

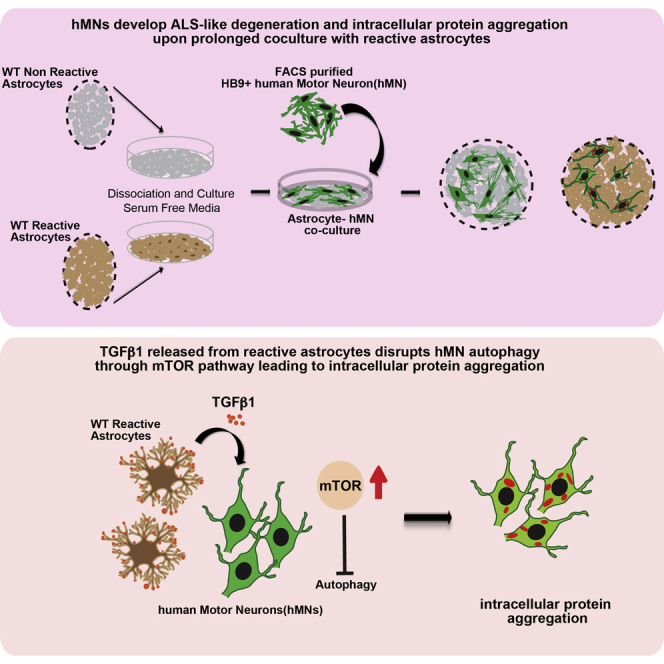
Summary
Amyotrophic lateral sclerosis (ALS) is a fatal and rapidly progressing motor neuron disease. Astrocytic factors are known to contribute to motor neuron degeneration and death in ALS. However, the role of astrocyte in promoting motor neuron protein aggregation, a disease hallmark of ALS, remains largely unclear. Here, using culture models of human motor neurons and primary astrocytes of different genotypes (wild-type or SOD1 mutant) and reactive states (non-reactive or reactive), we show that reactive astrocytes, regardless of their genotypes, reduce motor neuron health and lead to moderate neuronal loss. After prolonged co-cultures of up to 2 months, motor neurons show increased axonal and cytoplasmic protein inclusions characteristic of ALS. Reactive astrocytes induce protein aggregation in part by releasing transforming growth factor β1 (TGF-β1), which disrupts motor neuron autophagy through the mTOR pathway. These results reveal the important contribution of reactive astrocytes in promoting aspects of ALS pathology independent of genetic influences.
Keywords: ALS, reactive astrocytes, autophagy, TGF-b, mTOR, human motor neurons
Graphical Abstract
Highlights
-
•
Reactive astrocytes induce ALS-like protein aggregation in human motor neurons
-
•
Reactive astrocytes have increased secretion of TGF-β1
-
•
TGF-β1 induces axonal and cytoplasmic protein aggregation in hMNs
-
•
TGF-β1 activates PI3K/AKT/mTOR pathway and impairs autophagy in hMNs
Using long-term co-cultures of reactive astrocytes and human motor neurons, Zhou and colleagues show that reactive astrocytes, regardless of their genetic makeup, are toxic to motor neurons and induce neuronal loss, neurite shrinkage, and axonal and cytoplasmic protein inclusions. Reactive astrocytes promote neuronal protein aggregation partly through enhanced TGF-β1 signaling, which activates mTOR pathway and impairs autophagy in motor neurons.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive degeneration of upper and lower motor neurons. This neurodegeneration is coupled with abnormal protein and neurofilament inclusions, which collectively lead to muscle atrophy and paralysis (Al-Chalabi and Hardiman, 2013, Rowland and Shneider, 2001, Strong et al., 2005). The majority of ALS cases (90%) are sporadic in nature (sALS), whereas the remaining 10% of cases are familial (fALS), with the majority of fALS associated with mutations in the C9ORF72 or the Superoxide dismutase 1 (SOD1) gene (40%–45% and 20%–25%, respectively) (Taylor et al., 2016).
The pathogenic mechanisms of ALS are complex but appear to be strongly influenced by astrocytes, the dominant glial cell type in the CNS (Boillee et al., 2006, Philips and Rothstein, 2014, Valori et al., 2014). Healthy astrocytes play an essential role in normal neuronal function, participating in metabolic support, ionic balance, blood-brain barrier maintenance, and immune modulation (Barres, 2008). Under disease or injury conditions, astrocytes change their morphology and properties and become “reactive astrocytes” (Hamby and Sofroniew, 2010, Parpura et al., 2012, Phatnani and Maniatis, 2015, Seifert et al., 2006). Reactive astrocytes can be recognized by their enhanced expression of glial fibrillary acidic protein (GFAP) and extracellular matrix proteins, including chondroitin sulfate proteoglycan, versican, and aggrecans (Sofroniew, 2005). Reactive astrocytes are also characterized by increased secretion of many factors, some of which are known inflammatory mediators such as cytokines and interferons (Sofroniew, 2005). The transition into a reactive state-reactive gliosis has been shown to be a prominent feature of ALS (Boillee et al., 2006, Ilieva et al., 2009, Philips and Robberecht, 2011, Valori et al., 2014). It is important to understand the role of reactive astrocytes in the neurodegenerative process, particularly because astrocytes can acquire a reactive state, not only in chronic neurodegenerative processes, but also under a variety of environmental conditions, such as traumatic CNS injury (Burda et al., 2016, Sofroniew, 2005, Sofroniew, 2009). Indeed, it is noteworthy that traumatic injuries have been associated with increased risks for ALS and other neurodegenerative diseases (Chen et al., 2007, Goldman et al., 2006). Therefore, understanding the role of reactive astrocytes in mediating motor neuron degeneration could shed light on both familial and sporadic ALS.
Many studies used mutant mouse models to investigate the influence of astrocytes in motor neuron degeneration in ALS. In chimeric studies, astrocytes carrying SOD1 mutant transgenes were toxic to their neighboring wild-type (WT) motor neurons (Clement et al., 2003). Conversely, astrocyte-specific deletion of the SOD1 transgene in SOD1 mutant mice delayed ALS progression (Ilieva et al., 2009, Wang et al., 2011, Yamanaka et al., 2008). Transplantation of SOD1 mutant astrocytes into WT rodents triggered motor neuron degeneration (Papadeas et al., 2011), whereas transplantation of WT astrocyte precursors delayed disease progression and extended survival of mutant SOD1 mice (Lepore et al., 2008). More recently, it was shown that astrocytes derived from sporadic human ALS patients led to motor neuron degeneration, disorganized neurofilaments, and aggregated ubiquitin after transplantation into mice (Qian et al., 2017). These studies established that mutant astrocytes contribute significantly to ALS pathogenesis.
In addition to in vivo genetic models, co-cultures of motor neurons and astrocytes have been used extensively to study astrocyte toxicity in ALS. Mutant mouse astrocytes carrying SOD1 transgenes showed a clear toxic effect in co-cultures with WT murine or human motor neurons (hMNs) (Di Giorgio et al., 2008, Di Giorgio et al., 2007, Marchetto et al., 2008, Nagai et al., 2007). Remarkably, astrocytes derived from both familial and sporadic human ALS spinal samples were also toxic to cultured murine motor neurons (Haidet-Phillips et al., 2011, Re et al., 2014). Extensive gene-profiling studies using SOD1 mice further showed a high level of concordance between in vivo spinal tissues and cultured astrocytes and motor neurons (Phatnani et al., 2013). Thus, the co-culture system faithfully recapitulates aspects of ALS pathogenesis and can serve as a valuable model to study astrocyte toxicity.
Significant progress has been made on understanding the molecular mechanisms by which astrocytes exert their detrimental influence on motor neurons. One well-studied mechanism is downregulation of glutamate transporters in astrocytes in both murine and human ALS and, consequently, excitotoxicity to motor neurons due to excessive postsynaptic stimulation (Rothstein et al., 1992, Rothstein et al., 1996). Increasing the transporter expression offers neuroprotection (Rothstein et al., 2005). In addition, a number of astrocytic factors have been proposed as potential toxic factors, including inflammatory cytokines, prostaglandins, and reactive oxygen species (de Boer et al., 2014, Di Giorgio et al., 2007, Drachman et al., 2002, Haidet-Phillips et al., 2011, Marchetto et al., 2008, Nagai et al., 2007). Other studies suggested involvement of increased Na/K ATPase-adducin complex or upregulation of the gap junction protein Connexin 43 in astrocyte-mediated toxicity (Almad et al., 2016, Gallardo et al., 2014). A recent study showed that diseased ALS astrocytes in both murine models and human patients potently downregulate expression of major histocompatibility complex 1 (MHC1) of motor neurons; reduction of MHC1 makes motor neurons susceptible to astrocyte-induced cell death (Song et al., 2016). Many of these studies employed pharmacological or genetic tools to inhibit the candidate astrocytic factors and showed efficacy in ameliorating pathology or extending survival of ALS mouse models. These advances provided important therapeutic candidates for further evaluation in human ALS disease.
The degenerative phenotypes of motor neurons in human ALS are complex and involve not only neuronal death but also neurite shrinkage and formation of a multitude of axonal and cytoplasmic inclusions that contain various proteins such as UBIQUITIN, TDP43, P62, and others (reviewed in Blokhuis et al., 2013). Although many studies have firmly established that astrocytic factors can promote motor neuron death, relatively little is known about astrocytic factors that promote formation of protein inclusions in motor neurons, a prominent disease feature of ALS. To address this question, we studied protein aggregation in long-term co-cultures of hMNs and primary mouse astrocytes of different genotypes (WT or mutant hSOD1-G93A) and reactive states (non-reactive or reactive). Our data indicate that the reactive state of astrocytes is an important contributor to motor neuron protein aggregation and neurite degeneration. In particular, in long-term cultures of up to 2 months, reactive astrocytes induced formation of cytoplasmic protein inclusions (UBIQUITIN, P62, and TDP43) and axonal pNF-H aggregates in hMNs. This induction is closely correlated with the reactive state of astrocytes and independent of their genotype (WT or SOD1 mutant). Our studies further indicate that reactive astrocytes exhibit enhanced secretion of TGF-β1, which activates the mammalian target of rapamycin (mTOR) signaling pathway and leads to defects in autophagy in hMNs. Inhibition of TGF-β1 signaling attenuates reactive astrocyte-induced protein aggregation in culture. These results provide insight on reactive astrocyte-mediated signaling pathways that contribute to protein aggregation phenotypes of motor neuron degeneration in ALS.
Results
Reactive Astrocytes Isolated from Adult hSOD1 Mutant Mice Impair hMN Survival and Induce Protein and Neurofilament Inclusions
Previous in vitro studies of astrocytes in ALS have often used astrocytes derived from proliferating glial progenitors isolated from early postnatal brains (Di Giorgio et al., 2008, Di Giorgio et al., 2007, Marchetto et al., 2008, Nagai et al., 2007). Such astrocytes have limited expression of classic “reactive astrocyte” genes. Indeed, prominent reactive gliosis does not appear until adulthood in ALS patients and animal models (Boillee et al., 2006). To model reactive astrocytes in ALS, we developed a method to directly culture astrocytes from spinal cords of 2-month-old SOD1 mutant mice, when reactive gliosis is present (Figure 1A). Our mutant adult astrocyte culture was highly enriched for GFAP+ cells (89% ± 6%) and devoid of TUJ1+ neurons, O4+ oligodendrocytes, and IBA1+ microglia, although a small number of NG2+ cells were present (<1% of total cultured cells) (Figures 1B and S1). qPCR showed that cultured adult astrocytes express many astrocytic genes including Gfap and S100b at levels comparable with postnatal astrocytes (Figure 1C). The mutant adult astrocytes, however, exhibit significant upregulation of multiple inflammatory and reactive factors (Figures 1D–1H). Using immunohistochemistry, we confirmed expression of one of the reactive factors, LCN2, in mutant adult spinal cord and cultured adult astrocytes (Figures 1E and 1G). In contrast, LCN2 was not detectable in mutant postnatal spinal cord or postnatal astrocyte cultures (Figures 1D and 1F).
Figure 1.

Astrocytes from Adult hSOD1G93A Mutant Mice Express Reactive Genes and Attenuate hMN Survival
(A) Schematic representation of astrocyte cultures from early postnatal (P3) or adult (P60) spinal cord of hSOD1G93A mutant mice.
(B) Adult astrocyte cultures are enriched for GFAP+ cells and devoid of Tuj1+ neurons, O4+ oligodendrocytes, and IBA1+ microglia. A small number of NG2+ cells were present. Data presented as mean ± SEM. N = 3 independent experiments.
(C) Adult astrocytes express astrocytic genes at comparable levels as postnatal astrocytes. Data presented as mean ± SEM. N = 3 independent experiments.
(D–G) Adult astrocytes express the reactive astrocyte marker LCN2 in vivo and in vitro (E and G). In contrast, postnatal astrocytes do not express LCN2 (D and F). CHAT labels motor neurons. Scale bars, 100 μM.
(H) Adult astrocytes express proinflammatory factors and reactive genes compared with early postnatal astrocytes. Data presented as mean ± SEM. N = 3 independent experiments.
(I) Experimental outline of our co-culture of mutant astrocytes with purified human wild-type motor neurons.
(J–L) Human motor neurons (hMNs) co-cultured with adult astrocytes showed reduced survival compared with those co-cultured with postnatal astrocytes. Data presented as mean ± SEM. N = 3 independent experiments. ∗p < 0.05. Mann-Whitney test. Scale bars, 100 μM.
To evaluate the interactions between mutant astrocytes and motor neurons, we carried out co-culture studies (Figure 1I). hMNs were generated by differentiating human embryonic stem (hES) cells carrying the Hb9-GFP transgene and purified by fluorescent-activated sorting (Figure S2). In a short-term 3-day culture, hMNs co-cultured with either postnatal astrocytes or adult astrocytes showed no significant difference in hMN number or branching (Figure S2). However, after an extended 30-day culture, there was a modest reduction in hMN number in the adult astrocyte co-culture (Figures 1J–1L), indicating toxicity from mutant adult astrocytes compared with postnatal astrocytes, consistent with prior in vitro studies (Di Giorgio et al., 2008, Di Giorgio et al., 2007, Haidet-Phillips et al., 2011, Marchetto et al., 2008, Nagai et al., 2007, Re et al., 2014).
A seminal pathological hallmark of ALS is the presence of cytoplasmic and axonal inclusions in motor neurons that perturb normal function and viability (Blokhuis et al., 2013). In both sporadic and familial ALS, for example, hMNs exhibit swelling neurites that contain neurofilament aggregates and cytoplasmic P62/UBIQUITIN inclusions (Blokhuis et al., 2013, Carpenter, 1968, Hirano et al., 1984, Leigh et al., 1991, Maekawa et al., 2009, Matsumoto et al., 1990, Mizuno et al., 2006, Strong et al., 2005). In co-cultures with mutant adult astrocytes, we observed bead-like swellings along neurites of hMNs as early as day 30 (9.5% ± 4% of all hMNs) (Figures 2D and S2). In comparison, only 0.75% ± 0.5% of the hMNs showed abnormal axonal swellings when cultured with mutant postnatal astrocytes (Figures 2A and S2). Phosphorylation of neurofilament heavy chain (pNF-H) is linked to disease progression in hSOD1 (G93A) mice and is considered an ALS biomarker (Calvo et al., 2012). Immunohistochemistry showed prominent pNF-H aggregates in the bead structures of hMNs co-cultured with mutant adult astrocytes (Figures 2D, 2G, and S2), but few in hMNs co-cultured with mutant postnatal astrocytes (Figures 2A, 2G, and S2). Cytoplasmic P62+ and UBIQUITIN+ inclusions were not detectable at day 30, but became prominent at day 60 in hMNs co-cultured with mutant adult reactive astrocytes (27% ± 2% and 12.2% ± 0.7% of all hMNs, respectively) (Figures 2E, 2F, 2H, 2I, and S2). In contrast, hMNs developed substantially fewer P62 and UBIQUITIN aggregates (6.3% ± 0.6% and 1.0% ± 0.7% of all hMNs, respectively) in postnatal astrocytes co-cultures (Figures 2B, 2C, 2H, 2I, and S2). These data indicate that reactive astrocytes derived from adult hSOD1 mutant mice have significantly enhanced toxic effects on cultured hMNs, compared with mutant postnatal astrocytes that do not show these reactive features. Our data are consistent with previous observations in ALS patients and animal models that reactive gliosis begins in adulthood and is associated with rapid deterioration of motor neuron health (Schiffer and Fiano, 2004).
Figure 2.

Astrocytes from Adult hSOD1G93A Mutant Mice Strongly Promote Protein and Neurofilament Inclusions in hMNs Compared with Postnatal hSOD1G93A Astrocytes
hMNs co-cultured with mutant postnatal astrocytes showed limited formation of pNF-H, P62, and UBIQUITIN inclusions (A, B, C, G, H, and I). In contrast, these inclusions were significantly increased in co-cultures with mutant adult astrocytes (D, E, F, G, H, and I). Quantifications are presented as mean ± SEM. Data collected from three independent experiments each with triplicates. ∗p < 0.05; ∗∗p < 0.01. Mann-Whitney test. Scale bars, 100 μM.
WT Reactive Astrocytes Reduce hMN Survival and Induce Protein and Neurofilament Inclusions
Our results thus far indicate enhanced motor neuron toxicity from SOD1 reactive astrocytes compared with non-reactive astrocytes. However, it is unclear whether the enhanced toxicity requires presence of the SOD1 transgene. To evaluate this, we obtained WT reactive astrocytes isolated from the cortex of a stab injury model of adult mice. As controls, we used WT postnatal astrocytes. We confirmed the high purity of the astrocytes and their elevated expression of reactive astrocyte markers (Figure S3). In co-culture assays, reactive astrocytes induced a modest decrease in hMN numbers after 30 days, compared with controls (Figures 3A–3D).
Figure 3.

Wild-Type Reactive Astrocytes Decrease hMN Survival and Significantly Induce Protein and Neurofilament Inclusions
(A) Experimental outline of our co-culture of hMNs with either wild-type non-reactive cortical astrocytes or reactive astrocytes activated by stab injury.
(B–D) Co-culture with WT reactive astrocytes reduced hMN survival compared with non-reactive astrocytes. Data presented as mean ± SEM. n = 3 experiments. ∗p < 0.05. Mann-Whitney test. Scale bars, 100 μM.
(E–V) hMNs co-cultured with WT non-reactive astrocytes showed limited or no cytoplasmic inclusions (E, F, K, L, and I–P). In contrast, hMNs in co-culture with reactive astrocytes developed intracellular inclusions that contain TDP43, UBIQUITIN, SOD1, and P62 (G–I and M–O). In addition, hMNs in non-reactive co-cultures developed few axonal swellings (Q, R, U, and V). In contrast, hMNs co-cultured with WT reactive astrocytes had numerous bead-like axonal swellings that were positive for pNF-H and occasionally for Tau (S–V). Data collected from three independent experiments each with triplicates and presented as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Mann-Whitney test. Scale bars, 100 μM.
Immunohistochemistry showed that hMNs co-cultured with non-reactive astrocytes rarely developed axonal abnormalities at day 30 (Figures 3Q, 3R, 3U, 3V, and S3). In contrast, hMNs co-cultured with reactive astrocytes had numerous bead-like axonal swellings that are positive for pNF-H and, occasionally, Tau protein at day 30 (Figures 3S–3V and S3). By day 60, hMNs in reactive cultures also developed significant numbers of cytoplasmic inclusions that are positive for TDP43, UBIQUITIN, hSOD1, and P62 (9.1% ± 0.8%, 4.3% ± 1%, 5% ± 1.1%, and 30.7% ± 2.1% of all hMNs, respectively) (Figures 3G–3I, 3M–3O, and S3). In contrast, hMNs in non-reactive cultures had few inclusions for TDP43, UBIQUITIN, hSOD1, or P62 (0.9% ± 0.8%, 0.5% ± 0.3%, 0% ± 0%, and 1.6% ± 0.3%, respectively) after 60 days (Figures 3E, 3F, 3K, 3L, 3I, 3J, 3O, 3P, and S3). It should be noted that the TDP43 and SOD1 aggregates appeared in a relatively small subset of hMNs and the aggregates were relatively small. In comparison, TDP43 and SOD1 aggregates in human pathology samples are larger in size and/or more abundant per neuron (Blokhuis et al., 2013). Nevertheless, our data indicate that reactive astrocytes can reduce hMN survival and induce axonopathy and proteinopathy in hMNs, which strikingly resembles the degenerative phenotype in ALS. Importantly, reactive astrocytes induced these phenotypes independent of any genetic mutations associated with ALS.
TGF-β1 Secreted by Reactive Astrocytes Induces Axonal and Cytoplasmic Protein Aggregation in hMNs
How do reactive astrocytes induce ALS-like motor neuron phenotype? It is well documented that reactive astrocytes secret proinflammatory factors, which could contribute to environmental stress experienced by hMNs. We evaluated this possibility by analyzing the formation of stress granules (SGs) in hMNs using the SG marker TIA1 (Anderson and Kedersha, 2008). In 3-day co-cultures of hMNs with WT reactive or non-reactive astrocytes, we did not detect any TIA1+ SGs, suggesting that reactive astrocytes did not confer acute stress on hMNs (Figure S4). Stimulation with the acute stress inducer arsenite, however, revealed that hMNs co-cultured with reactive astrocytes were more prone to SG formation, with increased number of neurons bearing SGs and increased number of SGs per neuron (Figure S4). These data indicate increased environmental stress for hMNs co-cultured with reactive astrocytes.
To identify the factors secreted by reactive astrocytes that could mediate hMN pathogenesis, we compiled a list of candidate factors from the existing literature. We confirmed using qPCR analysis that 13 factors were significantly upregulated in our adult reactive astrocyte cultures. To evaluate these factors, hMNs cultured on PDL/laminin-coated dishes were individually treated with these 13 factors at 20 ng/mL for 14 days and analyzed for pNF-H and P62 aggregation (Figure 4A). Treatment with TGF-β1, but not the other factors, resulted in a significant increase of pNF-H and P62 aggregates in hMNs (Figures 4B–4I and S5). At high concentrations (200 ng/mL), other cytokines including IL6, CXCL1, and CXCL12 also induced modest P62 aggregation in subsets of pMNs (Figure S5). Lastly, addition of TGF-β1 in co-cultures of hMNs and non-reactive astrocytes for 60 days led to increased P62 aggregates in hMNs (8% ± 3%) (Figures 4J–4L).
Figure 4.

TGF-β1 Secreted by Reactive Astrocytes Induces Protein Aggregation in hMNs
(A) Schematic diagram of treating human wild-type motor neurons with secreted factors enriched in reactive astrocytes.
(B–I) Of the 14 factors tested, TGF-β1 significantly suppressed hMN branching (B) and induced P62 inclusions (C and E) compared with controls (B, C, and D). TGF-β1 treatment also led to significant axonal swelling and bead-like structures in neurites that often contain pNF-H aggregates (G, H, and I). In contrast, control hMNs have less these swollen, bead structures (F, H, and I). Scale bars, 100 μM.
(J–L) Addition of TGF-β1 protein in co-cultures of hMNs and wild-type non-reactive astrocytes for 60 days led to induction of P62 inclusions in the hMNs.
Data collected from three independent experiments each with triplicates. ∗p < 0.05; ∗∗p < 0.01. Mann-Whitney test. Scale bars, 100 μM.
To further evaluate the effect of astrocyte-derived TGF-β1 at inducing P62 and pNF-H aggregations, we treated hMNs cultured on PDL/laminin-coated dishes with conditioned media from reactive astrocytes (ACM) for 14 days, which consistently induced pNF-H and P62 aggregates (Figures 5A, 5D, 5C, and 5F). Addition of the TGF-β receptor1 inhibitor, RepSox in ACM significantly suppressed aggregate formation (Figures 5B, 5E, 5C, and 5F). Together, these data identified TGF-β1 as an inducer of P62 and pNF-H protein aggregation for hMNs.
Figure 5.

The TGF-β1 Inhibitor Repsox Reduces Protein Aggregation in hMNs Induced by Reactive Astrocytes
Conditioned medium from adult reactive astrocyte cultures (ACM) can induce P62 and pNF-H aggregation in hMNs (A, D, C, and F). Addition of the TGF-β1 inhibitor Repsox to ACM significantly ameliorated this effect (B, E, C, and F), suggesting an important role of TGF-β1 in inducing P62 and pNF-H aggregation. Data collected from three independent experiments each with triplicates. ∗p < 0.05; ∗∗p < 0.01. Mann-Whitney test. Scale bars, 100 μM.
TGF-β1 Induces Autophagy Defects in hMNs through the mTOR Pathway
It is notable that P62 aggregation is significantly enhanced by TGF-β1 treatment or by co-culture with reactive astrocytes. P62 functions as a cargo-recognition protein for autophagy, a cellular pathway that mediates lysosomal degradation of proteins and organelles, but it is also an autophagy substrate (Zaffagnini and Martens, 2016). P62 aggregation and accumulation therefore suggests that TGF-β1 impairs autophagy function in hMNs.
Given that mTOR activation can strongly suppress autophagy (Ganley et al., 2009, Hosokawa et al., 2009, Jung et al., 2009), we examined whether excessive TGF-β1 could stimulate the mTOR pathway and block autophagy in hMNs. Western blot analysis on hMNs treated with TGF-β1 for 14 days revealed a significant upregulation of phosphorylated S6 and P70 compared with controls (Figures 6A–6D), indicating that the mTOR pathway was indeed activated in hMNs upon TGF-β1 treatment. To test whether mTOR activation was impairing autophagy activity, we measured autophagy flux by treating hMNs with a cocktail of lysosomal activity inhibitors, or the autophagy inducer rapamycin, for 8 hr in the presence or absence of TGF-β1. We observed that the increase in the amount of LC3-II upon treatment with lysosomal inhibitors was notably reduced in TGF-β1-treated hMNs (fold increase 1.78 ± 0.03) compared with DMSO control-treated ones (fold increase 4.23 ± 0.3), suggesting an impairment of autophagy activity (Figures 6E and 6F). Consistent with this observation, the increase in LC3-II observed in control hMNs exposed to rapamycin was also higher than in the TGF-β1-treated hMNs (Figures 6E and 6F). These data together indicate reduced LC3-II turnover in hMNs exposed to TGF-β1, suggesting a partial blockage of the autophagy flux. Furthermore, TGF-β1-treated hMNs also displayed elevated P62 protein levels (Figure 6E), but not elevated P62 mRNA levels (data not shown), consistent with a blockage in autophagy activity. Addition of an mTOR inhibitor, rapamycin (200 nM), in ACM-treated hMNs reduced the amount of P62 aggregates in hMNs (Figures 6G–6I), suggesting that TGF-β1 works through the TOR pathway to induce P62 aggregation.
Figure 6.

TGF-β1 Activates mTOR Pathway in Motor Neurons
(A) Schematic diagram of treating purified hMNs with TGF-β1.
(B–D) Western blot analysis revealed significantly increased phosphorylation of P70 (p-P70) and S6 (p-S6) in TGF-β1 treated hMNs compared with control experiments, indicating activation of mTOR pathway by TGF-β1.
(E and F) hMNs were treated with lysosomal inhibitors or the autophagy inducer rapamycin for 8 hr. Western blot analysis showed reduced LC3-II turnover in hMNs that were exposed to TGF-β1, suggesting a partial blockage of the autophagy flux. Quantification of LC3-II levels are shown relative to control-treated hMNs (DMSO or TGF-β1-treated only). Further, hMNs exposed to TGF-β1 showed elevated levels of P62 compared with control-treated hMNs, confirming a defect in autophagy activity.
(G–I) Addition of an mTOR inhibitor (rapamycin) to hMNs with ACM significantly attenuated P62 aggregation, suggesting that TGF-β1 induces P62 aggregation through the mTOR pathway.
Data collected from three independent experiments each with triplicates. ∗p < 0.05. Mann-Whitney test.
Lastly, we examined whether the mTOR pathway is also activated in vivo in the hSOD1-G93A mutant mouse model (Figure S6). The ventral part of the spinal cord, where lower motor neurons reside, was micro-dissected from hSOD1-G93A transgenic mice and WT littermate controls. Western blot analysis showed a significant upregulation of P62 and UBIQUITIN in hSOD1-G93A mice compared with littermate controls, indicating protein degradation defects (Figure S6). We also observed upregulation of phosphorylated S6, phosphatidylinositol 3-kinase (PI3K), and AKT, upstream activators of the mTORC1 pathway (Figure S6). Collectively, these data suggest that TGF-β1 activation of the PI3K-AKT-mTOR pathway could potentially lead to autophagy defects in motor neurons and contribute to abnormal protein aggregation and cellular toxicity.
Discussion
Reactive gliosis is a prominent feature of ALS. Prior studies have noted that the reactive state of astrocytes could be a contributor to ALS pathogenesis (Di Giorgio et al., 2008, Di Giorgio et al., 2007, Haidet-Phillips et al., 2011, Marchetto et al., 2008, Nagai et al., 2007, Re et al., 2014). However, the mechanism by which reactive astrocytes (particularly WT reactive astrocytes) contributes to ALS is not fully understood. Our data strongly suggest that the reactive state of astrocytes, independent of genetic mutations, is important in motor neuron degeneration. In particular, WT reactive astrocytes can induce characteristic ALS-like degenerative phenotypes in hMNs that include axonal swellings containing neurofilament aggregates, cytoplasmic inclusions containing P62, UBIQUITIN, and TDP43 aggregates, and reduced neuronal survival. Based on these observations, we propose that reactive astrocytes can potentially play a significant role in ALS induction and/or progression for both familial and sporadic cases. In familial ALS, it is possible that genetic defects lead to progressive tissue damage over time and trigger chronic reactive gliosis; this gliosis, in turn, could accelerate motor neuron degeneration and death via secretion of many factors. Likewise, reactive astrocytes can also potentially serve as an important mediator of sporadic ALS. There is emerging evidence that traumatic injuries may trigger or accelerate ALS (Chen et al., 2007). It is also well documented that traumatic injuries can lead to chronic reactive gliosis (Sofroniew, 2005). It is conceivable, therefore, that reactive gliosis serves as a link between trauma and the induction and/or progression of sporadic ALS.
To date several in vitro astrocyte-motor neuron co-culture models have been established to identify neurotoxic factors secreted by astrocytes (Bilsland et al., 2008, Di Giorgio et al., 2008, Di Giorgio et al., 2007, Ferraiuolo et al., 2011, Haidet-Phillips et al., 2011, Marchetto et al., 2008, Nagai et al., 2007, Phatnani et al., 2013, Re et al., 2014). Many of these studies documented relatively rapid motor neuron death, which often occurs within the initial few weeks of culture time. We observed modest neuronal loss in the first 4 weeks of culture. In addition, we were able to maintain the co-culture for up to 8 weeks, providing a chronic stress environment for motor neurons that mimics aspects of the slow degenerative conditions in vivo. After prolonged stress, the surviving motor neurons developed prominent protein aggregations in axons and cell bodies of hMNs. Thus, our model revealed two phases of motor neuron pathology when co-cultured with reactive astrocytes: an initial phase where a subpopulation of neurons was lost without developing prominent protein inclusions, and a second phase in which the surviving neurons developed protein inclusions and axonopathy. Our culture model therefore could be a particularly useful tool to investigate mechanisms of protein aggregation in ALS disease.
The toxic astrocytic factor(s) that kill motor neurons in our culture system remain unclear, but past studies have identified a number of toxic mechanisms and toxic factors, including glutamate transporters, prostaglandins, reactive oxygen species, proinflammatory cytokines, gap junction proteins, among others, which induce motor neuron death (Boillee et al., 2006, Di Giorgio et al., 2007, Gallardo et al., 2014, Haidet-Phillips et al., 2011, Ilieva et al., 2009, Marchetto et al., 2008, Nagai et al., 2007, Philips and Rothstein, 2014, Re et al., 2014). Comparatively less is known about astrocytic factors that contribute to protein aggregation in hMNs. In this study, we identified TGF-β1 as a reactive astrocyte-released factor that can induce P62 aggregation and pNF-H aggregation. Many studies in the past have suggested TGF-β1 as a pro-survival factor for motor neurons (Bottner et al., 2000, Day et al., 2005, Katsuno et al., 2010, Katsuno et al., 2011). However, the role of TGF-β1 in inducing protein aggregation during chronic stress conditions is unclear. Our data are consistent with reports of pSmad aggregation in spinal motor neurons in autopsied ALS patients (Nakamura et al., 2008), and a recent study in SOD1 mutant mice suggesting that astrocytic TGF-β1 overproduction in astrocytes reduces motor neuron survival (Endo et al., 2015). It has also been reported that chronic production of TGF-β1 in brain astrocytes of transgenic mice promoted amyloid deposition and microvascular degeneration in the brain (Wyss-Coray et al., 2000). These observations together suggest that the role of TGF-β1 may be complex in a chronic degenerative disease setting, and that certain levels of TGF-β1 may well be pro-survival, whereas chronic exposure to high levels of TGF-β1, perhaps in combination with other proinflammatory cytokines, could induce protein aggregation and lead to certain disease features.
Experimental Procedures
Astrocyte Culture and Animal Surgery
Neonatal astrocytes (mutant/WT) were derived from proliferating neural progenitors from both spinal cord and brain. After removal of the meninges, gray matter tissue from the cerebral cortex or spinal cord of mutant and WT P1-P2 mice was dissected and dissociated mechanically. Subsequently, cells were centrifuged for 5 min at 1,000 rpm, resuspended, and plated in a medium consisting of DMEM/F12 (Gibco), 3.5 mM glucose (Sigma), 10% fetal calf serum (Gibco), 5% horse serum (Gibco), and penicillin/streptomycin (Gibco), supplemented with B27 (Gibco), 10 ng/mL epidermal growth factor (EGF, Roche), and 10 ng/mL fibroblast growth factor 2 (FGF2, Roche). Contaminating oligodendrocyte precursor cells were removed by brusquely shaking the culture flasks several times. Cells were passaged after a week using trypsin/EDTA (Gibco) and plated on poly-D-lysine- and laminin-coated 96-well plates in the same medium. The vast majority of the cells (>90%) in these cultures were positive for GFAP.
To derive adult astrocytes from the mutant hSOD1-G93A line, spinal cords were harvested at the presymptomatic phase (2 months). Spinal cords were dissected and the meninges were removed in ice-cold Hank’s balanced salt solution (HBSS) supplemented with HEPES, NAC (N-acetylcysteine), and glucose. Neural tissues were dissected, minced, and treated with an enzyme mixture containing papain (40 U), DNAse I (1,000 U), elastase (20 U), and dispase (20 U) for 30 min at 3°C. Tissues were mechanically triturated with a fire-polished Pasteur pipette several times during the enzyme treatment. The enzyme activity was stopped by addition of an equal volume of solution containing 4% BSA in EBSS. The resulting mixture was then filtered, resuspended in 0.9 M sucrose in HBSS, and centrifuged for 20 min at 2,000 rpm. The cell pellet was resuspended in Earle's balanced salt solution containing 4% BSA and centrifuged again at 1,500 rpm for 10 min. The final cell pellet was resuspended in serum-free culture medium (50% neurobasal, 50% DMEM:F12, Glutamax, P/S, 1 mM sodium pyruvate, 10 μg/mL N-acetylcysteine, 1× SATO, 10 μg/mL heparin-binding EGF ) and plated onto poly-ornithine- and laminin-coated 6-well plates (tissues from ten dissected injury regions per plate). After 3 days in serum-free medium, the culture was changed to serum-plus medium for 7–10 days (serum-free medium plus 10% fetal bovine serum, 5% horse serum, supplemented with EGF and basic FGF, every other day) before replating at confluence for subsequent co-culture experiments.
To harvest WT reactive astrocytes, we performed a cortical stab injury and harvested tissues surrounding the injury site (within 1–2 mm of cut site). This was followed by dissociation and plating, as described above. We routinely obtained approximately one million cultured astrocytes from 20 micro-dissected brain injury samples.
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Harvard University. The use of human embryonic cell lines was approved by the Committee on the Use of Human Subjects of Harvard University.
Differentiation of hES Cells into Motor Neurons
A WT hES line carrying the Hb9-GFP transgene was used for derivation of hMNs. Confluent hES cells were washed with PBS and dissociated with accutase into single cells. Cells (6–9 × 105) were resuspended into mTeSR supplemented with 10 μM Rho-associated kinase inhibitor Y27632 (Sigma) and cultured in Matrigel-coated 6-well plates. At day 1, 50% more mTeSR was added, and the final volume was supplemented with 10 μM SB431542 (Sigma) and 1 μM Dorsomorphin (EMD Biosciences, no. 171261). At day 3, medium was changed into 1:1 mTeSR and KOSR medium, supplemented with 10 μM SB431542 and 1 μM Dorsomorphin. KOSR medium consisted of 15% KOSR in DMEM/F12 (Gibco) with 10,000 U penicillin and 1 mg/mL streptomycin (PS; Gibco), L-glutamine, and nonessential amino acids (NEAA; Invitrogen). The next day, the medium was changed into 1× KOSR medium supplemented with 10 μM SB431542 and 1 μM Dorsomorphin. On day 5 medium was switched to Neural Induction Medium (NIM) supplemented with 10 μM SB431542, 1 μM Dorsomorphin, 0.2 μg/mL ascorbic acid (AA; Sigma), 1 μM retinoic acid (RA; Sigma), and 1 μM Smoothened Agonist 1.3 (SAG; EMD Biosciences). NIM medium consisted of DMEM/F12 with P/S, L-glutamine, NEAA, 0.16% D-(+)-glucose (Sigma), 2 μg/mL heparin sulfate, and 1% N2 supplement (Gibco). From days 7 to 24, the medium was changed every other day with NIM medium supplemented with 0.2 μg/mL AA (Sigma), 1 μM RA (Sigma), and 1 μM SAG (EMD Biosciences).
FACS Purification of Motor Neuron
At day 14 of stem cell differentiation, cell clusters were dissociated and centrifuged at 1,000 rpm for 5 min. The cells were washed with PBS and incubated for 45 min at 37°C with 20 U of papain and 1,000 U of DNase I (Worthington Biochemical Corporation) in PBS. GFP-positive motor neurons were purified using a BD Biosciences LSRII flow cytometer.
Motor Neuron and Astrocyte Co-Culture
Twenty thousand postnatal (mutant/WT) and adult (mutant/WT) astrocytes were plated per well in 384-well plates pre-coated with poly-ornithine and laminin to generate a confluent astrocyte monolayer. Purified motor neurons were plated onto the astrocyte monolayer at 2,000 cells per well. Co-cultures were carried out in hMN medium, N2B27 medium, containing DMEM:F-12, 1% N2 supplement (Invitrogen), 1% B27 supplement (Invitrogen), 0.2 mM AA (Sigma-Aldrich), 0.16% D-glucose (Sigma-Aldrich), 1% NEAA (Invitrogen), 2 mM GlutaMAX-I (Invitrogen), 50 U/mL penicillin, and 50 g/mL streptomycin (Invitrogen). The N2B27 medium was supplemented with neurotrophic factors (GDNF, BDNF, CNTF, 10 ng/mL, R&D Systems) each time while changing the medium (every other day).
Motor Neuron Treatment with Various Cytokines and Inhibitors
hMNs were fluorescence-activated cell sorting (FACS) purified and plated on PDL/laminin-coated 384-well plates at a density of 5,000 cells/well. One day after cell plating, motor neurons were treated with 0.1% BSA in PBS or cytokines (TGF-β1, R&D Systems, IL-6, CXCL1, CXCL12, INF-ϒ, IL-1β, TNF-α, GDNF, BDNF, CNTF, LCN2, SERPINA3 [Peprotech]) at three concentrations, 2, 20, and 200 ng/mL. The media containing the various cytokines were changed every other day for 14 days. For adult conditioned medium (ACM) experiments, TGF-β1 inhibitor (RepSox; 8 μm) or mTOR inhibitor (rapamycin; 200 nM, R&D Systems) were added to ACM 1 day after plating FACS-purified hMNs. After 14 days of culture, motor neurons with various treatments were either fixed for analysis or used for western blot analysis. For autophagy flux analysis the cocktail of lysosomal inhibitors contained E-64-D (10 mg/mL, Enzo), pepstatin A (10 mg/mL, Tocris), and leupeptin (100 μM, Fisher).
Western Blot
Cells were lysed in 50 mM Tris-HCl (pH 6.8) containing glycerol 10%, 2% SDS, and protease and phosphatase inhibitors, and boiled at 95°C for 15 min. Protein quantification was performed with a BCA Protein Assay Kit (Thermo Scientific) following the manufacturer's instructions. Protein lysates were resolved on an SDS-PAGE gels (Bio-Rad), transferred to polyvinylidene fluoride membranes (Trans-Blot Turbo Transfer Packs, Bio-Rad) for 10 min at 2.5 V using the Trans-Blot Turbo Transfer System (Bio-Rad), blocked in 5% nonfat dry milk (Quality Biological) at room temperature for 1 hr, and probed for the indicated antibodies. Densitometric analysis was performed on scanned autoradiographs using the Quantity One software (Bio-Rad).
Motor Neuron Treatment with ACM and Inhibitors
For ACM experiments, the TGF-β1 inhibitor RepSox (8 μm) or the mTOR inhibitor rapamycin (20 nm) were added to ACM 1 day after plating FACS-purified hMNs. The medium was changed every other day. After 14 days of culture, motor neurons with various treatments were either fixed for analysis or used for western blot analysis.
Arsenite Treatment
To induce SG formation, 0.5 mM sodium arsenite (Sigma) was added to neuronal medium without growth factors for 1 hr. Cells were fixed immediately after treatment for analysis.
Immunocytochemistry
Cultured cells were fixed with 4% paraformaldehyde. The primary antibodies that were used include GFP (Invitrogen; A10262), GFAP (Dako; Z0334), S100B (Sigma; AMAB91038), pNF-H (Millipore; MAB1592), LCN2 (Millipore; AB2267), MAP2 (Millipore; AB5622), TAU (Millipore; T9450-200UL), UBIQUITIN (Dako; Z0458), UBIQUITIN (Santa Cruz, SC-9133; for western), hSOD1 (Sigma; HPA001401), and TIA1(Santa Cruz; sc1751), P62 (Abcam, ab56416), CHAT (Millipore; AB143), LC3 (Cell Signaling, 2775), p-P70S6K1 (Thr389; Cell Signaling, 9234P), P70S6K1(Cell Signaling, 2708S), p-S6 (Ser235/236; Cell Signaling, 2211s), S6 (Cell Signaling, 2217S), Phospho-Akt (Ser473; Cell Signaling, 9271S), Akt (Cell Signaling, 4685S), Phospho-PI3K p85 ((Tyr458)/p55(Tyr199); Cell Signaling, 4228S), β-actin (Cell Signaling, 3700S), and β-tubullin (Abcam, ab6046). Secondary antibodies were obtained from Jackson ImmunoResearch. Pictures were taken with a Nikon Eclipse fluorescence microscope.
qRT-PCR
Total RNA was isolated from primary astrocytes using RNeasy Micro Kit (QIAGEN) or Trizol (Invitrogen), and treated with DNaseI (Life Technologies) to remove the DNA contamination. RNA was converted to cDNA with a cDNA Synthesis Kit (Bio-Rad). qRT-PCR was performed with SYBR green master mix (Bio-Rad) on Bio-Rad iCycler RT-PCR detection system according to the manufacturer’s instructions.
Author Contributions
P.T. and Q.Z. designed the experiments; P.T., N.R.M., J.R.K., A.S.B., S.A., J.S., and W.L. performed the experiments; P.T., N.R.M., and S.A. analyzed the data; P.T., N.R.M., and Q.Z. interpreted the data; P.T. and Q.Z. wrote the manuscript.
Acknowledgments
We thank Adrian Zumsteg for sharing plasmids and reagents and the Bauer FACS core facility for FACS analysis. This study was supported by awards from the Harvard Stem Cell Institute (DP-0065-10-00) and a collaborative agreement with GlaxoSmithKline to Q.Z.
Published: July 13, 2017
Footnotes
Supplemental Information includes six figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.06.008.
Supplemental Information
References
- Al-Chalabi A., Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- Almad A.A., Doreswamy A., Gross S.K., Richard J.P., Huo Y., Haughey N., Maragakis N.J. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia. 2016;64:1154–1169. doi: 10.1002/glia.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P., Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Barres B.A. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Bilsland L.G., Nirmalananthan N., Yip J., Greensmith L., Duchen M.R. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J. Neurochem. 2008;107:1271–1283. doi: 10.1111/j.1471-4159.2008.05699.x. [DOI] [PubMed] [Google Scholar]
- Blokhuis A.M., Groen E.J., Koppers M., van den Berg L.H., Pasterkamp R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:777–794. doi: 10.1007/s00401-013-1125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bottner M., Krieglstein K., Unsicker K. The transforming growth factor-betas: structure, signaling, and roles in nervous system development and functions. J. Neurochem. 2000;75:2227–2240. doi: 10.1046/j.1471-4159.2000.0752227.x. [DOI] [PubMed] [Google Scholar]
- Burda J.E., Bernstein A.M., Sofroniew M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016;275 Pt 3:305–315. doi: 10.1016/j.expneurol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo A.C., Manzano R., Atencia-Cibreiro G., Olivan S., Munoz M.J., Zaragoza P., Cordero-Vazquez P., Esteban-Perez J., Garcia-Redondo A., Osta R. Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS One. 2012;7:e32632. doi: 10.1371/journal.pone.0032632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology. 1968;18:841–851. doi: 10.1212/wnl.18.9.841. [DOI] [PubMed] [Google Scholar]
- Chen H., Richard M., Sandler D.P., Umbach D.M., Kamel F. Head injury and amyotrophic lateral sclerosis. Am. J. Epidemiol. 2007;166:810–816. doi: 10.1093/aje/kwm153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement A.M., Nguyen M.D., Roberts E.A., Garcia M.L., Boillee S., Rule M., McMahon A.P., Doucette W., Siwek D., Ferrante R.J. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Day W.A., Koishi K., Nukuda H., McLennan I.S. Transforming growth factor-beta 2 causes an acute improvement in the motor performance of transgenic ALS mice. Neurobiol. Dis. 2005;19:323–330. doi: 10.1016/j.nbd.2005.01.010. [DOI] [PubMed] [Google Scholar]
- de Boer A.S., Koszka K., Kiskinis E., Suzuki N., Davis-Dusenbery B.N., Eggan K. Genetic validation of a therapeutic target in a mouse model of ALS. Sci. Transl. Med. 2014;6:248ra104. doi: 10.1126/scitranslmed.3009351. [DOI] [PubMed] [Google Scholar]
- Di Giorgio F.P., Carrasco M.A., Siao M.C., Maniatis T., Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio F.P., Boulting G.L., Bobrowicz S., Eggan K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Drachman D.B., Frank K., Dykes-Hoberg M., Teismann P., Almer G., Przedborski S., Rothstein J.D. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann. Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- Endo F., Komine O., Fujimori-Tonou N., Katsuno M., Jin S., Watanabe S., Sobue G., Dezawa M., Wyss-Coray T., Yamanaka K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015;11:592–604. doi: 10.1016/j.celrep.2015.03.053. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L., Higginbottom A., Heath P.R., Barber S., Greenald D., Kirby J., Shaw P.J. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:2627–2641. doi: 10.1093/brain/awr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo G., Barowski J., Ravits J., Siddique T., Lingrel J.B., Robertson J., Steen H., Bonni A. An alpha2-Na/K ATPase/alpha-adducin complex in astrocytes triggers non-cell autonomous neurodegeneration. Nat. Neurosci. 2014;17:1710–1719. doi: 10.1038/nn.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I.G., Lam du H., Wang J., Ding X., Chen S., Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S.M., Tanner C.M., Oakes D., Bhudhikanok G.S., Gupta A., Langston J.W. Head injury and Parkinson's disease risk in twins. Ann. Neurol. 2006;60:65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips A.M., Hester M.E., Miranda C.J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M.J., Foust K.D. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby M.E., Sofroniew M.V. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A., Nakano I., Kurland L.T., Mulder D.W., Holley P.W., Saccomanno G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984;43:471–480. doi: 10.1097/00005072-198409000-00002. [DOI] [PubMed] [Google Scholar]
- Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S., Natsume T., Takehana K., Yamada N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H., Polymenidou M., Cleveland D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.H., Jun C.B., Ro S.H., Kim Y.M., Otto N.M., Cao J., Kundu M., Kim D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M., Adachi H., Minamiyama M., Waza M., Doi H., Kondo N., Mizoguchi H., Nitta A., Yamada K., Banno H. Disrupted transforming growth factor-beta signaling in spinal and bulbar muscular atrophy. J. Neurosci. 2010;30:5702–5712. doi: 10.1523/JNEUROSCI.0388-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M., Adachi H., Banno H., Suzuki K., Tanaka F., Sobue G. Transforming growth factor-beta signaling in motor neuron diseases. Curr. Mol. Med. 2011;11:48–56. doi: 10.2174/156652411794474356. [DOI] [PubMed] [Google Scholar]
- Leigh P.N., Whitwell H., Garofalo O., Buller J., Swash M., Martin J.E., Gallo J.M., Weller R.O., Anderton B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114:775–788. doi: 10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- Lepore A.C., Rauck B., Dejea C., Pardo A.C., Rao M.S., Rothstein J.D., Maragakis N.J. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 2008;11:1294–1301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S., Leigh P.N., King A., Jones E., Steele J.C., Bodi I., Shaw C.E., Hortobagyi T., Al-Sarraj S. TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology. 2009;29:672–683. doi: 10.1111/j.1440-1789.2009.01029.x. [DOI] [PubMed] [Google Scholar]
- Marchetto M.C., Muotri A.R., Mu Y., Smith A.M., Cezar G.G., Gage F.H. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Matsumoto S., Hirano A., Goto S. Ubiquitin-immunoreactive filamentous inclusions in anterior horn cells of Guamanian and non-Guamanian amyotrophic lateral sclerosis. Acta Neuropathol. 1990;80:233–238. doi: 10.1007/BF00294639. [DOI] [PubMed] [Google Scholar]
- Mizuno Y., Amari M., Takatama M., Aizawa H., Mihara B., Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006;249:13–18. doi: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- Nagai M., Re D.B., Nagata T., Chalazonitis A., Jessell T.M., Wichterle H., Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M., Ito H., Wate R., Nakano S., Hirano A., Kusaka H. Phosphorylated Smad2/3 immunoreactivity in sporadic and familial amyotrophic lateral sclerosis and its mouse model. Acta Neuropathol. 2008;115:327–334. doi: 10.1007/s00401-007-0337-z. [DOI] [PubMed] [Google Scholar]
- Papadeas S.T., Kraig S.E., O'Banion C., Lepore A.C., Maragakis N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:17803–17808. doi: 10.1073/pnas.1103141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V., Heneka M.T., Montana V., Oliet S.H., Schousboe A., Haydon P.G., Stout R.F., Jr., Spray D.C., Reichenbach A., Pannicke T. Glial cells in (patho)physiology. J. Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani H., Maniatis T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015;7:a020628. doi: 10.1101/cshperspect.a020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani H.P., Guarnieri P., Friedman B.A., Carrasco M.A., Muratet M., O'Keeffe S., Nwakeze C., Pauli-Behn F., Newberry K.M., Meadows S.K. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA. 2013;110:E756–E765. doi: 10.1073/pnas.1222361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T., Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- Philips T., Rothstein J.D. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2014;262 Pt B:111–120. doi: 10.1016/j.expneurol.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian K., Huang H., Peterson A., Hu B., Maragakis N.J., Ming G.L., Chen H., Zhang S.C. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports. 2017;8:843–855. doi: 10.1016/j.stemcr.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re D.B., Le Verche V., Yu C., Amoroso M.W., Politi K.A., Phani S., Ikiz B., Hoffmann L., Koolen M., Nagata T. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein J.D., Martin L.J., Kuncl R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein J.D., Dykes-Hoberg M., Pardo C.A., Bristol L.A., Jin L., Kuncl R.W., Kanai Y., Hediger M.A., Wang Y., Schielke J.P. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein J.D., Patel S., Regan M.R., Haenggeli C., Huang Y.H., Bergles D.E., Jin L., Dykes Hoberg M., Vidensky S., Chung D.S. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Rowland L.P., Shneider N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Schiffer D., Fiano V. Astrogliosis in ALS: possible interpretations according to pathogenetic hypotheses. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004;5:22–25. doi: 10.1080/14660820310016822. [DOI] [PubMed] [Google Scholar]
- Seifert G., Schilling K., Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat. Rev. Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Sofroniew M.V. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sofroniew M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Miranda C.J., Braun L., Meyer K., Frakes A.E., Ferraiuolo L., Likhite S., Bevan A.K., Foust K.D., McConnell M.J. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 2016;22:397–403. doi: 10.1038/nm.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong M.J., Kesavapany S., Pant H.C. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy? J. Neuropathol. Exp. Neurol. 2005;64:649–664. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- Taylor J.P., Brown R.H., Jr., Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valori C.F., Brambilla L., Martorana F., Rossi D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2014;71:287–297. doi: 10.1007/s00018-013-1429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Gutmann D.H., Roos R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2011;20:286–293. doi: 10.1093/hmg/ddq463. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T., Lin C., Sanan D.A., Mucke L., Masliah E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am. J. Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H., Takahashi R., Misawa H., Cleveland D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini G., Martens S. Mechanisms of selective autophagy. J. Mol. Biol. 2016;428:1714–1724. doi: 10.1016/j.jmb.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.