

Abstract
Background
Atopic status of the mother and maternal exposure to environmental factors is associated with increased asthma risk. Moreover, animal models demonstrate that exposure to allergens in strongly sensitized mothers influences offspring asthma development, suggesting that in utero exposures can influence offspring asthma. However, it is unclear whether maternal exposure to common human allergens like house dust mite (HDM), in the absence of additional adjuvants, influences offspring asthma development.
Objective
To determine if maternal HDM exposure influences asthma development in offspring.
Methods
Pregnant female mice were exposed to PBS or HDM during pregnancy. Using offspring of PBS or HDM-exposed mothers, the magnitude of HDM or Aspergillus fumigatus (AF) extract-induced airway hyperresponsiveness (AHR), airway inflammation, immunoglobulin production, Th2-associated cytokine synthesis and pulmonary dendritic cell activity was assessed.
Results
Compared to offspring of PBS-exposed mothers, offspring of HDM-exposed mothers demonstrate increased AHR, airway inflammation, Th2 cytokine production, immunoglobulin levels and a modest decrease in the phagocytic capacity of pulmonary macrophage populations following HDM exposure. Increased sensitivity to AF-induced airway disease was not observed. Offspring of HDM-exposed B cell deficient mothers also demonstrated increased HDM-induced AHR, suggesting transfer of maternal immunoglobulins is not required.
Conclusions
Our data demonstrate that maternal exposure to HDM during pregnancy increases asthma sensitivity in offspring in an HDM-specific manner, suggesting that vertical transmission of maternal immune responses may be involved. These findings have important implications for regulation of asthma risk, and suggest that exposure to HDM in the developed world may have under-appreciated influences on the overall prevalence of allergic asthma.
Keywords: Asthma, Allergen, Pregnancy, Maternal Exposures, Environment
Introduction
Allergic asthma is one of the most common immunological disorders of children in developed nations1 and more than 300 million people suffer from allergic asthma worldwide2. The incidence of asthma has increased over the past few decades3. While there is a genetic component to asthma heritability4–7, the rise in asthma incidence is too rapid to be ascribed a purely genetic basis, suggesting that changes in the environment are influencing disease development in genetically susceptible populations8–10. Recent studies suggest that exposure to specific environmental stimuli during critical early life periods influence the development of asthma later in life. For example, childhood exposure to microbial products11, cigarette smoke12, 13, or environmental pollutants14 influences asthma development later in life. This critical developmental window appears to extend into the prenatal period as well, as in utero exposures to these factors also influence asthma risk13, 15–18, as do alterations in maternal diet19, 20. Similarly, infection with the helminthic parasite Schistosoma mansoni increases development of asthma in offspring if pregnancy was initiated during the “Th2 phase” of the anti-parasitic immune response21. Consistent with the existence of such a “prenatal window”, maternal asthma is a risk factor for the development of asthma, whereas paternal asthma does not confer as great a risk22–26 suggesting that maternal exposures/factors can contribute to asthma development. However, the precise nature of these factors remains unclear.
One important risk factor for the development of asthma is allergen sensitization27, 28. Given that asthma development can be influenced by maternal asthmatic status22–26, and exposures occurring in the prenatal period exert a profound influence on asthma development, we speculate maternal exposure to allergens themselves may alter asthma development in offspring. Supporting this, pre-sensitization of female mice to ovalbumin (OVA) in the presence of alum, and OVA re-exposure during pregnancy increased asthma development in offspring of exposed mothers29–32. Prior sensitization of female mice to Aspergillus fumigatus extract, and re-exposure during early pregnancy increased airway eosinophilia, and IL-4 promoter hypomethylation in grandoffspring of exposed mothers33. Intra-uterine injection of OVA or house dust mite (HDM)-derived related proteins (Der P2) at very high concentrations (>5 μg) triggered the development of fatal anaphylaxis and pronounced airway dysfunction in exposed fetuses34. However, OVA is not an aeroallergen, A. fumigatus exposure is not predictive of asthma development in humans35, the co-administration of alum makes it difficult to determine whether the observed effects are allergen or adjuvant-driven and while allergen has been detected in the amniotic fluid in humans, it is typically present at ng/ml concentrations36. As such, it remains unclear whether maternal exposure to common allergens can also influence asthma development in offspring.
In humans, exposure to HDM is regarded as a powerful driver of asthma as 1) there is a dose-response relationship between HDM exposure and asthma development that does not exist for other common indoor aeroallergens (e.g. cat or dog dander)37–39; 2) 50 – 80% of asthmatics demonstrate evidence of a HDM-specific immune response40; and 3) locales where HDM responses are not strongly linked to the development of allergic asthma are regions with low relative humidity (New Mexico, Northern Sweden41, 42), which naturally limits the growth of dust mite species. Given the strong association between HDM and allergic sensitization in humans, we sought to determine if maternal exposure during pregnancy to a common human allergen, house dust mite (HDM), in the absence of additional adjuvants, could influence the development of asthma in offspring of exposed mothers using a mouse model of experimental asthma.
Methods
For a complete description of the materials and methods used in the murine and in vitro experiments, please see the Methods section in this article’s Online Repository at www.jacionline.org
Mice
Male and female A/J mice (5–6 weeks old) were bred and housed in a specific pathogen-free facility at Cincinnati Children’s Hospital (Cincinnati, OH). Cincinnati Children’s Hospital IACUC approved all animal protocols.
Mating, and allergen treatment protocols
As A/J mice are relatively poor breeders, naïve, age-matched colony reared A/J females were mated, and allowed to rear one litter to gain experience in rearing pups prior to being selected for further study. A/J mice were chosen as we have demonstrated that this strain develops severe AHR associated with a mixed Th2/Th17 profile43, 44. Age-matched, experienced mothers were subsequently mated in pairs or trios. For delivery, and during the nursing period, mothers were individually housed. After setting up mating cages, females were checked for vaginal plugs daily. The day a mating plug was first observed was considered day 0. At this time mothers were randomized into PBS- or HDM-exposed groups. Mothers were exposed to 100 μl PBS or 20 μg HDM (Greer Labs, Lenoir NC) i.p. on days 0, 4 and 10 of pregnancy. PBS- and HDM-exposed mothers were housed singly, or in pairs, by group. These days were selected to ensure a robust HDM-specific immune response was ongoing during initial lung and immune (thymic) development (days 9–12 of gestation). Offspring of exposed mothers were weaned at 28 days of life, and subsequently housed by maternal exposure. At 6, 8 and 9 weeks of age offspring were anaesthetized with ketamine/xylazine and exposed to PBS (40 μl), HDM (200 μg) as described elsewhere59, or 1 μg Aspergillus fumigatus extract (Greer Labs, Lenoir NC) i.t. as indicated. As C57Bl/6 mice (and μMT mice which are on a C57Bl/6 background) are considered refractory to induction of asthma, a more vigorous regimen of allergen exposure consisting of i.p. HDM at 6 and 7 weeks of life, and i.t. HDM at 8 and 9 weeks of life was used. Mice were sacrificed 72 hours after final allergen/PBS exposure for assessment of airway function and DC recruitment. Where indicated, AlexaFluor405 (Invitrogen, Carlsbad, CA) labeled HDM (AF405-HDM) was used.
Isolation of fetal tisue, placental tissue and amniotic fluid
To isolate fetal tissue, placental tissue and amniotic fluid, pregnant females were euthanized with sodium pentobarbital, the fetuses and placentas were individually excised and amniotic fluid was carefully extracted through a 28 gauge needle and samples were pooled by maternal exposure. Placentas and fetuses were digested and minced as described for the lung (see online repository). Single cell suspensions were generated for flow cytometric analysis.
Statistical Analysis
To determine differences between multiple groups, analysis of variance (ANOVA) was used with post hoc comparisons using Tukey’s test. Significance was assumed at p < 0.05. Titers of HDM-specific IgG1 and IgG2a were Log10 transformed prior to analysis.
Results
Maternal exposure to house dust mite allergen exacerbates asthma in offspring
Given that prenatal exposures to environmental stimuli can influence offspring asthma, and HDM sensitization is a powerful risk factor for asthma development, we sought to determine if maternal exposure to HDM during pregnancy impacted offspring asthma. Female A/J mice (a strain that develops severe AHR associated with a mixed Th2/Th17 response43, 44) were exposed to 20 μg HDM extract on days 0, 4 and 10 of pregnancy to ensure a strong HDM-specific immune response was ongoing during initial pulmonary and immune (thymic) development (days 9–12 of gestation). At 6, 8 and 9 weeks of age, offspring were exposed to 200 μg of intratracheal (i.t.) HDM. AHR was assessed 72 hours after the final allergen exposure. Pooled male and female offspring of HDM-exposed mothers demonstrated a marked increase in AHR (~90% increase over offspring of PBS-exposed mothers - Fig 1A). As maternal asthma status was recently suggested to differentially influence asthma development in male and female offspring45, we also compared HDM-induced AHR in male and female offspring of HDM-exposed mothers. Both male and female offspring of HDM-exposed mothers demonstrated significantly increased AHR compared to offspring from PBS-exposed mothers (Supplementary Figure 1). Increased AHR in offspring of HDM-exposed animals was accompanied by increased recruitment of inflammatory cells in BAL fluid (Fig 1B). Significantly increased numbers of lymphocytes (Fig 1C) and eosinophils (Fig 1D) were observed in offspring of HDM-exposed mothers. While we also observed a trend towards increased the numbers of neutrophils (Fig 1E) and macrophages (Fig 1F) in offspring of HDM-exposed mothers this did not reach statistical significance.
Figure 1.
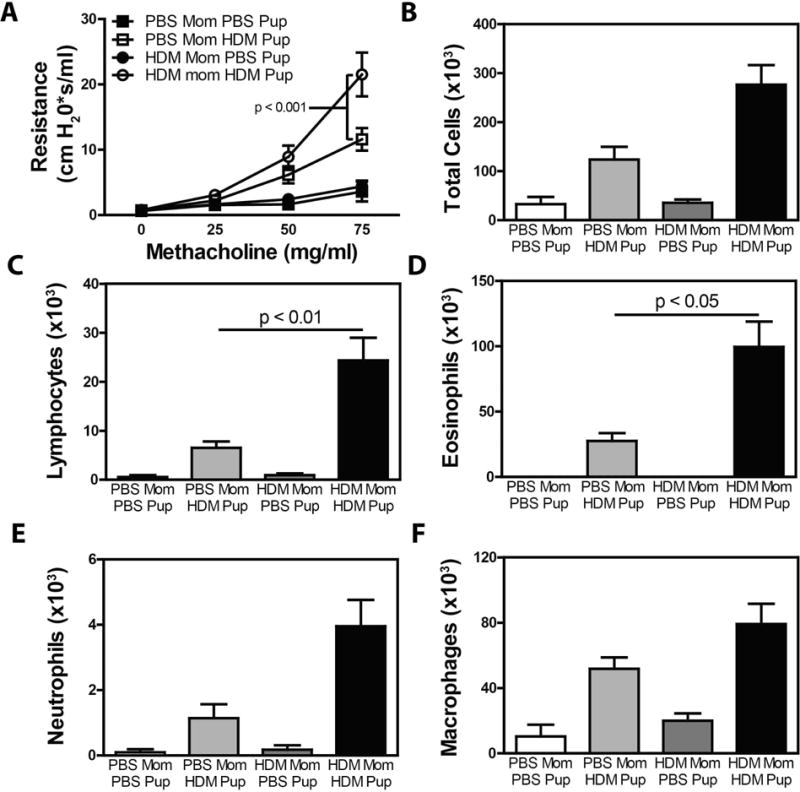
Maternal HDM exposure increases the severity of AHR and airway inflammation in HDM-exposed offspring. Pregnant female mice were exposed to HDM or PBS on days 0, 4 and 10 of pregnancy. Offspring of PBS- and HDM-exposed mothers were exposed to PBS or HDM at 6, 8 and 9 weeks of age and development of AHR was assessed (A). After sacrifice, BAL fluid was collected to assess (B) total BAL cellularity, and the number of (C) lymphocytes, (D) eosinophils, (E) neutrophils and (F) macrophages. Mean + SEM shown. Data represent 7 – 18 mice pooled from 4 experiments.
Increased AHR in offspring of HDM-exposed mothers is associated with increased Th2 cytokine production and HDM-specific immunoglobulin production
As development of asthma is the result of excessive Th2 cytokine production, we also assessed cytokine production in HDM-restimulated lung cell cultures from PBS or HDM-exposed offspring of PBS- or HDM-exposed mothers. HDM exposure induced production of the Th2 cytokines IL-4, IL-5, IL-13, as well as the Th17-associated cytokine IL-17A (Fig 2A–D). There was no appreciable production of IFNγ (data not shown). Consistent with in vivo data, elevated levels of IL-4, IL-5, and IL-13 (Fig 2A, 2B, 2C) were observed in cells isolated from HDM-exposed offspring of HDM-exposed mothers compared to cultures of cells from HDM-exposed offspring of PBS-exposed mothers. In contrast, production IL-17A was comparable in HDM-exposed offspring of PBS- and HDM-exposed mothers (Fig 2D). Moreover accumulation of pulmonary CD4+ T cells expressing IL-13 (Fig 2E) and IL-17A (Fig 2F) was observed in HDM-challenged offspring, and maternal HDM exposure significantly increased the frequency of IL-13+ CD4+ T cells (Fig 2E) but did not influence the frequency of IL-17A+ CD4+ T (Fig 2F) following HDM exposure of offspring. MFI of IL-13 or IL-17A in cytokine expressing cells was not elevated (Supplementary Figure 2), suggesting that the increase in cytokine production observed in HDM-stimulated lung cell cultures was due to increased frequency of cytokine producing cells, and not an increased capacity for cytokine production in individual cells.
Figure 2.
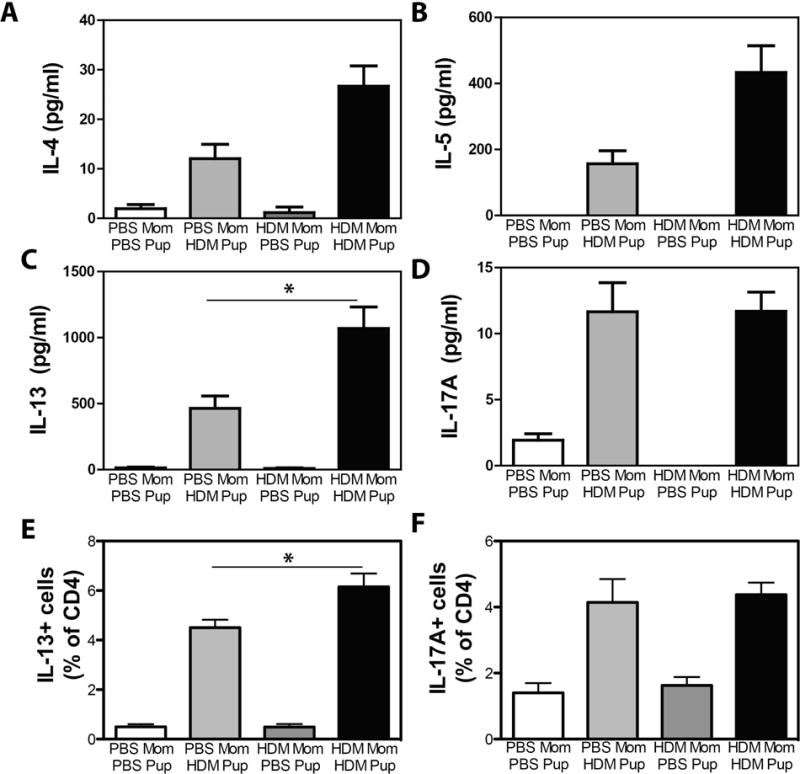
Increased Th2 cytokine production in HDM-exposed offspring of HDM-exposed mothers. Offspring of PBS- and HDM-exposed mothers were treated with PBS or HDM as described in Methods. Single cell suspensions of lung cells were restimulated with HDM, and production of (A) IL-4, (B) IL-5, (C) IL-13 and (D) IL-17A were assessed by ELISA. The frequency of CD4+ cells producing (E) IL-13 and (F) IL-17A was assessed by flow cytometry. Mean + SEM shown. * indicates p < 0.05. Data represent 7 – 18 mice pooled from 4 experiments.
Finally, as development of allergy is also associated with an allergen-specific humoral response, we also examined Ig levels in animals from the various groups. HDM exposure induced substantial increases in levels of total IgE (a reliable surrogate for HDM-specific IgE)46, HDM-specific IgG1, and HDM-specific IgG2a (Fig 3). Offspring of HDM-exposed mothers tended to have higher levels of total IgE (Fig 3A) although this did not reach statistical significance. HDM-specific IgG1 (Fig 3D) and HDM-specific IgG2a levels (Fig 3E) were significantly elevated in HDM-exposed offspring of HDM-exposed mothers compared to HDM-exposed offspring of PBS-exposed mothers. Interestingly, we observed significant levels of HDM-specific IgG1 and IgG2a in PBS-exposed offspring of HDM-exposed mothers, presumably a result of vertical transmission of immunoglobulins from mother to child. Moreover, while differences in the amounts of Der p allergens, HDM and serine protease activity in different batches of commercially available HDM can contribute to differential capacity to induce airway inflammation and AHR, we observed similar results when using two separate batches of HDM (one providing 1.5 μg Der P1 and 12.9 EU LPS per maternal exposure, and another providing 0.65 μg Der p1 and 1.6 EU LPS per maternal exposure). As such, it is unlikely that the observed results were HDM lot-specific. Collectively these data demonstrate that HDM-exposure of mothers during pregnancy is sufficient to drive the development of more intense humoral responses, Th2 cytokine production, pulmonary inflammation and AHR following HDM exposure of offspring.
Figure 3.
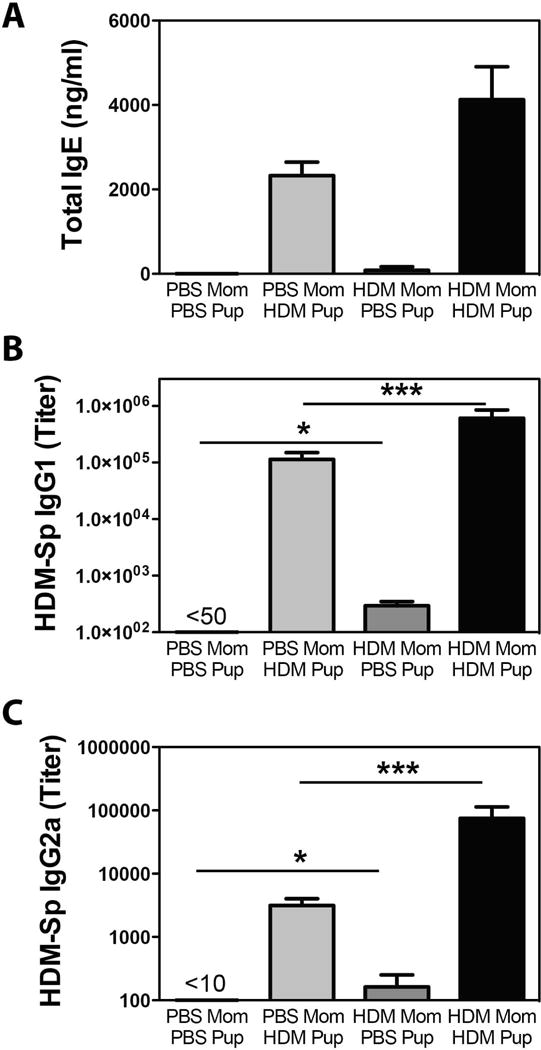
Increased total and HDM-specific IgG production in HDM-exposed offspring of HDM-exposed mothers. Offspring of PBS- and HDM-exposed mothers were treated with PBS or HDM as described in Methods. Serum isolated at time of sacrifice was assayed for (A) total IgE, (B) HDM-specific IgG1 and (C) HDM-specific IgG2a by ELISA. Lowest dilution on plate indicated. Mean + SEM shown. * and *** indicate p < 0.05 and p < 0.001. Data represent 7 – 18 mice pooled from 4 experiments.
Offspring of HDM-exposed mothers display decreased allergen uptake by pulmonary macrophages
As maternal exposure to allergens in the context of adjuvants globally alters DC function in offspring29, we also examined the impact of maternal HDM exposure on in vivo pulmonary DC function in offspring. HDM exposure increased the numbers of most DC subsets in the lung (with the exception of pDCs), but there was no difference in the numbers of DCs recruited to the lungs in offspring of control, or HDM-exposed mothers (Supplementary Fig 3). To assess the functional capacity of different DC subsets to take up exogenous allergen, mice were exposed to fluorescently labeled HDM on the final allergen exposure, and allergen uptake was monitored through gating on HDM+ cells. Maternal HDM exposure did not alter the phagocytic capacity of pulmonary CD103+ DCs, inflammatory DCs, or pDCs (data not shown). A significant decrease in phagocytic capacity of mDCs (measured by HDM mean fluorescence intensity (MFI)) and a decreased phagocytic capacity and frequency of HDM-bearing alveolar macrophages was observed in offspring of HDM-exposed mothers (Fig 4). We observed no significant differences in co-stimulatory molecule expression in pulmonary DC subsets (CD80, CD86, PD-L1, PD-L2, MHC Class II) between HDM-exposed offspring of HDM-exposed mothers and HDM-exposed offspring of PBS-exposed mothers (data not shown). Interestingly, when bone marrow derived dendritic cells (BMDCs) generated from control, and offspring of HDM-exposed dams were stimulated with HDM in vitro, we observed no differences in either allergen uptake, or costimulatory molecule expression (Supplementary Fig 4), suggesting that the observed decrease in pulmonary mDC phagocytic activity may be related to alteration in the local inflammatory milieu in the lungs of offspring of HDM-exposed mothers and not intrinsic differences in DC populations following maternal HDM exposure.
Figure 4.
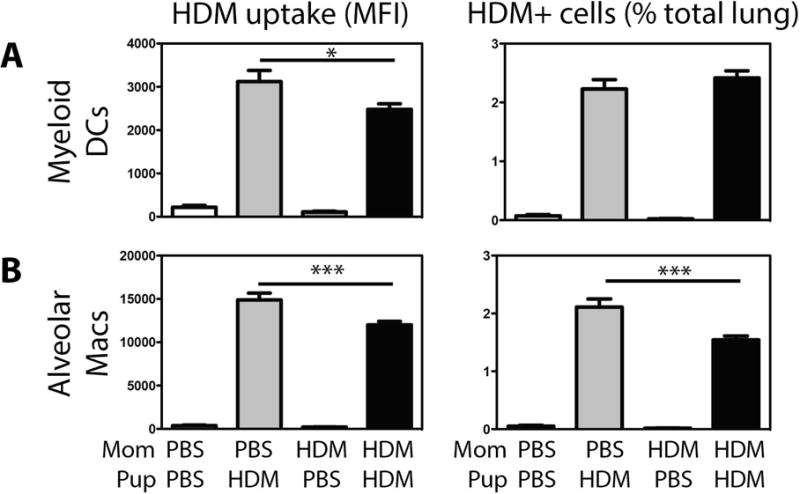
Minimal changes in allergen uptake by pulmonary DC populations were observed in HDM-exposed offspring of HDM-exposed mothers. Offspring of PBS- and HDM-exposed mothers were treated with PBS or HDM as described in Methods. Amount of HDM phagocytosed (MFI) and frequency of HDM-bearing (% of total lung cells) (A) myeloid DCs, and (B) alveolar macrophages were assessed by flow cytometry. Mean + SEM shown. * and *** indicate p < 0.05 and p < 0.001. Data represent 7 – 18 mice pooled from 4 experiments.
Maternal HDM exposure does not influence the magnitude Aspergillus fumigatus-induced airway inflammation
Prior maternal sensitization with OVA (alum), and subsequent OVA challenge in pregnancy has been demonstrated to drive the development of asthma to unrelated allergens in offspring of exposed mothers30. To assess the influence of maternal HDM exposure on the development of asthma to an unrelated allergen, mothers were exposed to HDM on days 0, 4 and 10 of pregnancy. At 6, 8 and 9 weeks of age, offspring (housed according to maternal exposure) were exposed to either intratracheal HDM or Aspergillus fumigatus (AF) extract. As expected, prior maternal HDM exposure increased the magnitude of HDM-induced airway inflammation (Fig. 5A). The effect was particularly striking for airway eosinophilia (Fig. 5B), although strong trends were observed for airway lymphocytes, neutrophils and macrophages (Fig 5C – E). In contrast, maternal HDM exposure had no impact on the severity of AF-induced airway inflammation as levels of total airway infiltrating cells, eosinophils, lymphocytes, neutrophils, and macrophages were all comparable in AF-exposed offspring of PBS-exposed, and HDM-exposed mothers.
Figure 5.
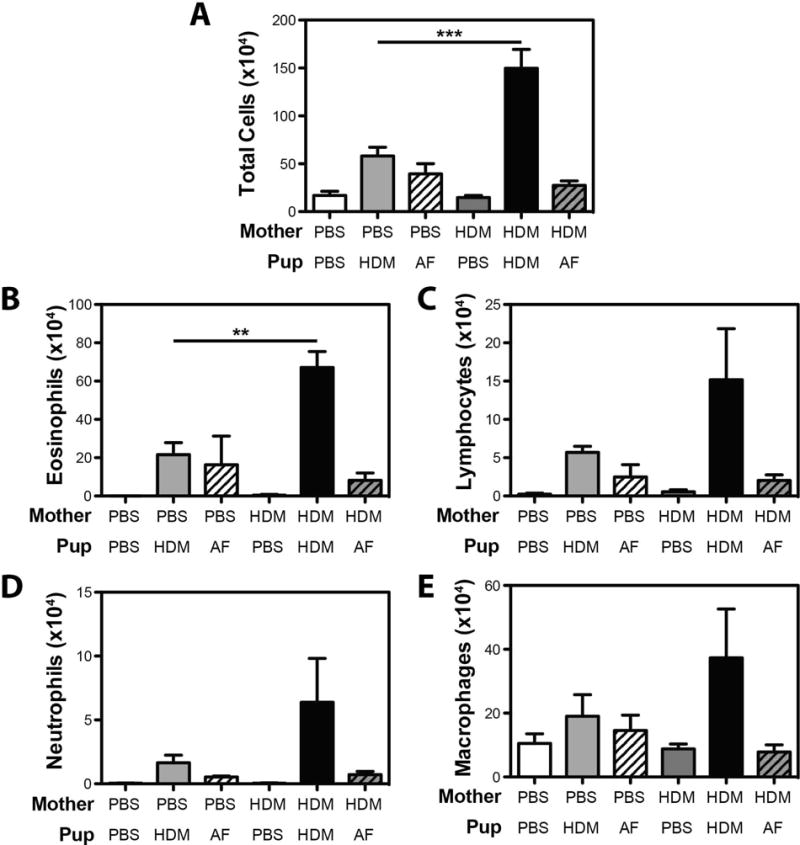
Maternal HDM exposure does not increase the severity of AHR and airway inflammation in Aspergillus fumigatus-exposed offspring. Offspring of PBS- and HDM-exposed mothers were exposed to PBS, HDM or AF extract at 6, 8 and 9 weeks of age. After sacrifice, BAL fluid was collected to assess (A) total BAL cellularity, and the number of (B) eosinophils, (C) lymphocytes, (D) neutrophils and (E) macrophages. Mean + SEM shown. Data represent 2 – 5 mice pooled from 2 experiments.
To further characterize the asthmatic response in AF-exposed offspring, we also examined production of Th2-associated cytokines in AF-exposed offspring. In these studies cells were stimulated with ConA to directly compare the changes in capacity for cytokine production induced by prior HDM or AF exposure. In HDM-exposed offspring prior maternal HDM exposure resulted in a trend towards increased production of the Th2 cytokines IL-4, IL-5 and IL-13, an increased frequency of IL-13+ T cells (Fig 6D), and increased levels of total IgE (Fig 6E). In contrast, we could discern no significant impact of maternal HDM exposure on ConA-induced cytokine production, the frequency of pulmonary IL-13+ CD4+ T cells, or IgE levels in AF-exposed offspring, (Fig 6A – E). Collectively these data suggest that the impact of maternal exposure to allergens in the absence of strong immune-skewing adjuvants is limited to the allergen to which the mother is exposed.
Figure 6.
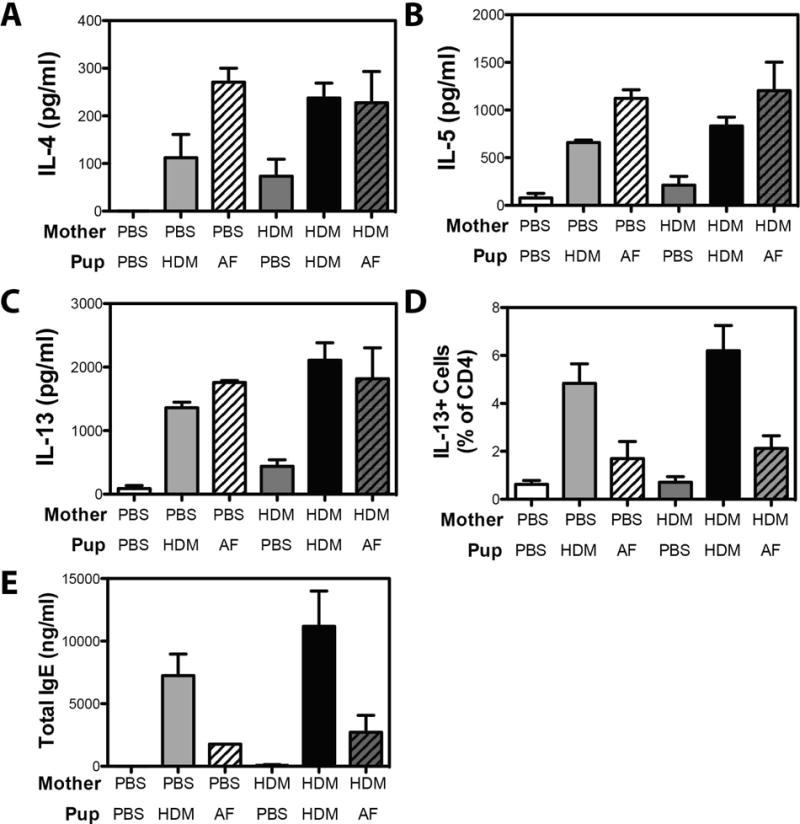
Maternal HDM exposure does not increased the magnitude of the Th2 response or magnitude of the Ig response following exposure of offspring to Aspergillus fumigatus. Offspring of PBS- and HDM-exposed mothers were exposed to PBS, HDM or AF extract at 6, 8 and 9 weeks of age. Single cell suspensions of lung cells were restimulated with ConA, and production of (A) IL-4, (B) IL-5, (C) IL-13 were assessed by ELISA. (D) The frequency of CD4+ cells producing IL-13 was assessed by flow cytometry. (E) Serum isolated at time of sacrifice was assayed for total IgE. Mean + SEM shown. Data represent 2 – 5 mice pooled from 2 experiments.
Exacerbation of offspring asthma following maternal HDM exposure is not associated with detectable allergen present in fetal tissues and does not require maternal immunoglobulins
To begin to explore the mechanisms whereby maternal allergen exposures influence the severity of offspring asthma, the final maternal HDM exposure (d10 of pregnancy) was replaced with AF405-labeled HDM. Mothers were sacrificed on day 12 of pregnancy, and we isolated the cells from the amniotic fluid, and both placental and fetal tissues to investigate the presence of AF405-labeled HDM. Both fetal and placental tissues showed unique populations of CD11b+, CD11c+, CD11b+CD11c+, and CD11b−CD11c− cell populations, although appreciable AF405 signals were not detected in any cell population in tissues harvested from HDM-exposed mothers (Supplementary Figure 5A – B). Cell numbers were much lower in samples of amniotic fluid, and analysis of pooled cell samples from HDM-exposed mothers demonstrated no increase in the AF405 channel (Supplementary Figure 5C). Finally, levels of major HDM allergens, DerP1 and DerP2, were assessed by ELISA in samples of pooled amniotic fluid from PBS- or HDM-exposed mothers. Again, levels of DerP1 and DerP2 were consistently below the level of detection (~1 ng/ml) in amniotic fluid of HDM-exposed mothers (data not shown). These data suggest there is not a strong transfer of HDM-derived proteins following maternal HDM exposure.
Finally, to determine if transfer of maternal immunoglobulins was required for the increased severity of allergic asthma observed in offspring of HDM-exposed mothers, we made use of immunoglobulin deficient μMT mice47. To this end, we mated female μMT mice with control C57Bl/6 mice to generate offspring in which no transfer of maternal HDM-specific immunoglobulins could occur, but which could themselves mount an immunoglobulin response following HDM sensitization and challenge. Pregnant female μMT mice were treated with HDM on days 0, 4 and 10 of pregnancy, as described above. As C57Bl/6 mice are more resistant to the development of AHR, a more robust protocol of HDM-induced AHR was utilized wherein mice are given 10 μg HDM i.p. (in the absence of additional adjuvant) at 6 and 7 weeks of life, and then challenged with 100 μg of HDM i.t. at 8 and 9 weeks of life. AHR was assessed 72 hours after the final allergen exposure. As observed in offspring of HDM-exposed A/J mothers, HDM exposure during pregnancy resulted significantly greater AHR in C57Bl/6 offspring (Fig 7A), suggesting that maternal HDM exposure-mediated exacerbation of asthma is not limited to A/J mice. Moreover, while the overall magnitude of AHR in offspring generated by mating μMT females with C57Bl/6 males was lower than that observed in offspring of pure C57Bl/6 matings, maternal exposure to HDM similarly exacerbated AHR in offspring of HDM-exposed μMT dams. Maternal HDM exposure was also associated with increased recruitment of all cell types into the BAL fluid (Fig 7B – F), as well as an increased recruitment of IL-13+ CD4+ T cells, but not IL-17A+ CD4+ T cells (Supplementary Figure 6). Additionally, maternal HDM exposure significantly increased the titer of total IgE and HDM-Specific IgG1 in offspring of C57Bl/6 mothers, and total IgE in offspring of HDM-exposed μMT mothers (Supplementary Figure 7)(IgG2a is not expressed in C57Bl/6 mice48, and as such, was not measured). As expected, PBS-treated offspring of HDM-exposed C57Bl/6 mothers displayed readily detectable levels of HDM-specific IgG1, while PBS-treated offspring of HDM-treated μMT mothers had undetectable levels of HDM-specific IgG1, suggesting that this immunoglobulin is likely maternally derived (Supplementary Figure 7). Thus, these data suggest that transfer of maternal HDM-specific immunoglobulins is not absolutely required for the development of more severe AHR after maternal HDM exposure.
Figure 7.
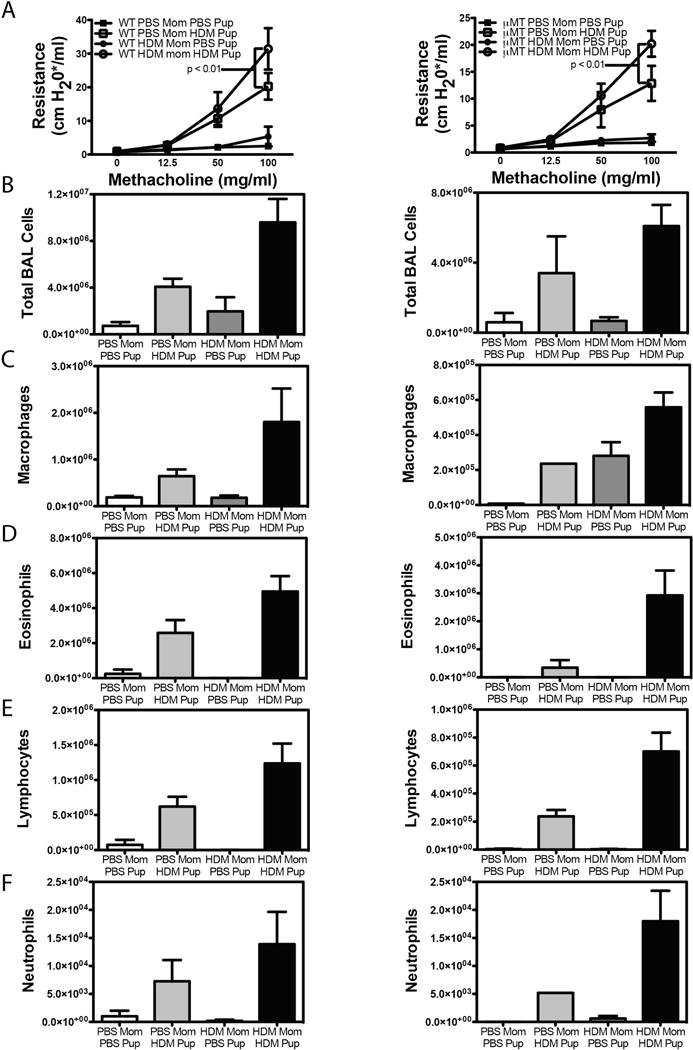
Offspring of HDM-exposed μMT dams demonstrate increased severity of AHR and airway inflammation. Male C57Bl/6 mice were mated with female C57Bl/6 dams (left column) or B cell deficient μMT dams (right columns) and pregnant female mice were exposed to HDM or PBS on days 0, 4 and 10 of pregnancy. Offspring of PBS- and HDM-exposed mothers were exposed to i.p. PBS or HDM at 6 and 7 weeks of age, and challenged with i.t. PBS or HDM at 8 and 9 weeks of age and development of AHR was assessed 72 hours later. (A). After sacrifice, BAL fluid was collected to assess (B) total BAL cellularity, and the number of (C) macrophages, (D) eosinophils, (E) lymphocytes and (F) neutrophils. Mean + SEM shown. Data represent 3 – 13 mice pooled from 3 experiments.
Discussion
Herein, we demonstrate that prenatal exposure to the relevant human aeroallergen, house dust mite (HDM), in the absence of powerful Th2-skewing adjuvants, is sufficient to exacerbate the development of HDM-driven allergic asthma in offspring later in life. Specifically, offspring of allergen-exposed mothers demonstrated more robust AHR and airway inflammation. The increased phenotypic measures of asthma were associated with a greater capacity for Th2 cytokine production and frequency of CD4+IL-13+ cells in the lung, increased immunoglobulin production, and a decreased capacity of allergen uptake by alveolar macrophages. Interestingly, the increased sensitivity to allergen-induced AHR in offspring of exposed mothers appeared to be unique to the allergen the mothers were exposed to, as offspring of HDM-exposed mothers did not demonstrate increased severity of Aspergillus fumigatus-induced airway inflammation.
As maternal asthma status, moreso than paternal asthma status, is linked to increased risk of asthma, our study focused on the ability of maternal exposures to influence offspring asthma. As such, we cannot ascertain whether paternal allergen exposures might similarly influence asthma development in offspring. However, maternal exposures have been reported to influence, both positively and negatively, the development of asthma in other animal models. For example, if Schistosoma mansoni-infected mothers were impregnated during the systemic Th1 phase of the infection, offspring were found to be protected from development of asthma21. Anti-IFNγ abrogated this protective effect of maternal Schistosoma mansoni infection, suggesting that maternal cytokines can influence asthma development in offspring21. Feeding female mice OVA prior to pregnancy to induce tolerance protects from the development of OVA-induced allergic airway disease in an FcRn and IFNγ-dependent manner49. Similarly, sensitization with OVA (CFA), and re-exposure to OVA during pregnancy limited the development of OVA-specific IgE and pulmonary eosinophilia in offspring of exposed mothers50. Contrary to this protective effect, our observations of increased asthma development in offspring of HDM-exposed mothers are consistent with reports showing increased asthma development in offspring following maternal OVA (alum) sensitization and challenge29–32, maternal Aspergillus fumigatus-exposure33, and in Schistosoma mansoni-infected mothers impregnated during the Th2 phase of the anti-helminthic response21. Blockade of IL-4 during pregnancy completely abrogated the increased sensitivity of offspring of following maternal OVA (alum) sensitization and challenge30, again suggesting maternal cytokines can influence offspring asthma. However, in their model of maternal OVA-exposure induced alterations of offspring asthma, Hamada et al also report that offspring are also more sensitive to asthma in a casein-driven model, suggesting that the vertical transmission of asthma sensitivity was not antigen specific. In contrast, we failed to observe increased airway inflammation, Th2 cytokine production or immunoglobulin production following Aspergillus fumigatus exposure in offspring of HDM-exposed mothers. Thus, while we do observe increased maternal IL-4 levels following HDM exposure (data not shown), it seems unlikely that circulating maternal IL-4 is responsible for increased development of asthma in offspring of HDM-exposed mothers in our study. It is possible that the failure to induce increased asthma development to an unrelated antigen in our model is due to a lower overall capacity of HDM to induce increased circulating IL-4 levels as compared to OVA (alum) sensitization and challenge performed by Hamada et al30. Alternatively, while HDM exposures in offspring of HDM-exposed mothers began at 6 weeks of age in our study, initial allergen exposure of offspring from OVA (alum) sensitized and challenged animals occurred at day 3 of life30. Thus it is also conceivable that the ability of maternal IL-4 to enhance the development of Th2 responses to unrelated allergens is lost between day 3 and week 6 of life.
Another mechanism whereby OVA exposure in pregnant OVA (alum) sensitized mothers influences offspring asthma is through modification of dendritic cell (DC) activity in offspring. Indeed, DCs isolated from the spleens of offspring of OVA (alum) sensitized and challenged mothers demonstrated an increased capacity to induce T cell proliferation in in vitro cultures despite no alterations in overall capacity to express MHC Class II or co-stimulatory molecules and transfer of DCs from offspring of OVA-exposed mothers to control offspring increased the development of both OVA and casein-induced AHR29. This increased capacity to drive T cell proliferation and development of asthma was associated with substantial changes in the DNA-methylation profile of splenic DCs from offspring of OVA (alum) sensitized and challenged mothers compared to splenic DCs from offspring of control mothers32. These results suggest that splenic DCs from offspring of allergen-exposed mothers were more “pro-asthmatic”. In contrast to these reports, we find that the natural allergen, HDM, in the absence of additional immune-activating adjuvants drives limited change in co-stimulatory molecule expression or function of DCs in offspring of HDM-exposed mothers, while actually decreasing phagocytic capacity of various pulmonary DC subsets. Interestingly, the observed decrease in phagocytic capacity was not observed in HDM-pulsed BMDCs generated from offspring of HDM-exposed mothers, suggesting that these alterations were not likely due to epigenetic changes within the DCs themselves, but rather related to differences in the inflammatory milieu of the lung. While we did not directly compare T cell stimulatory capacity of DCs from control offspring, and offspring of HDM-exposed animals our observations that HDM-specific, but not AF-specific immune responses are impacted following maternal HDM-exposure is not consistent with the induction of an intrinsically more “pro-asthmatic” population of pulmonary dendritic cells.
The key distinguishing feature of our model from other published reports is the antigen specific nature of our observed effects. Interestingly, there are readily detectable levels of HDM-specific IgG1 and IgG2a in the serum of PBS-exposed offspring of HDM-exposed mothers, and levels can persist up to 7 months after birth (Supplementary Figure 8), suggesting that maternal-derived immunoglublins may drive more severe AHR in offspring of HDM-exposed mothers. However, we find that exposure of B cell deficient μMT dams to HDM following mating with WT C57Bl/6 animals still drives significantly enhanced HDM-induced AHR, airway inflammation and pulmonary Th2 recruitment in offspring. This result suggests that transfer of maternal immunoglobulins is not required for exacerbation of the asthma phenotype following maternal HDM exposure. However, it was recently demonstrated that B-1B cells present in the lungs of μMT mice can produce antigen non-specific IgE and IgG1, IgG2c following allergen challenge51. As B-1B cell-derived Abs typically recognize polysaccharide antigens with low affinity52, and HDM contains glycosylated proteins53, it is conceivable that B1-derived maternal Igs are either sufficient to transfer increased HDM-reactivity to offspring, or they facilitate the transplacental passage of antigens in Ig:antigen complexes54–57, allowing for priming of HDM-specific T cells in offspring of exposed mothers. Subsequent inhalational exposure in offspring of HDM-exposed mothers may then represent additional “boosts” not present in offspring of PBS-exposed mothers, and drive the development of a more robust response. Finally, it is possible that vertical transmission of maternal T cells (either through transplacental passage or via breast milk) passively sensitizes the fetus to HDM through a maternal microchimerism-dependent mechanism as has been observed in model of maternal tuberculosis of Candida infection58. These possibilities are currently being considered in our laboratory.
In conclusion, we report that maternal HDM exposure during pregnancy is sufficient to enhance neonatal sensitivity to HDM. This is associated with increased AHR, airway inflammation, Th2 cytokine production, immunoglobulin levels and a modest decrease in the phagocytic capacity of pulmonary macrophage populations following HDM exposure in offspring. Interestingly, in contrast to previous reports of allergen-independent influence of maternal OVA (alum) sensitization and challenge on offspring asthma, these effects are wholly allergen dependent, as exposure of offspring to an unrelated allergen, Aspergillus fumigatus, does not result in the development of more severe airway inflammation, Th2 cytokine production or immunoglobulin synthesis. These findings have important implication for regulation of asthma risk, and suggest that increased HDM burden in the developed world may have under-appreciated influences on the overall prevalence of allergic asthma.
Supplementary Material
Key Messages.
Maternal exposure to a common human allergen, house dust mite, during pregnancy increases severity of house dust mite-induced asthma in murine offspring.
Maternal exposure to house dust mite does not exacerbate Aspergillus fumigatus-induced asthma in murine offspring of house dust mite-exposed mothers, suggesting that the effect of house dust mite is allergen-specific.
These findings have important implications for regulation of asthma risk, and suggest that exposure to house dust mite in the developed world may have under-appreciated influences on the overall prevalence of allergic asthma.
Acknowledgments
Funded by NIEHS P30 ES006096 (IPL), NHLBI R01 HL122300 (IPL), the CCHMC Perinatal Infection and Inflammation Collaborative (CAC, IPL), and NIAID R21 AI119385 (IPL).
Abbreviations
- AF
Aspergillus fumigatus
- AHR
Airway hyperresponsiveness
- ANOVA
Analysis of Variance
- BAL
bronchoalveolar lavage
- CFA
complete Freund’s adjuvant
- DC
dendritic cell
- HDM
house dust mite
- Ig
immunoglobulin
- i.p.
intraperitoneal
- i.t.
intratracheal
- MFI
mean fluorescence intensity
- PBS
Phosphate buffered saline
- SEM
Standard error of the mean
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wong GW, Chow CM. Childhood asthma epidemiology: insights from comparative studies of rural and urban populations. Pediatr Pulmonol. 2008;43:107–16. doi: 10.1002/ppul.20755. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. 2007 [Google Scholar]
- 3.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–35. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 4.Vercelli D. Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol. 2008;8:169–82. doi: 10.1038/nri2257. [DOI] [PubMed] [Google Scholar]
- 5.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sleiman PM, Flory J, Imielinski M, Bradfield JP, Annaiah K, Willis-Owen SA, et al. Variants of DENND1B associated with asthma in children. N Engl J Med. 2010;362:36–44. doi: 10.1056/NEJMoa0901867. [DOI] [PubMed] [Google Scholar]
- 7.Mathias RA, Grant AV, Rafaels N, Hand T, Gao L, Vergara C, et al. A genome-wide association study on African-ancestry populations for asthma. J Allergy Clin Immunol. 2010;125:336–46 e4. doi: 10.1016/j.jaci.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee AB, Zhang Z. Allergic asthma: influence of genetic and environmental factors. J Biol Chem. 2011;286:32883–9. doi: 10.1074/jbc.R110.197046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol. 2009;123:3–11. doi: 10.1016/j.jaci.2008.10.046. [DOI] [PubMed] [Google Scholar]
- 10.Vercelli D. Gene-environment interactions: the road less traveled by in asthma genetics. J Allergy Clin Immunol. 2009;123:26–7. doi: 10.1016/j.jaci.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 11.Riedler J, Braun-Fahrlander C, Eder W, Schreuer M, Waser M, Maisch S, et al. Exposure to farming in early life and development of asthma and allergy: a cross-sectional survey. Lancet. 2001;358:1129–33. doi: 10.1016/S0140-6736(01)06252-3. [DOI] [PubMed] [Google Scholar]
- 12.Stein RT, Holberg CJ, Sherrill D, Wright AL, Morgan WJ, Taussig L, et al. Influence of parental smoking on respiratory symptoms during the first decade of life: the Tucson Children’s Respiratory Study. Am J Epidemiol. 1999;149:1030–7. doi: 10.1093/oxfordjournals.aje.a009748. [DOI] [PubMed] [Google Scholar]
- 13.Singh SP, Gundavarapu S, Pena-Philippides JC, Rir-Sima-ah J, Mishra NC, Wilder JA, et al. Prenatal secondhand cigarette smoke promotes Th2 polarization and impairs goblet cell differentiation and airway mucus formation. J Immunol. 2011;187:4542–52. doi: 10.4049/jimmunol.1101567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt EB, Kovacic MB, Lee GB, Gibson AM, Acciani TH, Le Cras TD, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol. 2013;132:1194–204 e2. doi: 10.1016/j.jaci.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One. 2009;4:e4488. doi: 10.1371/journal.pone.0004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–7. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuman A, Hohmann C, Orsini N, Pershagen G, Eller E, Kjaer HF, et al. Maternal smoking in pregnancy and asthma in preschool children: a pooled analysis of eight birth cohorts. Am J Respir Crit Care Med. 2012;186:1037–43. doi: 10.1164/rccm.201203-0501OC. [DOI] [PubMed] [Google Scholar]
- 18.Chiu YH, Coull BA, Sternthal MJ, Kloog I, Schwartz J, Cohen S, et al. Effects of prenatal community violence and ambient air pollution on childhood wheeze in an urban population. J Allergy Clin Immunol. 2014;133:713–22 e4. doi: 10.1016/j.jaci.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J Clin Invest. 2008;118:3462–9. doi: 10.1172/JCI34378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Haberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements in pregnancy and early childhood respiratory health. Arch Dis Child. 2009;94:180–4. doi: 10.1136/adc.2008.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straubinger K, Paul S, Prazeres da Costa O, Ritter M, Buch T, Busch DH, et al. Maternal immune response to helminth infection during pregnancy determines offspring susceptibility to allergic airway inflammation. J Allergy Clin Immunol. 2014;134:1271–9 e10. doi: 10.1016/j.jaci.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz RG, Kemeny DM, Price JF. Higher risk of infantile atopic dermatitis from maternal atopy than from paternal atopy. Clin Exp Allergy. 1992;22:762–6. doi: 10.1111/j.1365-2222.1992.tb02816.x. [DOI] [PubMed] [Google Scholar]
- 23.Litonjua AA, Carey VJ, Burge HA, Weiss ST, Gold DR. Parental history and the risk for childhood asthma. Does mother confer more risk than father? Am J Respir Crit Care Med. 1998;158:176–81. doi: 10.1164/ajrccm.158.1.9710014. [DOI] [PubMed] [Google Scholar]
- 24.Liu CA, Wang CL, Chuang H, Ou CY, Hsu TY, Yang KD. Prenatal prediction of infant atopy by maternal but not paternal total IgE levels. J Allergy Clin Immunol. 2003;112:899–904. doi: 10.1016/j.jaci.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 25.Moffatt MF, Cookson WO. The genetics of asthma. Maternal effects in atopic disease. Clin Exp Allergy. 1998;28(Suppl 1):56–61. doi: 10.1046/j.1365-2222.1998.0280s1056.x. discussion 5–6. [DOI] [PubMed] [Google Scholar]
- 26.Lim RH, Kobzik L, Dahl M. Risk for asthma in offspring of asthmatic mothers versus fathers: a meta-analysis. PLoS One. 2010;5:e10134. doi: 10.1371/journal.pone.0010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peat JK. The epidemiology of asthma. Curr Opin Pulm Med. 1996;2:7–15. [PubMed] [Google Scholar]
- 28.Squillace SP, Sporik RB, Rakes G, Couture N, Lawrence A, Merriam S, et al. Sensitization to dust mites as a dominant risk factor for asthma among adolescents living in central Virginia. Multiple regression analysis of a population-based study. Am J Respir Crit Care Med. 1997;156:1760–4. doi: 10.1164/ajrccm.156.6.9704026. [DOI] [PubMed] [Google Scholar]
- 29.Fedulov AV, Kobzik L. Allergy risk is mediated by dendritic cells with congenital epigenetic changes. Am J Respir Cell Mol Biol. 2011;44:285–92. doi: 10.1165/rcmb.2009-0400OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada K, Suzaki Y, Goldman A, Ning YY, Goldsmith C, Palecanda A, et al. Allergen-independent maternal transmission of asthma susceptibility. J Immunol. 2003;170:1683–9. doi: 10.4049/jimmunol.170.4.1683. [DOI] [PubMed] [Google Scholar]
- 31.Hubeau C, Apostolou I, Kobzik L. Adoptively transferred allergen-specific T cells cause maternal transmission of asthma risk. Am J Pathol. 2006;168:1931–9. doi: 10.2353/ajpath.2006.051231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikhaylova L, Zhang Y, Kobzik L, Fedulov AV. Link between epigenomic alterations and genome-wide aberrant transcriptional response to allergen in dendritic cells conveying maternal asthma risk. PLoS One. 2013;8:e70387. doi: 10.1371/journal.pone.0070387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niedzwiecki M, Zhu H, Corson L, Grunig G, Factor PH, Chu S, et al. Prenatal exposure to allergen, DNA methylation, and allergy in grandoffspring mice. Allergy. 2012;67:904–10. doi: 10.1111/j.1398-9995.2012.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen JC, Chan CC, Wu CJ, Ou LS, Yu HY, Chang HL, et al. Fetal Phagocytes Take up Allergens to Initiate T-Helper Cell Type 2 Immunity and Facilitate Allergic Airway Responses. Am J Respir Crit Care Med. 2016;194:934–47. doi: 10.1164/rccm.201508-1703OC. [DOI] [PubMed] [Google Scholar]
- 35.Reponen T, Lockey J, Bernstein DI, Vesper SJ, Levin L, Khurana Hershey GK, et al. Infant origins of childhood asthma associated with specific molds. J Allergy Clin Immunol. 2012;130:639–44 e5. doi: 10.1016/j.jaci.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holloway JA, Warner JO, Vance GH, Diaper ND, Warner JA, Jones CA. Detection of house-dust-mite allergen in amniotic fluid and umbilical-cord blood. Lancet. 2000;356:1900–2. doi: 10.1016/S0140-6736(00)03265-7. [DOI] [PubMed] [Google Scholar]
- 37.Platts-Mills TA, Rakes G, Heymann PW. The relevance of allergen exposure to the development of asthma in childhood. J Allergy Clin Immunol. 2000;105:S503–8. doi: 10.1016/S0091-6749(00)90051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Platts-Mills T, Vaughan J, Squillace S, Woodfolk J, Sporik R. Sensitisation, asthma, and a modified Th2 response in children exposed to cat allergen: a population-based cross-sectional study. Lancet. 2001;357:752–6. doi: 10.1016/S0140-6736(00)04168-4. [DOI] [PubMed] [Google Scholar]
- 39.Ownby DR, Johnson CC, Peterson EL. Exposure to dogs and cats in the first year of life and risk of allergic sensitization at 6 to 7 years of age. JAMA. 2002;288:963–72. doi: 10.1001/jama.288.8.963. [DOI] [PubMed] [Google Scholar]
- 40.Platts-Mills TAE, de Weck AL. Dust mite allergens and asthma–a worldwide problem. J Allergy Clin Immunol. 1989;83:416–27. doi: 10.1016/0091-6749(89)90128-0. [DOI] [PubMed] [Google Scholar]
- 41.Ronmark E, Perzanowski M, Platts-Mills T, Lundback B. Four-year incidence of allergic sensitization among schoolchildren in a community where allergy to cat and dog dominates sensitization: report from the Obstructive Lung Disease in Northern Sweden Study Group. J Allergy Clin Immunol. 2003;112:747–54. doi: 10.1016/S0091. [DOI] [PubMed] [Google Scholar]
- 42.Sporik R, Ingram JM, Price W, Sussman JH, Honsinger RW, Platts-Mills TA. Association of asthma with serum IgE and skin test reactivity to allergens among children living at high altitude. Tickling the dragon’s breath. Am J Respir Crit Care Med. 1995;151:1388–92. doi: 10.1164/ajrccm.151.5.7735590. [DOI] [PubMed] [Google Scholar]
- 43.Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol. 2010;11:928–35. doi: 10.1038/ni.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewkowich IP, Lajoie S, Clark JR, Herman NS, Sproles AA, Wills-Karp M. Allergen uptake, activation, and IL-23 production by pulmonary myeloid DCs drives airway hyperresponsiveness in asthma-susceptible mice. PLoS One. 2008;3:e3879. doi: 10.1371/journal.pone.0003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arshad SH, Karmaus W, Raza A, Kurukulaaratchy RJ, Matthews SM, Holloway JW, et al. The effect of parental allergy on childhood allergic diseases depends on the sex of the child. J Allergy Clin Immunol. 2012;130:427–34 e6. doi: 10.1016/j.jaci.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewkowich IP, Rempel JD, HayGlass KT. In vivo IgE levels in exogenous antigen stimulated responses: measurement of total IgE as a valid, simple surrogate for Ag-specific IgE. J Immunol Methods. 2004;286:123–32. doi: 10.1016/j.jim.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 47.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–6. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 48.Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods. 1998;212:187–92. doi: 10.1016/s0022-1759(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 49.Polte T, Hennig C, Hansen G. Allergy prevention starts before conception: maternofetal transfer of tolerance protects against the development of asthma. J Allergy Clin Immunol. 2008;122:1022–30 e5. doi: 10.1016/j.jaci.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Matson AP, Zhu L, Lingenheld EG, Schramm CM, Clark RB, Selander DM, et al. Maternal transmission of resistance to development of allergic airway disease. J Immunol. 2007;179:1282–91. doi: 10.4049/jimmunol.179.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh S, Hoselton SA, Schuh JM. mu-chain-deficient mice possess B-1 cells and produce IgG and IgE, but not IgA, following systemic sensitization and inhalational challenge in a fungal asthma model. J Immunol. 2012;189:1322–9. doi: 10.4049/jimmunol.1200138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 53.Al-Ghouleh A, Johal R, Sharquie IK, Emara M, Harrington H, Shakib F, et al. The glycosylation pattern of common allergens: the recognition and uptake of Der p 1 by epithelial and dendritic cells is carbohydrate dependent. PLoS One. 2012;7:e33929. doi: 10.1371/journal.pone.0033929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol. 2006;168:1210–26. doi: 10.2353/ajpath.2006.050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.May K, Grube M, Malhotra I, Long CA, Singh S, Mandaliya K, et al. Antibody-dependent transplacental transfer of malaria blood-stage antigen using a human ex vivo placental perfusion model. PLoS One. 2009;4:e7986. doi: 10.1371/journal.pone.0007986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szepfalusi Z, Pichler J, Elsasser S, van Duren K, Ebner C, Bernaschek G, et al. Transplacental priming of the human immune system with environmental allergens can occur early in gestation. J Allergy Clin Immunol. 2000;106:530–6. doi: 10.1067/mai.2000.108710. [DOI] [PubMed] [Google Scholar]
- 57.Szepfalusi Z, Loibichler C, Pichler J, Reisenberger K, Ebner C, Urbanek R. Direct evidence for transplacental allergen transfer. Pediatr Res. 2000;48:404–7. doi: 10.1203/00006450-200009000-00024. [DOI] [PubMed] [Google Scholar]
- 58.Ghosh MK, Nguyen V, Muller HK, Walker AM. Maternal Milk T Cells Drive Development of Transgenerational Th1 Immunity in Offspring Thymus. J Immunol. 2016;197:2290–6. doi: 10.4049/jimmunol.1502483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewkowich IP, Herman NS, Schleifer KW, Dance MP, Chen BL, Dienger KM, et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J Exp Med. 2005;202:1549–61. doi: 10.1084/jem.20051506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.