

Abstract
Transforming growth factor (TGF) β1, β2, and β3 (TGF-β1–TGF-β3, respectively) are small secreted signaling proteins that each signal through the TGF-β type I and type II receptors (TβRI and TβRII, respectively). However, TGF-β2, which is well-known to bind TβRII several hundred-fold more weakly than TGF-β1 and TGF-β3, has an additional requirement for betaglycan, a membrane-anchored nonsignaling receptor. Betaglycan has two domains that bind TGF-β2 at independent sites, but how it binds TGF-β2 to potentiate TβRII binding and how the complex with TGF-β, TβRII, and betaglycan undergoes the transition to the signaling complex with TGF-β, TβRII, and TβRI are not understood. To investigate the mechanism, the binding of the TGF-βs to the betaglycan extracellular domain, as well as its two independent binding domains, either directly or in combination with the TβRI and TβRII ectodomains, was studied using surface plasmon resonance, isothermal titration calorimetry, and size-exclusion chromatography. These studies show that betaglycan binds TGF-β homodimers with a 1:1 stoichiometry in a manner that allows one molecule of TβRII to bind. These studies further show that betaglycan modestly potentiates the binding of TβRII and must be displaced to allow TβRI to bind. These findings suggest that betaglycan functions to bind and concentrate TGF-β2 on the cell surface and thus promote the binding of TβRII by both membrane-localization effects and allostery. These studies further suggest that the transition to the signaling complex is mediated by the recruitment of TβRI, which simultaneously displaces betaglycan and stabilizes the bound TβRII by direct receptor–receptor contact.
Betaglycan is a coreceptor for the transforming growth factor β (TGF-β) family of signaling proteins, which have numerous essential roles in regulating cellular growth and differentiation, in both developing embryos and adults.1−3 Betaglycan is expressed in many cell types and is typically present at levels much higher than those of the type I and type II signaling receptors of the family,4,5 which in contrast to betaglycan are required for signaling.6 Betaglycan binds several ligands of the TGF-β family, including the TGF-β isoforms TGF-β1–TGF-β3, as well as inhibins, and in cultured cells enhances their association with their type II receptors, TβRII and ActRII or ActRIIB.4,7 Betaglycan binds TGF-β2 with the highest affinity,8 which is important for the function of this ligand, as TGF-β2 binds TβRII 200–300-fold more weakly than TGF-β1 and TGF-β3.4,9,10 Cells that do not express betaglycan do not respond to TGF-β2 as robustly as they do to TGF-β1 and TGF-β3, requiring in some cases as much as 100–500-fold higher concentrations to achieve the same response.9−11 Cells that naturally express betaglycan or that do not but exhibit ectopic expression respond to TGF-β2 with potencies similar to those of TGF-β1 and TGF-β3.4,8,12 Betaglycan also enhances the binding of inhibin A to the type II receptors, ActRII and ActRIIB, which inhibits the response of activin by sequestering its type II receptors, ActRII and ActRIIB, in a dead-end complex incapable of recruiting a type I receptor.7,13,14 Thus, in some instances, betaglycan functions to enhance the signaling of TGF-β family ligands, while in other instances, it is inhibitory.
Betaglycan is a transmembrane proteoglycan with heparan and chondroitin sulfate chains, but these are not required for binding of TGF-β ligands.8,15 Betaglycan has a large extracellular domain, comprised of two subdomains, a membrane distal orphan domain and a membrane proximal zona pellucida domain16 (Figure 1A). The zona pellucida domain binds inhibins and TGF-βs, while the orphan domain binds only TGF-βs.8,14,17−19 Cross-linking studies have demonstrated TGF-β/TβRII/betaglycan complexes on the cell surface.4 Furthermore, Esparza-Lopez and colleagues reported that while both orphan and zona pellucida domains are capable of independently promoting TGF-β2-mediated Smad-2 phosphorylation, only full-length betaglycan or the betaglycan orphan domain increases the level of TGF-β2 radiolabeling of TβRII.8 Thus, both domains are capable of independently promoting TGF-β2-mediated signaling, while only the orphan domain appears to be sufficient for enhancing TGF-β2/TβRII complex formation.
Figure 1.

Betaglycan’s domain structure and isolation of these domains. (A) Schematic diagram of the betaglycan domain structure, with the N-terminal orphan domain (BGO) colored cyan and the N- and C-terminal zona pellucida domains (BGZP-N and BGZP-C, respectively) colored red and green, respectively. Glycosaminoglycan chains attached to two residues in the ZP-N subdomain are shown schematically as beads on a string. Disulfide bonds are represented by S–S, while free cysteines are represented by -SH. (B–E) SDS–PAGE analysis of the purified betaglycan constructs run under nonreducing conditions. Predicted masses for the protein core are shown along the top of each gel. Proteins produced in mammalian cells (B–D) were run either as isolated (−) or as isolated but treated with a catalytic amount of the deglyocosidase, endoglycosidase H (EndoH) (+).
Betaglycan also functions as an inhibin coreceptor by enhancing its binding to ActRII.7 Complexes of betaglycan with inhibin A and ActRII can be found on the cell surface.7 The major difference between TGF-β and inhibin is that both domains of betaglycan bind TGF-β, while only the zona pellucida domain binds inhibin.8,14,15,19 Makanji et al. reported the betaglycan binding site on inhibin A, which lies on finger 2 of the betaglycan binding α subunit.20 Inhibin A’s P51, V108, S112, and K119 contribute to binding of betaglycan, with V108 and K119 being the most important. Interestingly, a corresponding set of residues is also present in the TGF-βs (P36, I88, T95, and K97), and these lie immediately adjacent to residues in TGF-β’s TβRII binding site, including R25, V92, and R94 in TGF-β1 and -β3 and K25, I92, and K94 in TGF-β2.9,21 Thus, it is conceivable the zona pellucida domain of betaglycan and TβRII have overlapping binding sites, which would be consistent with the report of Esparza-Lopez that the zona pellucida domain alone does not increase the level of TGF-β2 labeling of TβRII on the cell surface.
Biophysical studies have begun to shed light on the mechanism by which betaglycan functions. By surface plasmon resonance (SPR)-based binding studies, it has been shown that the betaglycan orphan and zona pellucida domains bind TGF-βs at independent sites.22 By deletion analysis and accompanying functional studies, it has been shown that betaglycan’s zona pellucida domain is comprised of tandem immunoglobulin-like domains and that the ability of this domain to bind to TGF-βs and inhibins resides exclusively in the C-terminal immunoglobulin-like domain.14,19 Recently, structures of the C-terminal immunoglobulin-like domain of rat and mouse betaglycan have been reported,23,24 and through accompanying functional studies, it has been suggested this domain binds TGF-βs through an extended loop region, known as an EHP motif.24
Beyond this, little is known about the precise nature of the complexes betaglycan forms with TGF-βs and how complex formation might potentiate receptor binding and signaling. Here, we report an in-depth study of the binding of the TGF-β isoforms to the betaglycan extracellular domain, as well as its two independent binding domains, either directly or in combination with the ectodomains of the TGF-β type I and type II receptors, TβRI and TβRII, respectively, using surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and size-exclusion chromatography (SEC). These studies show that betaglycan binds TGF-β homodimers with a 1:1 stoichiometry, but in a manner that allows one molecule of TβRII to bind. These studies further show that betaglycan modestly potentiates the binding of TβRII but must be displaced to allow TβRI to be recruited. These findings suggest that betaglycan functions to bind and concentrate TGF-β2 on the cell surface and thus promote the binding of TβRII by membrane-localization effects and allostery. These studies further suggest that the transition to the signaling complex is mediated by the recruitment of TβRI, which simultaneously displaces betaglycan and stabilizes the bound TβRII by direct receptor–receptor contact.
Experimental Procedures
Protein Preparation
Recombinant human TGF-β2 and the TGF-β2TM variant bearing Lys25 → Arg, Ile92 → Val, and Lys94 → Arg substitutions9 were expressed in Escherichia coli as insoluble inclusion bodies and refolded and purified as described previously.25 TβRI-ED and TβRII-ED were expressed in E. coli as insoluble inclusion bodies and refolded and purified as described previously.26,27
To produce BGO, bacterial T7 expression vector pET32a (EMD Millipore, Billerica, MA) was modified so that the coding sequence for a thrombin cleavage site (LVPRGS) downstream of the thioredoxin-hexahistidine tag coding cassette was replaced with the coding sequence for a tobacco etch virus (TEV) protease cleavage site (ENLYFQG). The coding sequence for residues 24–384 of rat betaglycan was inserted downstream of the TEV cleavage site and modified using site-directed mutagenesis (Quikchange, Agilent, Santa Clara, CA) so that Cys225 was substituted with serine. The entire length of the coding cassette was verified by DNA sequencing.
The thioredoxin–BGO fusion protein was overexpressed in BL21(DE3) cells cultured in LB medium at 37 °C containing 150 μg/mL ampicillin. Expression of thioredoxin-BGO was induced with 1 mM IPTG when the absorbance at 600 nm was 0.6. Cells were harvested by centrifugation and resuspended in 100 mM Tris, 10 mM EDTA, and 1 mM phenylmethanesulfonyl fluoride (PMSF) (pH 8.0) and lysed by sonication. Inclusion bodies containing the overexpressed fusion protein were isolated by washing the insoluble fraction with lysis buffer with 500 mM NaCl and 0.5% Triton X-100 and solubilized in 8 M urea, 25 mM Tris, and 7.5 mM imidazole (pH 8.0). Solubilized inclusion bodies were then loaded onto a Ni-NTA column (Qiagen, Valencia, CA) equilibrated with solubilization buffer. The resin was washed in solubilization buffer, and the histidine-tagged fusion protein was eluted with solubilization buffer with 300 mM imidazole. The eluted protein was reduced with 50 mM reduced glutathione (Sigma, St. Louis, MO) and added to folding buffer [20 mM Tris, 5% glycerol, and 0.5 mM oxidized glutathione (pH 9.0)] (Sigma-Aldrich, St. Louis, MO) such that the final protein concentration was 0.1 mg/mL and the final reduced glutathione concentration was 2 mM. After being stirred overnight at 4 °C, the folding mixture was adjusted to pH 8.0 by adding solid Na2HPO4 and then loaded onto a Ni-NTA column equilibrated with 25 mM Tris-HCl and 5% glycerol (pH 8.0). The resin was washed with equilibration buffer and eluted with equilibration buffer with 300 mM imidazole. The thioredoxin and hexahistidine tag were removed by treating the isolated fusion protein with TEV protease. BGO was separated from the thioredoxin by passing the digestion mixture over a Ni-NTA column equilibrated with 25 mM Tris (pH 8.0) and 5% glycerol and by binding the eluate to a Source Q ion exchange column (GE Healthcare, Piscataway, NJ) equilibrated with 25 mM Tris (pH 8.0) and 5% glycerol. BGO was isolated by eluting the ion exchange column with a linear 0 to 0.25 M NaCl gradient. BGO produced by this method was used for all of the measurements shown, except the ITC measurements shown in Figure 7, which used a sample produced in mammalian cells (described below).
Figure 7.

Effect of BGO on binding of TβRII to TGF-β2TM and effect of TβRII on binding of BGZP to TGF-β2TM. (A and B) SPR sensorgrams for binding of TβRII to TGF-β2TM in the absence and presence of 800 nM BGO, respectively. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series of TβRII from 4 to 0.008 μM. (C) Plot of the equilibrium response for binding of TβRII to TGF-β2TM in the absence (black) or presence (blue) of 800 nM BGO. Equilibrium binding constants were obtained by fitting the equilibrium response as a function of concentration to a standard binding isotherm. The fitted curve is shown as a solid line, black or blue in the absence or presence of BGO, respectively. (D and E) SPR sensorgrams for binding of BGZP to TGF-β2TM in the absence and presence of 4 μM TβRII, respectively. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series of BGZP from 4 to 0.008 μM. Other details are as described for panels A and B. (F) Plot of the equilibrium response for binding of BGZP to TGF-β2TM in the absence (black) or presence (blue) of 4 μM TβRII. Other details are as described for panel C. (G and H) Schematic depiction showing the manner of binding of BGO and BGZP/BGZP-C, respectively, by the SPR binding data shown in Figures 6 and 7.
The full-length betaglycan extracellular domain (BGO-ZP) and the orphan, ZP, and ZP-C subdomains (BGO, BGZP, and BGZP-C, respectively) were expressed as secreted proteins in a Chinese hamster ovary (CHO) cell line (CHO-lec3.2.8.1) using the method previously described for TGF-β128 (BGO-ZP, BGZP, and BGZP-C) or HEK-293 expi cells (Invitrogen, Carlsbad, CA) (BGO). This was accomplished by modifying the previously described pcDNA3.1+ expression vector for TGF-β128 to include a NotI restriction site immediately following the last residue of the rat serum albumin signal peptide. DNA fragments encoding the different domains of rat betaglycan [residues 24–761 for the full-length betaglycan extracellular domain, residues 24–383 for the orphan domain (BGO), residues 450–761 for the full-length zona pellucida domain (BGZP), and residues 589–761 for the C-terminal portion of the zona pellucida domain (BGZP-C)] together with a C-terminal hexahistidine sequence were generated using polymerase chain reaction (PCR) primers that introduced NotI and ApaI restriction sites on the 5′ and 3′ ends, respectively. PCR products were digested with NotI and ApaI and then ligated into the modified form of the TGF-β1 expression vector described above.
Stably transfected CHO cells expressing BGO-ZP, BGZP, and BGZP-C were generated by culturing CHO-lec3.2.8.1 cells to near confluence in a T-25 flask maintained in nonselective medium, DMEM/F12 (Gibco, Gaithersburg, MD), containing 5% fetal bovine serum (FBS) (GE Healthcare). Prior to transfection, the medium was replaced with 4 mL of fresh DMEM/F12 supplemented with 5% FBS. Lipofectamine 2000 (Invitrogen) (30 μL) and the betaglycan pcDNA3.1+ plasmid DNA (10 μg) were diluted with 500 μL each of OPTI-MEM I (Gibco) medium and then combined and incubated at room temperature for 20 min. The mixture in OPTI-MEM I medium was then added to the flask of confluent cells. After 24 h, the medium was replaced with fresh DMEM/F12 supplemented with 5% FBS, and 2 days post-transfection, the cells were trypsinized and seeded in 10 96-well plates and cultured in 150 μL/well of selection medium, glutamine-free GMEM-S (SAFC Biosciences) supplemented with 5% FBS, GS supplement (Sigma-Aldrich), and 30 μM methionine sulfoximine (MSX) (Sigma-Aldrich). After 3 weeks, the medium from wells containing colonies was assayed for protein expression by an enzyme-linked immunosorbent assay (ELISA) using a rabbit-derived anti-betaglycan IgG. The 24 most strongly expressing clones were transferred into a 24-well plate containing 500 μL of selection medium and assayed again by an ELISA. The clone with the highest level of expression was expanded into six T-225 flasks in 50 mL of selection medium; once confluent, the cells were washed with PBS, and the medium was replaced with 50 mL of CHO-S-SFM II per flask (Gibco).
The CHO-S-SFM was collected every 2–4 days for five or six cycles and stored at −20 °C. The collected medium was thawed, centrifuged at 6000g, filtered with a 0.22 μm poly(ether sulfone) filter, and diluted with 1 volume of loading buffer [25 mM Tris (pH 8.0), 150 mM NaCl, and 10 mM imidazole]. The diluted medium was passed over a column of Ni-NTA (Qiagen, Valencia, CA) equilibrated with loading buffer. The resin was washed in loading buffer, and the histidine-tagged protein was eluted with loading buffer with 300 mM imidazole. The proteins were further purified on a Superdex 200 16/60 size-exclusion column (GE Healthcare) equilibrated in 25 mM Tris and 50 mM NaCl (pH 8.0). To test for glycosylation, 1 unit of endoglycosidase H (New England Biolabs, Ipswich, MA) per microgram of BGO-ZP, BGZP, or BGZP-C was incubated at 37 °C in 0.5 M sodium citrate (pH 5.5).
The betaglycan orphan domain, BGO, was expressed by transient transfection of HEK293 expi cells (Invitrogen, Carlsbad, CA) grown in suspension in Expi 293 medium at 8% CO2 and 80% humidity and rotating at 125 rpm. The HEK-293 expi cells were grown to a density of 2.5 × 106 cells/mL and incubated with 1.5 μg of cesium chloride gradient-purified plasmid DNA and 3.0 μg of polyethylenimine (Polysciences, Warrington, PA) per milliliter of cells. Sixteen hours later, valproic acid (Sigma-Aldrich) was added to a final concentration of 2.2 mM.29 Conditioned media were collected by centrifugation 4 days after the transfection, and BGO was purified as described above for BGO-ZP, BGZP, or BGZP-C.
SPR Binding Measurements
SPR binding analyses were performed with a Biacore 3000 surface plasmon resonance instrument (GE Healthcare). All SPR experiments, except those reported in Table 1 of the Supporting Information, were performed using TGF-βs biotinylated in 25 mM MES (pH 4.8) with a 100-fold molar excess of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide·HCl (EDC), a 25-fold molar excess of N-hydroxysulfosuccinimide (Sulfo-NHS), and a 100-fold molar excess of EZ-Link Amine-PEG3-biotin (Pierce, Rockford, IL). SPR experiments, reported in Table 1 of the Supporting Information, were performed using TGF-β2 biotinylated by prebinding it to BGO-ZP in 10 mM sodium phosphate and 140 mM NaCl (pH 7.5) followed by treatment with 1 molar equivalant of sulfo-NHS-LC-LC-Biotin (Pierce). The biotinylation reactions were quenched with 10 volumes of 100 mM acetic acid, and the biotinylated TGF-βs were isolated by ion exchange chromatography (Source S, GE Healthcare) at pH 4.0 in 25 mM NaOAc and 30% isopropanol. Streptavidin was coupled to a CM5 sensor chip (GE Healthcare) by activation with EDC/NHS to 3000–5000 resonance units (RUs). Biotinylated TGF-βs were captured on the streptavidin surface at a density of 150–200 RUs. All experiments were performed in HBS-EP buffer [10 mM Hepes (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20].
Equilibrium experiments were performed for TβRII, BGO, BGZP, BGZP-C, and BGO-ZP binding to TGF-β2 and TGF-β2TM. A series of 2-fold dilutions (from 4 to 0.002 μM) were injected and allowed to associate and reach equilibrium for 15 min at a flow rate of 10 μL/min. The protein was then allowed to dissociate for 5 min. The injections were performed in duplicate. The surface was then regenerated with a brief injection of 4 M guanidinium hydrochloride (10 s, 100 μL/min). In experiments with saturating protein, the protein was present throughout the experiment, i.e., in both the buffer and the injected samples. The concentrations of protein used for saturation were 4 μM for TβRII, 80 nM for BGO-ZP, and 800 nM for BGO. In all cases, the equilibrium data were processed and analyzed using the software package Scrubber 2 and double referencing was used to remove background binding and instrument noise. The equilibrium response was normalized by dividing the response by the molecular weight of the analyte in daltons and multiplying by 100000. A standard binding curve [y = (Rmax[conc])/(KD + [conc])] was used to fit the normalized equilibrium response at the end of the injection as a function of concentration to derive Rmax and KD (KaleidaGraph, Synergy Software, Reading, PA).
Competition experiments were performed by first injecting 1.0 μM receptor (1.0 μM TβRII alone or 1.0 μM TβRII with 1.0 μM BGO or 1.0 μM BGO-ZP) at a flow rate of 10 μL/min to saturate the TGF-β surface, followed by the same receptor at the same concentration and flow rate, but with increasing concentrations of TβRI (0.063, 0.13, 0.25, 0.50, 1.0, or 2.0 μM). The injections were performed in duplicate and randomized, with a 15 s pulse of 0.85% phosphoric acid to regenerate the TGF-β surfaces at the end of each injection cycle. All of the sensorgrams were referenced to the blank control surface and normalized to the start of the TβRI injection for comparison using BiaEval version 3.2 (GE Healthcare).
SEC and SEC–MALS
Protein complexes for SEC were prepared in two steps. First, a 2.5:1 TβRII/TGF-β2TM binary complex was formed by holding the pH at 7.0 as a concentrated stock of TGF-β2TM in 100 mM acetic acid was added to TβRII in 0.2 M Tris (pH 7.0). Second, after the 2.5:1 TβRII/TGF-β2TM binary complex had been dialyzed into column buffer [25 mM Tris, 100 mM NaCl, and 0.05% NaN3 (pH 7.0)], the complex was combined with a concentrated stock of BGO-ZP or BGO in column buffer to achieve the desired molar ratio (0.75 equiv of BGO-ZP/equiv of TβRII/TGF-β2TM binary complex and 3 equiv of BGO/equiv of TβRII/TGF-β2TM binary complex). Samples were then concentrated to a volume of ≤0.5 mL and loaded onto a Superdex 200 16/60 column (GE Healthcare) equilibrated in buffer containing 25 mM Hepes and 150 mM arginine (pH 7.4) and run at a flow rate of 0.5 mL/min. Partition coefficients, Kav, were calculated by the equation Kav = (Ve – Vo)/(Vt – Vo), where Ve corresponds to the elution volume for the species of interest and Vo and Vt correspond to the column void and total volumes, respectively.
SEC–MALS measurements on protein complexes were taken using a Superdex 200 Increase 10/300 GL column (GE Healthcare) in line with the multiwavelength UV detector of the Agilent high-performance liquid chromatography system (Agilent), multiangle light scattering (HELEOS, Wyatt Technology, Santa Barbara, CA), and refractive index detector (Optilab rEX, Wyatt Technology). Protein complexes for SEC–MALS were prepared in a manner identical to that described for the SEC samples, except the amount and volume of material injected were reduced by 5-fold. Typically, 100 μL of a protein solution was injected onto the SEC column at a flow rate of 0.5 mL/min in a buffer containing 25 mM Hepes and 150 mM arginine (pH 7.4). Instrument control and data analysis were performed with the Astra software package (Wyatt Technology).
Native Gel Electrophoresis
Protein samples were mixed under nonreducing conditions with an equal volume of native gel sample buffer [20% glycerol and 3.0 M Tris (pH 8.4)] at room temperature and immediately loaded onto a native polyacrylamide gel. Native gels were cast with a short (1 cm) 4% stacking gel buffered with 0.25 M Tris-HCl (pH 6.8) followed by a long (7 cm) 12% running gel buffered with 0.38 M Tris-HCl (pH 8.8) and run at 125 V for approximately 2 h.
Isothermal Titration Calorimetry
ITC data were generated using a Microcal PEAQ-ITC instrument (Malvern Instruments, Westborough, MA). In Table 3, a listing is provided of the buffers used and the proteins included in the syringe and sample cell (and their concentrations). In the two experiments performed without the detergent {3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)} in the buffer, the proteins to be included in both the syringe and sample cell were dialyzed exhaustively against the buffer and concentrated as necessary prior to being transferred to the syringe or sample cell. In the experiment with CHAPS in the buffer, the protein to be included in the syringe was dialyzed and concentrated without CHAPS in the buffer. Immediately prior to the sample being loaded into the calorimetry syringe, CHAPS was added from a concentrated stock prepared in buffer to a final concentration of 30 mM. In this experiment, the protein to be included in the calorimeter cell (TGF-β2) was dialyzed into 100 mM acetic acid, lyophilized, and resuspended in dialysis buffer supplemented with 30 mM CHAPS. Titrations were performed at 25 °C. Twenty 2 μL injections were performed with an injection duration of 4 s, a spacing of 150 s, and a reference power of 6. Data analysis was performed using the PEAQ-ITC software provided with the instrument.
Table 3. ITC Binding Data.
| sample cell component | TGF-β2 | TGF-β2TM/BGO-ZP | TGF-β2TM/TβRII |
| syringe component | BGO-ZP | TβRII | BGO |
| sample cell concentration (μM) | 5.40 | 16.7 | 10.0 |
| syringe concentration (μM) | 58.0 | 263 | 161 |
| buffer | 10 mM NaH2PO4 and 30 mM CHAPS (pH 7.4) | 10 mM NaH2PO4 (pH 7.4) | 25 mM glycine and 50 mM NaCl (pH 8.5) |
| N (sites) | 1.04 ± 0.04 | 1.04 ± 0.04 | 1.07 ± 0.02 |
| KD (nM) | 109 ± 56 | 510 ± 212 | 82 ± 26 |
| ΔH (kJ mol–1) | –52.6 ± 4.1 | –29.3 ± 1.8 | –38.8 ± 1.1 |
| ΔG (kJ mol–1) | –39.8 | –36.0 | –40.5 |
| –TΔS (kJ mol–1) | 12.8 | –6.7 | –1.7 |
| –8.6 |
Results
Expression of Betaglycan and Its Subdomains, BGO, BGZP, and BGZP-C
Betaglycan is a proteoglycan with a large extracellular domain (82.1 kDa without glycoslyation) and a single membrane-anchoring helix, as depicted in Figure 1A. On the basis of the secondary structure prediction and plasmin or BMP1 digestion,22,30 betaglycan’s extracellular domain can be divided into two subdomains, the membrane-distal orphan domain of approximately 42 kDa (BGO) and the zona pellucida domain (BGZP) of approximately 36 kDa. BGZP can also be further subdivided into N- and C-terminal domains termed BGZP-N and BGZP-C, respectively.14,23 Both BGO and BGZP bind TGF-βs, although as shown previously, BGZP-C includes all of the residues within the zona pellucida domain responsible for binding both TGF-β and inhibin A.14,23
Previously, the full-length betaglycan extracellular domain (BGO-ZP) and its subdomains were expressed in insect cells.8 However, the isolated protein was highly glycosylated and contained large amounts of disulfide-linked aggregates, which made the protein difficult to purify, particularly the high-molecular weight BGO-ZP. To improve the homogeneity, BGO-ZP, BGZP, and BGZP-C were expressed in CHO-lec3.2.8.1 cells that have four mutations that almost entirely eliminate O-linked glycans and severely truncate N-linked glycans.31Figure 1B–D shows that treatment of CHO-lec3.2.8.1 cell-expressed BGO-ZP, BGZP, and BGZP-C with endoglycosidase H, which cleaves mannose oligosaccharides linked to asparagines, reduced them to the expected size of their core protein. BGO, in contrast, was expressed as an insoluble protein in E. coli and renatured into the native receptor by oxidative refolding (Figure 1E). Recombinant BGO produced in E. coli bound TGF-β2 in a manner identical to that of recombinant BGO produced in insect cells as assessed by a sandwich ELISA with immobilized BGO (Figure S1).
TGF-β2 Binds Betaglycan but Only Weakly Binds TβRII
TGF-β2 is well-known to bind betaglycan with high affinity,22,32 but it only weakly binds TβRII.9,10,33 SPR was used to quantitate the relative affinities of these two receptors for TGF-β2 as shown in panels A and D of Figure 2. The individual sensorgrams were normalized to the molecular weight of the analyte. The binding affinity (KD) and maximal response (Rmax) were obtained by fitting the normalized equilibrium response (Req) as a function of concentration to the equation Req = (Rmax[conc])/(KD + [conc]) (Figure 2C,F). The affinity of BGO-ZP for TGF-β2 is 4.2 ± 0.6 nM, and the affinity of TβRII for TGF-β2 is 2.9 ± 1.1 μM (Table 1). Although we were able to calculate a KD and Rmax for binding of TβRII to TGF-β2, the KD is close to the highest concentration measured (4 μM), and therefore, the KD and Rmax provide only very approximate estimates of the actual values.
Figure 2.

Binding of full-length betaglycan (BGO-ZP) to TGF-β2 and TGF-β2TM and estimation of its binding stoichiometry by SPR. (A and B) SPR sensorgrams for binding of BGO-ZP to immobilized TGF-β2 and TGF-β2TM, respectively. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series of BGO-ZP from 1 to 0.002 μM. Normalized responses were calculated by dividing the measured response by the molecular weight of the analyte in daltons and multiplying by 106. (C) Plot of the normalized equilibrium response for binding of BGO-ZP to TGF-β2 (orange) or TGF-β2TM (black) as a function of the concentration of BGO-ZP. Equilibrium binding constants were obtained by fitting the normalized equilibrium response as a function of concentration to a standard binding isotherm (fitted curve shown as a solid dashed orange line for TGF-β2 and solid black line for TGF-β2TM). (D and E) SPR sensorgrams for binding of TβRII to immobilized TGF-β2 and TGF-β2TM, respectively. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series of TβRII from 4 to 0.008 μM. Other details are as described for panels A and B. (F) Plot of the normalized equilibrium response for binding of TβRII to TGF-β2 (orange) or TGF-β2TM (black) as a function of the concentration of TβRII. Other details are as described for panel C. (G) Schematic depiction of 1:1 TGF-β/BGO-ZP complexes suggested by the binding data shown in panels C and F.
Table 1. Binding Constants for Binding of TGF-β2 and TGF-β2TM to BGO-ZP and TβRII.
| surface | analyte | KD (nM) | Rmax (RUa) |
|---|---|---|---|
| TGF-β2 | BGO-ZP | 4.2 ± 0.6 | 180 ± 4 |
| TGF-β2TM | BGO-ZP | 5.5 ± 0.6 | 182 ± 4 |
| TGF-β2 | TβRII | 2900 ± 1100 | 105 ± 22 |
| TGF-β2TM | TβRII | 148 ± 8 | 386 ± 5 |
Normalized to molecular weight.
The crystal structure of the TGF-β ternary complex21,34,35 shows each TGF-β homodimer binds two molecules of TβRII and two molecules of TβRI. The stoichiometry with which betaglycan binds TGF-β has not, however, been rigorously established. Pepin et al. reported that deletion mutants of betaglycan could form dimers and oligomers when chemically cross-linked to radiolabeled TGF-β2,19 and Vilichis-Landeros et al. reported that betaglycan ecotodomains form stable noncovalent dimers.32 However, a more recent study showed that upon TGF-β stimulation, betaglycan did not form dimers on the cell surface.36
To investigate the stoichiometry directly, the SPR measurements described above were repeated, but using TGF-β2TM, a variant of TGF-β2 that binds TβRII with an affinity comparable to that of TGF-β1 and TGF-β3 because of substitution of three residues in the TβRII binding site (K25R, I92V, and K94R).9,10 TGF-β2TM was shown to bind BGO-ZP in a manner indistinguishable from that of TGF-β2, with KDs of 5.5 ± 0.6 nM for TGF-β2TM and 4.2 ± 0.6 nM for TGF-β2 and similar kinetics (Figure 2A,B and Table 1). TβRII was shown to bind TGF-β2TM with an affinity (KD of 148 ± 8 nM) significantly greater than that for TGF-β2 (KD of nearly ≥3 μM) (Figure 2B,E and Table 1), consistent with earlier reports that TGF-β2TM bound TβRII with an affinity comparable to that of TGF-β1 and TGF-β3.10 Therefore, the three substitutions significantly increase the binding affinity for TβRII but do not affect the affinity for BGO-ZP. The maximal SPR response (Rmax), normalized for the molecular weight, for binding of BGO-ZP to TGF-β2 and TGF-β2TM is near 200 RU, while the normalized maximal response for binding of TβRII to TGF-β2TM is near 400 RU (Figure 2C,F and Table 1). This indicates that half the number of BGO-ZP molecules bind each TGF-β homodimer compared to TβRII, suggesting that BGO-ZP binds TGF-β homodimers with a 1:1 stoichiometry. This finding, together with the previous finding that BGO and BGZP bind TGF-βs without cooperating or competing with one another,22 suggests that BGO and BGZP bind at independent sites and that BGO-ZP binds TGF-β homodimers in the manner shown in Figure 2G.
Effect of Betaglycan on TβRII Binding
Cross-linked complexes have been detected between the TGF-β isoforms and TβRII and betaglycan on the cell surface,4 suggesting that such complexes exist and that they play a role in the potentiation of TGF-β signaling by betaglycan. To assess whether the betaglycan ectodomain could potentiate the binding of TβRII, SPR was used to measure the affinity of TβRII for TGF-β2TM or TGF-β2 in the presence or absence of 80 nM BGO-ZP, which should be sufficient to almost completely saturate the immobilized TGF-β2TM or TGF-β2. The SPR sensorgrams show that BGO-ZP appears to have two effects on the binding of TβRII. The first is a slight potentiation of the binding affinity as shown by an approximate 3–8-fold enhancement of the concentration dependence of the equilibrium response for binding of TβRII to TGF-β2TM or TGF-β2 (Figure 3A,B,D,E and Table 2). The second is a decrease in the SPR maximal response for binding of TβRII to TGF-β2TM or TGF-β2 in the presence of BGO-ZP by a factor of approximately 2.5 (Figure 3C,F and Table 2). This suggests that the full-length betaglycan extracellular domain binds TGF-β dimers in a manner that blocks one of the TβRII binding sites. The fact that TβRII binds TGF-β2 or TGF-β2TM with a higher affinity in the presence of BGO-ZP suggests either that betaglycan induces small changes in ligand structure and/or dynamics that indirectly enhance the binding of TβRII or that the two receptors bind in such a way that they directly contact one another. These findings are consistent with the earlier cell-based cross-linking studies that demonstrated the existence of TGF-β/TβRII/betaglycan ternary complexes on the cell surface4 and suggest that betaglycan-bound TGF-β retains the ability to bind one molecule of TβRII and forms a 1:1:1 ternary complex, as shown in Figure 3G.
Figure 3.

Effect of betaglycan binding on TβRII binding to TGF-β2 and TGF-β2TM. (A and B) SPR sensorgrams for binding of TβRII to TGF-β2TM in the absence and presence of 80 nM BGO-ZP, respectively. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series of TβRII from 4 to 0.008 μM. (C) Plot of the equilibrium response for binding of TβRII to TGF-β2TM in the absence (black) or presence (red) of 80 nM BGO-ZP. Equilibrium binding constants were obtained by fitting the equilibrium response as a function of concentration to a standard binding isotherm. The fitted curve is shown as a solid black or red line in the absence or presence of BGO-ZP, respectively. (D and E) SPR sensorgrams for binding of TβRII to TGF-β2 in the absence and presence of 80 nM BGO-ZP, respectively. Other details are as described for panels A and B. (F) Plot of the equilibrium response for binding of TβRII to TGF-β2 in the absence (black) or presence of 80 nM BGO-ZP (red). Other details are as described for panel C. (G) Schematic depiction of the 1:1:1 TGF-β/TβRII/BGO-ZP ternary complex suggested by the SPR binding data shown in Figures 2 and 3.
Table 2. Binding Constants for Binding of TGF-β2 and TGF-β2TM to TβRII.
| surface | analyte | KD (nM) | Rmax (RU) |
|---|---|---|---|
| TGF-β2 | TβRII | 9400 ± 2200 | 56 ± 10 |
| TGF-β2 | TβRII (80 nM BGO-ZP) | 1070 ± 160 | 21 ± 1 |
| TGF-β2TM | TβRII | 129 ± 11 | 158 ± 3 |
| TGF-β2TM | TβRII (80 nM BGO-ZP) | 43 ± 8 | 60 ± 2 |
Betaglycan Binding in Solution
The SPR results presented in Figures 2 and 3 suggest that TGF-β, TβRII, and BGO-ZP form a 1:1:1 complex. To assess whether such a complex could form in solution, a 1:2.5 TGF-β2TM/TβRII binary complex (1.0 equiv) was prepared and subjected to size-exclusion chromatography (SEC), either alone (Figure 4A) or with a substoichiometric amount of BGO-ZP added (0.75 equiv relative to 1.0 equiv of 1:2.5 TGF-β2TM/TβRII binary complex) (Figure 4B). Three peaks were eluted for the TGF-β2TM/TβRII:BGO-ZP sample, the first of which (peak a) had the highest UV absorbance and as shown by SDS–PAGE corresponded to the TGF-β2TM/BGO-ZP/TβRII ternary complex (inset). The intensities of the second and third peaks (peaks b and c, respectively) were much lower; these eluted at the same volume as the first and second peaks (peaks a and b) present in the TGF-β2TM/TβRII sample and corresponded to excess TGF-β2TM/TβRII binary complex and excess TβRII, respectively (inset). To determine whether the three proteins in Figure 4B peak a corresponded to that of a stable stoichiometric ternary complex, an aliquot was analyzed by native PAGE, alongside a ternary complex assembled from individual components. The native gel revealed a sharp band that migrated like that of the ternary complex assembled from individual components, but no band that corresponded to excess TGF-β2TM/TβRII binary complex or BGO-ZP (Figure S2A). To estimate the molecular mass of the TGF-β2TM/TβRII/BGO-ZP complex, BG, BGO, and TβRII, which are of known size, were analyzed alone by SEC, and their partition coefficients, Kav, were plotted as a function of the log of their molecular weight (Figure 4D). The three data points for BG, BGO, and TβRII could be readily fit to a straight line, which in turn was used to estimate the molecular mass of the TGF-β2TM/TβRII/BGO-ZP complex based on its Kav value. This line predicted a near perfect match with the predicted mass for the 1:2 TGF-β2TM/TβRII and 1:1:1 TGF-β2TM/TβRII/BGO-ZP complexes (54 and 132 kDa, respectively) (Figure 4D), confirming the known stoichiometry of the 1:2 TGF-β2TM/TβRII complex21,34,35 and tentatively confirming the 1:1:1 stoichiometry inferred from the SPR measurements of the TGF-β2TM/TβRII/BGO-ZP complex.
Figure 4.

Complexes formed between BGO-ZP and BGO with TGF-β and TβRII in solution as assessed using SEC and SEC–MALS. (A–C) Superdex 200 16/60 SEC chromatograms for complexes formed by adding 2.5 equiv of TβRII to 1.0 equiv of TGF-β2TM, 0.75 equiv of BGO-ZP to 1.0 equiv of 2.5:1 TGF-β2TM/TβRII binary complex, and 3.0 equiv of BGO to 1.0 equiv of 2.5:1 TGF-β2TM/TβRII binary complex, respectively. Peaks labeled “el” and “tv” on the chromatograms correspond to the exclusion limit and total volume for the column, respectively. Shown in the inset is a nonreducing SDS–PAGE gel of the major peaks that eluted. (D) Plot of the SEC partition coefficient, Kav, as a function of the logarithm of the molecular weight for three proteins studied alone, TβRII, BGO, and BGO-ZP (black triangles). The red line corresponds to a fit of the data for the proteins alone (TβRII, BGO, and BGO-ZP), which are of known size, to a straight line. Green circles shown on the plot correspond to the Kav values for the TGF-β2TM/TβRII, TGF-β2TM/TβRII/BGO, and TGF-β2TM/TβRII/BGO-ZP complexes plotted as a function of the molecular weights of the complexes assuming the stoichiometries inferred from the SPR measurements (1:2 TGF-β2TM:TβRII, 1:2:1 TGF-β2TM:TβRII:BGO, and 1:1:1 TGF-β2TM:TβRII:BGO-ZP). (E) Superdex 200 Increase 10/300 GL SEC–MALS chromatograms obtained for the same three complexes shown in panels A–C. Complexes are labeled as follows: BG-TC (TGF-β2TM/TβRII/BGO-ZP), BGO-TC (TGF-β2TM/TβRII/BGO), and TβRII-BC (TGF-β2TM/TβRII). Estimated molecular weights derived from the multiangle light scattering measurements are shown below the peak for the TGF-β2TM/TβRII binary complex (blue traces) and above the peaks for the TGF-β2TM/TβRII/BGO and TGF-β2TM/TβRII/BGO-ZP ternary complexes (green and red traces, respectively). One unexpected observation is that the peak corresponding to the excess TGF-β2TM/TβRII binary complex present in the TGF-β2TM/TβRII/BGO-ZP sample eluted at a volume (panel E, red trace, 13.6 mL) slightly larger than that of the peak for the TGF-β2TM/TβRII binary complex sample (panel E, blue trace, 12.6 mL). Multiple runs performed with decreasing amounts of the TGF-β2TM/TβRII complex loaded show that this is due to a loading effect, with larger amounts loaded (and thus higher concentrations) eluting earlier (Figure S3). Most likely, the earlier elution at higher loading concentrations is the result of the preponderance of 1:2 TGF-β2TM/TβRII binary complexes, while at lower loading concentrations, there is a preponderance of 1:1 TGF-β2TM/TβRII binary complexes.
To directly assess the mass and stoichiometry, SEC–MALS and ITC experiments were performed. To perform the SEC–MALS measurements, the TβRII/TGF-β2TM and TβRII/TGF-β2TM/BGO-ZP samples were prepared in an identical manner and analyzed by SEC–MALS. The chromatograms obtained were very similar to those obtained before, and the estimated molecular masses for the TGF-β2TM/TβRII and TGF-β2TM/TβRII/BGO-ZP complexes were between 52 and 59 kDa and between 116 and 125 kDa, respectively (Figure 4E). The former is in close agreement with the mass expected for the 2:1 TGF-β2TM/TβRII complex (54 kDa),21,34,35 while the latter is in close agreement with the mass of 132 kDa estimated for the 1:1:1 TGF-β2TM/TβRII/BGO-ZP complex.
To further confirm the 1:1:1 stoichiometry for the TGF-β2TM/TβRII/BGO-ZP complex, ITC was performed in which BGO-ZP was titrated into TGF-β2. To accomplish this, CHAPS was included in the buffer used to prepare TGF-β2 (as well as BGO-ZP) because TGF-βs are practically insoluble over the entire pH range (4.5–9.5), where BGO-ZP is expected to be natively folded and bind.37 The ITC data showed a readily detectable binding curve with a negative enthalpy that could be fit to a binding model with a stoichiometry of 1.04 ± 0.04 and a KD of 109 ± 56 nM (Figure 5A,B and Table 3). The observed stoichiometry is consistent with the stoichiometry estimated from the SPR data shown in Figure 2, although the KD is roughly 10–30-fold higher. To investigate whether the increase in KD might have been caused by the different solution conditions used for the SPR and ITC experiments (namely, the presence of 30 mM CHAPS for the ITC experiments, but not the SPR), an additional direct binding SPR experiment was performed with BGO-ZP, BGO, and BGZP-C in the presence of increasing concentrations of CHAPS. The SPR results clearly show that CHAPS diminishes the binding affinity of BGO-ZP for TGF-β2 by ∼6-fold and that most of the decrease stems from the orphan domain (Table S1). Thus, the presence of CHAPS accounts for a large part of the decrease in affinity, though other factors might also contribute, such as immobilization of TGF-β on a hydrogel in the SPR experiment but not in the ITC experiment.
Figure 5.

Assessment of binding stoichiometry using ITC. (A and B) ITC raw heats for injection of BGO-ZP into TGF-β2 at pH 7.0 in the presence of 30 mM CHAPS and integrated heat values (black data points) as a function of the BGO-ZP:TGF-β2 molar ratio fitted to a standard binding isotherm (smooth red curve), respectively. (C and D) ITC raw heats and integrated heat values, respectively, for injection of TβRII into the TGF-β2TM/BGO-ZP binary complex at pH 7.0 in the absence of CHAPS. (E and F) ITC raw heats and integrated heat values, respectively, for injection of BGO into the TGF-β2TM/TβRII complex at pH 7.0 in the absence of CHAPS. Other details in panels C–F are the same as those in panels A and B.
To directly assess the effect of betaglycan on TβRII binding stoichiometry, an additional ITC experiment was performed in which TβRII was titrated into the preformed 1:1 TGF-β2TM/BGO-ZP complex. This experiment was performed in the absence of CHAPS as the TGF-β2TM/BGO-ZP complex is soluble at neutral pH. The ITC data showed a readily detectable binding transition with a negative enthalpy for binding of TβRII to the TGF-β2TM/BGO-ZP complex at an approximate 1:1 molar ratio (Figure 5C). The fitted value for the stoichiometry is 1.04 ± 0.04 (Figure 5D and Table 3), which is consistent with the 1:1 binding stoichiometry estimated from the SPR data shown in Figure 3. The fitted value for the KD was 510 ± 212 nM (Table 3), which after taking into account experimental error is roughly 5-fold higher than that measured by SPR (Table 2). The buffer conditions used for the two experiments had the same pH; however, the buffer and salt concentrations were slightly different (10 mM Hepes and 150 mM NaCl for SPR vs 10 mM phosphate for ITC), so this might be partially responsible for these differences. Other differences, such as immobilization of TGF-β on a hydrogel in the SPR experiment, but not the ITC experiment, might also contribute. Together, these ITC experiments demonstrate that BGO-ZP binds the TGF-β dimer with a 1:1 stoichiometry, and in contrast to TGF-β alone, BGO-ZP-bound TGF-β binds TβRII with a 1:1 stoichiometry.
BGO, BGZP, and BGZP-C Binding Stoichiometry
To further dissect how betaglycan binds, the binding of the isolated domains of betaglycan, BGO, BGZP, and BGZP-C, together with TβRII, to TGF-β2TM was assessed by SPR. The SPR sensorgrams for binding of BGO, BGZP, BGZP-C, and TβRII to TGF-β2TM are shown in Figure 6A–D, respectively, and plots of the mass-normalized equilibrium response as a function of concentration are shown in Figure 6E. The data show that the isolated orphan domain binds TGF-β2TM with an affinity slightly greater than that for TβRII, while the zona pellucida domain, BGZP, and the C-terminal portion of the zona pellucida domain, BGZP-C, bind TGF-β2TM with an affinity roughly 2-fold weaker than that for TβRII (Table 4). The similar affinity of BGZP and BGZP-C for TGF-β2TM is consistent with earlier reports that only the C-terminal portion of the zona pellucida domain is required for binding TGF-β.14,19 The normalized maximal responses for BGZP and BGZP-C are comparable to that of TβRII (Figure 6E and Table 4), suggesting that BGZP and BGZP-C each bind TGF-β homodimers with a 2:1 stoichiometry. The normalized response for BGO was found to be variable; in some experiments, it was found to be less than half the response for TβRII, BGZP, and BGZP-C, while in other experiments, such as the one shown, the maximal response was 60–65% percent of that of TβRII, BGZP, and BGZP-C. This suggested that BGO might bind TGF-β2TM with a 1:1 stoichiometry; however, this is not definitive, and other approaches, including SEC, SEC–MALS, and ITC, were used to further investigate the binding stoichiometry for this domain.
Figure 6.

Binding of TβRII, BGO, BGZP, and BGZP-C to TGF-β2TM and estimation of their binding stoichiometries. (A–D) SPR sensorgrams for binding of TβRII, BGO, BGZP, and BGZP-C, respectively, to immobilized TGF-β2TM. Black lines over sensorgrams denote the period of injection of a 2-fold dilution series (from 4 to 0.008 μM for TβRII, BGZP, and BGZP-C and from 1 to 0.008 μM for BGO). SPR data for TβRII, BGO, BGZP, and BGZP-C were all collected on the same SPR sensor chip; normalized responses were calculated by dividing the measured response by the molecular weight of the analyte in daltons and multiplying by 106. (E) Plot of the normalized equilibrium response for binding of TβRII, BGO, BGZP, and BGZP-C to TGF-β2TM as a function of their concentration. Equilibrium binding constants were obtained by fitting the normalized equilibrium response as a function of concentration to a standard binding isotherm (fitted curve shown as a solid line, red for TβRII, purple for BGO, greeen for BGZP, and black for BGZP-C).
Table 4. Binding of BGO, BGZP, BGZP-C, and TβRII to TGF-β2TM.
| surface | analyte | KD (nM) | Rmax (RUa) |
|---|---|---|---|
| TGF-β2TM | TβR-II | 148 ± 8 | 280 ± 4 |
| TGF-β2TM | BGO | 98 ± 7 | 172 ± 9 |
| TGF-β2TM | BGZP | 287 ± 37 | 265 ± 10 |
| TGF-β2TM | BGZP-C | 325 ± 40 | 290 ± 10 |
Normalized to molecular weight.
Effect of BGO on TβRII binding
Esparza-Lopez previously showed that the membrane-bound orphan domain promoted the cross-linking of TGF-β2 to TβRII in a manner similar to that of the full-length betaglycan.8 To assess whether the isolated orphan domain could potentiate the binding of TβRII to TGF-β2, the binding of TβRII to TGF-β2TM in the absence and presence of 800 nM BGO was measured using SPR. The sensorgrams show that BGO increases the affinity for binding of TβRII to TGF-β2TM by approximately 5-fold, while its effects on the Rmax are more modest, with an approximate 1.4-fold increase (Figure 7A–C and Table 5). The 5-fold potentiation of binding of TβRII by BGO is comparable to that previously observed for BGO-ZP, suggesting that the orphan domain alone is capable of potentiating the binding of TβRII. The lack of a decrease in Rmax indicates that BGO does not compete with TβRII binding to either site on the dimeric ligand. The 1.4-fold increase in Rmax may in fact be reflective of binding of BGO and TβRII to the ligand in a cooperative manner, i.e., because the concentration of BGO used was not saturating; if its affinity for the ligand was increased by TβRII, an increase in Rmax is expected. The same experiment performed with TGF-β2 showed a 35-fold potentiation of TβRII binding affinity by BGO and an approximate 2-fold increase in the Rmax (Figure S4A–C and Table 5). The 7-fold stronger potentiation of TβRII affinity for TGF-β2 by BGO (compared to that for TGF-β2TM) likely results from the influence of BGO on TβRII binding being more evident when the affinity of the TβRII/ligand interaction is lower. The 2-fold increase in Rmax probably occurs for the same reasons mentioned above for TGF-β2TM. Together, these results indicate that, in contrast to BGO-ZP, BGO and TβRII do not compete for binding to TGF-β and in fact exhibit cooperative binding. This indicates that BGO binds TGF-β dimers somewhere between the two bound TβRIIs, as shown schematically in Figure 7G.
Table 5. Binding Constants for Binding of TGF-β2 and TGF-β2TM to TβRII in the Presence and Absence of BGO.
| surface | analyte | KD (nM) | Rmax (RU) |
|---|---|---|---|
| TGF-β2 | TβRII | 4600 ± 700 | 142 ± 13 |
| TGF-β2 | TβRII (800 nM BGO) | 130 ± 100 | 320 ± 13 |
| TGF-β2TM | TβRII | 145 ± 17 | 720 ± 20 |
| TGF-β2TM | TβRII (800 nM BGO) | 30 ± 5 | 1000 ± 30 |
Effect of TβRII on BGZP Binding
The data of Makanji et al. have shown that the residues in inhibin A responsible for binding the betaglycan zona pellucida domain reside on the edge of the ligand fingers and that these are also highly conserved in the TGF-βs.20 This suggests that the betaglycan zona pellucida domain might bind near the ligand fingertips at a position that partially overlaps with that of TβRII. To assess this, binding of BGZP to TGF-β2TM, in the absence and presence of a nearly saturating level of TβRII (4 μM), was measured using SPR. The sensorgrams show that in the absence of TβRII, BGZP binds with a KD of 290 nM, while in the presence of 4 μM TβRII, there is a dramatic drop in the amplitudes and the apparent KD for binding is increased ∼17-fold to 5000 ± 1300 nM (Figure 7D–F and Table 6). The increase in the apparent KD for binding of BGZP to TGF-β2TM in the presence of 4 μM TβRII is consistent with competitive binding; KD,app = KD(1 + [competitor]/Ki), which predicts that KD,app would increase by 1 + 4 μM/0.13 μM, or ∼30-fold. The same experiment performed with TGF-β2 showed that the presence of 4 μM TβRII had little effect on the apparent affinity or response amplitude for binding of BGZP (Figure S4D–F and Table 6). This is expected because the concentration of the competitor, TβRII, is not saturating but rather is close to its KD, and thus, an at most 2-fold increase in KD,app is expected. The same experiments described above were also performed with BGZP-C, and as shown by the results presented in Table 6, TβRII inhibits the binding of BGZP-C in the same manner as it does BGZP. These results demonstrate that the zona pellucida domain of betaglycan binds at a site that partially overlaps with that of TβRII but requires that TβRII be displaced to allow BGZP/BGZP-C to bind (Figure 7H). These results also suggest that the ability of BGO-ZP to reduce the TβRII binding stoichiometry from 2:1 to 1:1 is due to the competitive effect of the zona pellucida domain. These measurements, together with those reported above for BGO, support the positioning of the orphan and ZP-C domains of betaglycan in the context of the 1:1:1 TGF-β/TβRII/BGO-ZP complex as shown in Figure 9 (stage I).
Table 6. Binding Constants for Binding of TGF-β2 and TGF-β2TM to BGZP in the Presence and Absence of TβRII.
| surface | analyte | KD (nM) | Rmax (RU) |
|---|---|---|---|
| TGF-β2 | BGZP | 450 ± 50 | 280 ± 10 |
| TGF-β2 | BGZP (4 μM TβRII) | 600 ± 70 | 220 ± 10 |
| TGF-β2TM | BGZP | 290 ± 40 | 310 ± 10 |
| TGF-β2TM | BGZP (4 μM TβRII) | 5000 ± 1300 | 130 ± 20 |
| TGF-β2 | BGZP-C | 450 ± 50 | 360 ± 10 |
| TGF-β2 | BGZP-C (2 μM TβRII) | 600 ± 50 | 340 ± 10 |
| TGF-β2TM | BGZP-C | 240 ± 30 | 620 ± 20 |
| TGF-β2TM | BGZP-C (2 μM TβRII) | 2400 ± 200 | 320 ± 10 |
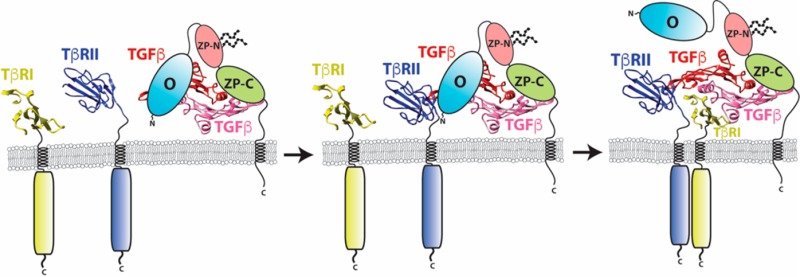
Figure 9.

Proposed mechanism by which betaglycan binds TGF-β homodimers to potentiate receptor complex assembly and signaling.
BGO Binding in Solution
The SPR results presented in Figure 7 show that BGO does not compete with TβRII for binding TGF-β; thus, any complexes that BGO forms with TGF-β and TβRII are likely to have the TGF-β and TβRII present in a 1:2 stoichiometry. The SPR data in Figure 6, however, did not definitively show whether BGO binds TGF-β homodimers with a 1:1 or 2:1 stoichiometry. To assess the binding stoichiometry in solution, excess BGO (3.0 equiv) was combined with 2.5:1 TβRII/TGF-β2TM binary complex (1.0 equiv) and subjected to size-exclusion chromatography (Figure 4C). Three peaks were eluted, the first of which (peak a) had the highest UV absorbance and as shown by SDS–PAGE corresponded to the TGF-β2TM/TβRII/BGO ternary complex (inset). The intensities of the second and third peaks (peaks b and c, respectively) were much lower, and they corresponded to excess BGO and TβRII, respectively (inset). To assess whether the three proteins in peak a corresponded to that of a stable stoichiometric ternary complex, an aliquot was analyzed by native PAGE, alongside the ternary complex assembled from individual components. The native gel revealed a well-defined band that migrated like a ternary complex assembled from individual components, but only very weak bands that corresponded to BGO or TβRII (Figure S2C).
To estimate the molecular mass of the TGF-β2TM/TβRII/BGO complex, the Kav versus molecular weight correlation established with BG, BGO, and TβRII was used to estimate the molecular mass of the TGF-β2TM/TβRII/BGO complex based on its Kav value. This predicted a near perfect match with the predicted mass for the 1:2:1 TGF-β2TM/TβRII/BGO complex (92 kDa) (Figure 4D), tentatively indicating that the stoichiometry is 1:2:1.
To directly assess the mass and stoichiometry, SEC–MALS and ITC experiments were performed. To perform the SEC–MALS measurements, a TGF-β2TM/TβRII/BGO sample was prepared in an identical manner and analyzed by SEC–MALS. The chromatogram obtained was very similar to that obtained before, and the molecular mass for the TGF-β2TM/TβRII/BGO complex was estimated to be 92–96 kDa (Figure 4E). This is in close agreement with the mass of 92 kDa estimated for the 1:2:1 TGF-β2TM/TβRII/BGO complex.
To further confirm the 1:1 stoichiometry with which BGO binds TGF-β2TM/TβRII complexes, an ITC experiment was performed in which BGO was titrated into a preformed 1:2 TGF-β2TM/TβRII complex. These experiments were performed in the absence of CHAPS as the TGF-β2TM/TβRII complex is highly soluble in the absence of CHAPS. The ITC raw heats showed a readily detectable binding curve with a negative enthalpy and could be fit to a binding model with a stoichiometry of 1.07 ± 0.02 and a KD of 82 ± 26 nM (Figure 5E−F and Table 3). The observed stoichiometry is consistent with the stoichiometry estimated from the SEC and SEC–MALS data shown in Figure 4, and the KD is comparable to that measured by SPR (Tables 3 and 4). The 1:1 stoichiometry with which BGO binds TGF-β homodimers is likely responsible for the overall 1:1 stoichiometry with which full-length betaglycan binds TGF-β homodimers.
Though attempts were also made to characterize the complexes formed between BGZP and TGF-β2 in solution using these approaches, this proved to be impractical because the BGZP/TGF-β2 complex is poorly soluble and it was not possible to identify solution conditions under which the complex was stably formed and soluble enough to be studied.
TβRI Binding to TGF-β2TM in the Presence of BGO and BGO-ZP
The previous cell-based studies established that betaglycan binds TGF-β2 and promotes the formation of a ternary complex with TβRII.4 This same study, however, failed to detect a quaternary complex of TGF-β2, TβRII, TβRI, and betaglycan, suggesting that TβRI might displace betaglycan as it binds to form the signaling complex with TβRI and TβRII. To investigate this, a SPR co-injection experiment was performed in which a saturating concentration of BGO-ZP (1 μM) was injected with a subsaturating concentration of TβRII (1 μM) onto a TGF-β2 surface until it approached equilibrium, followed by an injection of the same two receptors at the same concentration, but with increasing concentrations (from 0.063 to 2 μM) of TβRI added. This co-injection experiment showed that betaglycan blocks binding of TβRI, as evidenced by the lack of a significant increase in the SPR response in the second part of the injection (Figure 8A). To confirm that the TβRII used for these experiments was capable of binding and recruiting TβRI, the same experiment was performed except BGO-ZP was omitted from both the first and second part of the injection. This yielded a readily detectable increase in the SPR response during the second part of the injection (Figure 8B), which is expected, because it is well-known that TβRI binds at a shared interface formed by TGF-β and TβRII, with the result that TβRII potentiates the binding of TβRI to ligand several hundred-fold.27,34,35 Thus, BGO-ZP evidently blocks the binding of TβRI, suggesting that one or both of its domains must be displaced to allow TβRI to be recruited into the complex. To determine whether one of betaglycan’s domains or both block the binding of TβRI, the same experiment shown in Figure 8A was performed, but by using 1 μM BGO in place of 1 μM BGO-ZP. In contrast to the experiment with BGO-ZP, there was a slight increase in the SPR response when TβRI was present during the second part of the injection (Figure 8C). In this case, the increase is roughly 25% of that observed in the absence of BGO-ZP or BGO. This is probably because the concentration to BGO used in the experiment (1000 nM) was only slightly greater than its Ki (700–900 nM), resulting in a 75%, but not complete, suppression of TβRI binding and recruitment [when BGO-ZP was used as a competitor, its concentration (1000 nM) was roughly 200-fold greater than its Ki (5 nM)]. These results show that betaglycan, in particular its orphan domain, competes for binding against TβRI (Figure 8D) and that displacement of this domain is required to allow TβRI to bind (Figure 8E).
Figure 8.

Effect of betaglycan on the binding and recruitment of TβRI. (A) SPR sensorgrams from a co-injection experiment in which a constant concentration of 1 μM TβRII and 1 μM BGO-ZP was injected over immobilized TGF-β2, followed immediately by an injection of 1 μM TβRII and 1 μM BGO-ZP with increasing concentrations of TβRI (serial 2-fold dilution from 2 to 0.063 μM TβRI). (B and C) SPR sensorgrams from a co-injection experiment performed in a manner identical to that described for panel A, but with no BGO-ZP present in either first or second injection (B) or 1 μM BGO used in place 1 μM BGO-ZP during the first and second injection (C). Other details are as described for panel A. (D and E) Schematic depiction of how BGO blocks the binding of TβRI and how it must be displaced to allow TβRI to bind, respectively.
To determine whether TβRI might in fact be capable of displacing bound BGO-ZP in the context of a TGF-β2TM/TβRII/BGO-ZP complex, preformed TGF-β2TM/TβRII/BGO-ZP complexes were incubated for an increasing period of time with excess TβRI and TβRII and subjected to native gel electrophoresis (Figure S5). This showed that TβRI rapidly displaced BGO-ZP to form TGF-β2TM/TβRII/TβRI complexes. This experiment was repeated using TGF-β2, but because TGF-β2/TβRII/TβRI complexes are too unstable to be detected by native gels,27 it could not be determined whether this type of handoff also occurs for this ligand. This does, however, not imply a handoff mechanism would not occur for TGF-β2 as this process normally occurs with membrane-attached receptors, which is likely to exert a strong influence on the assembly mechanism.
Discussion
TGF-βs signal by binding and bringing together two cell surface receptors, TβRI and TβRII. The early work of Laiho6 and Wrana,38 and more recently that of Zúñiga27 and Groppe,34 has helped to define how TGF-βs assemble their signaling complex. TGF-βs first bind TβRII with a high affinity to form a stable binary complex. This creates a composite TGF-β/TβRII interface, to which TβRI is recruited.34,35 The recruitment of TβRI and assembly of the TβRII/TβRI heterotetramer initiate a phosphorylation cascade that elicits TGF-β signaling.38 TGF-β1 and TGF-β3 bind TβRII with high affinity and can therefore assemble the signaling complex in this manner, but TGF-β2 differs in that it binds TβRII with an affinity that is roughly 200-fold lower.9−11
The TGF-β family coreceptor betaglycan, also known as the TGF-β type III receptor, binds TGF-β1–TGF-β3 with high affinity (Kd = 5–20 nM) by simultaneously contacting TGF-βs at independent sites through its two component binding domains.22 The effects of betaglycan are nonetheless the strongest for TGF-β2, which because of its low intrinsic affinity for TβRII signals at only supraphysiological concentrations in betaglyan’s absence.4,9,11 Betaglycan has been shown by cross-linking to form a ternary complex on the cell surface with TGF-β2 and TβRII,4 but the nature of this complex and how it promotes the transition to the signaling complex with TβRI and TβRII are not understood. The importance of betaglycan for potentiation of TGF-β2 signaling in vivo is demonstrated by betaglycan knockout mice, which are embryonic lethal39 and share many of the phenotypic characteristics of the TGF-β2 knockout mice,40 including pronounced cardiac and liver defects.
The binding studies presented here show that the full-length betaglycan extracellular domain, encompassing both its N-terminal orphan and C-terminal zona pellucida domains, binds TGF-β homodimers with a 1:1 stoichiometry in a manner that allows one molecule of TβRII to bind. This suggests that the TGF-β2/TβRII/betaglycan complex previously detected in the cross-linking studies by López-Casillas and co-workers4 likely has a stoichiometry of 1:1:1. The binding studies presented here further show that the full-length betaglycan ectodomain leads to a modest (5–9-fold) potentiation of TβRII binding. This suggests at least two possible mechanisms by which betaglycan might potentiate the binding of TβRII to TGF-β2. The first is by binding and sequestering TGF-β2 on the cell surface, which should promote TβRII binding by increasing the local concentration and diminishing the unfavorable translational entropy that must be overcome to bind. The second is by increasing the favorable enthalpy of binding, either indirectly by altering the conformation of TGF-β2 to improve contacts with TβRII or, alternatively, by directly contacting TβRII to reinforce its binding.
The binding studies presented here further showed that the full-length betaglycan extracellular domain (BGO-ZP) and the betaglycan orphan domain alone (BGO) competed with TβRI for binding TGF-β2. This suggests that for TβRI to be recruited, betaglycan must be at least partially displaced by TβRI. This suggests a possible “handoff” mechanism in which the recruitment of TβRI functions not only to displace the orphan domain of the coreceptor but also to stabilize the weakly bound TβRII through direct receptor–receptor contact. It should be noted that this direct receptor–receptor contact has been demonstrated in crystal structures of the TGF-β1/TβRII/TβRI and TGF-β3/TβRII/TβRI ternary complexes.21,34,35 In accompanying functional studies, the direct receptor–receptor contact has been shown to be responsible for the several-hundred fold higher affinity with which TβRI binds the TGF-β/TβRII complex compared to that of TGF-β alone.27,34,35,41 Importantly, if TβRII potentiates the binding of TβRI several-hundred fold, then it must also hold that TβRI stabilizes the binding of TβRII.
The precise nature of the TGF-β/TβRII/betaglycan complex must await the direct determination of this structure using crystallography or other methods, but one model consistent with the observations in this paper is shown in Figure 9 (stages I and II). One interesting aspect of this model is that it predicts the existence of a TGF-β2/TβRII/TβRI/betaglycan quaternary complex (Figure 9, stage III), which may represent a functional signaling complex based on the previous observation that artificial TGF-β3 heterodimers capable of binding only one TβRII and one TβRI retain nearly half the signaling activity of TGF-β3 homodimers.42 However, even if this quaternary complex is capable of signaling, it is likely short-lived, as previous cell-based studies detected the TGF-β/TβRII/betaglycan4 and TGF-β/TβRII/TβRI ternary complexes,43−47 but not quaternary complexes with TGF-β2, TβRII, TβRI, and betaglycan. Importantly, the complete displacement of betaglycan may be due to its lowered affinity as it undergoes a transition from binding TGF-β homodimers in a bivalent (Figure 9, stages I and II) to monovalent manner (Figure 9, stage III).
The overall 1:1 stoichiometry for binding of the full-length betaglycan extracellular domain to TGF-β homodimers is somewhat unprecedented as TGF-β family homodimers have been shown to bind type I and type II receptor signaling domains, as well as most monomeric TGF-β family modulator proteins, such follistatin,48−50 RGMs,51,52 and DAN family antagonists,53 with 1:2 stoichiometries. Thus, one obvious question is why betaglycan might bind TGF-β homodimers with a 1:1 stoichiometry whereas most other nondimeric TGF-β family accessory proteins bind with a 1:2 stoichiometry. The definitive answer to this question will clearly have to await determination of the structure of the betaglycan orphan domain, which appears to be responsible for dictating the 1:1 stoichiometry, bound to TGF-β, but it is nonetheless tempting to speculate that this is because of two distinctive features of the betaglycan orphan domain. The first is that it has a monomeric size that is large compared to that of TGF-β (more than 1–1.5 times the size of TGF-β) as well as the individual domains of most other modulator proteins; the other is that it binds near the center of the TGF-β homodimer, which is inferred by the known positioning of TβRII on the distal ends of the growth factor homodimer on the ligand fingertips and the fact that BGO and TβRII do not compete for binding TGF-β (Figure 7G). Thus, even though TGF-β homodimers are in principle capable of symmetrically binding the betaglycan orphan domain, they may be unable because of steric overlap with the first bound orphan domain. This possibility is not without precedent as 1:1 stoichiometries have been reported for two other TGF-β family modulator proteins, GASP-154 and chordin.55,56 Though the structure of GASP-1 with its cognate ligand, myostatin, has not been reported, it has been nonetheless shown that C-terminal truncations alter the binding stoichiometry from 1:1 to 1:2. Thus, the 1:1 stoichiometry for GASP-1 may be achieved in the same manner as that of betaglycan, via occlusion of the binding of the second molecule at the symmetry-related site by steric overlap from the first bound molecule.
The transmembrane protein endoglin is homologous to betaglycan and has been shown to directly bind other TGF-β family ligands, particularly BMP-9 and BMP-10, and to affect the signaling of these ligands.57 Through SPR-based binding studies, it has been shown that endoglin’s abilty to bind BMP-9 and BMP-10 is derived solely from its orphan domain.58,59 These studies further showed that the endoglin orphan domain competes for binding with type II receptors that bind BMP-9 and -10, namely, ActRII, ActRIIB, and BMPRII, but not with type I receptors that BMP-9 and BMP-10 bind, namely Alk1. These observations may seem at odds with those reported here in which the betaglycan orphan domain was shown not to compete with TβRII for binding TGF-β, but to compete with TβRI. This, however, assumes that endoglin and betaglycan bind their cognate ligands in the same overall manner and that these two family ligands bind their type I and type II receptors in the same overall manner. There are currently no structures reported for either endoglin or betaglycan bound to their cognate ligands; thus, it is not possible to draw any conclusions regarding differences in coreceptor binding. There are, however, structures available for both TGF-βs bound to TβRI and TβRII21,34,35 and for BMP-9 bound to ActRIIB and Alk1,60 and these reveal very significant differences in the manner by which the receptors bind, particularly for the type II receptor, but also for the type I receptor. The TGF-β type II receptor, TβRII, binds to the TGF-β fingertips through an edge β-strand, whereas the BMP-9 type II receptor, ActRIIB, binds to the BMP-9 knuckle through the exposed face of its central three-stranded β-sheet. The type I receptor for TGF-β (Alk5) and the type I receptor for BMP-9 (Alk1) both use the same β4−β5 loop region and adjacent sheet to bind their cognate ligands. Nonetheless, the two type I receptors are positioned differently on the ligand, with the type I receptor for TGF-β shifted toward the fingertips where it contacts TβRII, the ligand monomer to which TβRII is bound, and, only to a limited extent, the other TGF-β monomer. The type I receptor Alk1, in contrast, has nearly equal contact with both BMP-9 monomers. TGF-β and BMP-9 therefore bind their type I and type II receptors in very different manners. While there might also be differences in the manner by which betaglyan and endoglin bind their cognate ligands, the differences in type I and type II signaling receptor binding alone are sufficient to account for the differences observed in competition studies.
Acknowledgments
The authors acknowledge Dr. Thomas Millstead, who generated the TEV cleavable thioredoxin-BGO construct, Dr. Tao Huang, who provided helpful guidance for the design and execution of the SPR experiments, and Dr. Jinwoo Ahn, who provided assistance with the SEC–MALS measurements and valuable comments on the manuscript.
Glossary
Abbreviations
- TGF-β
transforming growth factor-β
- TGF-β2TM
variant of TGF-β2 bearing K25R, I92V, and K94R substitutions
- TβRI
TGF-β type I receptor extracellular domain
- TβRII
TGF-β type II receptor extracellular domain
- BG
betaglycan
- BGO-ZP
full-length betaglycan extracellular domain
- BGO
betaglycan orphan domain
- BGZP
betaglycan zona pellucida domain
- BGZP-C
C-terminal IgG-like domain of the betaglycan zona pellucida domain
- BGZP-N
N-terminal IgG-like domain of the betaglycan zona pellucida domain
- CHO
Chinese hamster ovary
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Tris
tris(hydroxymethyl)aminomethane
- PMSF
phenylmethanesulfonyl fluoride
- IPTG
isopropyl β-d-thiogalactopyranoside
- Sulfo-NHS
N-hydroxysulfosuccinimide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide·HCl
- SPR
surface plasmon resonance
- ITC
isothermal titration calorimetry
- SEC
size-exclusion chromatography
- SEC–MALS
size-exclusion chromatography–multiangle light scattering
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00566.
Five figures and one table. Figures show a comparison of the binding properties of insect cell- and E. coli-derived BGO, an analysis of the complexes isolated by SEC by native gel electrophoresis, SEC profiles for TGF-β2TM/TβRII complexes as a function of the amount of material loaded, SPR binding data for binding of TβRII and BGZP to TGF-β2, and the conversion of the TGF-β2TM/TβRII/BGO-ZP ternary complex to the TGF-β2TM/TβRII/TβRI ternary complex by native gel electrophoresis. Table lists SPR binding constants for binding of BGO-ZP, BGO, and BGZP-C to TGF-β2 as a function of CHAPS concentration (PDF)
Author Present Address
¶ (M.A.H.) Faculty of Pharmacy, Mansoura University, Egypt.
This research was supported by the National Institutes of Health (GM58670 and CA172886 to A.P.H.), the Robert A. Welch Foundation (AQ1842 to A.P.H.), and the Consejo National de Ciencia y Tecnología (254046 to F.L.-C.). Additional support was provided by the University of Texas Health Science Center CTRC Macromolecular Structure and Interactions Core supported by National Cancer Institute Grant P30 CA54174 and the University of Texas Health Science Center at San Antonio Center for Macromolecular Interactions Core Facility supported by the University of Texas Health Science Center UCRF.
The authors declare the following competing financial interest(s): A.P.H. is the co-inventor of U.S. Patent 7,795,389, which includes protein-based TGF-beta inhibitors constructed from various domains of the TGF-beta receptors. M.D.O.-M. is presently Chief Scientific Officer for Formation Biologics.
Supplementary Material
References
- Massague J. (1998) TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753–791. 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Bier E.; De Robertis E. M. (2015) EMBRYO DEVELOPMENT. BMP gradients: A paradigm for morphogen-mediated developmental patterning. Science 348, aaa5838. 10.1126/science.aaa5838. [DOI] [PubMed] [Google Scholar]
- Morikawa M.; Derynck R.; Miyazono K. (2016) TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harbor Perspect. Biol. 8, a021873. 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F.; Wrana J. L.; Massague J. (1993) Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73, 1435–1444. 10.1016/0092-8674(93)90368-Z. [DOI] [PubMed] [Google Scholar]
- Stenvers K. L.; Tursky M. L.; Harder K. W.; Kountouri N.; Amatayakul-Chantler S.; Grail D.; Small C.; Weinberg R. A.; Sizeland A. M.; Zhu H. J. (2003) Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Mol. Cell. Biol. 23, 4371–4385. 10.1128/MCB.23.12.4371-4385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiho M.; Weis F. M.; Boyd F. T.; Ignotz R. A.; Massague J. (1991) Responsiveness to transforming growth factor-beta (TGF-beta) restored by genetic complementation between cells defective in TGF-beta receptors I and II. J. Biol. Chem. 266, 9108–9112. [PubMed] [Google Scholar]
- Lewis K. A.; Gray P. C.; Blount A. L.; MacConell L. A.; Wiater E.; Bilezikjian L. M.; Vale W. (2000) Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404, 411–414. 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- Esparza-Lopez J.; Montiel J. L.; Vilchis-Landeros M. M.; Okadome T.; Miyazono K.; Lopez-Casillas F. (2001) Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. J. Biol. Chem. 276, 14588–14596. 10.1074/jbc.M008866200. [DOI] [PubMed] [Google Scholar]
- De Crescenzo G.; Hinck C. S.; Shu Z.; Zuniga J.; Yang J.; Tang Y.; Baardsnes J.; Mendoza V.; Sun L.; Lopez-Casillas F.; O’Connor-McCourt M.; Hinck A. P. (2006) Three key residues underlie the differential affinity of the TGFbeta isoforms for the TGFbeta type II receptor. J. Mol. Biol. 355, 47–62. 10.1016/j.jmb.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Baardsnes J.; Hinck C. S.; Hinck A. P.; O’Connor-McCourt M. D. (2009) TbetaR-II discriminates the high- and low-affinity TGF-beta isoforms via two hydrogen-bonded ion pairs. Biochemistry 48, 2146–2155. 10.1021/bi8019004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S.; Hernandez H.; Laiho M.; ten Dijke P.; Iwata K. K.; Massague J. (1990) Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J. Biol. Chem. 265, 20533–20538. [PubMed] [Google Scholar]
- Sankar S.; Mahooti-Brooks N.; Centrella M.; McCarthy T. L.; Madri J. A. (1995) Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor beta 2. J. Biol. Chem. 270, 13567–13572. 10.1074/jbc.270.22.13567. [DOI] [PubMed] [Google Scholar]
- Wiater E.; Vale W. (2003) Inhibin is an antagonist of bone morphogenetic protein signaling. J. Biol. Chem. 278, 7934–7941. 10.1074/jbc.M209710200. [DOI] [PubMed] [Google Scholar]
- Wiater E.; Harrison C. A.; Lewis K. A.; Gray P. C.; Vale W. W. (2006) Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actions. J. Biol. Chem. 281, 17011–17022. 10.1074/jbc.M601459200. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F.; Payne H. M.; Andres J. L.; Massague J. (1994) Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J. Cell Biol. 124, 557–568. 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F.; Cheifetz S.; Doody J.; Andres J. L.; Lane W. S.; Massague J. (1991) Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell 67, 785–795. 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- Fukushima D.; Butzow R.; Hildebrand A.; Ruoslahti E. (1993) Localization of transforming growth factor beta binding site in betaglycan. Comparison with small extracellular matrix proteoglycans. J. Biol. Chem. 268, 22710–22715. [PubMed] [Google Scholar]
- Pepin M. C.; Beauchemin M.; Collins C.; Plamondon J.; O’Connor-McCourt M. D. (1995) Mutagenesis analysis of the membrane-proximal ligand binding site of the TGF-beta receptor type III extracellular domain. FEBS Lett. 377, 368–372. 10.1016/0014-5793(95)01378-4. [DOI] [PubMed] [Google Scholar]
- Pepin M. C.; Beauchemin M.; Plamondon J.; O’Connor-McCourt M. D. (1994) Mapping of the ligand binding domain of the transforming growth factor beta receptor type III by deletion mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 91, 6997–7001. 10.1073/pnas.91.15.6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makanji Y.; Walton K. L.; Wilce M. C.; Chan K. L.; Robertson D. M.; Harrison C. A. (2008) Suppression of inhibin A biological activity by alterations in the binding site for betaglycan. J. Biol. Chem. 283, 16743–16751. 10.1074/jbc.M801045200. [DOI] [PubMed] [Google Scholar]
- Hart P. J.; Deep S.; Taylor A. B.; Shu Z.; Hinck C. S.; Hinck A. P. (2002) Crystal structure of the human TbetaR2 ectodomain--TGF-beta3 complex. Nat. Struct. Biol. 9, 203–208. 10.1038/nsb766. [DOI] [PubMed] [Google Scholar]
- Mendoza V.; Vilchis-Landeros M. M.; Mendoza-Hernandez G.; Huang T.; Villarreal M. M.; Hinck A. P.; Lopez-Casillas F.; Montiel J. L. (2009) Betaglycan has two independent domains required for high affinity TGF-beta binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 48, 11755–11765. 10.1021/bi901528w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. J.; Hu Y.; Zhu J.; Woodruff T. K.; Jardetzky T. S. (2011) Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-beta binding. Proc. Natl. Acad. Sci. U. S. A. 108, 5232–5236. 10.1073/pnas.1010689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diestel U.; Resch M.; Meinhardt K.; Weiler S.; Hellmann T. V.; Mueller T. D.; Nickel J.; Eichler J.; Muller Y. A. (2013) Identification of a Novel TGF-beta-Binding Site in the Zona Pellucida C-terminal (ZP-C) Domain of TGF-beta-Receptor-3 (TGFR-3). PLoS One 8, e67214. 10.1371/journal.pone.0067214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.; Hinck A. P. (2016) Production, Isolation, and Structural Analysis of Ligands and Receptors of the TGF-beta Superfamily. Methods Mol. Biol. 1344, 63–92. 10.1007/978-1-4939-2966-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck A. P.; Walker K. P. 3rd; Martin N. R.; Deep S.; Hinck C. S.; Freedberg D. I. (2000) Sequential resonance assignments of the extracellular ligand binding domain of the human TGF-beta type II receptor. J. Biomol. NMR 18, 369–370. 10.1023/A:1026775321886. [DOI] [PubMed] [Google Scholar]
- Zuniga J. E.; Groppe J. C.; Cui Y.; Hinck C. S.; Contreras-Shannon V.; Pakhomova O. N.; Yang J.; Tang Y.; Mendoza V.; Lopez-Casillas F.; Sun L.; Hinck A. P. (2005) Assembly of TbetaRI:TbetaRII:TGFbeta ternary complex in vitro with receptor extracellular domains is cooperative and isoform-dependent. J. Mol. Biol. 354, 1052–1068. 10.1016/j.jmb.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Zou Z.; Sun P. D. (2004) Overexpression of human transforming growth factor-beta1 using a recombinant CHO cell expression system. Protein Expression Purif. 37, 265–272. 10.1016/j.pep.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Backliwal G.; Hildinger M.; Kuettel I.; Delegrange F.; Hacker D. L.; Wurm F. M. (2008) Valproic acid: a viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnol. Bioeng. 101, 182–189. 10.1002/bit.21882. [DOI] [PubMed] [Google Scholar]
- Delolme F.; Anastasi C.; Alcaraz L. B.; Mendoza V.; Vadon-Le Goff S.; Talantikite M.; Capomaccio R.; Mevaere J.; Fortin L.; Mazzocut D.; Damour O.; Zanella-Cleon I.; Hulmes D. J.; Overall C. M.; Valcourt U.; Lopez-Casillas F.; Moali C. (2015) Proteolytic control of TGF-beta co-receptor activity by BMP-1/tolloid-like proteases revealed by quantitative iTRAQ proteomics. Cell. Mol. Life Sci. 72, 1009–1027. 10.1007/s00018-014-1733-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. (1989) Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Mol. Cell. Biol. 9, 377–383. 10.1128/MCB.9.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchis-Landeros M. M.; Montiel J. L.; Mendoza V.; Mendoza-Hernandez G.; Lopez-Casillas F. (2001) Recombinant soluble betaglycan is a potent and isoform-selective transforming growth factor-beta neutralizing agent. Biochem. J. 355, 215–222. 10.1042/bj3550215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S. W.; Burmester J. K.; Tsang M. L.; Weatherbee J. A.; Hinck A. P.; Ohlsen D. J.; Sporn M. B.; Roberts A. B. (1996) Binding affinity of transforming growth factor-beta for its type II receptor is determined by the C-terminal region of the molecule. J. Biol. Chem. 271, 30656–30662. 10.1074/jbc.271.48.30656. [DOI] [PubMed] [Google Scholar]
- Groppe J.; Hinck C. S.; Samavarchi-Tehrani P.; Zubieta C.; Schuermann J. P.; Taylor A. B.; Schwarz P. M.; Wrana J. L.; Hinck A. P. (2008) Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 29, 157–168. 10.1016/j.molcel.2007.11.039. [DOI] [PubMed] [Google Scholar]
- Radaev S.; Zou Z.; Huang T.; Lafer E. M.; Hinck A. P.; Sun P. D. (2010) Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 285, 14806–14814. 10.1074/jbc.M109.079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Yuan J.; Yang Y.; Xu L.; Wang Q.; Zuo W.; Fang X.; Chen Y. G. (2010) Monomeric type I and type III transforming growth factor-beta receptors and their dimerization revealed by single-molecule imaging. Cell Res. 20, 1216–1223. 10.1038/cr.2010.105. [DOI] [PubMed] [Google Scholar]
- Pellaud J.; Schote U.; Arvinte T.; Seelig J. (1999) Conformation and self-association of human recombinant transforming growth factor-beta3 in aqueous solutions. J. Biol. Chem. 274, 7699–7704. 10.1074/jbc.274.12.7699. [DOI] [PubMed] [Google Scholar]
- Wrana J. L.; Attisano L.; Wieser R.; Ventura F.; Massague J. (1994) Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Stenvers K. L.; Tursky M. L.; Harder K. W.; Kountouri N.; Amatayakul-Chantler S.; Grail D.; Small C.; Weinberg R. A.; Sizeland A. M.; Zhu H. J. (2003) Heart and liver defects and reduced transforming growth factor beta2 sensitivity in transforming growth factor beta type III receptor-deficient embryos. Molecular and cellular biology 23, 4371–4385. 10.1128/MCB.23.12.4371-4385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford L. P.; Ormsby I.; Gittenberger-de Groot A. C.; Sariola H.; Friedman R.; Boivin G. P.; Cardell E. L.; Doetschman T. (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga J. E.; Ilangovan U.; Mahlawat P.; Hinck C. S.; Huang T.; Groppe J. C.; McEwen D. G.; Hinck A. P. (2011) The TbetaR-I pre-helix extension is structurally ordered in the unbound form and its flanking prolines are essential for binding. J. Mol. Biol. 412, 601–618. 10.1016/j.jmb.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.; David L.; Mendoza V.; Yang Y.; Villarreal M.; De K.; Sun L.; Fang X.; Lopez-Casillas F.; Wrana J. L.; Hinck A. P. (2011) TGF-beta signalling is mediated by two autonomously functioning TbetaRI:TbetaRII pairs. EMBO J. 30, 1263–1276. 10.1038/emboj.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis Y. I.; Moustakas A.; Lin H. Y.; Lodish H. F. (1994) The types II and III transforming growth factor-beta receptors form homo-oligomers. J. Cell Biol. 126, 139–154. 10.1083/jcb.126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A.; Lin H. Y.; Henis Y. I.; Plamondon J.; O’Connor-McCourt M. D.; Lodish H. F. (1993) The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 268, 22215–22218. [PubMed] [Google Scholar]
- Weis-Garcia F.; Massague J. (1996) Complementation between kinase-defective and activation-defective TGF-beta receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 15, 276–289. [PMC free article] [PubMed] [Google Scholar]
- Wells R. G.; Gilboa L.; Sun Y.; Liu X.; Henis Y. I.; Lodish H. F. (1999) Transforming growth factor-beta induces formation of a dithiothreitol-resistant type I/Type II receptor complex in live cells. J. Biol. Chem. 274, 5716–5722. 10.1074/jbc.274.9.5716. [DOI] [PubMed] [Google Scholar]
- Yamashita H.; ten Dijke P.; Franzen P.; Miyazono K.; Heldin C. H. (1994) Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-beta. J. Biol. Chem. 269, 20172–20178. [PubMed] [Google Scholar]
- Cash J. N.; Rejon C. A.; McPherron A. C.; Bernard D. J.; Thompson T. B. (2009) The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 28, 2662–2676. 10.1038/emboj.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler R.; Keutmann H. T.; Sidis Y.; Kattamuri C.; Schneyer A.; Thompson T. B. (2008) The structure of FSTL3.activin A complex. Differential binding of N-terminal domains influences follistatin-type antagonist specificity. J. Biol. Chem. 283, 32831–32838. 10.1074/jbc.M801266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T. B.; Lerch T. F.; Cook R. W.; Woodruff T. K.; Jardetzky T. S. (2005) The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 9, 535–543. 10.1016/j.devcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Bell C. H.; Healey E.; van Erp S.; Bishop B.; Tang C.; Gilbert R. J.; Aricescu A. R.; Pasterkamp R. J.; Siebold C. (2013) Structure of the repulsive guidance molecule (RGM)-neogenin signaling hub. Science 341, 77–80. 10.1126/science.1232322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey E. G.; Bishop B.; Elegheert J.; Bell C. H.; Padilla-Parra S.; Siebold C. (2015) Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein. Nat. Struct. Mol. Biol. 22, 458–465. 10.1038/nsmb.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan K.; Kattamuri C.; Rankin S. A.; Read R. J.; Zorn A. M.; Thompson T. B. (2016) Structure of Gremlin-2 in Complex with GDF5 Gives Insight into DAN-Family-Mediated BMP Antagonism. Cell Rep. 16, 2077–2086. 10.1016/j.celrep.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. G.; Angerman E. B.; Kattamuri C.; Lee Y. S.; Lee S. J.; Thompson T. B. (2015) Alternative binding modes identified for growth and differentiation factor-associated serum protein (GASP) family antagonism of myostatin. J. Biol. Chem. 290, 7506–7516. 10.1074/jbc.M114.624130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troilo H.; Zuk A. V.; Tunnicliffe R. B.; Wohl A. P.; Berry R.; Collins R. F.; Jowitt T. A.; Sengle G.; Baldock C. (2014) Nanoscale structure of the BMP antagonist chordin supports cooperative BMP binding. Proc. Natl. Acad. Sci. U. S. A. 111, 13063–13068. 10.1073/pnas.1404166111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. L.; Huang Y.; Qiu L. Y.; Nickel J.; Sebald W. (2007) von Willebrand factor type C domain-containing proteins regulate bone morphogenetic protein signaling through different recognition mechanisms. J. Biol. Chem. 282, 20002–20014. 10.1074/jbc.M700456200. [DOI] [PubMed] [Google Scholar]
- David L.; Mallet C.; Mazerbourg S.; Feige J. J.; Bailly S. (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–1961. 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Alt A.; Miguel-Romero L.; Donderis J.; Aristorena M.; Blanco F. J.; Round A.; Rubio V.; Bernabeu C.; Marina A. (2012) Structural and functional insights into endoglin ligand recognition and binding. PLoS One 7, e29948. 10.1371/journal.pone.0029948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay R.; Werner E. D.; Matthews R. G.; Presman E.; Mulivor A. W.; Solban N.; Sako D.; Pearsall R. S.; Underwood K. W.; Seehra J.; Kumar R.; Grinberg A. V. (2011) Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 286, 30034–30046. 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson S. A.; Martinez-Hackert E.; Greppi C.; Lowden P.; Sako D.; Liu J.; Ucran J. A.; Liharska K.; Underwood K. W.; Seehra J.; Kumar R.; Grinberg A. V. (2012) Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 287, 27313–27325. 10.1074/jbc.M112.377960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.