

ABSTRACT
Understanding the computations that take place in neural circuits requires identifying how neurons in those circuits are connected to one another. In addition, recent research indicates that aberrant neuronal wiring may be the cause of several neurodevelopmental disorders, further emphasizing the importance of identifying the wiring diagrams of brain circuits. To address this issue, several new approaches have been recently developed. In this review, we describe several methods that are currently available to investigate the structure and connectivity of the brain, and discuss their strengths and limitations.
KEYWORDS: brain circuit, connectomics, neuron, synapse, wiring diagram
Introduction
In recent years, there has been a resurgence in interest in the analysis of the wiring diagrams of nervous systems, or the so-called connectomics. The study of the connectome originated with the pioneering reconstruction of the entire C. elegans nervous system by electron microscopy (EM) in 1980, and has expanded to additional organisms, such as Drosophila and mice. The C. elegans nervous system, comprised of just over 1000 neurons, is relatively simple to analyze. In addition, due to its fast generation time and reproductive peculiarities, C. elegans is an outstanding model for genetic analysis. However, there are a few issues that limit the usefulness of C. elegans for studies of the nervous system. First, C. elegans has a very limited behavioral repertoire. Second, C. elegans neurons lack voltage-gated sodium channels and consequently do not fire action potentials. Neurotransmitter release in C. elegans occurs via graded depolarizations mediated by voltage-gated calcium channels, a mechanism that is not shared by vertebrates or most invertebrates. Finally, electrophysiological recordings in C. elegans are notoriously challenging. Consequently, it is difficult to establish correlations between synaptic activity and behavior in C. elegans.
The mouse is an attractive model for neuroscience. It is the most genetically tractable mammal, and there is strong conservation of physiology and anatomy between the rodent and human brain. In addition, despite its relatively slow generation time, techniques for genetic manipulation and electrophysiological recordings in mice are highly sophisticated. However, the mouse brain has more than 100,000,000 neurons, making it very difficult to analyze and understand the structure and dynamics of its nervous system.
The Drosophila brain, by comparison, only has around 100,000 neurons. The simplicity of its brain combined with the sophisticated tools available for genetic manipulation allow scientists to map entire circuits in Drosophila at the cellular level. At the same time, Drosophila exhibit more sophisticated behavior and circuitry than do C. elegans, making the study of its nervous system pertinent to understanding our own. In fact, some of its brain circuits, such as the olfactory circuit, show significant overlaps in organization and function with equivalent circuits in the mammalian brain. Ultimately, the Drosophila brain is an excellent model system for understanding how the connectome gives rise to function and behavior. As a result, many of the new methods to investigate the connectivity of neurons have been developed for use in Drosophila. These methods can be broadly divided into 2 groups: those based on electron microscopy (EM) analysis of brain tissue, and those based on genetic techniques.
Analysis of synaptic organization by EM
EM represents the gold standard for analysis of brain structure. Due to its high resolution, EM can unambiguously identify synapses. EM uses accelerated electron beams instead of light as its source of illumination and can consequently reveal the ultrastructure of biologic tissue with xy-resolution as high as 2 nm. Chemical synapses can be visually identified from EM image because synaptic vesicles are concentrated at the presynaptic site and most Drosophila presynaptic sites have a morphological specialization called the T-bar.
There are several variations of EM that can be used to study brain connectivity, but the one that has been used most extensively by neuroanatomists is serial-section transmission electron microscopy (ssTEM). For this technique, biologic tissue is fixed, stained, embedded in resin, and then serially cut into sections around 40 nm thick with an ultramicrotome. The resulting slices are collected and visualized by transmission electron microscopy (TEM), which measures the electrons that pass through the sample. TEM offers the best xy-resolution of all EM methods. After images are generated from serial sections, they are first aligned along the xy-axis. Then, cellular membranes and organelles are identified and marked in a process known as segmentation. Finally, the segmented images are linked across the z-axis for 3D-reconstruction of neurons. Though the initial steps (sectioning, imaging, alignment and segmentation) can be automated with varying degrees of success, reconstructing and proofreading a 3D-model from serial images requires hundreds of hours of skilled labor and represents the limiting step. Ongoing work seeks to streamline this process.1,2
The resolution of EM reconstructions offer 2 main advantages over other connectomic techniques. First, EM reconstructions can yield a comprehensive understanding of a small neuronal circuit. For example, the EM volume generated from the antennal lobe of Drosophila larva identified over 38,000 synapses among 160 neurons.3 Analysis of this connectome allowed the authors to build a circuit-level model that suggests how larva could move toward food sources while staying vigilant to predator-related olfactory cues. Additionally, the high resolution of EM leads to a quantitative assessment of neural circuits. Because EM reconstructions can reveal the size and the number of the synapses that exist between neurons, EM can provide an estimation of the strength of connection between neurons. This can be subsequently used to support theories of circuit function.4 Although understanding circuit mechanisms rarely comes from its wiring diagram alone, the detail of connectomes generated by EM allow for models that can be subsequently tested with functional experiments.
EM is an extremely powerful method, but it has several important limitations. First, for tissues to be analyzed by EM, they need to be fixed and dehydrated. This process kills the cells in the tissue, preventing the direct combination of EM with functional analysis by electrophysiological or optical recordings. Second, EM is extremely time and labor-intensive. For reference, the seminal reconstruction of the C. elegans connectome took several years to complete for one single specimen. Similarly, EM reconstructions of the larval antennal lobe,3 the adult optic medulla,4 and the A2 and A3 segments of VNC5 provide exhaustive connectomic data but only in localized circuits and for single samples. Even with the continuing optimization of automated reconstructions, EM will likely remain unsuitable for studying the connectivity of neurons with long-range projections. For example, when analyzing the structure of a brain circuit in Drosophila, it is impossible to identify the origin of the axons that belong to neurons located in the ventral nerve cord (VNC). Additionally, the variability of neural connections across time, between individuals, or with respect to directed mutations is unsuitable for analysis by EM.
Future advances for EM
New advances in EM techniques promise to address some of the bottlenecks associated with traditional ssTEM. For example, automatic tape-collecting ultramicrotome scanning electron microscopy (ATUM-SEM) automates the process of collecting the serial sections created with an ultramicrotome. Serial block-face scanning microscopy (SBEM), on the other hand, utilizes an electron microscope with a built-in ultramicrotome such that the sample is imaged as the top layer is serially sectioned and removed. Similarly, in focused ion beam (FIB) SBEM, the surface of the sample is imaged, and then vaporized with a focused ion-beam to allow for imaging of the next layer. SBEM techniques lead to flawlessly automated alignment across the z-axis. Though all of these techniques provide lower xy-resolution than ssTEM, FIB-SEM offers higher z-resolution than does ssTEM (10 nm vs. 40 nm). The superior z-resolution enables scientists to generate EM volumes with isotropic voxels that can be digitally resectioned for best graphical representation of neurites.6 Such advancements in EM techniques, combined with ongoing efforts to automate 3D-reconstructions, offer the possibility of applying EM to larger volumes and larger sample size in the future.
Additionally, there is great interest in developing strategies for Correlative Light Electron Microscopy (CLEM).7 Traditionally, sample preparations for EM and LM have been incompatible, preventing the combination of EM and LM for the same sample. Several methods now exist to allow for CLEM. Optimized freeze-substitution embedding methods allow fluorescence imaging and EM imaging of the same sample in succession.8 In addition, a gene called miniSOG has been designed to act as a genetically encoded tag that can be visualized by LM and EM. miniSOG is a fluorescent protein that can be directly imaged by fluorescence microscopy, and can be induced to produce singlet oxygens that generate an electron-dense material detectable by EM.9 Similarly, neurons of interest can be tagged with GFP, imaged in vivo with 2-photon microscopy, and marked by etching with a near-infrared laser for identification at the EM level.10 Parallel efforts in EM chemistry led to the development of multicolor EM.11 In this strategy, conjugated antibodies are used to locally deposit lanthanides that have distinct energy-loss spectra. The distribution of each lanthanide in the sample can be independently recorded with energy-filtered TEM to create pseudocolored images that are overlaid on a traditional EM image. Currently, there are 3 lanthanides that can be used to orthogonally mark molecules on an EM image. Application of multicolor EM to connectomic studies, in conjunction with advancements in CLEM, would allow scientists to conveniently integrate ultrastructural information about synapses with genetic tools that can readily label distinct subpopulations of neurons.
Analysis of synaptic organization by LM
As mentioned above, the main limitations of EM are that it cannot be used to examine live tissue, trace long distance connections, or investigate large numbers of samples. These bottlenecks of EM can be overcome by LM, which is considerably less time- and labor-intensive. Unfortunately, the resolution of LM is restricted to the wavelength of light used to illuminate the samples (400–600 nm). However, recently developed techniques such as STED (STimulated Emission Depletion) microscopy,12,13 PALM (PhotoActivatable Light Microscopy), and STORM (STochastic Optical Reconstruction Microscopy)14,15 allow investigators to image the brain at subdiffraction limit resolution. STED uses patterned illumination to restrict light emission to a small region, while PALM and STORM use spare photoactivation of fluorophores to achieve a similar goal.16 At this resolution, subcellular structures such as individual neurotransmitter vesicles or T-bars can be discerned. Although the resolution of these methods is still lower than that of TEM, LM offers 2 major advantages. First, LM can be directly combined with genetic methods for labeling cells. The wealth of genetic tools in flies makes it possible to direct expression of fluorescent proteins or other transgenes to neuronal subpopulations. In conjunction with thousands of promoter elements that are widely available, there are 3 modular systems (Gal4/UAS, LexA/LexAop, and QF/QUAS) that can be used orthogonally to express pre- and postsynaptic markers in distinct sets of neurons, but also combinatorially to direct expression to highly precise subpopulations of neurons.17 Additionally, LM can be used in live animals with parallel electrophysiological and optical imaging techniques. This allows researchers to combine structural and functional information in a way that EM cannot.
Several methods, including STaR (Synaptic Tagging with Recombination), use genetic labeling of synaptic sites to identify putative synaptic contacts between neurons.18-21 These methods have been used to study the variation in synaptic organization among individuals in different conditions and at different developmental stages, and to compare the changes in connectivity caused by mutations.19-23 In these methods, pre- and postsynaptic markers need to be genetically fused with different tags, such as fluorescent proteins, or tags that can be detected by immunocytochemical staining, such as the V5, HA, or OLLAS tag (Fig. 1). Therefore, by analyzing the proximity of pre- and postsynaptic markers, putative synaptic sites can be revealed directly by LM imaging. Moreover, as in most methods that depend on LM, fluorescent protein-tagged synaptic markers allow for the monitoring of changes in synapses in the same animal over time by live imaging using methods such as 2-photon microscopy.20
Figure 1.
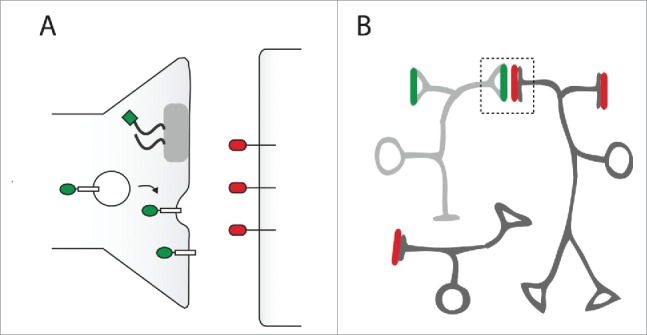
Visualization of synapse by genetically tagging synaptic proteins. (A) Fluorescent proteins or epitope tags can be targeted to synaptic membranes by fusion to synaptically localized proteins. For example, tags fused to Brp (curly black line) or synaptic vesicular proteins (white rectangle) will localize to the presynaptic-terminal (left). There are no pan-neuronal postsynaptic markers, but tags (red oval) fused to neurotransmitter receptors such as Ort have been successfully used in the past. (B) The axon terminal (triangle) of the presynaptic neuron of interest (light gray) is marked with tags that are fused to presynaptic proteins (green). A different tag (red) is expressed at the postsynaptic sites of candidate partner neurons (dark gray). The proximity of these 2 distinct tags (green, red) is assessed to verify synaptic connection.
The most common tagged marker to locate presynaptic sites is Bruchpilot (Brp), a large protein that selectively localizes to most active zones in the presynaptic site. However, it is important to note that several EM studies have shown that some presynaptic sites in the Drosophila brain, such as the calyx of the mushroom body (MB), are devoid of the T-bar structure, and the Brp protein level in some of the T-bars is either too low to be detected or completely absent.5,24,25 Therefore, it is necessary to validate the expression of the tagged Brp marker in different types of synapses in different brain regions before using this marker. As an alternative, tagged synaptic vesicular proteins, such as synaptobrevin and synaptotagmin, can be used to mark presynaptic sites.26-29 Because these 2 markers are located in the synaptic vesicular pools, they would likely label the entire presynaptic boutons, not just the active zones where the neurotransmitter release occurs. There is currently no pan-postsynaptic marker available for Drosophila. However, tagged neurotransmitter receptors can be used to label postsynaptic sites. Examples include the histamine-gated chloride channel, Ort, for some neurons in the optic lobe20 and the acetylcholine receptor subunit, nAChRα7, in the AL and MB.19,30 However, it is important to note that using these neurotransmitter receptors will only detect the synapses that are mediated by those receptors. Thus, these strategies are useful to confirm the presence or absence of synaptic connections that are known (or suspected) a priori, but are not suitable to discover synaptic connections in an unbiased way.
Additionally, it is important to note that overexpression of full length Brp or neurotransmitter receptors might disturb normal synaptic properties and neural circuits.31-36 Close attention should be paid to the possible influences on native function of synapses when synaptic markers are overexpressed. Alternatively, using endogenous regulatory promoters of synaptic markers allows investigators to recapitulate normal expression levels and avoid overexpression. For example, for STaR, Chen et al.20 used the full length of genomic locus of Brp and Ort incorporated into a bacterial artificial chromosome (BAC) that included their endogenous regulatory promoter to keep the expression at physiologic levels. In this case, a stop cassette flanked by FRT sites followed by the tag epitopes was replaced the stop codon of Brp or placed after the signal peptide of Ort. By crossing these flies to flies expressing the Flp recombinase in the desired neuronal types, the stop cassette was removed, and the tagged synaptic markers were selectively expressed in those neurons. Finally, instead of using the full-length tagged Brp, which is a very large protein, it is possible to use the C-terminal fragment of Brp fused with a fluorescent protein (Brp-short-FP) to label presynaptic sites.19,21,37-39 The advantage of using Brp-short-FP is that it will bind to the endogenous Brp clusters in T-bars without forming aggregation, minimizing any potential changes to native synaptic properties.18,39,40
To precisely analyze the proximity of pre- and postsynaptic markers, especially in regions with dense synapses, super-resolution microscopy methods (see above) may be required. Recently, these technologies have been used in Drosophila brain thin sections (8 µm) or whole brains.41 By using super-resolution microscopy in Drosophila brain sections, individual active zones labeled with Brp-short-mCherry could be clearly recognized. Moreover, combined with the proper image analysis methods, the spatial organization of the tagged synapses can be analyzed with resolving powers that approach those of EM. However, because of the influence of light scattering and spherical aberrations, only the top 1–3 µm of the sections can be imaged to obtain the optimal resolution. For this reason, reconstructing the connectome from a large piece of fly brain by super-resolution microscopy would still be time-consuming. If super-resolution microscopy could be combined with methods that allow for imaging in thick tissues, such as tissue clarification techniques, this would benefit the processes to analyze the synaptic organization in large brain regions or even whole brains in Drosophila. Recently, Ke et al.42 used super-resolution microscopy to image the optic lobe neurons in whole Drosophila brains treated by a tissue-clearing method called SeeDBD2. Their approach allowed them to reconstruct the morphology of entire axon terminals of Mi1 medullary neurons at the resolution of 50–150nm.
GRASP (GFP reconstitution across synaptic partners)
GRASP is a genetic method used to identify cell contacts and synapses in living animals.43 It was initially developed in C. elegans, but it has been subsequently applied to the study of Drosophila brain connectivity. GRASP labels synapses based on the proximity of the pre- and postsynaptic plasma membranes. In CNS (central nervous system) synapses, the membranes of 2 synaptic partners are typically separated by less than 100 nm of extracellular space, which is known as the synaptic cleft. This distance can be spanned by transmembrane proteins expressed in the 2 interacting neurons. As the name implies, GRASP is based on the reconstitution of 2 fragments of the split-GFP across the synapses of interacting neurons. Each of the 2 non-fluorescent split-GFP fragments are added to carrier transmembrane proteins. The 2 fragments of the split GFP assemble into a fluorescent form only when the membranes are sufficiently close to permit carrier proteins to span the intercellular gap. One fragment of the split GFP, spGFP1–10, is 214 aa long while the second fragment, spGFP11, is just 16 residues long. The spGFP11 fragment can therefore be inserted into many different proteins without affecting their function.
In initial experiments in C. elegans, each fragment of GFP was fused to the extracellular domain of the cell adhesion molecule CD443. However, this molecule is homogeneously distributed throughout the plasma membrane, without any specificity for synaptic sites. As a result, this implementation of GRASP led to GFP reconstitution throughout the membrane including at sites of non-synaptic contact (Fig. 2A). To improve the specificity of the system, one or both of the GRASP components were fused to synaptically localized proteins, restricting GFP reconstitution to synapses (Fig. 2B).
Figure 2.
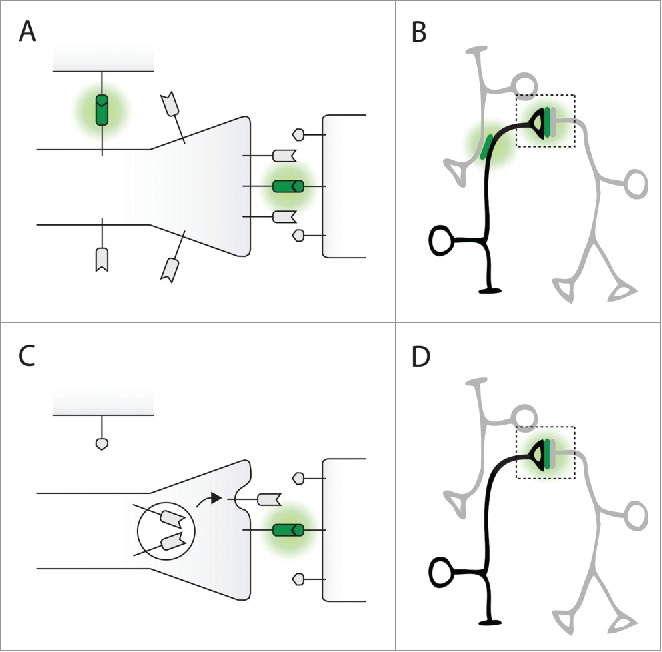
GFP Reconstitution Across Synaptic Partners (GRASP). (A) CD4::spGFP1–10 (dented rectangle) is distributed homogeneously throughout the membrane of the presynaptic neuron. As a result, GFP reconstitution (green) can occur at the synapse (right) as well as at sites of non-synaptic contact (top). (B) A presynaptic neuron (black) expressing CD4::spGFP1–10 makes contact with 2 neurons (gray) expressing CD4::spGFP11. GFP reconstitution occurs at all sites of membrane contact and is not specific to synapses. (C) NSyb::spGFP1–10 (dented rectangle) is localized to synaptic vesicles. Upon synaptic release, NSyb::spGFP1–10 is exposed specifically to the synaptic cleft. As a result, GFP reconstitution (green) only occurs at synaptic sites when synaptic vesicles fuse. (D) A presynaptic neuron (black) expressing NSyb::spGFP1–10 makes contact with 2 neurons (gray) expressing CD4::spGFP11. GFP reconstitution occurs at the synapse but not at the site of nonsynaptic contact.
Gordon and Scott44 demonstrated that GRASP works efficiently in flies by using it to detect cell-cell contact at the synapses between olfactory receptor neurons (ORNs) and projection neurons (PNs). The power of fly genetics has allowed the GRASP system to be expanded for studying neuron-muscle connections,45,46 the visual circuit,47 the circadian rhythm circuit,48,49 and the olfactory circuit.50 A recent modification of the GRASP system has taken advantage of the properties of some synaptic proteins that are displayed on the membrane only after the release of neurotransmitters.51 To limit GFP reconstitution to active synapses, the investigators fused spGFP1–10 to the extracellular domain of N-synaptobrevin (NSyb), which is exposed to the synaptic cleft only after vesicle release (NSyb::spGFP1–10). They tested this by expressing NSyb::spGFP1–10 in ORNs and its GRASP partner CD4::spGFP11 broadly in PNs. GFP was reconstituted at synapses after artificial stimulation with KCl or natural stimulation with cognate odorants, demonstrating that NSyb::GRASP can preferentially label active synapses in the Drosophila brain.
Many useful fluorescent proteins, such as GFP itself or the calcium indicator GCaMP,52 cannot be used simultaneously with GRASP since initially it was only available in green. Recently developed variants such as CRASP (cyan)53 or YRASP (yellow)51 allow investigators more flexibility because they do not overlap with GCaMP signal. In addition, the 3 reconstitution techniques can be used in parallel to study multiple synaptic connections orthogonally.
The GRASP system is very powerful, but it has 3 important limitations. First, the reconstituted fluorescence is often weak, making it difficult to detect in vivo. The reconstitution of GFP can be detected in fixed tissue using immunofluorescence with different GFP antibodies that can selectively detect spGFP1–10, spGFP11, or the reconstituted GFP. However, immunostaining requires fixing the tissue, and thus it precludes the combination with functional methods such as electrophysiological or optical recordings. Second, the interaction between the sp11 and sp1–10 fragments is irreversible, and can artificially render permanent cell-cell contacts that are natively transient. Third, GRASP reveals the point of contact between cells, but it does not allow for genetic manipulation of synaptically connected neurons, a feature that would be invaluable for functional analysis of circuitry.
Photoactivatable fluorescent protein
When using GRASP or STaR to analyze synaptic contacts, putative pre- and postsynaptic neurons have to be known a priori so that the transgenic proteins can be expressed precisely in these neurons. Therefore, these strategies cannot be used to discover novel synaptic partners in an unbiased manner. In contrast, photoactivatable green fluorescent protein (PA-GFP) is an ideal tool for discovering novel synapses, and has been successfully used in Drosophila.54-57 To implement this tool, a neuron of interest (first-order neuron) must be marked with dye or fluorescent proteins to outline its neurites. PA-GFP is genetically expressed pan-neuronally, or more narrowly in candidate subsets of neurons that are hypothesized to be connected to the first-order neuron. By spatially restricting the application of UV light to specific dendrite or axon arborizations of the first-order neuron, only the PA-GFP in neurites of partner neurons is converted from a weakly fluorescent state to a strongly fluorescent state (∼100 fold increase).58 Gradually, the activated PA-GFP diffuses from the neurites to the cell body to highlight the morphology of the entire neuron (Fig. 3A). Recently, 2 new enhanced variants of PA-GFP, SPA-GFP and C3PA-GFP, have been generated, with the latter having the strongest fluorescence.54
Figure 3.
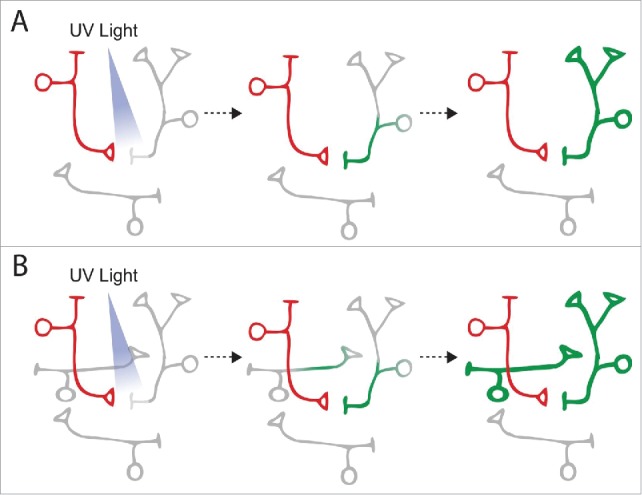
Detection of synaptic partners by PhotoActivatable GFP (PA-GFP). (A) A neuron of interest (red) is first highlighted with dye or fluorescent proteins to mark its neurites while PA-GFP is expressed in all candidate partner neurons (gray). When UV light is applied just outside the neurite of the highlighted neuron, PA-GFP in its synaptic partner (green) is converted to a strongly fluorescent state. The converted PA-GFP then diffuses throughout the partner neuron, highlighting its entire morphology. (B) One of the pitfalls of this technique is that UV light stimulation can also activate PA-GFP in bystander neurons. In this scenario, PA-GFP in 2 neurons (green) is activated but only one of these is synaptically connected to the neuron of interest (red). In practice, functional studies need to be performed to confirm the candidate synaptic connections identified with PA-GFP.
One caveat of this approach is that it can highlight any neuron that has arborizations within the illuminated region, including neurons that have passing axons or dendrites which do not synapse with the first-order neuron (Fig. 3B). Therefore, electrophysiological recordings or genetically encoded calcium indicators (GECIs) are required to confirm the existence of synaptic connections between the highlighted neurons and the first-order neuron. The recently developed PA-GECIs have the features of PA-GFP (high-contrast of fluorescence intensity after photoactivation) and GECIs (high-sensitivity of calcium detection).59 PA-GECIs could provide a more convenient way of investigating connectivity by allowing investigators to initially identify the potential synaptic partners and subsequently test their functional connectivity in the same animals.
Transsynaptic tracers
Transneuronal tracing in mammals has benefited from the availability of specific neurotropic viruses that are selectively transported across synapses. Herpes simplex virus type 1, pseudorabies virus, and rabies virus have all successfully been used to elucidate neural connections in mice.60 Unfortunately, none of the transsynaptic viruses that have been used in mammals work in Drosophila, presumably due to the lack of receptors or host cellular machinery that these viruses require for entry and replication. Non-viral tracers such as C-fragment of tetanus toxin or wheat germ agglutinin (WGA), can be expressed as transgenes into specific “source” neurons, and transferred across their synapses.61 The transfer of tracing agents may occur in an anterograde (from the source neuron's axon to its postsynaptic partner) or retrograde (from the source neuron's dendrite to its presynaptic partner) manner. WGA, in particular, has been used successfully as a transneuronal tracer in multiple species including Drosophila.62
WGA is a lectin protein that binds certain sugar moieties of glycoproteins covering eukaryotic cell membranes. WGA is transported preferentially to the axon terminal, where it is secreted and endocytosed by the postsynaptic partner of the source neuron. WGA can then be visualized by staining or by conjugation to horseradish peroxidase. After being produced by a source neuron, secreted into the synaptic cleft and endocytosed by a postsynaptic partner, WGA can be repeatedly passed along a circuit of connected neurons. However, this multi-synaptic transport of WGA may not give a clear interpretation of data. Because only a small fraction of the WGA from the source neurons jumps across the synapse, and WGA does not replicate, the little WGA that reaches the synaptic partner can be difficult to detect. To solve this problem, adeno-associated viruses (AAV) encoding WGA fused with Cre recombinase, WGA-Cre, has been used in combination with Cre-dependent reporters in mice to amplify the WGA signal.63,64 However, this strategy does not seem to work robustly in mammals, and it remains to be tested whether parallel strategies such as WGA-Flp or WGA-GAL4 can be used in Drosophila.
Tango
Several recently developed genetic tools have adopted the molecular mechanisms of the Delta-Notch signaling pathway to monitor and modify interacting cells. The Delta-Notch pathway plays significant roles in controlling cell fate during development, through cell-cell interactions. The Notch receptor includes a set of EGF motif repeats at its N-terminus, followed by a key structural element called the Notch regulatory region (NRR), which is located in the extracellular domain (ECD), just outside of the transmembrane domain (TMD).65 Without Delta binding, the NRR is folded in such a way that a cleavage site (called S2) is inaccessible to the action of ubiquitous metalloproteases such as Kuzbanian (in Drosophila) or TACE (tumor necrosis factor-α-converting enzyme, or ADAM17, in vertebrates).66 Upon Delta-Notch binding, it is hypothesized that the mechanical force generated by the endocytosis of Delta partially unfolds the NRR, exposing the S2 site for cleavage by metalloproteases.66-69 After S2 cleavage, a subsequent cleavage (called S3) by the ubiquitous metazoan gamma-secretase complex occurs in the TMD, within the lipid bilayer,70,71 which releases the intracellular domain (ICD). The released ICD then translocates to the nucleus where it regulates transcription of cell fate-related genes.72,73
The Tango assay is one such tool derived from the molecular logic of Delta-Notch signaling. It was initially developed to monitor the interaction of a receptor of interest with its ligand or agonist in the extracellular space. In the Tango assay, 3 exogenous genetic elements are introduced into a “receiver” cell. The first element is a protein fusion consisting of a transmembrane receptor fused to an intracellular transcription factor. The transmembrane domain and intracellular domain are separated by a cleavage site that is targeted by a site-specific protease, such as tobacco etch virus (TEV) protease. The second element is a protein fusion consisting of the aforementioned protease linked to a protein that associates with the receptor upon agonist-binding. The third element is a reporter gene that can be activated by the transcription factor. The binding of the agonist recruits the protease to the receptor. The protease then frees the transcription factor from its membrane anchoring, allowing it to reach the nucleus and activate transcription of the reporter cassette. This results in a visual indication of receptor activation.
Barnea et al.74 initially developed Tango assays that could be used to monitor receptor activation in G protein-coupled receptors (GPCRs), receptor tyrosine kinases, and steroid hormone receptors, in vitro. In designing the Tango assay for GPCRs, the human arginine vasopressin receptor 2 (AVPR2) was used as a model. The receptor was fused to a transcription factor by a TEV Protease Cleavage Site (TCS), and the TEV protease was fused to human β-arrestin2, which associates with AVPR2 upon receptor activation. When these 2 chimeric constructs and a reporter gene were introduced into a cell line, the Tango-modified AVPR2 was able to induce the reporter gene expression upon binding to its respective agonist. The GPCR Tango assay has since been adopted for use in the Drosophila nervous system. The assay can be used to both screen which circuits are responsive to a specific neurotransmitter (Tango-map) and identify the postsynaptic partners of known neurons (Tango-Trace) that use GPCRs as neurotransmitter receptors.
Inagaki et al.75 applied the Tango-map assay to dopamine receptors in the Drosophila nervous system to investigate the neural circuits on which dopamine acts. In transgenic flies, they expressed the Tango-modified dopamine receptor, arrestin-TEV protease, and a reporter transgene in all neurons, to assess its functionality as a mapping system (Tango-map) (Fig. 4). They were able to detect expression of the GFP reporter in receiver cells that expressed the modified dopamine receptor upon their activation by dopamine. Next, they used this technique to explore which neurons received dopamine signaling following starvation periods, to determine how hunger affects the action of neuromodulators, like dopamine.
Figure 4.
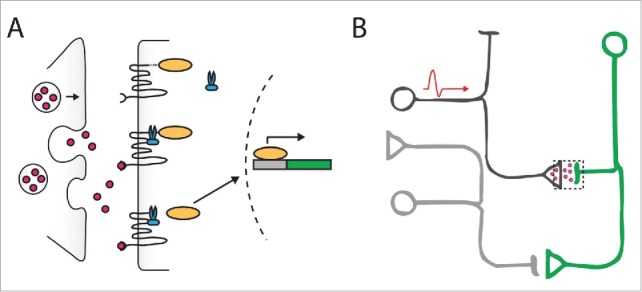
Identifying functional synaptic contact using Tango-modified GPCRs. (A) When neurotransmitters (red) are released from the active zone of the presynaptic neuron (left), they bind Tango-modified GPCRs on the postsynaptic neuron (right). The binding of neurotransmitter recruits an intracellular protease (blue) that cleaves the transcription factor (orange) which is tethered to the GPCR. This transcription factor then translocates to the nucleus to activate transcription of reporter genes such as GFP (green rectangle). (B) After the presynaptic neuron of interest (dark gray) fires an action potential, it releases neurotransmitters (pink) that bind to the Tango-modified GPCRs expressed in the postsynaptic neuron. This induces GFP expression in the postsynaptic neuron (green) but not in the bystander neuron (light gray).
The Tango approach was next used by Jagadish et al.,76 to identify synaptic partners of neurons in Drosophila. They developed a Tango-based technique called Tango-Trace to trace the synaptic connections of photoreceptor neurons in the Drosophila visual system. Their goal was to identify the synaptic partners of the 4 inner photoreceptors in the different layers of the optic lobe. Jagadish et al. adapted the Tango assay to the histamine (HA) receptor, since Drosophila photoreceptor neurons use HA as their main neurotransmitter. As in the previous study, 2 fusion proteins were constructed (HA receptor-TCS-Gal4 and Arrestin-TEV protease) and expressed in flies with a reporter. In this variant of the Tango method, there was very little activation of the receptor by the endogenous levels of HA in the brain. However, by artificially stimulating HA release in specific photoreceptor neurons with a temperature-sensitive cation channel (trpA1), they were able to visualize the postsynaptic partners to the trpA1-expressing cells.
The Tango assay as described here has several benefits that make it a versatile, useful tool for visually detecting cell-cell interactions. The assay can be applied to 89 members of the GPCR class, making it suitable for many different cell types. In addition, since all 3 components of the receptor and reporter are exogenous, there is very little risk that endogenous proteases or transcription factors will cross-react with the system. Also, although the native receptor is modified with the protease cleavage site and transcription factor intracellular domain, the specificity of the ligand binding is unaffected, allowing receptor activation to remain specific to its respective ligand. Additionally, if neurotransmitter release can be controlled specifically in presynaptic neurons of interest, as with the trpA1 channel, Tango can allow for identification of its functional postsynaptic partners.
Tango also has several limitations. Although Tango can be adapted for many members of the GPCR family, it is currently restricted to the study of cell-cell interactions mediated by GPCRs. This precludes the use of Tango for studying synapses mediated by ionotropic receptors. Furthermore, Tango requires a priori knowledge of the neurotransmitter used by the presynaptic neuron of interest, preventing its application for unbiased identification of uncharacterized synapses. Second, Tango has caveats that could possibly lead to false identification of connectivity. Theoretically, if the neurotransmitters diffuse outside of the synaptic cleft, neighboring neurons that express the receptor, but are not synaptically connected to the source neuron may be activated. To overcome these limitations, Tango-based systems used for future applications could be modified such that a membrane-tethered ligand localized to a presynaptic site could activate a Tango-modified receptor located in its postsynaptic partner.
TRanscellular Activation of Transcription (TRACT)
In contrast to Tango, which was designed to monitor interactions of a receptor to its soluble ligand or agonist, other applications of the Delta-Notch system have aimed at studying the contact between the plasma membranes of neighboring cells. The Delta-Notch system can be engineered to monitor cell-cell contact without interfering with endogenous signaling. By replacing the EGF repeats with a single-chain antibody domain (SCAD), a synthetic version of Notch can be made to recognize orthogonal ligands, such as CD19 or GFP, instead of Delta. In addition, the ICD of Notch can be replaced with an orthogonal transcriptional regulator such as Gal4 or tTA. Ultimately, “emitter” cells expressing the artificial ligand can trigger proteolysis of the modified Notch receptor in “receiver” cells by membrane-membrane contact. Used in conjunction with UAS or tetO, membrane-membrane contact can be observed by expression of a reporter gene such as GFP (Fig. 5A).
Figure 5.
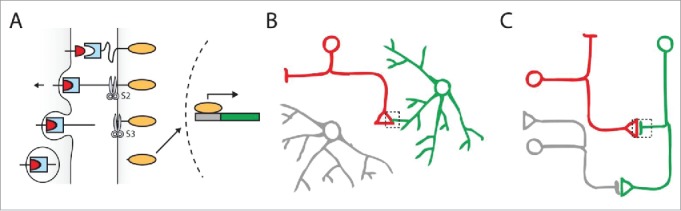
Visualizing cell-cell contact using the TRACT method. (A) The ligand (red) is localized to the plasma membrane in the cell of interest (left). When the ligand binds to the receptor (light blue) on a neighboring cell (right), it partially unfolds the NRR to allow for proteolysis of the receptor (S2). S2 induces another proteolysis (S3), liberating the transcription factor (orange oval) that is fused to the receptor. This transcription factor then translocates to the nucleus to activate transcription of reporter genes such as GFP (green rectangle). (B) TRACT can be used in Drosophila to detect neuron-astrocyte contact in vivo. The artificial ligand is expressed in the neuron of interest (red) while the receptor is expressed in all astrocytes (green, gray). Only the astrocyte that is in contact with the ligand-expressing neuron is induced to express GFP (green). (C) With optimization, TRACT could also be used to detect neuron-neuron contact. For anterograde tracing, the ligand needs to be localized to the presynaptiC-terminal in the neuron of interest (red). If the receptor is expressed at the postsynaptic sites of all candidate neurons (green, gray), only the neuron postsynaptic to the neuron of interest (red) will be induced to express GFP (green). Alternatively, for retrograde tracing, the ligand would be localized at the postsynaptic sites in the neuron of interest, and the receptor would be localized at the presynaptic sites of all candidate neurons.
This strategy has been used in vitro to engineer cell-cell interactions based on their contact77 and to enhance T cell activity against tumor cells in mice.78 Our group has developed a parallel method called TRACT, which we have used to investigate neuron-glia interactions in the Drosophila nervous system (Fig. 5B).79 As a proof of concept, we expressed the CD19 ligand in 2 different subsets of glial cells under either the alrm or repo driver and the receptor in all neurons. The spatial pattern of each of these drivers is distinct: whereas the repo driver leads to strong expression of the ligand in many types of glial cells, the alrm driver is relatively specific to astrocytes. Consistent with our expectations, depending on which glial cells were expressing the ligand, different populations of neurons were induced to transcribe GFP. This suggested that TRACT was capable of detecting glia-neuron contact in vivo. We tested this in subsequent experiments by restricting ligand expression to few cells. When the receptor was expressed in all astrocytes (under the alrm driver) and the ligand in subsets of neurons with a restricted localization in the brain, we observed GFP expression only in the astrocytes that contact the ligand-expressing neurons. For example, expressing the ligand in the ORNs (under the orco driver) led to GFP expression in the astrocytes surrounding the ORN axons in the antennal lobe. Similarly, expressing the ligand in neurons that produce the PDF peptide (under the pdf driver) led to GFP induction in a small set of astrocytes bordering PDF+ neurons. Our results clearly demonstrate that TRACT is capable of detecting neuron-glia contact in Drosophila. In addition, this application of TRACT serves as a much-needed tool for selectively labeling groups of glial cells. Though systems such as MARCM or Flip-Out can be used to sparsely label glial cells in mosaic clones, both systems are inherently stochastic and cannot be used to control gene expression in a stereotypical set of cells. With TRACT, we have shown that it is now possible to readily identify and consistently label subpopulations of glial cells based on their contact with known neuronal types.
TRACT can also be used in neurons to identify synaptic partners. In preliminary experiments, expression of the ligand in ORNs, and the receptor in all neurons (under the elav driver) resulted in GFP induction the antennal lobe PNs and LNs, the established postsynaptic targets of ORNs (unpublished). Although this observation indicates that TRACT can be used to detect membrane-membrane contact between interacting neurons, it does not guarantee that TRACT can specifically identify synaptic connections - at least in its current implementation. As was the case for GRASP, it would be necessary to localize the molecular components to pre- and postsynaptic sites. Theoretically, targeting the ligand and/or receptor to synaptic sites will prevent the possibility of receptor activation by membrane-membrane contact at non-synaptic sites (for instance, between fasciculating axons).
We anticipate that with optimization TRACT will become useful as a neural tracer. Because it does not require prior knowledge of the neurotransmitters used or putative synaptic partners, TRACT would be ideal for unbiased identification of novel synaptic connections. In addition, TRACT offers investigators the power to regulate expression of transgenes such as GECIs,80 TRPA1, or light-sensitive channels81 based on synaptic contact for functional analysis of newly identified circuits.
Concluding remarks
Each of the methods described here has its advantages and disadvantages (Table 1). In choosing the appropriate method for tracing nervous system connectivity, it is crucial to first identify the nature of the question at hand. EM is impractical for investigating how synapses change over time or in response to genetic perturbations. But when resources allow, EM is an extremely valuable tool to build systems-level models of complex microcircuits. To directly test if 2 subsets of neurons are synaptically connected, straightforward LM techniques such as synaptic tagging or GRASP will suffice. If the connectivity of the neuron of interest has never been characterized, PA-GFP may be the best place to begin. On the other hand, if the neurotransmitter is already known and it is feasible to selectively stimulate the neuron of interest, Tango is a useful option. Tango additionally provides the option to genetically manipulate neurons based on their synaptic input. Similarly to Tango, TRACT allows for genetic manipulation based on cell-cell contact. With optimization, TRACT may be better suited for unbiased identification of novel synapses, since it does not require prior knowledge of the neurotransmitter and can theoretically be used for both anterograde and retrograde tracing. As connectomic methods continue to develop for higher organisms, there is a lesson to be learned from the precedent in C. elegans: the connectome alone, no matter how complete, cannot explain how brain circuits work in real time. The most efficacious use of the connectomic methods described in this review, therefore, is in synergy with each other, and more importantly, with electrophysiological or optical recordings to establish an understanding of the dynamics of neural circuits in parallel.
Table 1.
An overview of the advantages and disadvantages associated with each method of identifying synaptic connections.
| Technique | Advantages | Disadvantages |
|---|---|---|
| Electron Microscopy | Provides detailed wiring diagram of small volumes. | Time and labor-intensive. Impractical for long-range connections or for many samples. |
| Synaptic Tagging | Can monitor synapses in vivo. | For regions dense with synapses, super-resolution microscopy may be required. Need genetic drivers for putative synaptic partners. |
| GRASP | Can monitor synapses in vivo. With NSyb::GRASP, can specifically detect active synapses. | May be not sensitive enough for in vivo detection. Can induce irreversible binding at natively transient synapses. |
| PA-GFP | Can be used for unbiased identification of novel synapses. | Not conclusive by itself. Requires functional studies to confirm. |
| WGA | Only requires a single transgene. Simple to implement. | Can result in labeling of higher-order synapses. The signal can be difficult to detect because it gets diluted. |
| Tango-Trace | Can identify functional synapses. Allows genetic manipulation based on synaptic input. | Requires prior knowledge of neurotransmitters. Requires a way of artificially stimulating the presynaptic neuron of interest. |
| TRACT | Allows genetic manipulation based on cell-cell contact. | Requires optimization to trace neuron-neuron connections. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Chklovskii DB, Vitaladevuni S, Scheffer LK. Semi-automated reconstruction of neural circuits using electron microscopy. Curr Opin Neurobiol 2010; 20:667-75; PMID:20833533; https://doi.org/ 10.1016/j.conb.2010.08.002 [DOI] [PubMed] [Google Scholar]
- [2].Schneider-Mizell CM, Gerhard S, Longair M, Kazimiers T, Li F, Zwart MF, Champion A, Midgley FM, Fetter RD, Saalfeld S, et al.. Quantitative neuroanatomy for connectomics in Drosophila. Elife 2016; 5:e12059; PMID:26990779; https://doi.org/ 10.7554/eLife.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Berck ME, Khandelwal A, Claus L, Hernandez-Nunez L, Si G, Tabone CJ, Li F, Truman JW, Fetter RD, Louis M, et al.. The wiring diagram of a glomerular olfactory system. Elife 2016; 5:e14859; PMID:27177418; https://doi.org/ 10.7554/eLife.14859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Takemura SY, Bharioke A, Lu Z, Nern A, Vitaladevuni S, Rivlin PK, Katz WT, Olbris DJ, Plaza SM, Winston P, et al.. A visual motion detection circuit suggested by Drosophila connectomics. Nature 2013; 500:175-81; PMID:23925240; https://doi.org/ 10.1038/nature12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Butcher NJ, Friedrich AB, Lu Z, Tanimoto H, Meinertzhagen IA. Different classes of input and output neurons reveal new features in microglomeruli of the adult Drosophila mushroom body calyx. J Comp Neurol 2012; 520:2185-201; PMID:22237598; https://doi.org/ 10.1002/cne.23037 [DOI] [PubMed] [Google Scholar]
- [6].Hayworth KJ, Xu CS, Lu Z, Knott GW, Fetter RD, Tapia JC, Lichtman JW, Hess HF. Ultrastructurally smooth thick partitioning and volume stitching for large-scale connectomics. Nat Methods 2015; 12:319-22; PMID:25686390; https://doi.org/ 10.1038/nmeth.3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Begemann I, Galic M. Correlative Light Electron Microscopy: Connecting Synaptic Structure and Function. Front Synaptic Neurosci 2016; 8:28; PMID:27601992; https://doi.org/ 10.3389/fnsyn.2016.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Collman F, Buchanan J, Phend KD, Micheva KD, Weinberg RJ, Smith SJ. Mapping synapses by conjugate light-electron array tomography. J Neurosci 2015; 35:5792-807; PMID:25855189; https://doi.org/ 10.1523/JNEUROSCI.4274-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol 2011; 9:e1001041; PMID:21483721; https://doi.org/ 10.1371/journal.pbio.1001041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blazquez-Llorca L, Hummel E, Zimmerman H, Zou C, Burgold S, Rietdorf J, Herms J. Correlation of two-photon in vivo imaging and FIB/SEM microscopy. J Microsc 2015; 259:129-36; PMID:25786682; https://doi.org/ 10.1111/jmi.12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adams SR, Mackey MR, Ramachandra R, Palida Lemieux SF, Steinbach P, Bushong EA, Butko MT, Giepmans BN, Ellisman MH, Tsien RY. Multicolor Electron Microscopy for Simultaneous Visualization of Multiple Molecular Species. Cell Chem Biol 2016; 23:1417-1427; PMID:27818300; https://doi.org/ 10.1016/j.chembiol.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 1994; 19:780-2; PMID:19844443; https://doi.org/ 10.1364/OL.19.000780 [DOI] [PubMed] [Google Scholar]
- [13].Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt Lett 1999; 24:954-6; PMID:18073907; https://doi.org/ 10.1364/OL.24.000954 [DOI] [PubMed] [Google Scholar]
- [14].Nienhaus K, Nienhaus GU. Fluorescent proteins for live-cell imaging with super-resolution. Chem Soc Rev 2014; 43:1088-106; PMID:24056711; https://doi.org/ 10.1039/C3CS60171D [DOI] [PubMed] [Google Scholar]
- [15].Yamanaka M, Smith NI, Fujita K. Introduction to super-resolution microscopy. Microscopy (Oxf) 2014; 63:177-92; PMID:24671128; https://doi.org/ 10.1093/jmicro/dfu007 [DOI] [PubMed] [Google Scholar]
- [16].Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell 2010; 143:1047-58; PMID:21168201; https://doi.org/ 10.1016/j.cell.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].del Valle Rodriguez A, Didiano D, Desplan C. Power tools for gene expression and clonal analysis in Drosophila. Nat Methods 2011; 9:47-55; PMID:22205518; https://doi.org/ 10.1038/nmeth.1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kremer MC, Christiansen F, Leiss F, Paehler M, Knapek S, Andlauer TF, Förstner F, Kloppenburg P, Sigrist SJ, Tavosanis G. Structural long-term changes at mushroom body input synapses. Curr Biol 2010; 20:1938-44; PMID:20951043; https://doi.org/ 10.1016/j.cub.2010.09.060 [DOI] [PubMed] [Google Scholar]
- [19].Christiansen F, Zube C, Andlauer TF, Wichmann C, Fouquet W, Owald D, Mertel S, Leiss F, Tavosanis G, Luna AJ, et al.. Presynapses in Kenyon cell dendrites in the mushroom body calyx of Drosophila. J Neurosci 2011; 31:9696-707; PMID:21715635; https://doi.org/ 10.1523/JNEUROSCI.6542-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Y, Akin O, Nern A, Tsui CY, Pecot MY, Zipursky SL. Cell-type-specific labeling of synapses in vivo through synaptic tagging with recombination. Neuron 2014; 81:280-93; PMID:24462095; https://doi.org/ 10.1016/j.neuron.2013.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mosca TJ, Luo L. Synaptic organization of the Drosophila antennal lobe and its regulation by the Teneurins. Elife 2014; 3:e03726; PMID:25310239; https://doi.org/ 10.7554/eLife.03726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Akin O, Zipursky SL. Frazzled promotes growth cone attachment at the source of a Netrin gradient in the Drosophila visual system. Elife 2016; 5:e20762; PMID:27743477; https://doi.org/ 10.7554/eLife.20762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu S, Liu Q, Tabuchi M, Wu MN. Sleep Drive Is Encoded by Neural Plastic Changes in a Dedicated Circuit. Cell 2016; 165:1347-60; PMID:27212237; https://doi.org/ 10.1016/j.cell.2016.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hamanaka Y, Meinertzhagen IA. Immunocytochemical localization of synaptic proteins to photoreceptor synapses of Drosophila melanogaster. J Comp Neurol 2010; 518:1133-55; PMID:20127822; https://doi.org/ 10.1002/cne.22268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meinertzhagen IA, Lee CH. The genetic analysis of functional connectomics in Drosophila. Adv Genet 2012; 80:99-151; PMID:23084874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 2011; 145:117-32; PMID:21458671; https://doi.org/ 10.1016/j.cell.2011.02.039 [DOI] [PubMed] [Google Scholar]
- [27].Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. Polarity and intracellular compartmentalization of Drosophila neurons. Neural Dev 2007; 2:7; PMID:17470283; https://doi.org/ 10.1186/1749-8104-2-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang YQ, Rodesch CK, Broadie K. Living synaptic vesicle marker: synaptotagmin-GFP. Genesis 2002; 34:142-5; PMID:12324970; https://doi.org/ 10.1002/gene.10144 [DOI] [PubMed] [Google Scholar]
- [29].Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature 2002; 418:336-40; PMID:12110845; https://doi.org/ 10.1038/nature00915 [DOI] [PubMed] [Google Scholar]
- [30].Leiss F, Groh C, Butcher N.J, Meinertzhagen I.A, Tavosanis G. Synaptic organization in the adult Drosophila mushroom body calyx. J Comp Neurol 2009; 517:808-24; PMID:19844895; https://doi.org/ 10.1002/cne.22184 [DOI] [PubMed] [Google Scholar]
- [31].Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, et al.. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 2006; 49:833-44; PMID:16543132; https://doi.org/ 10.1016/j.neuron.2006.02.008 [DOI] [PubMed] [Google Scholar]
- [32].Andretic R, Kim Y.C, Jones F.S, Han K.A, Greenspan R.J. Drosophila D1 dopamine receptor mediates caffeine-induced arousal. Proc Natl Acad Sci U S A 2008; 105:20392-7; PMID:19074291; https://doi.org/ 10.1073/pnas.0806776105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].DiAntonio A. Learning something ORIGINal at the Drosophila neuromuscular junction. Neuron 1999; 23:1-2; PMID:10402184; https://doi.org/ 10.1016/S0896-6273(00)80743-6 [DOI] [PubMed] [Google Scholar]
- [34].Petersen S.A, Fetter R.D, Noordermeer J.N, Goodman C.S, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 1997; 19:1237-48; PMID:9427247; https://doi.org/ 10.1016/S0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- [35].Tsurudome K, Tsang K, Liao EH, Ball R, Penney J, Yang JS, Elazzouzi F, He T, Chishti A, Lnenicka G, et al.. The Drosophila miR-310 cluster negatively regulates synaptic strength at the neuromuscular junction. Neuron 2010; 68:879-93; PMID:21145002; https://doi.org/ 10.1016/j.neuron.2010.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu X, Buchanan ME, Han KA, Davis RL. The GABAA receptor RDL suppresses the conditioned stimulus pathway for olfactory learning. J Neurosci 2009; 29:1573-9; PMID:19193904; https://doi.org/ 10.1523/JNEUROSCI.4763-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Berger-Muller S, Sugie A, Takahashi F, Tavosanis G, Hakeda-Suzuki S, Suzuki T. Assessing the role of cell-surface molecules in central synaptogenesis in the Drosophila visual system. PLoS One 2013; 8:e83732; PMID:24386266; https://doi.org/ 10.1371/journal.pone.0083732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sugie A, Hakeda-Suzuki S, Suzuki E, Silies M, Shimozono M, Möhl C, Suzuki T, Tavosanis G. Molecular Remodeling of the Presynaptic Active Zone of Drosophila Photoreceptors via Activity-Dependent Feedback. Neuron 2015; 86:711-25; PMID:25892303; https://doi.org/ 10.1016/j.neuron.2015.03.046 [DOI] [PubMed] [Google Scholar]
- [39].Schmid A, Hallermann S, Kittel RJ, Khorramshahi O, Frölich AM, Quentin C, Rasse TM, Mertel S, Heckmann M, Sigrist SJ. Activity-dependent site-specific changes of glutamate receptor composition in vivo. Nat Neurosci 2008; 11:659-66; PMID:18469810; https://doi.org/ 10.1038/nn.2122 [DOI] [PubMed] [Google Scholar]
- [40].Fouquet W, Owald D, Wichmann C, Mertel S, Depner H, Dyba M, Hallermann S, Kittel RJ, Eimer S, Sigrist SJ. Maturation of active zone assembly by Drosophila Bruchpilot. J Cell Biol 2009; 186:129-45; PMID:19596851; https://doi.org/ 10.1083/jcb.200812150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Spuhler IA, Conley GM, Scheffold F, Sprecher SG. Super Resolution Imaging of Genetically Labeled Synapses in Drosophila Brain Tissue. Front Cell Neurosci 2016; 10:142.; PMID:27303270; https://doi.org/ 10.3389/fncel.2016.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ke MT, Nakai Y, Fujimoto S, Takayama R, Yoshida S, Kitajima TS, Sato M, Imai T. Super-Resolution Mapping of Neuronal Circuitry With an Index-Optimized Clearing Agent. Cell Rep 2016; 14:2718-32; PMID:26972009; https://doi.org/ 10.1016/j.celrep.2016.02.057 [DOI] [PubMed] [Google Scholar]
- [43].Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, Bargmann CI. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 2008; 57:353-63; PMID:18255029; https://doi.org/ 10.1016/j.neuron.2007.11.030 [DOI] [PubMed] [Google Scholar]
- [44].Gordon M.D, Scott K. Motor control in a Drosophila taste circuit. Neuron 2009; 61:373-84; PMID:19217375; https://doi.org/ 10.1016/j.neuron.2008.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hasegawa E, Truman J.W, Nose A. Identification of excitatory premotor interneurons which regulate local muscle contraction during Drosophila larval locomotion. Sci Rep 2016; 6:30806; PMID:27470675; https://doi.org/ 10.1038/srep30806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Itakura Y, Kohsaka H, Ohyama T, Zlatic M, Pulver SR, Nose A. Identification of Inhibitory Premotor Interneurons Activated at a Late Phase in a Motor Cycle during Drosophila Larval Locomotion. PLoS One 2015; 10:e0136660; PMID:26335437; https://doi.org/ 10.1371/journal.pone.0136660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lin TY, Luo J, Shinomiya K, Ting CY, Lu Z, Meinertzhagen IA, Lee CH Mapping chromatic pathways in the Drosophila visual system. J Comp Neurol 2016; 524:213-27; PMID:26179639; https://doi.org/ 10.1002/cne.23857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gorostiza EA, Depetris-Chauvin A, Frenkel L, Pirez N, Ceriani MF. Circadian pacemaker neurons change synaptic contacts across the day. Curr Biol 2014; 24:2161-7; PMID:25155512; https://doi.org/ 10.1016/j.cub.2014.07.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cavanaugh DJ, Geratowski JD, Wooltorton JR, Spaethling JM, Hector CE, Zheng X, Johnson EC, Eberwine JH, Sehgal A. Identification of a circadian output circuit for rest:activity rhythms in Drosophila. Cell 2014; 157:689-701; PMID:24766812; https://doi.org/ 10.1016/j.cell.2014.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Masuda-Nakagawa LM, Ito K, Awasaki T, O'Kane CJ. A single GABAergic neuron mediates feedback of odor-evoked signals in the mushroom body of larval Drosophila. Front Neural Circuits 2014; 8:35; PMID:24782716; https://doi.org/ 10.3389/fncir.2014.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Macpherson LJ, Zaharieva EE, Kearney PJ, Alpert MH, Lin TY, Turan Z, Lee CH, Gallio M. Dynamic labelling of neural connections in multiple colours by trans-synaptic fluorescence complementation. Nat Commun 2015; 6:10024; PMID:26635273; https://doi.org/ 10.1038/ncomms10024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al.. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013; 499:295-300; PMID:23868258; https://doi.org/ 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li Y, Guo A, Li H. CRASP: CFP reconstitution across synaptic partners. Biochem Biophys Res Commun 2016; 469:352-6; PMID:26682922; https://doi.org/ 10.1016/j.bbrc.2015.12.011 [DOI] [PubMed] [Google Scholar]
- [54].Ruta V, Datta SR, Vasconcelos ML, Freeland J, Looger LL, Axel R. A dimorphic pheromone circuit in Drosophila from sensory input to descending output. Nature 2010; 468:686-90; PMID:21124455; https://doi.org/ 10.1038/nature09554 [DOI] [PubMed] [Google Scholar]
- [55].Lai JS, Lo SJ, Dickson BJ, Chiang AS. Auditory circuit in the Drosophila brain. Proc Natl Acad Sci U S A 2012; 109:2607-12; PMID:22308412; https://doi.org/ 10.1073/pnas.1117307109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fisek M, Wilson RI. Stereotyped connectivity and computations in higher-order olfactory neurons. Nat Neurosci 2014; 17:280-8; PMID:24362761; https://doi.org/ 10.1038/nn.3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Clowney EJ, Iguchi S, Bussell JJ, Scheer E, Ruta V. Multimodal Chemosensory Circuits Controlling Male Courtship in Drosophila. Neuron 2015; 87:1036-49; PMID:26279475; https://doi.org/ 10.1016/j.neuron.2015.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002; 297:1873-7; PMID:12228718; https://doi.org/ 10.1126/science.1074952 [DOI] [PubMed] [Google Scholar]
- [59].Berlin S, Carroll EC, Newman ZL, Okada HO, Quinn CM, Kallman B, Rockwell NC, Martin SS, Lagarias JC, Isacoff EY. Photoactivatable genetically encoded calcium indicators for targeted neuronal imaging. Nat Methods 2015; 12:852-8; PMID:26167640; https://doi.org/ 10.1038/nmeth.3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nassi JJ, Cepko CL, Born RT, Beier KT. Neuroanatomy goes viral! Front Neuroanat 2015; 9:80; PMID:26190977; https://doi.org/ 10.3389/fnana.2015.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Huh Y, Oh MS, Leblanc P, Kim KS. Gene transfer in the nervous system and implications for transsynaptic neuronal tracing. Expert Opin Biol Ther 2010; 10:763-72; PMID:20367126; https://doi.org/ 10.1517/14712591003796538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tabuchi K, Sawamoto K, Suzuki E, Ozaki K, Sone M, Hama C, Tanifuji-Morimoto T, Yuasa Y, Yoshihara Y, Nose A, et al.. GAL4/UAS-WGA system as a powerful tool for tracing Drosophila transsynaptic neural pathways. J Neurosci Res 2000; 59:94-9; PMID:10658189; https://doi.org/ 10.1002/(SICI)1097-4547(20000101)59:1%3c94::AID-JNR11%3e3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- [63].Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 2010; 141:154-65; PMID:20303157; https://doi.org/ 10.1016/j.cell.2010.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Libbrecht S, Van den Haute C, Malinouskaya L, Gijsbers R, Baekelandt V. Evaluation of WGA-Cre-dependent topological transgene expression in the rodent brain. Brain Struct Funct 2017; 222:717-33; https://doi.org/10.1007/s00429-016-1241-x. [DOI] [PubMed] [Google Scholar]
- [65].Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol 2007; 14:295-300; PMID:17401372; https://doi.org/ 10.1038/nsmb1227 [DOI] [PubMed] [Google Scholar]
- [66].Tiyanont K, Wales TE, Aste-Amezaga M, Aster JC, Engen JR, Blacklow SC. Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure 2011; 19:546-54; PMID:21481777; https://doi.org/ 10.1016/j.str.2011.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Stephenson NL, Avis JM. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc Natl Acad Sci U S A 2012; 109:E2757-65; PMID:23011796; https://doi.org/ 10.1073/pnas.1205788109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Meloty-Kapella L, Shergill B, Kuon J, Botvinick E, Weinmaster G. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev Cell 2012; 22:1299-312; PMID:22658936; https://doi.org/ 10.1016/j.devcel.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ, et al.. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev Cell 2015; 33:729-36; PMID:26051539; https://doi.org/ 10.1016/j.devcel.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israël A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 2000; 5:207-16; PMID:10882063; https://doi.org/ 10.1016/S1097-2765(00)80417-7 [DOI] [PubMed] [Google Scholar]
- [71].Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell 2000; 5:197-206; PMID:10882062; https://doi.org/ 10.1016/S1097-2765(00)80416-5 [DOI] [PubMed] [Google Scholar]
- [72].Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell 1998; 93:649-60; PMID:9604939; https://doi.org/ 10.1016/S0092-8674(00)81193-9 [DOI] [PubMed] [Google Scholar]
- [73].Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 2000; 6:625-36; PMID:11030342; https://doi.org/ 10.1016/S1097-2765(00)00061-7 [DOI] [PubMed] [Google Scholar]
- [74].Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci U S A 2008; 105:64-9; PMID:18165312; https://doi.org/ 10.1073/pnas.0710487105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Inagaki HK, Ben-Tabou de-Leon S, Wong AM, Jagadish S, Ishimoto H, Barnea G, Kitamoto T, Axel R, Anderson DJ. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell 2012; 148:583-95; PMID:22304923; https://doi.org/ 10.1016/j.cell.2011.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jagadish S, Barnea G, Clandinin TR, Axel R. Identifying functional connections of the inner photoreceptors in Drosophila using Tango-Trace. Neuron 2014; 83:630-44; PMID:25043419; https://doi.org/ 10.1016/j.neuron.2014.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016; 164:780-91; PMID:26830878; https://doi.org/ 10.1016/j.cell.2016.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, Lim WA. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016; 167:419-432 e16; PMID:27693353; https://doi.org/ 10.1016/j.cell.2016.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Huang TH, Velho T, Lois C. Monitoring cell-cell contacts in vivo in transgenic animals. Development 2016; 143:4073-4084; PMID:27660327; https://doi.org/ 10.1242/dev.142406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Tian L, Hires SA, Looger LL. Imaging neuronal activity with genetically encoded calcium indicators. Cold Spring Harb Protoc 2012; 2012:647-56 [DOI] [PubMed] [Google Scholar]
- [81].Yizhar O, Fenno L, Zhang F, Hegemann P, Diesseroth K. Microbial opsins: a family of single-component tools for optical control of neural activity. Cold Spring Harb Protoc 2011; 2011:top102; PMID:21363959; https://doi.org/ 10.1101/pdb.top102 [DOI] [PubMed] [Google Scholar]