

Nano- and microparticles that are composed of biodegradable polymers have demonstrated unprecedented potential as drug carriers.[1, 2] Recent research activities have emphasized that controlled degradation of polymeric particles[2] triggered by external stimuli, such as pH,[3] temperature,[4] ultrasound,[5] and light[6] are of increasing interest for therapeutic applications.[7] In particular, optical stimuli offer the potential for spatial and temporally controlled drug release. However, light-sensitive biodegradable polymers as a drug carriers typically require elaborate designs and their experimental realization may be synthetically demanding.[8]
Polysaccharides are biocompatible and find use for a wide range of biological applications. Moreover, straightforward chemical modification can further augment their utility.[9] Fréchet and coworkers synthesized acetal-modified dextran (Ac-Dex) by reacting hydroxyl groups of dextran, a homopolysaccharide of glucose, with 2-methoxypropene under acidic conditions (Figure 1a).[10] The high density of acetal groups makes Ac-Dex insoluble in water, while the unsubstituted biopolymer is readily soluble in water. To deliver drugs, genes, and antigens,[11] acid-catalyzed hydrolysis of Ac-Dex can be used to regenerate dextran, leading to dissolution of the particles in water.[10–13] While these systems respond to changes in pH, they still do not offer on-demand control over drug release triggered by the application of an external stimulus, such as light.
Figure 1.
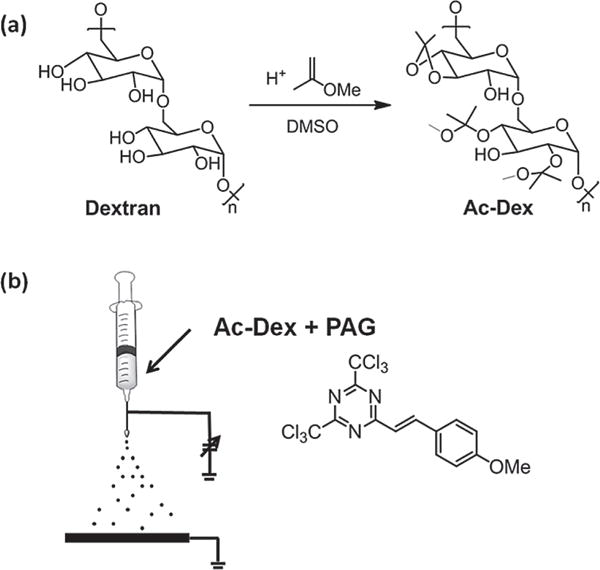
(a) Synthesis of acetal-modified dextran (Ac-Dex).[10] Illustration of (b) preparation of photodegradable Ac-Dex nanoparticles by electrospraying a solution of Ac-Dex (7.5 w/v%) and 2-(4-methoxystyryl)-4,6-bis(trichloromethyl)-1,3,5-triazine (3 wt% based on Ac-Dex) as a photoacid generator (PAG).
In search of novel responsive delivery systems, we envisioned strategies where light is used to initiate the degradation of Ac-Dex particles. We tested our hypothesis by formulating polymer microparticles comprised of blends of Ac-Dex and caged photoacids. The photolabile acids generate free acid upon illumination with UV light. Such caged acids are widely used in the photocationic polymerization of vinyl ethers and play an important role in photoinduced curing systems, technical coatings, adhesives, and inks.[14]
Using an electrospraying technique, submicron polymer particles were prepared that contained Ac-Dex as well as caged photoacids. Illumination of the nanoparticles can then trigger deprotection of Ac-Dex and ultimately leads to the dissolution of the particles under physiological conditions. In addition, we demonstrate that the control of illumination times allows for on-demand particle degradation and controlled release.
In principle, polymer nano- and microparticles can be made by a wide range of synthesis methods such as emulsion polymerization,[15] self-assembly,[16] or template-assisted fabrication.[17] However, these approaches may require changes in materials and tedious optimization of experimental parameters to ensure effective encapsulation of dissimilar cargo molecules. Such orthogonal solubility has been used for Ac-Dex based microparticles to deliver drugs, genes, and antigens. The fact that Ac-Dex is only soluble in organic solvents and completely insoluble in water enables processing into microparticles via double emulsion and single emulsion methods. On the other hand, acid-catalyzed hydrolysis of acetal protecting groups yields water-soluble dextran leading to dissolution of particles in water and releasing any encapsulated payload. Alternatively, electrospraying of dilute polymer solutions is a straightforward method for fabrication of nano- and microparticles with controlled sizes and narrow size distributions.[18–22] Because dilute polymer solutions can be processed, relatively high loadings of the active ingredients, such as drugs or dyes, can be achieved by simply blending these components into the jetting solutions.[23–26]
Prior to particle fabrication, acetal-modified dextran (Ac-Dex) was synthesized according to previously described protocols.[10] Briefly, dexran was reacted with 2-methoxypropene with catalytic pyridinium p-toluenenesulfonate in anhydrous DMSO for 3 h under Ar (Figure 1a). To estimate the degree of protection, Ac-Dex was hydrolyzed in D2O containing traces of DCl, and 1H NMR spectra were recorded.[10] Based on the integration of the 1H NMR signals for acetone and methanol at 2.2 and 3.35 ppm, 73% of the available hydroxyl groups were reacted and the ratio of cyclic to acyclic acetal groups was estimated to be 1.1:1 (see Supporting Information for further details). Control experiments confirmed that this polymer is initially unsoluble in water, but readily dissolves at pH values below 5.
Next, we attempted to formulate nanoparticles from the Ac-Dex polymer. We decided to explore the use of electrospraying, because (i) it can be conducted under completely anhydrous conditions, (ii) can lead to particles with desirable sizes, and (iii) it minimizes the exposure of the particles to ambient light. Figure 1b shows a schematic illustration of the electrospraying setup. The jetting solution was prepared by dissolving Ac-Dex (75 mg) in a mixture of THF and DMF (1:1, 1.0 mL). Next, 2-(4-methoxystyryl)-4,6-bis(trichloromethyl)-1,3,5-triazine (PAG, 2.25 mg) was added in the dark to avoid activation of the caged acids. The solution was drawn into a 1 mL syringe (Becton-Dickinson, USA) and the needle (diameter: 26 G, length: 8.2 cm) was connected to the cathode of a DC voltage source (range: 0–30 kV, Gamma High Voltage Research, USA). The flow rate was controlled by a syringe pump (Kd Scientific, USA), and a square piece of aluminum foil was used as a collecting substrate (anode). The distance between needle tip and aluminum foil was maintained in the range of 20 cm. Electrospraying was performed at a flow rate of 0.08 mL h−1 and a spraying voltage of 6–7 kV, resulting in a stable cone-jet mode. Because PAG may respond to room light, all jetting processes were performed in the dark.
Figure 2 shows scanning electron microscope (SEM) and transmission electron microscope (TEM) images of particles that were collected on an aluminum substrate. The electrospraying of an Ac-Dex solution created nanoparticles with rough surfaces. Such morphologies are likely caused by the combination of several factors including polymer, solvent, and jetting solution viscosity and conductivity. On the other hand, a stable Taylor cone provided relatively uniform particles in size, and particle size distribution was calculated from the SEM images (Figure 2c) assuming the heavily wrinkled particles are spherical; the estimated mean diameter is 520 ± 63.4 nm. In addition, a dynamic light scattering measurement of the particles dispersed in water including Tween-20 (1.0 v/v%) presented 626 ± 88.0 nm of mean hydrodynamic diameter (Figure 2d).
Figure 2.
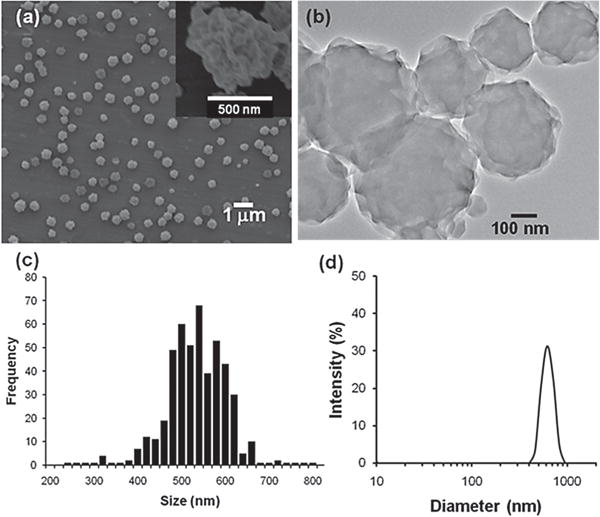
(a) SEM and (b) TEM images of photoresponsive Ac-Dex particles that were electrosprayed. (c) Particle size distribution calculated from SEM images and the mean diameter is 520 ± 63.4 nm. (d) Dynamic light scattering measurement. The estimated mean hydrodynamic diameter is 626 ± 88.0 nm.
Once well-defined particles were routinely prepared, we tested whether the photoacid was able to trigger Ac-Dex particle degradation. For comparison, PAG-free Ac-Dex particles were fabricated by electrospraying a polymer solution without PAG and suspended in water in parallel with Ac-Dex particles encapsulating PAG; both particles led to milky suspensions due to the insolubility of acetalated dextran in water. Figure 3a displays a digital image of the particle suspensions after irradiation with a portable UV lamp (345 nm, 4 W) for 20 min. While the suspension of Ac-Dex particles without PAG remained turbid, that of the Ac-Dex particles containing PAG turned clear after UV light exposure. As direct excitation of PAG leads to photoclevage of one of the carbon-chlorine bonds and subsequent hydrogen abstraction by the chlorine atom generates HCl (Figure 3b),[27] UV illumination on Ac-Dex particles encapsulating PAG leads to the deprotection of the acetal groups by photo-generated HCl within the particles and concomitantly dissolution of deprotected dextran in water as illustrated in Figure 3c.
Figure 3.
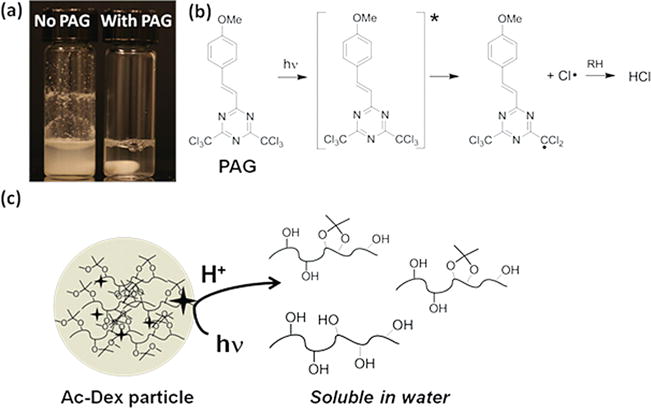
(a) Digital image after illuminating a PAG-free Ac-Dex particle suspension (left) and a PAG-encapsulating Ac-Dex particle suspension (right) with a UV-lamp (345 nm, 4 W) for 20 min (image contains stir bar). (b) Photoacid generation scheme by exciting 2-(4′-methoxystyryl)-4,6-bis(trichloromethyl)-1,3,5-triazine (PAG). (c) Schematic illustration of photoacid triggered deprotection of Ac-Dex and subsequent degradation of particles by dissolution.
The photodegradation of Ac-Dex was also investigated with NMR spectroscopy. AcDex particles (~5 mg) were suspended in D2O (~0.3 mL) and the suspension was exposed to UV light for 20 min, leading to a clear solution. The NMR spectrum after photoreaction is displayed in Figure 4 and reveals that (i) the deprotection of the acetal groups resulted in the manifestation of methanol and acetone peaks,[10, 11] (ii) a persistent methyl signal at 1.5 ppm indicates that deprotection was not completely achieved, and (iii) nevertheless the degree of deprotection (~70% estimated based on the NMR spectra) was sufficient to dissolve the partially deacetalated particles in water.
Figure 4.
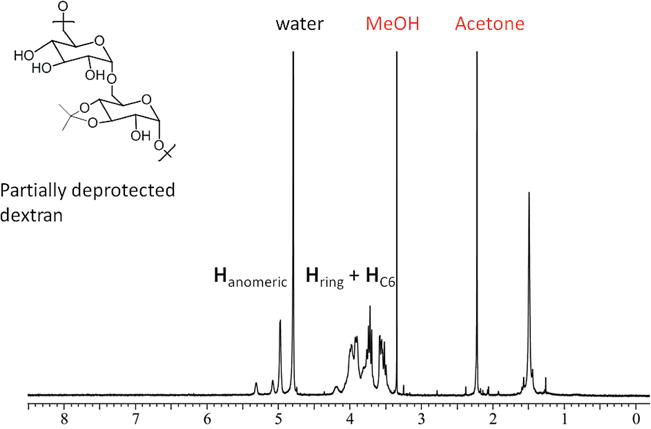
1H NMR spectrum obtained after photo-triggered degradation of Ac-Dex nanoparticles in D2O.
Next, we wanted to confirm the on-demand degradation of the Ac-Dex particles. Two batches of triplicated Ac-Dex particles were suspended in dd-H2O at a concentration of 2 mg/mL and gently agitated in the dark. UV light (345 nm) was applied for 1 min at certain time points that are indicated as lightning bolt symbols in Figure 4, whereas the reference sample was kept in the dark. At various time points, the suspensions were centrifuged and 0.12 mL aliquots were collected for further analysis. The supernatants were diluted with dd-water (total volume 1 mL) and stored at −20 °C. The pellets were redispersed and agitated after dd-water (0.12 mL) was replenished. The collected supernatant samples were analyzed for the presence of reducing polysaccharides using a bicinchoninic acid based assay[28] according to the Test Tube Procedure of the Micro BCA Protein Assay Kit (Thermo Scientific, USA). Figure 5 displays that Ac-Dex particles incubated in the dark remained an opaque suspension and the assay detected ~10% release of soluble reducing polysccha-rides after 40 min. In contrast, the periodic irradiations of UV light yielded a dextran release profile that was indicative of a stepwise dissolution of dextran. Notably, the initial release behavior of both batches in the dark (up to ~15 min, Figure 5) was similar. Because PAG may contain or generate traces of acid during particle preparation, slight deprotection of the Ac-Dex particles is possible, even though they were kept in the dark. Nevertheless, the suspension was opaque and only ~10% of dextran was detected after 20 min. After that, further release was insignificant as long as the samples were stored in the dark. On the other hand, after the first irradiation on the particle suspension for 1 min, a substantial amount of soluble dextran was detected compared to control samples. The second and third exposure to light further released soluble dextran, but the net amount released became smaller. The reason for such reduction in dextran release is likely that the first UV light illumination consumed a large portion of fresh PAG and released the acid very efficiently; later illuminations, however, utilized smaller amount of active PAG, considerably decreasing efficiency of deprotection.
Figure 5.
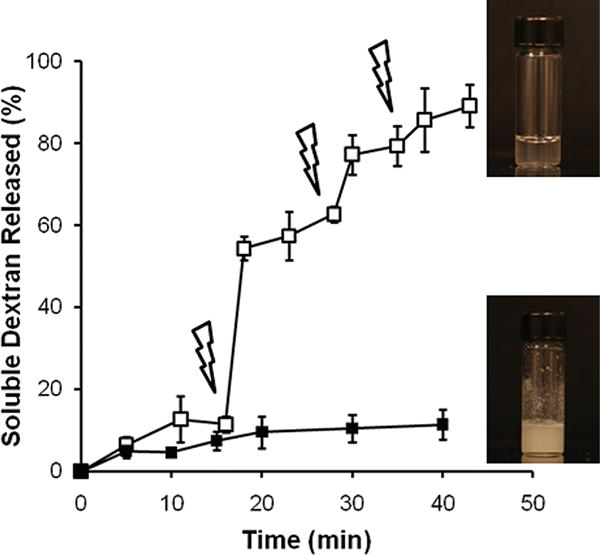
Photoacid triggered dissolution of dextran from Ac-Dex particles either in the dark (closed squares) or exposed to a UV light (open squares) in H2O. The particle suspensions were illuminated using a UV-lamp (345 nm, 4 W) for 1 min at the points indicated by lightning bolt symbols. The digital images of both suspensions were taken after a reaction time of 45 min.
The controlled degradation of photo-responsive Ac-Dex particles was further studied by generating a release profile of a fluorescent payload (Figure 6). Rhodamine B-labeled PEG (m-PEG-RB, 7 wt% based on Ac-Dex) was added to the jetting solution, which was used to prepare the nanoparticles. Electrospraying of this solution allowed for the encapsulation of PEG-RB within the photoresponsive Ac-Dex particles. The harvested particles were washed with water in the dark. The supernatants became red and fluorescence measurements revealed that nearly 16% of m-PEG-RB was released in form of a burst release. Although Ac-Dex particles still remained in the suspensions without significant rupture, the burst-like, initial release was attributed to leaching of the dye molecules from the hydrophobic surface areas of the particles, further contributing to the small degree of early dextran release observed in Figure 5. The release behavior of m-PEG-RB was then measured, and closely followed the degradation profiles measured for dextran release of the Ac-Dex particles. In control experiments conducted in the dark, the particle suspension presented slow but moderate m-PEG-RB release over time. The rough particle surfaces could increase the contact area to water and facilitate not only initial burst-like unloading of hydrophilic cargo but also alter the steady-state release kinetics of m-PEG-RB in the dark. On the other hand, the photo-triggered degradation of Ac-Dex particles led to a clear and sudden jump in release of m-PEG-RB after the first UV irradiation. As expected, the release amount of m-PEG-RB became smaller with subsequent illuminations, as previously observed for the release profile of dextran (Figure 5).
Figure 6.
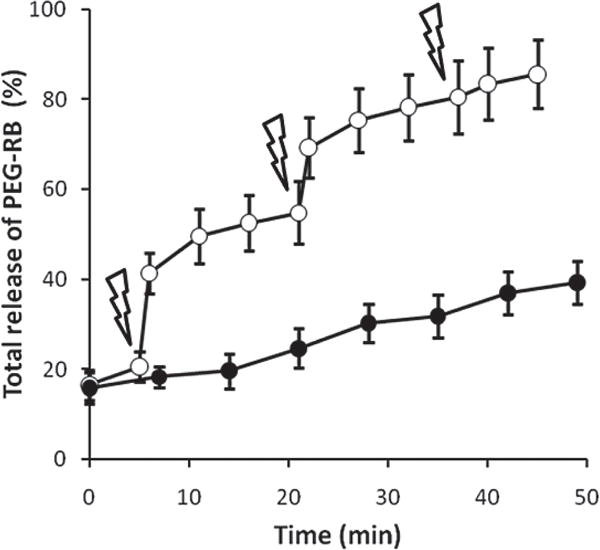
Release profile of m-PEG-RB encapsulated in photoresponsive Ac-Dex particles either in the dark (closed circles) or exposed to a UV light (open squares; exposure time: 1 min, lightning bolts). The amount of PEG-RB in the each supernatant sample was determined by measuring the emission at 580 nm (λex = 540 nm) and fitting to a calibration curve.
In order to demonstrate the controlled delivery of drugs using photoresponsive Ac-Dex particles, we loaded Ac-Dex particles with the anti-cancer drug irinotecan, a topoisomerase-I inhibitor. Irinorecan was simply added to the jetting solution (10 wt% based on Ac-Dex) that was used to synthesize the particles. The direct assessment of on-demand, triggered irinotecan release was done in a colorectal carcinoma model. For this purpose HT-29 cells, a well-characterized epithelial colorectal adenocarcinoma line that undergoes apoptosis upon exposure to irinotecan,[29] were incubated with drug loaded particles. Colorectal cancer is a serious and debilitating disease which was responsible for an estimated 51 370 US deaths in 2010. This is second only to lung cancer.[30] Iriniotecan (trade name Camptosar) has been used effectively to treat colorectal cancer and plays a key role in the chemotherapeutic mixture/regimen FOLFIRI.[31–33] Upon uptake by the body, irinotecan is hydrolyzed to 7-ethyl-10-hydroxycamptothecin (SN-38) via carboxylesterase present in the liver and the intestines.[31, 34] The molecule SN-38 is the actual metabolite that is responsible for the majority of its cytotoxic effects of irinotecan. The development of novel methods for spatial and temporal controlled release of this drug could be significant, as irinotecan has severe systemic side effects (e.g. diarrhea, neutropina, etc.) that limit the effective allowable concentration.[35]
To test the light-triggered release of irinotecan, we first exposed Ac-Dex particles loaded with drug to UV light for 20 min after triplicate PBS washing/centrifuging in order to remove drug that was not securely encapsulated within the particles. Next, we incubated pre-plated HT-29 with 0.2 mg mL−1 of drug-loaded particles with/without UV exposure, along with a soluble drug control (0.02 mg mL−1, the theoretical maximal release from the particles) and particles containing no drug. As irinotecan has been measured to have a solubility of < 2.5 mg mL−1 in water at 20 °C, this lower concentration was felt to be a safe choice, and observed to be more than cytotoxic enough towards the chosen cell model and timeframe.[36] The XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) cell proliferation assay was used to assess cell viability after 48 h, and pictures were taken with a brightfield microscope after 54 h (Figure 7). Compared to a control, in which no particles or drug were added to the cells, the drug-loaded and drug-free Ac-Dex particles without UV led to an approximate 24 − 36% reduction respectively in metabolic activity as measured by the XTT assay. A low level of innate toxicity may be associated with the particles themselves, perhaps due to the presence of PAG. The slightly cytotoxic effect observed with non-irradiated particles can thus be ascribed to low levels of innate particle toxicity. Importantly, there is no statistically significant difference in toxicity between drug-loaded particles and drug-free particles (significance set at p < 0.001), indicating that the drug remains mostly encapsulated in the particles and displays minimal leaching even after 48 hours of culture with the cells. However, upon particle exposure to UV light, an astonishing drop in cell viability by ~83% was observed compared to the control. The effect was so drastic, that there was no statistical difference (set as p < 0.001) to a positive control that contained a theoretical concentration of the free drug (assuming all drug is released from the particles). This demonstrates that the drug released by light-triggered degradation of Ac-Dex particles reaches the same effective toxicity as the pure soluble drug and furthermore indicates that drug-loaded particles unexposed to UV have only low levels of release in vitro. Particles without drug were also exposed to UV for 20 min and incubated with cells; but no toxic effect was observed (data not shown), indicating that cell death was mainly due to the released drug and not to the UV treatment. Furthermore, particles without drug almost completely degraded in UV light after 20 min, yet showed less toxicity than particles unexposed to UV. Bright-field microscopy was used to confirm the XTT results, clearly showing far fewer viable cells in the presence of UV-exposed drug-loaded particles and soluble drug control in comparison to other experimental conditions.
Figure 7.
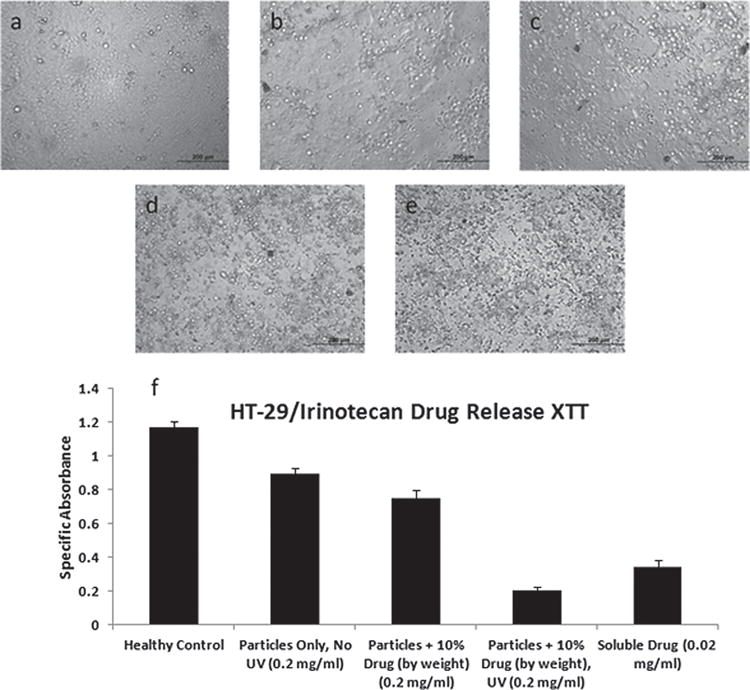
HT-29 incubated for 2 days with (a) media only (no particles or drug), (b) particles only unexposed to UV, (c) irinotecan-loaded particles (10% by weight) unexposed to UV, (d) irinotecan-loaded particles (10% by weight) previously exposed to 20 min of UV, and (e) soluble irinotecan (no particles) at the theoretical maximal release concentration. (f) Specific absorbance from XTT assay.
We have shown that electrospraying acetalated dextran (Ac-Dex) solution containing 2-(4-methoxystyryl)-4,6-bis(trichloromethyl)-1,3,5-triazine, a photoacid generator (PAG), can yield photoswitchable nanoparticles for triggered release. The exposure of the particles to UV light creates photoacids that trigger the demasking of hydroxyl groups on dextran and lead to the dissolution of Ac-Dex particles in water. In addition, the versatility of the electrospraying process allows for the encapsulation of various molecules such as fluorescent macromolecules and anti-cancer drugs within photo-responsive Ac-Dex particles. We demonstrate that controlled illumination with UV light for relatively short periods of time allows for on-demand degradation of Ac-Dex particles and release of payloads. This simple approach may allow for the utilization of a wide range of acid-sensitive biocompatible and biodegradable polymers that synthetically are more flexible than light-sensitive corresponding polymers for remote controllable drug delivery carriers. To circumvent the potential issues of using UV light, recent developments of two-photon photoacid generators may be utilized in the future that allow for the use of red and near infrared light,[37] thereby providing the added benefit of penetrating the tissue deeper than UV and visible light.
Experimental Section
Materials
Dextran from Leuconostoc mesenteroides (average MW 9000–11 000 g mol−1), pyridinium p-toluenesulfonate, 2-methoxypropene, tetrahydrofuran (THF), N, N-dimethylformamide (DMF), anhydrous dimethyl sulfoxide (DMSO), and Tween-20 were purchased from Sigma-Aldrich. Rhodamine B-labeled PEG (m-PEG-RB, MW 10 000) was purchased from NANOCS. All reagents were used without further purification unless otherwise specified. Acetalated Dextran (Ac-Dex) was synthesized according to a previously reported procedure.[10] The NMR characterization is found in the Supporting Information.
Electron Microscopy
Scanning electron microscopy (SEM, FEI Nova Nanolabs) and transmission electron microscopy (TEM, JEOL 3011) were used to characterize particles electrosprayed on the top of an aluminum sheet or a copper TEM grid coated with a carbon film (Ted Pella). SEM images were obtained at 5 kV after coating particles with gold sputtering and TEM images were obtained with an accelerating voltage of 300 kV. Average diameters of the particles were determined by measuring approximately 400–500 particles in randomly selected areas of the SEM images.
m-PEG-RB Release from Ac-Dex Particles
The procedure used to perform this experiment was similar to that used for particle degradation experiment. m-PEG-RB loaded particles were used. After centrifugation, whole supernatants were collected and 1 mL of dd-H2O was replenished. The quantity of m-PEG-RB was determined by measuring the emission at 580 nm with an excitation of 540 nm. The amount loaded in samples was calculated using a calibration curve.
Cell Proliferation Assay
HT-29 at passage 2 were incubated with McCoy’s 5A media (ATCC) + 10% FBS (ATCC) + 10% Pen/Strep (Gibco) on a 96-well plate at 30 000 cells/well and allowed to attach and spread for 48 h. Particles were triplicate washed/centrifuged before use, then particles loaded with irinotecan (10% bw) were exposed to UV light for 20 min. Cells were then incubated with 0.2 mg mL−1 particles only (no irinotecan, no UV), 0.02 mg/ml soluble irinotecan, 0.2 mg mL−1 particles + 10% by weight irinotecan exposed to UV, and 0.2 mg mL−1 particles + 10% by weight irinotecan unexposed to UV. After 48 h, the plate was assayed for cell viability using XTT (ATCC) and a plate reader. Specific absorbance is calculated as SA = Experimental Absorbance (475 nm) – Blank Absorbance (475 nm) – Experimental Absorbance (660 nm), and error bars are standard error (standard deviation/sqrt(n)), with n = 17 (where n counts each individual well over two independent trials – data presented is combined). Statistical analysis based on a T-test was performed with a 99.9% confidence interval. Bright-field microscopy was performed after 54 h of particle/drug incubation using a Leica DMIRB inverted microscope.
Supplementary Material
Acknowledgments
The authors thank the American Cancer Society (RSG–08–284–01–CDD) for financial support and acknowledge funding from the Multidisciplinary University Research Initiative of the Department of Defense and the Army Research Office (W911NF-10–1–0518). The authors would like to thank the Microscopy and Image Analysis Lab (MIL) at the University of Michigan for use of the CLSM and SEM.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Tae-Hong Park, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA.
Thomas W. Eyster, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
Joshua M. Lumley, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
Dr. Sangyeul Hwang, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
Dr. Kyung Jin Lee, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
Asish Misra, Biomedical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA.
Sahar Rahmani, Biomedical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA.
Prof. Joerg Lahann, Department of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA Department of Materials Science and Engineering, University of Michigan, Ann Arbor, 48109, USA; Department of Macromolecular Science and Engineering, University of Michigan, Ann Arbor, 48109, USA; Department of Biomedical Engineering, University of Michigan, Ann Arbor, 48109, USA.
References
- 1.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Science. 1994;263:1600. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 2.Mitragotri S, Lahann J. Adv Mater. 2012;24:3717. doi: 10.1002/adma.201202080. [DOI] [PubMed] [Google Scholar]
- 3.Murthy N, Xu M, Schuck S, Kunisawa J, Shastri N, Fréchet JMJ. Proc Natl Acad Sci USA. 2003;100:4995. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung JE, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T. J Control Release. 1999;62:115. doi: 10.1016/s0168-3659(99)00029-2. [DOI] [PubMed] [Google Scholar]
- 5.Gao ZG, Fain HD, Rapoport N. J Control Release. 2005;102:203. doi: 10.1016/j.jconrel.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 6.Fomina N, McFearin C, Sermsakdi M, Edigin O, Almutairi A. J Am Chem Soc. 2010;132:9540. doi: 10.1021/ja102595j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timko BP, Dvir T, Kohane DS. Adv Mater. 2010;22:4925. doi: 10.1002/adma.201002072. [DOI] [PubMed] [Google Scholar]
- 8.Jin Q, Mitschang F, Agarwal S. Biomacromolecules. 2011;12:3684. doi: 10.1021/bm2009125. [DOI] [PubMed] [Google Scholar]
- 9.Sinha VR, Kumria R. Int J Pharmaceut. 2001;224:19. doi: 10.1016/s0378-5173(01)00720-7. [DOI] [PubMed] [Google Scholar]
- 10.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Fréchet JMJ. J Am Chem Soc. 2008;130:10494. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Fréchet JMJ. Proc Natl Acad Sci USA. 2009;106:5497. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen JA, Beaudette TT, Cohen JL, Brooders KE, Bachelder EM, Fréchet JMJ. Adv Mater. 2010;22:3593. doi: 10.1002/adma.201000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen JL, Schubert S, Wich PR, Cui L, Cohen JA, Mynar JL, Fréchet JMJ. Bioconjug Chem. 2011;22:1056. doi: 10.1021/bc100542r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirai M, Tsunooka M. Bull Chem Soc Jpn. 1998;71:2483. [Google Scholar]
- 15.Kawaguchi H. Prog Polym Sci. 2000;25:1171. [Google Scholar]
- 16.Tyrrell ZL, Shen YQ, Radosz M. Prog Polym Sci. 2010;35:1128. [Google Scholar]
- 17.Wang Y, Angelatos AS, Caruso F. Chem Mater. 2007;20:848. [Google Scholar]
- 18.Jaworek A. Powder Technology. 2007;176:18. [Google Scholar]
- 19.Xie J, Lim LK, Phua Y, Hua J, Wang CH. J Colloid Interf Sci. 2006;302:103. doi: 10.1016/j.jcis.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 20.Hogan CJ, Jr, Yun KM, Chen D-R, Lenggoro IW, Biswas P, Okuyama K. Colloids Surf, A. 2007;311:67. [Google Scholar]
- 21.Wu Y, MacKay JA, McDaniel JR, Chilkoti A, Clark RL. Biomacromolecules. 2008;10:19. doi: 10.1021/bm801033f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almería B, Deng W, Fahmy TM, Gomez A. J Colloid Interf Sci. 2010;343:125. doi: 10.1016/j.jcis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Valo H, Peltonen L, Vehviläinen S, Karjalainen M, Kostiainen R, Laaksonen T, Hirvonen J. Small. 2009;5:1791. doi: 10.1002/smll.200801907. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi K, Ono K, Suzuki H, Sawada M, Moriya M, Sakamoto W, Yogo T. Small. 2010;6:2384. doi: 10.1002/smll.201000399. [DOI] [PubMed] [Google Scholar]
- 25.Hwang S, Roh KH, Lim DW, Wang GY, Uher C, Lahann J. Phys Chem Chem Phys. 2010;12:11894. doi: 10.1039/c0cp00264j. [DOI] [PubMed] [Google Scholar]
- 26.Lim DW, Hwang S, Uzun O, Stellacci F, Lahann J. Macromol Rapid Commun. 2010;31:176. doi: 10.1002/marc.200900597. [DOI] [PubMed] [Google Scholar]
- 27.Pohlers G, Scaiano JC, Sinta R. Chem Mater. 1997;9:3222. [Google Scholar]
- 28.Doner LW, Irwin PL. Anal Biochem. 1992;202:50. doi: 10.1016/0003-2697(92)90204-k. [DOI] [PubMed] [Google Scholar]
- 29.Pavillard V, Agostini C, Richard S, Charasson V, Montaudon D, Robert J. Cancer Chemother Phamacol. 2002;49:329. doi: 10.1007/s00280-001-0416-0. [DOI] [PubMed] [Google Scholar]
- 30.Jemal A, Siegel R, Xu J, Ward E. CA: Cancer J Clin. 2010;60:277. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 31.Ramesh M, Ahlawat P, Srinivas NR. Biomed Chromatogr. 2010;24:104. doi: 10.1002/bmc.1345. [DOI] [PubMed] [Google Scholar]
- 32.Dodds HM, Robert J, Rivory LP. J Pharm Biomed Anal. 1998;17:785. doi: 10.1016/s0731-7085(97)00256-2. [DOI] [PubMed] [Google Scholar]
- 33.Colucci G, Gebbia V, Paoletti G, Giuliani F, Caruso M, Gebbia N, Carteni G, Agostara B, Pezzella G, Manzione L, Borsellino N, Misino A, Romito S, Durini E, Cordio S, Di Seri M, Lopez M, Maiello E. J Clin Oncol. 2005;23:4866. [Google Scholar]
- 34.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Cancer Res. 1991;51:4187. [PubMed] [Google Scholar]
- 35.Abigerges D, Armand J-P, Chabot GG, Costa LD, Fadel E, Cote C, érait PH, Gandia D. J Natl Cancer Inst. 1994;86:446. doi: 10.1093/jnci/86.6.446. [DOI] [PubMed] [Google Scholar]
- 36.Meyer-Losic F, Nicolazzi C, Quinonero J, Ribes F, Michel M, Dubois V, de Coupade C, Boukaissi M, Chéné A-S, Tranchant I, Arranz V, Zoubaa I, Fruchart J-S, Ravel D, Kearsey J. Clin Cancer Res. 2008;14:2145. doi: 10.1158/1078-0432.CCR-07-4580. [DOI] [PubMed] [Google Scholar]
- 37.Zhou W, Kuebler SM, Braun KL, Yu T, Cammack JK, Ober CK, Perry JW, Marder SR. Science. 2002;296:1106. doi: 10.1126/science.296.5570.1106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.