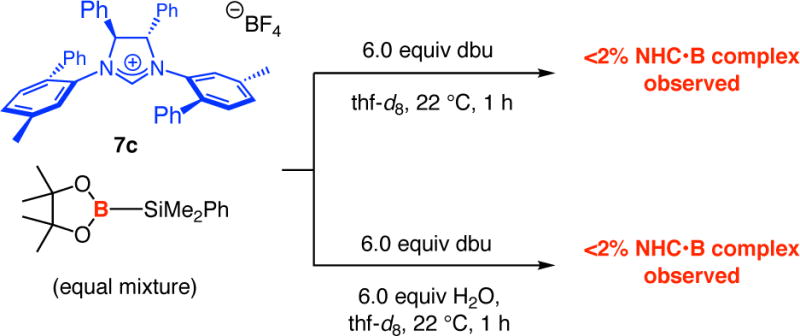
Probing the Association of Chiral NHC with (pin)B–SiMe2Ph
To gain insight regarding the possible influence of water on NHC-borosilane association, we first examined the fate of (pin)B–SiMe2Ph when treated with 6.0 equivalents of dbu and H2O in thf at 22 °C. Within 30 minutes, the reagent was entirely consumed and two types of B-containing species were generated (Scheme 10a). A signal at 21.5 ppm is likely due to formation of boronate 31, Me2PhSiH (characteristic peak in 1H NMR spectrum at 4.4 ppm) and complex 32 [in equilibrium with (pin)B–SiMe2Ph and dbu], while another at 7.6 ppm is because of the tetravalent B atom of 32b; these complexes may form by pathways similar to those shown in Scheme 7a [involving B2(pin)2 and MeOH].
Scheme 10.
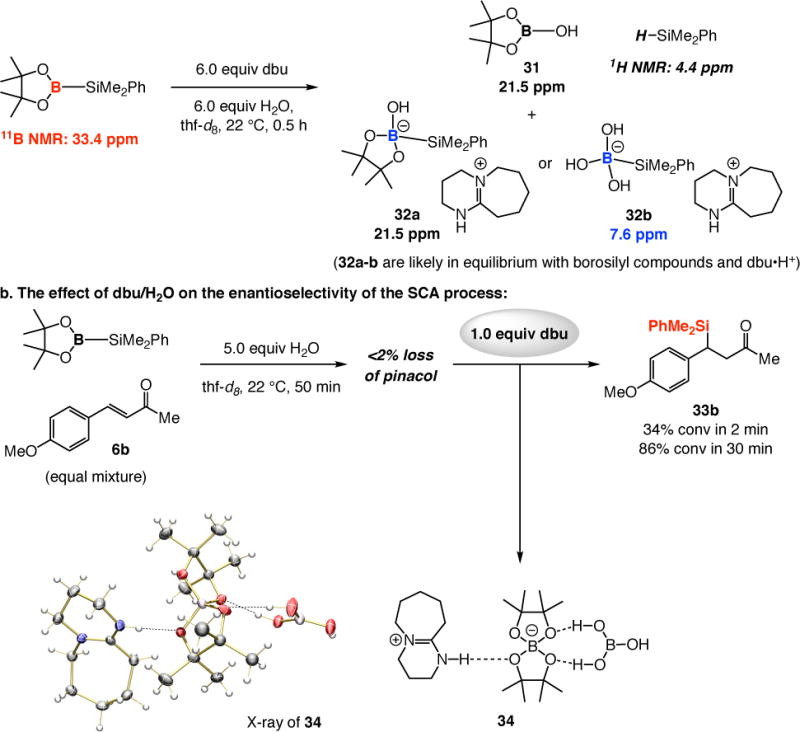
The Influence of Base and H2O on (pin)B–SiMe2Ph and SCA Reactionsa
aSee the Supporting Information for details.
Through another experiment, summarized in Scheme 10b, we established that, although (pin)B–SiMe2Ph is relatively stable in aqueous media, soon after the addition of dbu, it is swiftly consumed and SCA takes place efficiently (34% conv in two min and 86% conv in 30 min to β-silyl ketone 33b). Moreover, we were able to isolate and obtain the X-ray crystal structure of borate complex 34, resulting from the loss of the pinacol group, an event that likely leads to a more reactive borosilane derivative (e.g., 32b) and facile SCA, regardless of whether an achiral (cf. 3b, Scheme 4) or a larger chiral NHC (cf. 7c, Scheme 4) is involved. These observations illustrate that a highly effective enantioselective SCA catalyst must be sufficiently active to overcome the somewhat rapid background processes that afford racemic β-silylcarbonyl compounds.
We resorted to spectroscopic analysis (11B NMR) in our quest to gain information regarding the details of borosilyl activation by a chiral NHC. A mixture of (pin)B–SiMe2Ph together with an equivalent of chiral imidazolinium salt 7c, 6.0 equivalents of dbu and 6.0 equivalents of H2O, shown in Figure 5, led to generation of different products, many of which contain an sp3-hybridized B atom (peaks a–b relating to 35a–b and 32b, respectively). The assignment of the signals at 4.4 and 7.7 ppm, are based on formerly reported values44 and the experiment performed in the absence of an NHC (cf. Scheme 10a), respectively. That is, a distinct signal that may be confidently attributed to NHC•borosilane complex 36 could not be formed. Considering the differences in the electron donor abilities of an OH and an NHC ligand and the aforementioned influence of such characteristics on the chemical shift for the signals of the associated boron atoms, the possibility that the signal for 36 might be hidden behind the peak at ~8 ppm is remote. It is noteworthy that the NHC•borosilane complex that is responsible for promoting SCA reactions with high enantioselectively in the face of a somewhat swift background process and at significantly lower concentrations (e.g., 5.0 mol % vs 100 mol %), is difficult to observe. It is reasonable to suggest, however, that the lifetime of the borosilyl reagent or 36 is substantially increased under biphasic conditions (i.e., 3:1 H2O/thf vs 6.0 equiv H2O in thf), as these species probably spend much of their time in the organic layer. Otherwise, extended reaction times (e.g., 12 h in Scheme 4) would not be productive.
Figure 5.
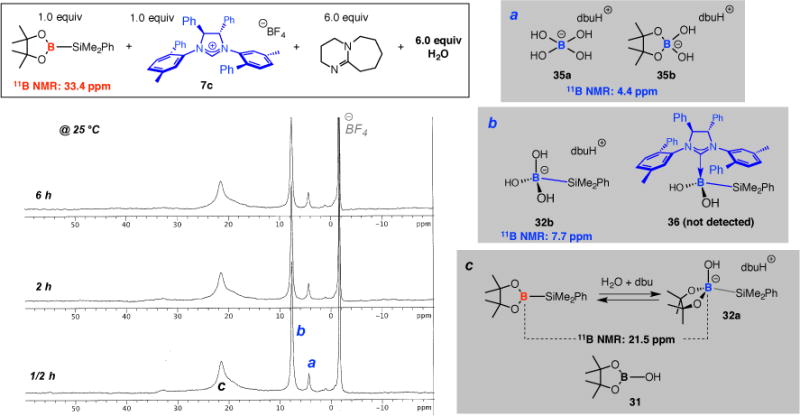
11B NMR spectra for reaction of (pin)B–SiMe2Ph with a chiral NHC in the presence of H2O and dbu (thf-d8, 25 °C).
To detect a chiral NHC•borosilane complex spectroscopically, we prepared dimethylphenylsilyl(dimethoxy)boron 37 (confirmed by HRMS), a species that is easier to synthesize and handle than the corresponding bis(hydroxy)boron derivative. Treatment of a sample of 37, after filtration to remove solid residues, with 1.0 equivalent of imidazolinium salt 7c and 1.0 equivalent of potassium hexamethyldisilazide (KHMDS, to ensure rapid and complete kinetic deprotonation) at 25 °C in thf-d8 for 30 minutes led to the appearance of a singlet in the 11B NMR spectrum at −0.4 ppm (Figure 6). This signal likely belongs to complex 39, and its broad shape indicates a rapid equilibrium between the chiral NHC•borosilane complex andthe unbound borosilane. To ensure that the aforementioned peak does not originate from association of the NHC with trimethoxyborane used for preparation of 37, we obtained the 11B NMR spectrum of the suspected complex (40), establishing that the associated peak appears at 1.4 ppm. Furthermore, when the dimethoxyborosilane reagent 37 was reacted with the achiral NHC derived from bis(mesityl)imidazolium salt 3b, the resulting NHC•boryl species (41) manifested itself with a signal at 0.1 ppm. Control experiments indicate that the reason for formation of the NHC•diboron complex is not the use of a stronger base (i.e., KHMDS vs dbu) as opposed to the availability of a more sterically accessible borosilane partner; treatment of 7c, KHMDS (1.0 equiv) and (pin)B–SiMe2Ph does not lead to any detectable amounts of a chiral NHC•borosilane complex (400 MHz 1H NMR analysis).
Figure 6.
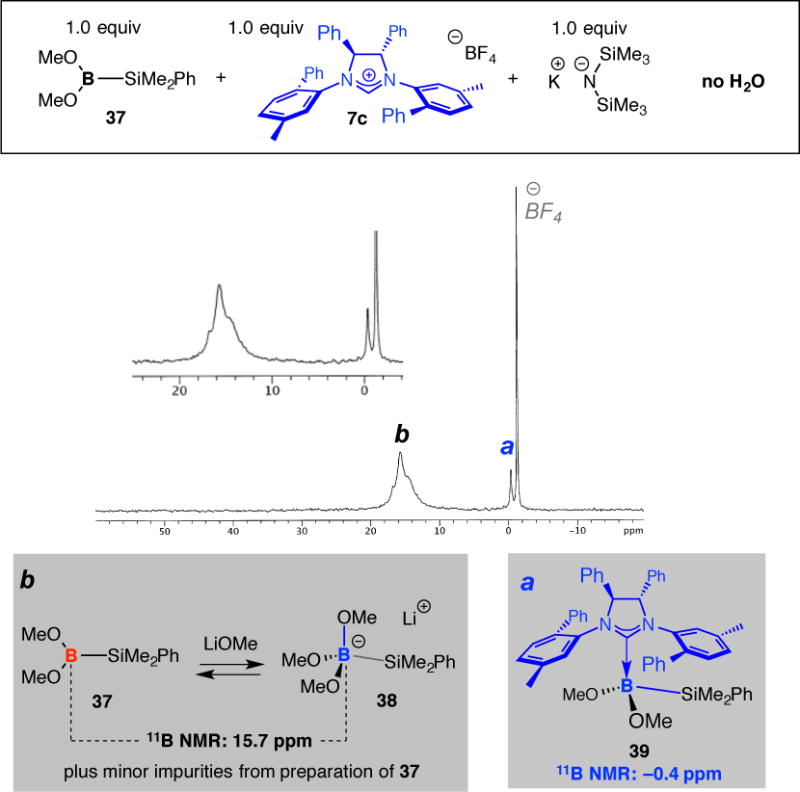
11B NMR spectrum for reaction of (MeO)2B–SiMe2Ph with a chiral NHC (thf-d8, 25 °C).
To explore the formation of the chiral NHC•borosilyl complex 39 further, we prepared a 13C-labeled sample of imidazolinium salt 7c and converted it to the corresponding NHC•BF3 complex 42 by the addition of 1.0 equivalent of KHMDS at 22 °C in thf-d8 for 30 minutes (Scheme 11). At ambient temperature, trifluoroboron species 42 may be identified by peaks at 164.2 ppm and −0.9 ppm in the 13C and 11B NMR spectra, respectively.45 The latter signal is broad, indicative of rapid equilibrium between the free carbene and the BF3 complex. Subsequent addition of 1.0 equivalent of bis(methoxy)borosilane 37 to the sample containing 42 resulted in the formation of 39, which was marked by the appearance of a new signal at 181.2 ppm in the 13C NMR spectrum (11B NMR signal at −0.4 ppm). The above findings imply that the catalytically active NHC•borosilane is probably formed and is capable of promoting substantial amounts of transformation prior to its decomposition.
Scheme 11.
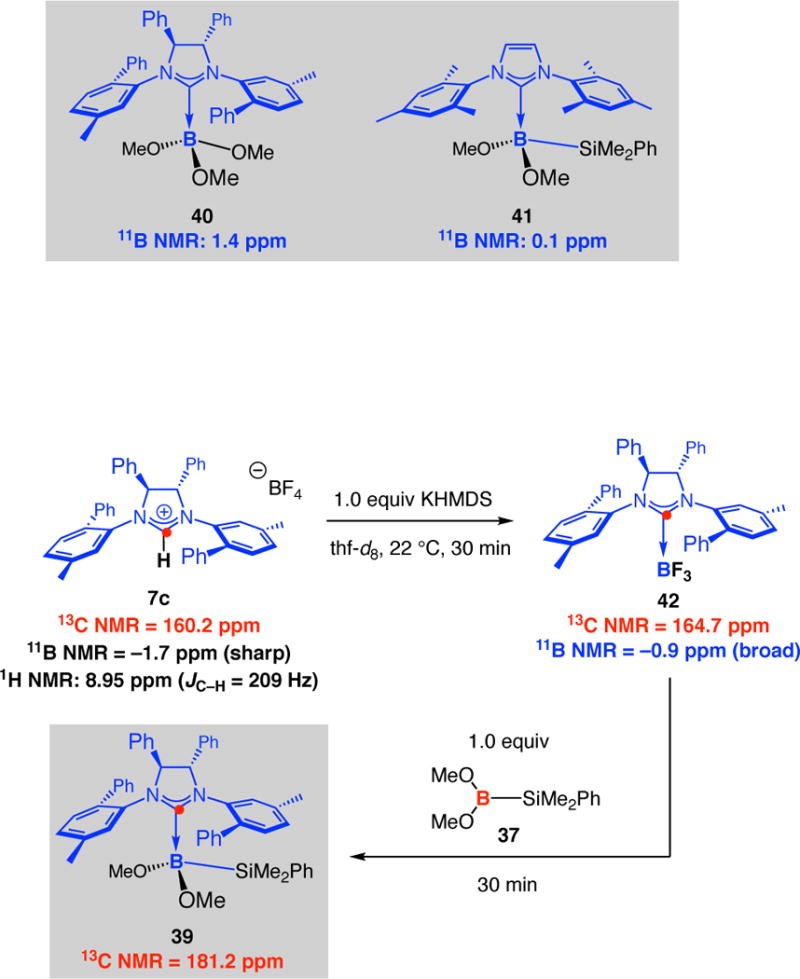
Probing the Formation of a Chiral NHC•Borosilane Complex by Spectroscopic Analysis of a 13C-Labeled Chiral NHCa
aSee the Supporting Information for details.
Imidazolinium salt 7c is stable in monophasic 3:1 H2O/thf solution (spectroscopic analysis). Incorporation of 22 equivalents of PhMe2Si–B(pin) (to emulate catalytic reaction conditions) caused the aqueous thf mixture to become biphasic and 7c remained unaltered (Scheme 12); however, when 3.0 equivalents of dbu were added, there was ~20% decomposition to acyclic formamide 43 (Scheme 12). When 7c was first treated with 1.0 equiv potassium hexamethyldisilazide (KHMDS, thf) before being placed in an aqueous thf solution that does not contain either the borosilane reagent or any dbu (monophasic), complete conversion to 43 occurred in less than two hours. The above experiments demonstrate that whereas the imidazolinium salt precursors are relatively stable, the NHC species, formed upon addition of dbu, can slowly decompose under the reaction conditions. Formamide 43 promotes SCA but non-enantioselectively (Scheme 12). Facile and highly enantioselective silyl additions therefore imply that the biphasic nature of the medium retards the rate of formamide generation and an NHC•borosilane complex is probably longer living than an unbound NHC.
Scheme 12.
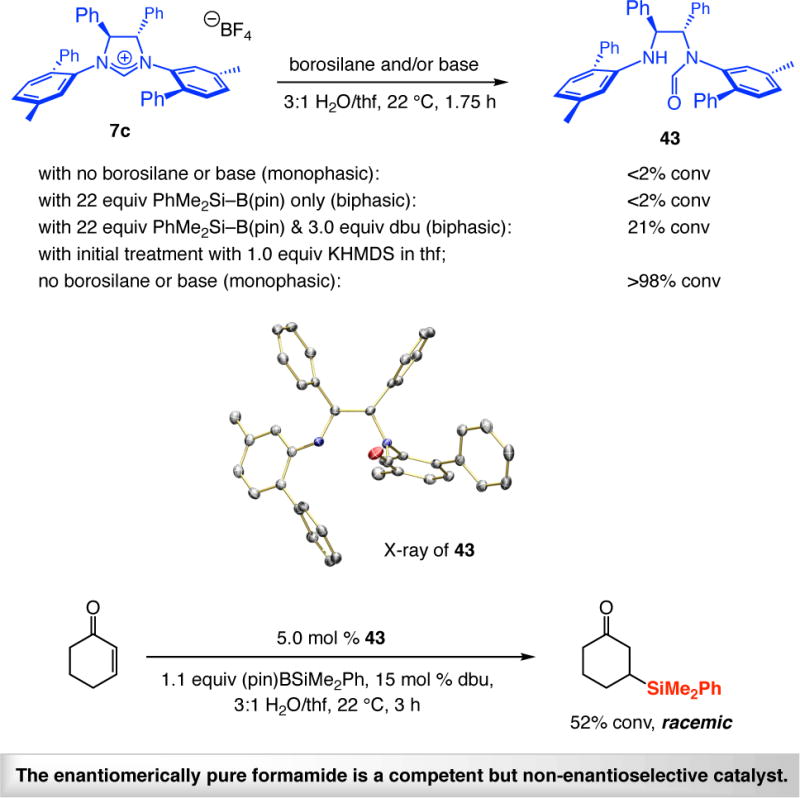
Decomposition of an Imidazolinium Salt in an Aqueous Mediuma
aSee the Supporting Information for details.
5. Effect of hydroxide ion permeability on efficiency and enantioselectivity of conjugate silyl additions
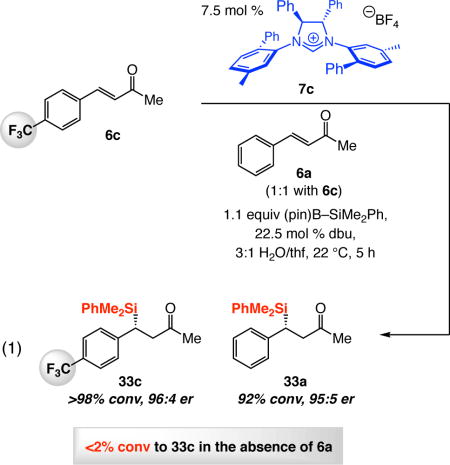
Through the studies described above we have demonstrated that the presence of water can have a positive [increasing activity by B(pin) hydrolysis] or a deleterious effect (NHC and/or borosilane decomposition) on NHC-catalyzed SCA reactions, and the degree and nature of the influence of water depends on the amount of dbu present (cf. Scheme 10b). With a better understanding of such factors, we can rationalize the origin of otherwise perplexing observations, and conditions may be modified leading to significant improvement in reaction outcomes. One noteworthy case relates to SCA to p-trifluoromethylphenyl-substituted enone 6c, which proved to be notoriously inefficient despite the substrate’s significant electrophilicity. Indeed, in our initial disclosure, we reported that the expected β-silyl ketone could not be observed after three hours in the biphasic 3:1 mixture of H2O and thf (Scheme 13).20 We reasoned that the presence of three fluorine atoms might cause enhanced phase separation, diminishing the concentration of the hydroxide in the organic phase (cf. Scheme 10b) needed to facilitate B(pin) removal, NHC•borosilane complex formation and efficient SCA. Indeed, as shown in Scheme 13, when reaction with 6c was performed with only 3.3 equivalents of H2O in thf (monophasic), we were able to obtain 33c in 86% yield after only three hours. We must note that the latter conditions do not translate to efficient SCA with other, less electrophilic, enones – it is in the case of highly active substrates that a relatively small amount of water is sufficient to generate enough B(pin) hydrolysis for efficient silyl conjugate addition.
Scheme 13.
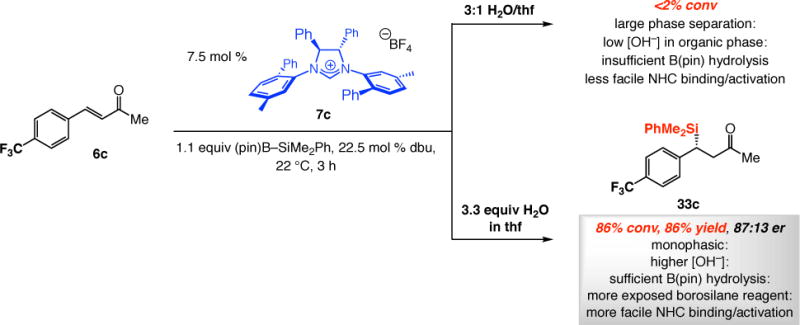
Influence of Ratio of Water/thf on Efficiency of NHC-Catalyzed SCA
Another notable implication of the biphasic reaction medium and the role of in situ generated hydroxide in facilitating a catalytic SCA process is that the optimal base must be adequately soluble in the aqueous as well as the organic phase. The base (e.g., dbu) can then penetrate the aqueous phase to generate the appropriate amount of hydroxide salt (e.g., H+dbu•− OH) that is lipophilic enough to enter the organic layer and promote B(pin) hydrolysis. The findings in Scheme 14 shed light on these issues.
Scheme 14.
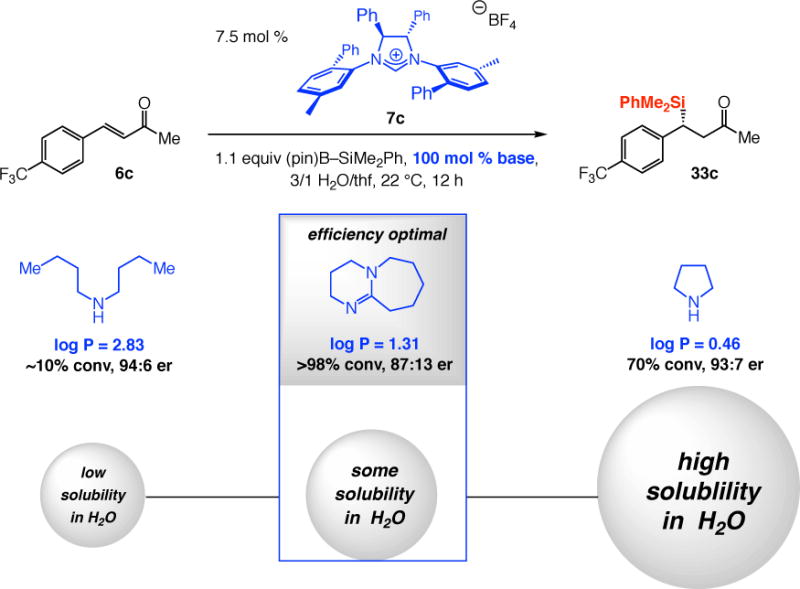
The Effect of Solubility of Different Amine Bases on Efficiency of Catalytic SCA Reactions
Firstly, with 100 mol % dbu, SCA with trifluorophenyl-substituted enone 6c proceeded to >98% conversion, affording 33c enantioselectively (87:13), even in biphasic 3:1 H2O/thf (Scheme 14); this is in stark contrast to <2% conversion when 22.5 mol % dbu was used (cf. Scheme 13). Secondly, efficiency variations with amine bases that possess different solubility properties indicate that with a more lipophilic base, or one that has higher hydrophilicity than dbu, SCA efficiency suffers [(~10% and 70% conv with (n-Bu)2NH (log P = 2.8346) and pyrrolidine (log P = 0.46), respectively, vs >98% conv with dbu (log P = 1.31)]. Thirdly, enantioselectivity is higher when an amine base that is less water soluble is present [94:6 er for (n-Bu)2NH] or when a base that is more hydrophilic than dbu is used (93:7 er for pyrrole vs 87:13 er with dbu). In either instance, the concentration of hydroxide in the organic layer, where 6c can be found, would be lower (vs with dbu), resulting in reduced generation of rac-33c (cf. Scheme 10b). For SCA with (n-Bu)2NH, there is insufficient base penetration into the aqueous layer, and with pyrrolidine there would be less partitioning of the ammonium hydroxide into the organic layer.
Higher efficiency of exchange between the phases is not caused by the nature of the amine base only. The presence of a suitably polar organic molecule can accelerate an otherwise inert enone as well. As previously mentioned (cf. Scheme 13), there was <2% conversion when 6c is subjected to 7.5 mol % 7c, 1.1 equivalents of (pin)B–SiMe2Ph, 22.5 mol % dbu in 3:1 H2O/thf after 12 hours. Nevertheless, with an equal amount of phenyl-substituted enone 6a simultaneously present (identical catalyst loading for total enone concentration; cf. eq 1), the more lipophilic trifluorophenyl-substituted enone 6c was fully consumed in five hours and 33c was obtained in 96:4 er. The higher er in the latter transformation (vs 87:13 er, Scheme 13) is due to the presence of 6a, which can increase hydroxide ion concentration enough so that SCA is facilitated but not to the extent that would lead to production of rac-33c (due to NHC decomposition; cf. Scheme 12).
6. Kinetic studies and stereochemical models
Initial rate measurements involving representative enantioselective NHC-catalyzed BCA and SCA reactions indicate that transformations are first-order in the NHC, the α,β-unsaturated carbonyl substrate, as well as B2(pin)2 and (pin)B–SiMe2Ph reagents, respectively; no dependence on dbu was observed.47 With the assumption that NHC•diboron and NHC•borosilane complex formation are rapidly reversible under the reaction conditions (22 °C; steady state approximation), a premise supported by the broad signals in the 11B NMR spectra (see above) that may be sharpened at lower temperatures,16b,32a the latter findings point to the C–B and C–Si bond forming steps as turnover-limiting. Accordingly, we set out to develop a rationale for the observed stereochemical outcomes of NHC-catalyzed enantioselective BCA and SCA processes.
To begin with, several general scenarios for BCA and SCA reactions were explored by means of DFT calculations [B97D/6−31+G(d,p) level of theory], most of which entailed initial B–B or B–Si bond activation by an NHC catalyst.37 Among the pathways examined were those proceeding via intermediates derived from NHC insertion into the B–B or B–Si bonds.48 These investigations indicated that the originally suggested route, namely the addition of polarized NHC•diboron or NHC•borosilane complexes to unsaturated carbonyl compounds, is energetically the more favorable possibility.
Model for additions to acyclic substrates
The lowest energy transition state structures for boryl and silyl conjugate additions to an acyclic enone are presented in Figure 7. For the BCA reactions, unlike I, complex II suffers from a destabilizing steric repulsion between the alkenyl substituent of the electrophile and the NAr moiety of the chiral NHC. This is consistent with experimental findings indicating that the size of the meta substituent of the NAr group influences BCA enantioselectivity (Me in I–II, i-Pr in the optimal catalyst derived from 7a; cf. Scheme 3). A larger ortho substituent within the NAr unit can cause the aromatic plane to tilt so that the coordinating substrate is more readily accommodated (e.g., Mes in 7a vs Ph in I–II).49
Figure 7.
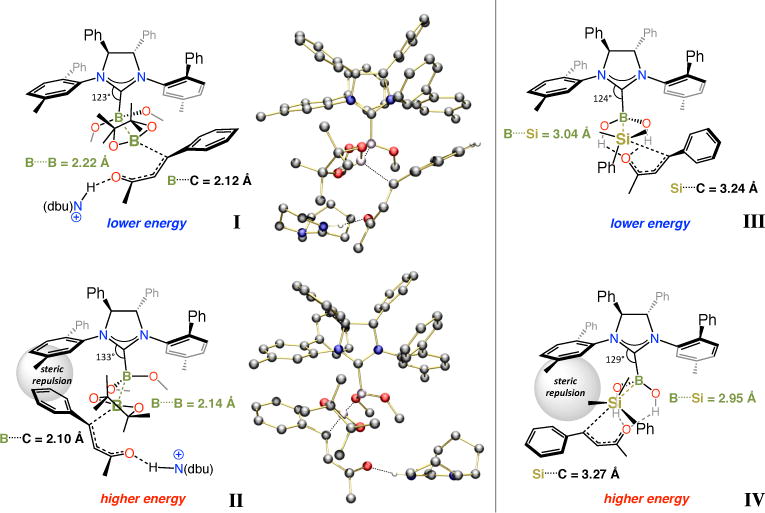
Transition state models for NHC-catalyzed enantioselective BCA and SCA reactions of acyclic enones obtained by DFT calculations.
DFT calculations point out that the N–C–B angle in the less favorable II is bent by ~10 degrees (133° vs 123° in I); this allows minimization of the destabilizing interactions between the aforementioned NAr group and the enone substituent (Ph in II). The transition states calculated for SCA transformations, III and IV, generally resemble those obtained for the BCA processes; the same arguments provided for I–II apply here as well. Widening of the N–C–B angle is less in the case of the borosilane complexes (124°/129° in III/IV vs 123°/133° in I/II) perhaps because the longer B….Si engenders a lesser degree of steric repulsion with the NHC. A difference between silyl and boryl additions is the possibility of H-bonding interactions between the B–OH within the NHC•borosilane ensemble and the substrate carbonyl group in III and IV. Such interactions alleviate the accumulation of electron density at the carbonyl oxygen that is developed after conjugate addition; the same type of stabilization may be provided externally by a protonated dbu or a MeOH molecule in the NHC-catalyzed boryl additions (cf. I–II); the zero-order rate dependence on dbu suggests that either MeOH, which is available in great excess, largely serves as the proton source, or association with Hdbu+ is fast and preceeds the turnover-limiting step.
Model for additions to cyclic substrates
As mentioned before, unlike transformations involving acyclic substrates, BCA and SCA reactions with cyclic enones proceed with the opposite sense of enantioselectivity. DFT calculations50 support the experimentally observed trend, indicating that whereas mode of addition V (Figure 8) is preferred for the C–B bond forming processes (vs VI), those that afford β-silyl carbonyl compounds probably proceed via VII (vs VIII). One reason for this distinction might be the size differences among the B(pin), the Hdbu-bound substrate and the especially sizeable Me2SiPh moieties. Another factor could be the shorter B….B versus B….Si distance in the transition state structure (e.g., 2.21 vs 3.37 Å in V and VII, respectively). It is therefore plausible that the B(pin) moiety would be preferentially situated in the vicinity of the sterically less demanding front/left hand quadrant of the chiral complex V. In the SCA process, on the other hand, the larger Me2SiPh group would be situated more distally from the aforementioned N-Ar group, reducing the degree of repulsion between the NHC’s N-Ar unit and the dbuH- or MeOH-bound enone. Another feature of the structures shown in Figure 8 is the absence of internal H-bonding between the B(OH)2 and the carbonyl unit (cf. III and IV in Figure 7);51 such interactions would be a great deal more geometrically demanding in cyclic α,β-unsaturated carbonyl compounds, which are locked into an s-cis conformation (vs s-trans conformers in Figure 7).
Figure 8.
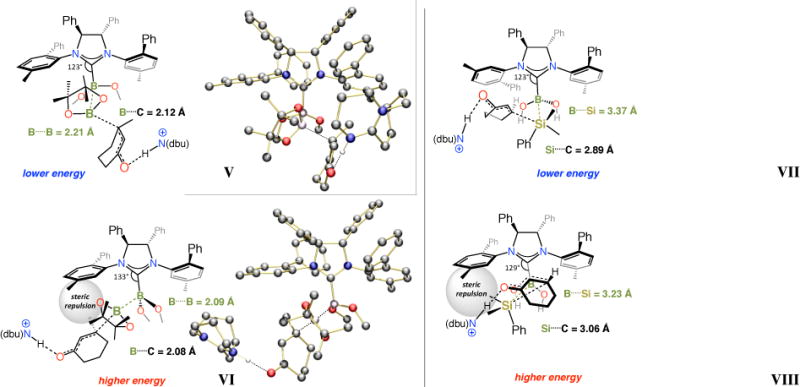
Transition state models for NHC-catalyzed enantioselective BCA and SCA reactions of cyclic enones obtained by DFT calculations.
Conclusions
The investigations detailed herein shed light on the following key mechanistic principles in regards to NHC-catalyzed enantioselective boryl and silyl conjugate addition reactions, which we show do not involve single-electron pathways:
Excess base MeOH and (dbu) are needed for BCA reactions promoted by the larger chiral NHC catalysts because conversion of the relatively hindered B(pin) unit to a more accessible B(OMe)2 or B(OMe)(OCMe2CMe2OH) facilitates diboron bond activation and NHC•diboron complex formation.
Initial rate studies and DFT calculations have resulted in the development of models that provide a plausible rationale for the different stereoselectivity profiles for boryl addition processes.
The lack of efficiency of silyl conjugate additions with the more diminutive achiral NHC (vs the corresponding BCA processes) in the absence of water is not because the necessary Lewis base•borosilane species cannot be generated. Rather, it might be due to the resulting NHC•B(pin) complex being too hindered, rendering approach by an α,β-unsaturated carbonyl compound energetically prohibitive.
The origin of high efficiency in SCA reactions in water with excess dbu is rapid hydrolysis of the sterically demanding B(pin) unit, allowing for the formation of a more sterically accessible and thus more reactive borosilane; this results in the more efficient NHC•borosilane complex generation and a more rapid initiation of a catalytic cycle.
For the silyl addition processes, the chiral NHC must be sufficiently active to overcome borosilane reagent decomposition as well as its own breakdown, which can yield a catalytically active but non-enantioselective acyclic formamide derivative.
The positive role of dbu in an NHC-catalyzed SCA is a culmination of a sensitive balance achieved regarding the solubility of the base in two phases of the reaction mixture (typically, 3:1 H2O/thf; biphasic nature caused by the borosilane reagent). Hydroxide ions must be transported into the organic layer in the form of Hdbu+•−OH in amounts that are sufficient for converting the B(pin) group of the silylboron reagent to its derived B(OH)2, which is critical for NHC activation of the B–Si bond. At the same time, the base must not cause too high an increase in hydroxide ion concentration in the organic layer or a substantial degree of reagent and catalyst decomposition results.
Initial rate studies and DFT calculations demonstrate that the opposite sense of enantioselectivity for BCA reactions involving cyclic enones versus SCA transformations are believed to originate from the larger size of a Me2SiPh moiety and a more elongated B–Si bond in the corresponding transition states.
Lewis base catalyzed reactions involving an NHC, a phosphine or an alkoxide species, particularly those that generate a C–B bond, have emerged as a potentially significant set of transformations that provide reactivity, functional group compatibility and stereoselectivity profiles and/or levels that are complementary to the more widely examined transition metal catalyzed systems. An understanding of the key mechanistic factors that facilitate such processes and/or render them highly selective allows for a more clear analysis of the existing data and is crucial if future initiatives in this burgeoning area are to achieve maximal success. The findings described above offer some of this needed insight.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM-57212).
Footnotes
Supporting Information Available. Experimental details for all reactions and analytic details for all enantiomerically enriched products. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.For enantioselective hydroborations of alkenes promoted by Rh or Ir complexes, see:; (a) Crudden CM, Edwards D. Eur J Org Chem. 2003:4695–4712. [Google Scholar]; (b) Carroll A-M, O’Sullivan TP, Guiry PJ. Adv Synth Catal. 2005;347:609–631. [Google Scholar]; For enantioselective Cu–B additions to/protonation of alkenes (protoboryl additions) and related compounds catalyzed by chiral Cu-based complexes, see:; (c) Lee Y, Hoveyda AH. J Am Chem Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee Y, Jang H, Hoveyda AH. J Am Chem Soc. 2009;131:18234–18235. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Corberán R, Mszar NW, Hoveyda AH. Angew Chem Int Ed. 2011;50:7079–7082. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]; (f) Meng F, Jang H, Hoveyda AH. Chem Eur J. 2013;19:3204–3214. doi: 10.1002/chem.201203803. [DOI] [PubMed] [Google Scholar]; (g) Matsuda N, Hirano K, Satoh T, Miura M. J Am Chem Soc. 2013;135:4934–4937. doi: 10.1021/ja4007645. [DOI] [PubMed] [Google Scholar]; (h) Jang H, Jung B, Hoveyda AH. Org Lett. 2014;16:4658–4661. doi: 10.1021/ol5022417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For enantioselective diboron additions to alkenes catalyzed by Pd or Pt complexes, see:; (a) Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J Am Chem Soc. 2004;126:16328–16329. doi: 10.1021/ja044167u. [DOI] [PubMed] [Google Scholar]; (b) Burks HE, Kliman LT, Morken JP. J Am Chem Soc. 2009;131:9134–9135. doi: 10.1021/ja809610h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kliman LT, Mlynarski SN, Morken JP. J Am Chem Soc. 2009;131:13210–13211. doi: 10.1021/ja9047762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schuster CH, Li B, Morken JP. Angew Chem, Int Ed. 2011;50:7906–7909. doi: 10.1002/anie.201102404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kliman LT, Mlynarski SN, Ferris GE, Morken JP. Angew Chem, Int Ed. 2012;51:521–524. doi: 10.1002/anie.201105716. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an overview, see:; (f) Takaya J, Iwasawa N. ACS Catal. 2012;2:1993–2006. [Google Scholar]
- 3.For reactions catalyzed by transition metal complexes and involving cyclic substrates to produce tertiary B-substituted carbon stereogenic centers, see:; Feng X, Yun J. Chem Commun. 2009:6577–6579. doi: 10.1039/b914207j. [DOI] [PubMed] [Google Scholar]
- 4.For BCA transformations promoted by transition metal based complexes and involving acyclic substrates to form quaternary B-substituted carbons stereogenic centers, see:; (a) O’Brien JM, Lee K-s, Hoveyda AH. J Am Chem Soc. 2010;132:10630–10633. doi: 10.1021/ja104777u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen IH, Kanai M, Shibasaki M. Org Lett. 2010;12:4098–4101. doi: 10.1021/ol101691p. [DOI] [PubMed] [Google Scholar]; (c) Feng X, Yun J. Chem Eur J. 2010;16:13609–13612. doi: 10.1002/chem.201002361. [DOI] [PubMed] [Google Scholar]
- 5.For BCA reactions facilitated by transition metal complexes and involving cyclic substrates to generate quaternary B-substituted carbons stereogenic centers, see:; Chen IH, Yin L, Itano W, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:11664–11665. doi: 10.1021/ja9045839. [DOI] [PubMed] [Google Scholar]
- 6.For allylic substitution reactions that generate tertiary or quaternary B-substituted stereogenic centers and are promoted by Cu-based complexes, see:; (a) Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M. J Am Chem Soc. 2007;129:14856–14857. doi: 10.1021/ja076634o. [DOI] [PubMed] [Google Scholar]; (b) Guzman-Martinez A, Hoveyda AH. J Am Chem Soc. 2010;132:10634–10637. doi: 10.1021/ja104254d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ito H, Okura T, Matsuura K, Sawamura M. Angew Chem, Int Ed. 2010;49:560–563. doi: 10.1002/anie.200905993. [DOI] [PubMed] [Google Scholar]; (d) Ito H, Kunii S, Sawamura M. Nat Chem. 2010;2:972–976. doi: 10.1038/nchem.801. [DOI] [PubMed] [Google Scholar]; (e) Park JK, Lackey HH, Ondrusek BA, McQuade DT. J Am Chem Soc. 2011;133:2410–2413. doi: 10.1021/ja1112518. [DOI] [PubMed] [Google Scholar]; (f) Park JK, McQuade DT. Angew Chem, Int Ed. 2012;51:2717–2721. doi: 10.1002/anie.201107874. [DOI] [PubMed] [Google Scholar]
- 7.For catalytic diastereo-or enantioselective diboron additions to imines, see:; (a) Beenen M, An C, Ellman JA. J Am Chem Soc. 2008;130:6910–6911. doi: 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]; (b) Zhang SS, Zhao YS, Tian P, Lin GQ. Synlett. 2013;24:437–442. [Google Scholar]; (c) Hong K, Morken JP. J Am Chem Soc. 2013;135:9252–9254. doi: 10.1021/ja402569j. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related additions to imines, see:; (d) Zhao C, Jiang C, Wang J, Wu C, Zhang QW, He W. Asian J Org Chem. 2014;3:851–855. [Google Scholar]; (e) Hensel A, Nagura K, Delvos LB, Oestreich M. Angew Chem, Int Ed. 2014;53:4964–4967. doi: 10.1002/anie.201402086. [DOI] [PubMed] [Google Scholar]; (f) Mita T, Sugawara M, Saito K, Sato Y. Org Lett. 2014;16:3028–3031. doi: 10.1021/ol501143c. [DOI] [PubMed] [Google Scholar]
- 8.For reviews regarding the use of organosilicon species in organic synthesis, see:; (a) Chan TH, Wang D. Chem Rev. 1992;92:995–1006. [Google Scholar]; (b) Jones GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]; (c) Fleming I, Barbero A, Walter D. Chem Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]; (d) Suginome M, Ito Y. Chem Rev. 2000;100:3221–3256. doi: 10.1021/cr9902805. [DOI] [PubMed] [Google Scholar]; For reviews on the utility of allylsilanes in organic synthesis, see:; (c) Masse CE, Panek JS. Chem Rev. 1995;95:1293–1316. [Google Scholar]; (d) Barbero A, Pulido FJ. Acc Chem Res. 2004;37:817–825. doi: 10.1021/ar0400490. [DOI] [PubMed] [Google Scholar]; (e) Chabaud L, James P, Landais Y. Eur J Org Chem. 2004:3173–3199. [Google Scholar]; For a review on the utility of β-silylcarbonyls in synthesis, see:; (f) Fleming I. Science of Synthesis; Thieme: Stuttgart, Germany. 2002;4:927–946. [Google Scholar]; For a review on the utility of borosilane reagents in organic chemistry, see:; (g) Oestreich M, Hartmann E, Mewald M. Chem Rev. 2013;113:402–441. doi: 10.1021/cr3003517. [DOI] [PubMed] [Google Scholar]
- 9.For synthesis of enantiomerically enriched allylsilanes by various catalytic protocols, see: Cross-coupling; (a) Hayashi T, Konishi M, Ito H, Kumada M. J Am Chem Soc. 1982;104:4962–4963. [Google Scholar]; (b) Hayashi T, Konishi M, Okamoto Y, Kabeta K, Kumada M. J Org Chem. 1986;51:3772–3781. [Google Scholar]; Hydrosilylation; (c) Hayashi T, Kabeta K, Yamamoto T, Tamao K, Kumada M. Tetrahedron Lett. 1983;24:5661–5664. [Google Scholar]; (d) Hayashi T, Han JW, Takeda A, Tang J, Nohmi K, Mukaide K, Tsuji H, Uozumi Y. Adv Synth Catal. 2001;343:279–283. [Google Scholar]; Allylic substitution, see:; (e) Hayashi T, Ohno A, Lu S-j, Matsumoto Y, Fukuyo E, Yanagi K. J Am Chem Soc. 1994;116:4221–4226. [Google Scholar]; (f) Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew Chem, Int Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841. [DOI] [PubMed] [Google Scholar]; (g) Takeda M, Shintani R, Hayashi T. J Org Chem. 2013;78:5007–5017. doi: 10.1021/jo400888b. [DOI] [PubMed] [Google Scholar]; (h) Delvos LB, Vyas DJ, Oestreich M. Angew Chem, Int Ed. 2013;52:4650–4653. doi: 10.1002/anie.201300648. [DOI] [PubMed] [Google Scholar]; Reduction of silyl-substituted allyl carbonates,; (i) Hayashi T, Iwamura H, Uozumi Y. Tetrahedron Lett. 1994;35:4813–4816. [Google Scholar]; Si–B addition to allenes,; (j) Ohmura T, Taniguchi H, Suginome M. J Am Chem Soc. 2006;128:13682–13683. doi: 10.1021/ja063934h. [DOI] [PubMed] [Google Scholar]; Olefin metathesis,; (k) Kiely AF, Jernelius JA, Schrock RR, Hoveyda AH. J Am Chem Soc. 2002;124:2868–2869. doi: 10.1021/ja012679s. [DOI] [PubMed] [Google Scholar]; (l) Adam JM, de Fays L, Laguerre M, Ghosez L. Tetrahedron. 2004;60:7325–7344. [Google Scholar]
- 10.For enantioselective silyl conjugate additions catalyzed by Pd or Rh complexes, see:; (a) Hayashi T, Matsumoto Y, Ito Y. J Am Chem Soc. 1988;110:5579–5581. [Google Scholar]; (b) Matsumoto Y, Hayashi T. Tetrahedron. 1994;50:335–346. [Google Scholar]; (c) Walter C, Auer G, Oestreich M. Angew Chem, Int Ed. 2006;45:5675–5677. doi: 10.1002/anie.200601747. [DOI] [PubMed] [Google Scholar]; (d) Walter C, Oestreich M. Angew Chem, Int Ed. 2008;47:3818–3820. doi: 10.1002/anie.200800361. [DOI] [PubMed] [Google Scholar]; (e) Walter C, Fröhlich R, Oestreich M. Tetrahedron. 2009;65:5513–5520. [Google Scholar]; (f) Hartmann E, Oestreich M. Angew Chem, Int Ed. 2010;49:6195–6198. doi: 10.1002/anie.201002916. [DOI] [PubMed] [Google Scholar]; (g) Hartmann E, Oestreich M. Org Lett. 2012;14:2406–2409. doi: 10.1021/ol300832f. [DOI] [PubMed] [Google Scholar]
- 11.For BCA reactions promoted by Cu complexes and involving acyclic substrates to produce tertiary B-substituted stereogenic carbon centers, see:; (a) Lee JE, Yun J. Angew Chem, Int Ed. 2008;47:145–147. doi: 10.1002/anie.200703699. [DOI] [PubMed] [Google Scholar]; (b) Sim HS, Feng X, Yun J. Chem Eur J. 2009;15:1939–1943. doi: 10.1002/chem.200802150. [DOI] [PubMed] [Google Scholar]; (c) Lillo V, Prieto A, Bonet A, Requejo MMD, Ramírez J, Pérez PJ, Fernández E. Organometallics. 2009;28:659–662. [Google Scholar]; (d) Weil DH, Abboud KA, Hong S. Chem Commun. 2010;46:7525–7527. doi: 10.1039/c0cc02211j. [DOI] [PubMed] [Google Scholar]; (e) Park JK, Lackey HH, Rexford MD, Kovnir K, Shatruk M, McQuade DT. Org Lett. 2010;12:5008–5011. doi: 10.1021/ol1021756. [DOI] [PubMed] [Google Scholar]; (f) Moure AL, Arrayás RG, Carretero JC. Chem Commun. 2011;47:6701–6703. doi: 10.1039/c1cc11949d. [DOI] [PubMed] [Google Scholar]; (g) Solé C, Whiting A, Gulyás H, Fernández E. Adv Synth Catal. 2011;353:376–384. [Google Scholar]; (h) Lee JCH, McDonald R, Hall DG. Nat Chem. 2011;3:894–899. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]; (i) Takatsu K, Shintani R, Hayashi T. Angew Chem, Int Ed. 2011;50:5548–5552. doi: 10.1002/anie.201008196. [DOI] [PubMed] [Google Scholar]; (j) Sole C, Bonet A, de Vries AHM, de Vries JG, Lefort L, Gulyás H, Fernández E. Organometallics. 2012;31:7855–7861. [Google Scholar]; (k) Kobayashi S, Xu P, Endo T, Ueno M, Kitanosono T. Angew Chem, Int Ed. 2012;51:12763–12766. doi: 10.1002/anie.201207343. [DOI] [PubMed] [Google Scholar]; (l) Calow ADJ, Batsanov AS, Pujol A, Solé C, Fernández E, Whiting A. Org Lett. 2013;15:4810–4813. doi: 10.1021/ol4022029. [DOI] [PubMed] [Google Scholar]; (m) Zhao L, Ma Y, He F, Duan W, Chen J, Song C. J Org Chem. 2013;78:1677–1681. doi: 10.1021/jo302694d. [DOI] [PubMed] [Google Scholar]; (n) Luo Y, Roy ID, Madec AGE, Lam HW. Angew Chem, Int Ed. 2014;53:4186–4190. doi: 10.1002/anie.201310380. [DOI] [PubMed] [Google Scholar]; (o) Kitanosono T, Xu P, Kobayashi S. Chem Asian J. 2014;9:179–188. doi: 10.1002/asia.201300997. [DOI] [PubMed] [Google Scholar]
- 12.For BCA reactions promoted by Ni, Pd or Rh complexes, see:; (a) Lillo V, Geier MJ, Westcott SA, Fernández E. Org Biomol Chem. 2009;7:4674–4676. doi: 10.1039/b909341a. [DOI] [PubMed] [Google Scholar]; (b) Shiomi T, Adachi T, Toribatake K, Zhou L, Nishiyama H. Chem Commun. 2009:5987–5989. doi: 10.1039/b915759j. [DOI] [PubMed] [Google Scholar]
- 13.For NHC–Cu-catalyzed enantioselective SCA reactions, see:; (a) Lee K-s, Hoveyda AH. J Am Chem Soc. 2010;132:2898–2900. doi: 10.1021/ja910989n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ibrahem I, Santoro S, Himo F, Córdova A. Adv Synth Catal. 2011;353:245–252. [Google Scholar]; (c) Lee K-s, Wu H, Haeffner F, Hoveyda AH. Organometallics. 2012;31:7823–7826. doi: 10.1021/om300790t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pace V, Rae JP, Harb HY, Procter DJ. Chem Commun. 2013;49:5150–5152. doi: 10.1039/c3cc42160k. [DOI] [PubMed] [Google Scholar]; (e) Plotzitzka J, Kleeberg C. Organometallics. 2014;33:6915–6926. [Google Scholar]; (f) Pace V, Rae JP, Procter DJ. Org Lett. 2014;16:476–479. doi: 10.1021/ol4033623. [DOI] [PubMed] [Google Scholar]
- 14.For a review on Cu-catalyzed ketone aldol processes, see:; (a) Shibasaki M, Kanai M. Chem Rev. 2008;108:2853–2873. doi: 10.1021/cr078340r. [DOI] [PubMed] [Google Scholar]; For a review on the use of catalytic enantioselective BCA and SCA to access aldol-type products, see:; (b) Hartmann E, Vyas DJ, Oestreich M. Chem Commun. 2011;47:7917–7932. doi: 10.1039/c1cc10528k. [DOI] [PubMed] [Google Scholar]; For an alternative Cu-catalyzed approach to generating ketone-aldol products, see:; (c) Meng F, Jang H, Jung B, Hoveyda AH. Angew Chem, Int Ed. 2013;52:5046–5051. doi: 10.1002/anie.201301018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For representative reviews on NHC-catalyzed processes in chemical synthesis, see:; (a) Enders D, Balensfiefer T. Acc Chem Res. 2004;37:534–541. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]; (b) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (c) Hahn FE, Jahnke MC. Angew Chem, Int Ed. 2008;47:3122–3172. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]; (d) Hopkinson MN, Richter C, Schedler M, Glorius F. Nature. 2014;510:485–496. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- 16.(a) Lee K-s, Zhugralin AR, Hoveyda AH. J Am Chem Soc. 2009;131:7253–7255. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee K-s, Zhugralin AR, Hoveyda AH. J Am Chem Soc. 2010;132:12766. [Google Scholar]
- 17.For NHC-catalyzed boryl conjugate additions to enals promoted by the same achiral NHC as used in ref 16a, followed by Wittig olefin synthesis, see:; Ibrahem I, Breistein P, Córdova A. Chem Eur J. 2012;18:5175–5179. doi: 10.1002/chem.201103572. [DOI] [PubMed] [Google Scholar]
- 18.(a) Wu H, Radomkit S, O’Brien JM, Hoveyda AH. J Am Chem Soc. 2012;134:8277–8285. doi: 10.1021/ja302929d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Radomkit S, Hoveyda AH. Angew Chem, Int Ed. 2014;53:3387–3391. doi: 10.1002/anie.201309982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Bonet A, Gulyás H, Fernández E. Angew Chem, Int Ed. 2010;49:5130–5134. doi: 10.1002/anie.201001198. [DOI] [PubMed] [Google Scholar]; For a related study involving Verkade’s base as catalyst, see:; (b) Pubill-Ulldemolins C, Bonet A, Bo C, Gulyás H, Fernández E. Chem Eur J. 2012;18:1121–1126. doi: 10.1002/chem.201102209. [DOI] [PubMed] [Google Scholar]
- 20.O’Brien JM, Hoveyda AH. J Am Chem Soc. 2011;133:7712–7715. doi: 10.1021/ja203031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solé C, Gulyás H, Fernández E. Chem Commun. 2012;48:3769–3771. doi: 10.1039/c2cc00020b. [DOI] [PubMed] [Google Scholar]
- 22.(a) Wen K, Chen J, Gao F, Bhadury PS, Fan E, Sun Z. Org Biomol Chem. 2013;11:6350–6356. doi: 10.1039/c3ob41499j. [DOI] [PubMed] [Google Scholar]; For phosphine-catalyzed B–B or B–Si additions to ynones, see:; (b) Nagao K, Ohmiya H, Sawamura M. Org Lett. 2015;17:1304–1307. doi: 10.1021/acs.orglett.5b00305. [DOI] [PubMed] [Google Scholar]
- 23.(a) Bonet A, Pubill-Ulldemolins C, Bo C, Gulyás H, Fernández E. Angew Chem, Int Ed. 2011;50:7158–7161. doi: 10.1002/anie.201101941. [DOI] [PubMed] [Google Scholar]; (b) Bonet A, Sole C, Gulyás H, Fernández E. Org Biomol Chem. 2012;10:6621–6623. doi: 10.1039/c2ob26079d. [DOI] [PubMed] [Google Scholar]
- 24.Blaisdell TP, Caya TC, Zhang L, Sanz-Marco A, Morken JP. J Am Chem Soc. 2014;136:9264–9267. doi: 10.1021/ja504228p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagashima Y, Hirano K, Takita R, Uchiyama M. J Am Chem Soc. 2014;136:8532–8535. doi: 10.1021/ja5036754. [DOI] [PubMed] [Google Scholar]
- 26.(a) Uematsu R, Yamamoto E, Maeda S, Ito H, Taketsugu T. J Am Chem Soc. 2015;137:4090–4099. doi: 10.1021/ja507675f. [DOI] [PubMed] [Google Scholar]; For an earlier study involving the use of significantly more Lewis basic alkyllithium reagents, see:; (b) Kawachi A, Minamimoto T, Tamao K. Chem Lett. 2001:1216–1217. [Google Scholar]
- 27.For discussions regarding the mechanism of phosphine-catalyzed enantioslective boryl conjugate additions, see:; Pubill-Ulldemolins C, Bonet A, Gulyás H, Bo C, Fernández E. Org Biomol Chem. 2012;10:9677–9682. doi: 10.1039/c2ob26899j. [DOI] [PubMed] [Google Scholar]
- 28.For key reports regarding the boryl conjugate addition processes catalyzed by Cu-based complexes with critical observations that shed light on different mechanistic issues, see:; (a) Takahashi K, Ishiyama T, Miyaura N. Chem Lett. 2000:982–983. [Google Scholar]; (b) Laitar DS, Müller P, Sadighi JP. J Am Chem Soc. 2005;127:17196–17197. doi: 10.1021/ja0566679. [DOI] [PubMed] [Google Scholar]; (c) Mun S, Lee JE, Yun J. Org Lett. 2006;8:4887–4889. doi: 10.1021/ol061955a. [DOI] [PubMed] [Google Scholar]; (d) Ref 11a.
- 29.The concept of Lewis base activation of Lewis acids was first introduced by Guttmann; see:; (a) Guttmann V. The Donor–Acceptor Approach to Molecular Interactions. Plenum Press; New York: 1978. Chapter 1. [Google Scholar]; For further analysis, see:; (b) Jensen WB. The Lewis Acid-Base Concepts. Wiley-Interscience; New York: 1980. pp. 135–142. [Google Scholar]; Thus, electron donation to a Cu–alkoxide or Cu–B(pin) species by an Lewis basic species (e.g., an NHC) results in increase in electron density at the more electronegative (vs the transition metal) C (including that of the NHC), B and oxygen ligands and diminution of the same at the Cu center. For an excellent in depth review regarding this important concept, see:; (c) Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. and references cited therein. [DOI] [PubMed] [Google Scholar]; For representative applications of the principle in catalyst and reaction development from these laboratories, see:; (d) Wieland LC, Deng H, Snapper ML, Hoveyda AH. J Am Chem Soc. 2005;127:15453–15456. doi: 10.1021/ja053259w. [DOI] [PubMed] [Google Scholar]; (e) Lee Y, Li B, Hoveyda AH. J Am Chem Soc. 2009;131:11625–11633. doi: 10.1021/ja904654j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Manville N, Alite H, Haeffner F, Hoveyda AH, Snapper ML. Nat Chem. 2013;5:768–774. doi: 10.1038/nchem.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) van Doorne W, Cordes AW, Hunt GW. Inorg Chem. 1973;12:1686–1689. [Google Scholar]; (b) Johnson Q, Kane J, Schaeffer R. J Am Chem Soc. 1970;92:7614–7615. [Google Scholar]; (c) Brock CP, Das MK, Minton RP, Niedenzu K. J Am Chem Soc. 1988;110:817–822. [Google Scholar]; (d) Haubold W, Hrebicek J, Sawitzki GZ. Naturforsch B: Chem Sci. 1984;39:1027–1031. [Google Scholar]; (e) Nguyen P, Dai C, Taylor NJ, Power WP, Marder TB. Inorg Chem. 1995;34:4290–4291. [Google Scholar]; (f) Clegg W, Dai C, Lawlor FJ, Marder TB, Nguyen P, Norman NC, Pickett NL, Power WP, Scott AJ. J Chem Soc, Dalton Trans. 1997:839–846. [Google Scholar]
- 31.To the best of our knowledge, the earliest mention of possible activation of the B–B bond in B2(pin)2 by a Lewis base appeared in a manuscript by Miyaura, et al. in the context of a process involving B2(pin)2, KOAc and CuCl. However, it is likely that the formation of Cu–B(pin) as the product entailed the intermediacy of a Cu(OAc) in the manner depicted in Scheme 1a (R = Ac; vs via an AcO•diboron complex). See:; Takahashi K, Ishiyama T, Miyaura N. J Organomet Chem. 2001;625:47–53. [Google Scholar]
- 32.(a) Kleeberg C, Crawford AG, Batsanov AS, Hodgkinson P, Apperley DC, Cheung MS, Lin Z, Marder TB. J Org Chem. 2012;77:785–789. doi: 10.1021/jo202127c. [DOI] [PubMed] [Google Scholar]; For another X-ray structure of an NHC•diboron complex, see:; (b) Bissinger P, Braunschweig H, Damme A, Dewhurst RD, Kupfer T, Radacki K, Wagner K. J Am Chem Soc. 2011;133:19044–19047. doi: 10.1021/ja208372k. [DOI] [PubMed] [Google Scholar]; For a report on phosphine adducts of a 1,2-dibromodiborane, see:; (c) Braunschweig H, Damme A, Jimenez-Halla JOC, Kupfer T, Radacki K. Angew Chem, Int Ed. 2012;51:6267–6271. doi: 10.1002/anie.201201673. [DOI] [PubMed] [Google Scholar]
- 33.For a recent application of NHC-catalyzed BCA in a total synthesis (neopeltolide), see:; Yu M, Schrock RR, Hoveyda AH. Angew Chem, Int Ed. 2015;54:215–220. doi: 10.1002/anie.201409120. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
34.Such observations (substantial difference between reactions promoted by NHC–Cu complexes and NHCs) indicate that BCA reactions are indeed NHC-catalyzed and do not involve trace amounts of transition metal impurities that might accompany a reagent, and in particular, sodium alkoxides (it should be noted that dbu is an equally effective base). It merits mention that altering the amount of NaOt-Bu does not lead to any change in reaction efficiency (see below for representative data); such would not be the case if the metal impurities contained within the alkali metal salt were playing an important role. The possibility of trace amounts of transition metal salts playing a role in phosphine-catalyzed BCA reactions (with Cs2CO3) has been addressed as well (cf. ref 19a).
- 35.For reports regarding SCA reactions promoted by Cu-based chiral complexes, see ref 13. For a review, see ref 14b.
- 36.For representative disclosures, see:; (a) Ueng S-H, Brahmi MM, Derat É, Fensterbank L, Lacôte E, Malacria M, Curran DP. J Am Chem Soc. 2008;130:10082–10083. doi: 10.1021/ja804150k. [DOI] [PubMed] [Google Scholar]; (b) Heng S-H, Solovyev A, Yuan X, Geib SJ, Fensterbank L, Lacôte E, Malacria M, Newcomb M, Walton J, Curran DP. J Am Chem Soc. 2009;131:11256–11262. doi: 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]; (c) Walton JC, Brahmi MM, Fensterbank LF, Lacôte E, Malacria M, Chu Q, Ueng S-H, Solovyev A, Curran DP. J Am Chem Soc. 2010;132:2350–2358. doi: 10.1021/ja909502q. [DOI] [PubMed] [Google Scholar]; (d) Walton JC, Brahmi MM, Monot J, Fensterbank L, Malacria M, Curran DP, Lacôte E. J Am Chem Soc. 2011;133:10312–10321. doi: 10.1021/ja2038485. [DOI] [PubMed] [Google Scholar]; (e) Pan X, Lacôte E, Lavelée J, Curran DP. J Am Chem Soc. 2012;134:5669–5674. doi: 10.1021/ja300416f. [DOI] [PubMed] [Google Scholar]; (f) Lavelée J, Telitel S, Tehfe MA, Fouassier JP, Curran DP, Lacôte E. Angew Chem, Int Ed. 2012;51:5958–5961. doi: 10.1002/anie.201200975. [DOI] [PubMed] [Google Scholar]; (g) Pan X, Vallet A-L, Schweizer S, Dahbi K, Delpech B, Blanchard N, Graff B, Geib SJ, Curran DP, Lavelée J, Lacôte E. J Am Chem Soc. 2013;135:10484–10491. doi: 10.1021/ja403627k. [DOI] [PubMed] [Google Scholar]; (h) Telitel S, Vallet A-L, Schweizer S, Delpech B, Blanchard N, Morlet-Savary F, Graff B, Curran DP, Robert M, Lacôte E, Lavelée J. J Am Chem Soc. 2013;135:16938–16947. doi: 10.1021/ja4066267. [DOI] [PubMed] [Google Scholar]; For a review, see:; Curran DP, Solovyev A, Brahmi MM, Fensterbank L, Malacria M, Lacôte E. Angew Chem, Int Ed. 2011;50:10294–10317. doi: 10.1002/anie.201102717. [DOI] [PubMed] [Google Scholar]
- 37.See the Supporting Information for further information on the DFT calculations.
- 38.Chatligialoglu C, Newcomb M. In: Advances in Organometallic Chemistry. West R, Hill AF, editors. Vol. 44. Academic Press; San Diego: 1999. pp. 67–112. [Google Scholar]
- 39.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 40.For representative cases where replacing a B(pin) group of a reagent to a less hindered derivative leads to reaction rate enhancement, see:; (a) Brown HC, Racherla US, Pellechia PJ. J Org Chem. 1990;55:1868–1874. [Google Scholar]; (b) Dhudshia B, Tiburcio J, Thadani AN. Chem Commun. 2005:5551–5553. doi: 10.1039/b511411j. [DOI] [PubMed] [Google Scholar]; (c) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660–12661. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (d) Ref 1h.
- 41.For representative cases where in situ rupture of a B(pin) group results in an increase reaction rates, see:; (a) Chen JLY, Scott HK, Hesse MJ, Willis CL, Aggarwal VK. J Am Chem Soc. 2013;135:5316–5319. doi: 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]; (b) Chen JLY, Aggarwal VK. Angew Chem, Int Ed. 2014;53:10992–10996. doi: 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- 42.See the Supporting Information for details.
- 43.Association of a Lewis basic, such as a methoxy group or the strongly σ-donating NHC, to a B(pin) unit can lead to a substantial increase in the electron density of the oxygen atoms of the pinacolato unit (see ref 29), increasing the probability of its protonation and subsequent rupture of the B-based heterocycle.
- 44.Two NHC•borosilane complexes have been prepared and characterized through X-ray crystallography; see:; Kleeberg C, Borner C. Eur J Inorg Chem. 2013:2799–2806. These investigators report that treatment of the NHC•borosilane complexes with benzyl bromide, methyl iodide and trimethylsilyl chloride does not lead to any reaction. We also find that NHC catalyzed boryl or silyl addition cannot be readily extended to alkylation processes. As stated in our original report in 2009 (ref 16a-b), the relatively high polarizability of an α,β-unsaturated carbonyl substrate is key to the efficiency of a Lewis base catalyzed C–B or C–Si addition process. [Google Scholar]
- 45.For spectral data on NHC•BF3 and NHC•B(Et)3 complexes, respectively, see:; (a) Arduengo AJ, Davidson F, Krafczyk R, Marshall WJ, Schmutzler R. Monatshefte Chem. 2000;131:251–265. [Google Scholar]; (b) Yamaguchi Y, Kashiwbara T, Ogata K, Miura Y, Nakamura Y, Kobayashi K, Ito T. Chem Commun. 2004:2160–2161. doi: 10.1039/b405459h. [DOI] [PubMed] [Google Scholar]
- 46.Log P, a partition-coefficient, is the ratio of concentrations of a species in a mixture of two immiscible phases at equilibrium. The log P values mentioned here involve water and n-octane; thus, log P = log Poct/H2O = log ([solute]oct/[solute]un-ionized water). For a comprehensive list of log P values, see:; Sangster J. J Phys Chem Ref Data. 1989;18:1111–1230. [Google Scholar]
- 47.See the Supporting Information for the details of the kinetic measurements and the calculation of rate equations leading to the conclusions regarding the identity of the turnover-limiting steps.
- 48.Although, to the best of our knowledge, there are no reports of compounds derived from NHC insertion into a B–B or a B–Si bond, entities resulting from reactions with H–H, C–Zr, B–H, Be–H, P–H, and Si–H bonds have been disclosed. See:; (a) Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439–441. doi: 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]; (b) Romain C, Miqueu K, Sotiropoulos JM, Bellemin-Laponnaz S, Dagorne S. Angew Chem, Int Ed. 2010;49:2198–2201. doi: 10.1002/anie.200906702. [DOI] [PubMed] [Google Scholar]; (c) Frey GD, Masuda JD, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2010;49:9444–9447. doi: 10.1002/anie.201005698. [DOI] [PubMed] [Google Scholar]; (d) Iversen KJ, Wilson DJD, Dutton JL. Organometallics. 2013;32:6209–6217. [Google Scholar]
- 49.For a detailed discussion regarding the influence of different substituents on the efficiency and enantioselctivity of various chiral C1-symmetric NHC compounds, see:; Lee K-s, Hoveyda AH. J Org Chem. 2009;74:4455–4462. doi: 10.1021/jo900589x. [DOI] [PubMed] [Google Scholar]
- 50.We selected to focus on carrying out DFT calculations on BCA reactions with cyclic enones that generate quaternary carbon stereogenic centers largely because such transformations generally proceed with higher enantioselectivity (see ref 18).
- 51.For examples of H-bonding between a B–OH group and a Lewis basic site in the course of a catalytic process, see:; (a) Gernigon N, Al-Zoubi RM, Hall DG. J Org Chem. 2012;77:8386–8400. doi: 10.1021/jo3013258. [DOI] [PubMed] [Google Scholar]; (b) Zi W, Wang YM, Toste FD. J Am Chem Soc. 2014;136:12864–12867. doi: 10.1021/ja507468u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.