

Abstract
Heparin-binding epidermal growth factor (EGF)-like growth factor (HBEGF) is a ligand for the epidermal growth factor receptor (EGFR), one of the most commonly amplified receptor tyrosine kinases (RTK) in glioblastoma. While HBEGF has been found to be expressed in a subset of malignant gliomas, its sufficiency for glioma initiation has not been evaluated. In this study, we demonstrate that HBEGF can initiate glioblastoma (GBM) in mice in the context of Ink4a/Arf and Pten loss, and that these tumors are similar to the classical GBM subtype observed in patients. Isogenic astrocytes from these mice showed activation not only of Egfr but also the RTK Axl in response to HBEGF stimulation. Deletion of either Egfr or Axl decreased the tumorigenic properties of HBEGF transformed cells; however only EGFR was able to rescue the phenotype in cells lacking both RTKs indicating that Egfr is required for activation of Axl in this context. Silencing of HBEGF in vivo resulted in tumor regression and significantly increased survival suggesting that HBEGF may be a clinically relevant target.
Keywords: HBEGF, Ink4a/Arf, glioma, mouse model, RCAS/TVA
Introduction
Malignant gliomas, including anaplastic astrocytomas and glioblastomas (GBM), are aggressive tumors that confer a poor prognosis upon diagnosis.1,2 Anaplastic astrocytomas are characterized by high mitotic activity and cytological atypia. Glioblastomas retain the features of lower grade gliomas but additionally present with microvascular proliferation and/or pseudopalisading necrosis.1 With treatment, which consists of surgery, radiotherapy, and chemotherapy, the average survival for patients diagnosed with GBM is about 14 months.2 Therefore, there is a strong need to understand the mechanisms underlying malignant glioma initiation and maintenance such that new therapies can be developed.
Glioblastoma was the first cancer to be systematically analyzed by The Cancer Genome Atlas (TCGA) network.3,4 Sequencing of these tumors revealed alterations in three core pathways. The receptor tyrosine kinase (RTK)/RAS/phosphatidylinositol-3-kinase (PI3K) pathway was altered in 88%, the retinoblastoma (RB) pathway was altered in 78%, and the p53 pathway was altered in 87% of samples.3,4 In the RTK/RAS/PI3K axis, the most common alterations were mutation or amplification of epidermal growth factor receptor (EGFR) and homozygous deletion of phosphatase and tensin homolog deleted on chromosome ten (PTEN). In the p53 and RB pathways, the most common lesion was homozygous deletion of cyclin dependent kinase 2A (CDKN2A), which encodes both p16INK4a and p14ARF. EGFR amplified tumors, which account for 65% of glioblastomas, are often associated with increased expression of EGFR ligands including heparin-binding epidermal growth factor (EGF)-like growth factor (HBEGF).5–7
HBEGF has been associated with the progression of many solid tumors including breast, pancreatic, and ovarian.8–10 In up to 40% of malignant gliomas, HBEGF has been shown to be co-expressed with EGFR by immunohistochemistry.5,7 Additionally, HBEGF mRNA transcripts were two to five-fold greater in GBM compared to normal brain tissue.5
HBEGF mRNA has been detected in the brainstem at E14, as well as in the developing cortical plate, hippocampus, thalamus, and Cerebellar Purkinje cells, suggestive of a role in neuronal and glial maturation.11 Additionally, administration of HBEGF in young and adult mice significantly increased the proliferation of neural progenitor cells in the subventricular zone and hippocampal dentate subgranular zone.12 HBEGF also stimulates proliferation of neural progenitor cells and astrocytes via activation of EGFR and the MAPK/ERK pathway.13 HBEGF is mitogenic in human glioma cell lines and this activity can be blocked with HBEGF neutralizing antibodies.5,7 In addition to activation of EGFR, HBEGF has been shown to have other pleiotropic properties in bladder cancer including induction of matrix metalloproteinase-9 (MMP9) and vascular endothelial growth factor (VEGF) expression, which allows for tumor invasion and proliferation.14 In chicken fibroblasts, expression of HBEGF induces transformation.15 In U251 glioma cells, expression of EGFRvIII, the most common variant of EGFR, induces expression of HBEGF. Moreover, silencing of HBEGF decreases EGFRvIII-tumorigenicity in this context.7,16 However, the ability of HBEGF to promote transformation in astrocytes in vitro and induce or sustain gliomas in vivo has not been assessed.
In the current study, we used the well-established RCAS/TVA glioma mouse model to assess the role of HBEGF in gliomagenesis and maintenance in vivo. Tumors generated using this model arise from a small number of cells within a normal microenvironment, which mimics the human disease. Furthermore, mammalian cells ectopically expressing the viral receptor TVA can be infected with several different RCAS viruses to allow the determination of the minimal components required for tumorigenesis. We demonstrate that HBEGF can promote tumor development in the context of either Ink4a/Arf and/or Pten loss. Furthermore, suppression of HBEGF expression in tumor-bearing mice resulted in significantly increased survival, which suggests it may be a relevant target for therapy. Interestingly, we discovered that HBEGF signals through both Egfr and Axl in this context. While AXL has been implicated in glioblastoma formation, it has not been previously reported to play a role in HBEGF signal transduction.17,18
Results
Overexpression of HBEGF correlates with decreased survival in GBM patients
To assess the effect of ligand overexpression on patient survival, we compared the median survival among cohorts of patients whose tumors overexpressed one of seven different EGFR ligands. We observed that patients harboring malignant gliomas that overexpressed HBEGF had a median survival of only 7.4 months, which was significantly shorter than the median survival of 13.6 months in patients with malignant gliomas that did not overexpress HBEGF (Table S1). This suggests that overexpression of HBEGF confers a worse prognosis in these patients. Further analysis of the relative expression levels of these EGFR ligands revealed that HBEGF is expressed at significantly higher levels in these tumors (Figure S1a and b).
HBEGF promotes anchorage-independent growth
Although HBEGF has been shown to increase the proliferation of human glioma cells in vitro,5 little is known about the role of this ligand in the biology of malignant gliomas. To evaluate the role(s) of specific genes in glioma initiation, progression and maintenance, we and others have used a somatic-cell gene delivery method based on the RCAS/TVA retroviral vector system and successfully induced malignant gliomas in vivo.19,20 Using this established model system, we initially assessed the ability of HBEGF to transform primary astrocytes derived from Nestin-TVA; Ink4a/Arflox/lox mice. These astrocytes were infected with RCAS-Cre alone or in combination with either RCAS-HBEGF and/or RCAS-EGFR. Following confirmation of HBEGF and EGFR expression (Figure 1a), the ability of the astrocytes to grow in an anchorage-independent manner was assessed by growth in soft agar. These astrocytes were immortalized by deletion of Ink4a/Arf to allow for ease of growth in culture but they are not transformed as evidenced by the lack of colonies detected in the Nestin-TVA; Ink4a/Arflox/lox astrocytes without EGFR or HBEGF infection (Figure 1b). Anchorage-independent growth was detected in astrocytes expressing HBEGF (P<0.0001). While colonies were also observed when EGFR was expressed in the absence of HBEGF, they were significantly fewer in number than was observed in astrocytes expressing HBEGF (P=0.01). No significant difference in colony number was observed between astrocytes expressing HBEGF alone or co-expressing EGFR (P=0.2). EGFR activity was confirmed by adding known quantities of HBEGF to astrocytes either without (−) or with exogenous receptor (EGFR) (Figure 1c). Dose dependent phosphorylation of EGFR (Y1068), as well as activation of downstream targets including Akt (S473) and Erk1/2 (T202/204), was observed. As expected, higher levels of phosphorylated EGFR were observed in cells expressing exogenous EGFR. For comparison with other EGFR ligands, the ability of AREG and EREG to promote the growth of immortal astrocytes in soft agar was also assessed. While both of these ligands conferred anchorage-independent growth, HBEGF was a more potent inducer of colony formation in this context (Figure S1c).
Figure 1. HBEGF promotes anchorage-independent growth of immortal astrocytes.
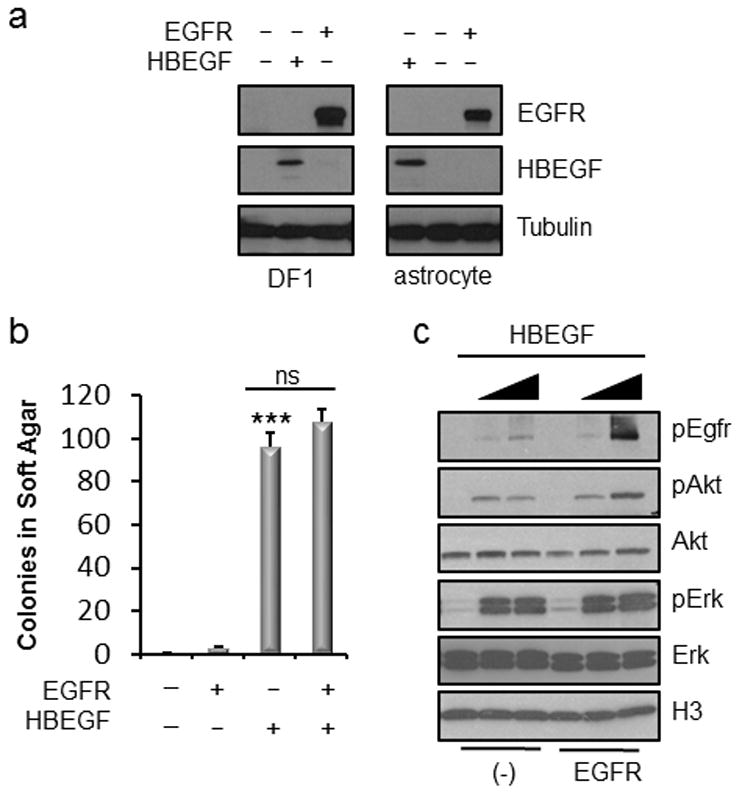
(a) Expression of EGFR and HBEGF in DF-1 cells and Nestin-TVAInk4a/Arf lox/lox mouse astrocytes. (b) Soft agar colony formation of Ink4a/Arf lox/lox astrocytes expressing exogenous EGFR and/or HBEGF compared with RCAS-Cre infected control cells. Data is represented as mean ± S.E.M. Using a two-tailed Student’s t test, the number of colonies formed were compared to control and statistical significance is shown by the following key: *** P < 0.0005 with not significant (ns) denoting P > 0.05. (c) Western blot analysis of downstream effector pathways in Ink4a/Arf lox/lox astrocytes expressing either endogenous (−) or exogenous (EGFR). HBEGF was added to serum-free media at two different concentrations (0.5 ng/ml and 5.0 ng/ml).
Expression of HBEGF results in reduced survival in vivo in the context of Ink4a/Arf or Pten loss
To evaluate the effect of HBEGF expression in nestin expressing cells in vivo, we utilized Nestin-TVA; Ink4a/Arflox/lox mice and generated both Nestin-TVA;Ptenlox/lox and Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox mice. Exons 2 and 3 of the Ink4a/Arf locus are flanked by LoxP sites such that in the absence of Cre these mice express normal levels of p16INK4a and p19ARF. Cre-mediated excision renders both p16Ink4a and p19Arf non-functional.21 Exon 5 of the Pten locus is flanked by LoxP sites such that in the absence of Cre, these mice express normal levels of Pten. Cre-mediated excision eliminates exon 5, which encodes for a region of Pten that is essential for its phosphatase activity.22 Delivery of either HBEGF or Cre alone did not result in a decrease in survival within the experimental time point of 16 weeks in any of the strains evaluated (Figure 2a, 2b and Table S2). Intracranial delivery of viruses containing HBEGF and Cre in Nestin-TVA;Ink4a/Arflox/lox mice resulted in a mean survival of 68.5±21.2 days whereas expression of HBEGF and Cre resulted in a mean survival of 56.1±11.8 days in Nestin-TVA;Ptenlox/lox mice and 28.7±2.3 days in Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox mice. A significant difference in mean survival was observed between each of these cohorts when compared with either the HBEGF or Cre alone cohorts (P<0.05) (Figure 2a and Table S2).
Figure 2. HBEGF cooperates with loss of Ink4a/Arf and Pten to promote glioma formation.
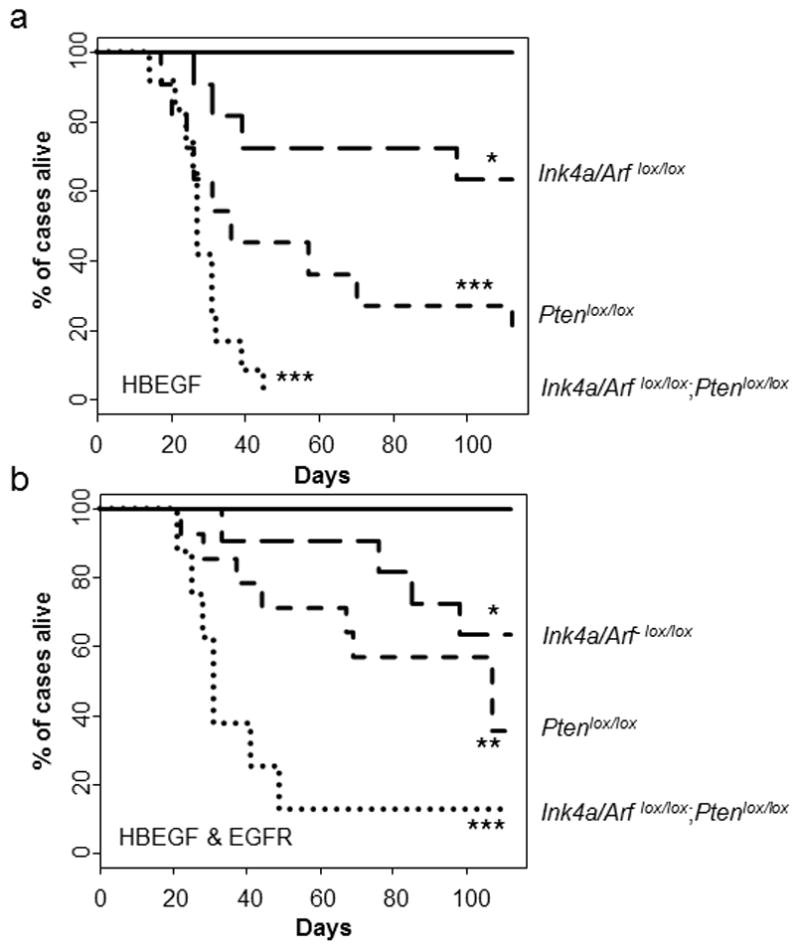
(a) Kaplan-Meier survival analysis of Nestin-TVA;Ink4a/Arflox/lox (n = 11, long dash), Nestin-TVA;Ptenlox/lox (n = 11, short dash), and Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox (n = 12, round dash) mice injected with viruses containing HBEGF and Cre was compared with injection of viruses containing HBEGF alone in Nestin-TVA;Ink4a/Arflox/lox (n = 10, solid line). Statistical significance is shown by the following key: * P < 0.05, ** P < 0.005, *** P < 0.0005. (b) Kaplan-Meier survival analysis of Nestin-TVA;Ink4a/Arflox/lox (n = 12, long dash), Nestin-TVA;Ptenlox/lox (n = 12, short dash), and Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox (n = 12, round dash) mice injected with viruses containing HBEGF, EGFR, and Cre compared with injection of viruses containing Cre alone in all three strains (n = 33 total: n = 11 Nestin-TVA;Ink4a/Arflox/lox, n = 14 Nestin-TVA;Ptenlox/lox, n = 8 Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox, solid line).
Addition of exogenous EGFR does not enhance the effect of HBEGF in vivo
Although a significant difference was not observed in soft-agar colony formation when exogenous EGFR was co-expressed with HBEGF, we evaluated the effect of exogenous EGFR in vivo by delivering RCAS-EGFR along with RCAS-Cre and RCAS-HBEGF in Nestin-TVA;Ink4a/Arflox/lox, Nestin-TVA;Ptenlox/lox, and Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox mice. The median survival for these cohorts was not significantly different than cohorts lacking exogenous EGFR (P>0.1) (Figure 2a, 2b, and Table S2).
HBEGF expressing mouse tumors histologically resemble human malignant gliomas
Brain sections were examined following necropsy and were scored as no tumor, low grade tumor, or high grade tumor based on histological features apparent on H&E sections. Low grade tumors demonstrated single cell infiltration with nuclear atypia including hyperchromasia and pleomorphism while high grade tumors also exhibited mitotic figures, vascular proliferation, and areas of pseudopalisading necrosis (Figure 3a and b). Seventy-five percent of the tumors were high grade when both Ink4a/Arf and Pten were lost while only 36% were high grade with Pten loss alone. No high-grade tumors were observed with loss of Ink4a/Arf alone but 45% of mice developed low grade tumors in this cohort. No tumors were observed in brain sections of mice injected with viruses carrying only HBEGF or Cre (Figure 3c and Table S2). Virally delivered HBEGF was detected in all tumors using immunohistochemistry (IHC) (Figure 3a). Loss of Ink4a/Arf and Pten were also confirmed with immunohistochemistry (Figure S2). All tumors also expressed the astrocyte marker glial fibrillary acidic protein (GFAP) and the neural progenitor cell marker nestin, though less staining was observed in the tumors with loss of Ink4a/Arf alone (Figure 3a). Assessment by IHC of the proliferative marker Ki67 showed that tumors lacking Pten or Pten and Ink4a/Arf were more proliferative than tumors lacking only Ink4a/Arf (P<0.002) (Figures 3b and d). Tumor penetrance correlated with mean survival; cohorts with combined Ink4a/Arf and Pten loss displayed the highest penetrance (100%), followed by those with Pten loss (90%) and Ink4a/Arf loss (45%) (Figures 2a and 3c).
Figure 3. Histological examination of brain sections from injected mice.
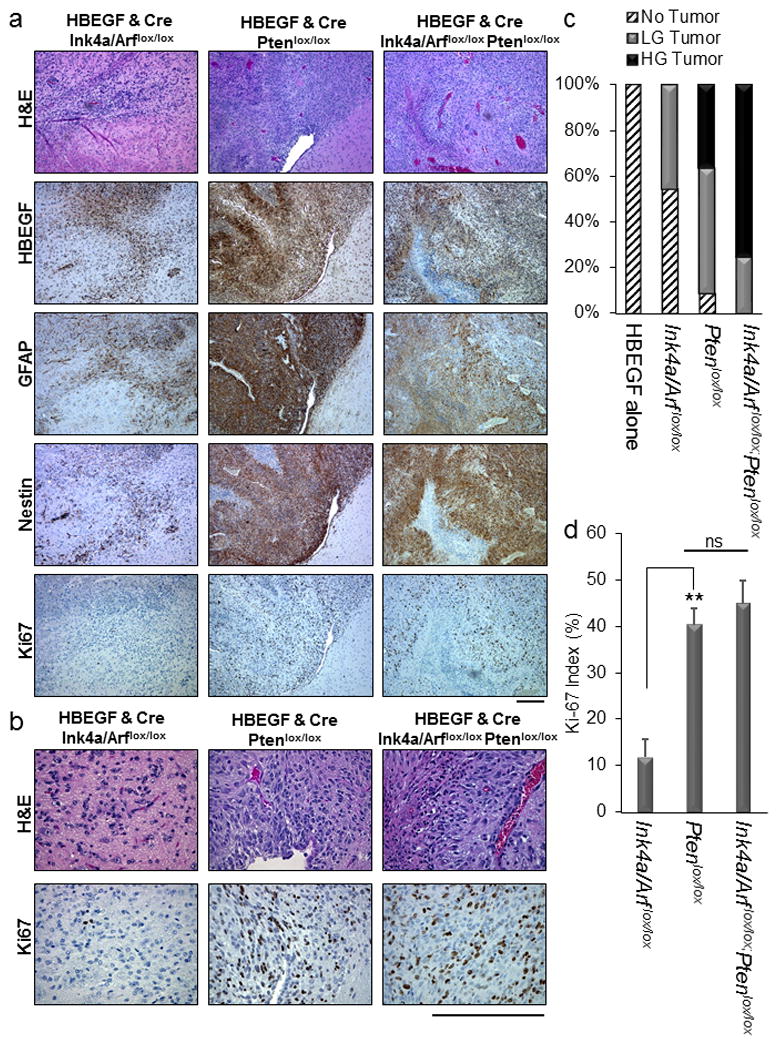
(a) Representative H&E and HBEGF, GFAP, Nestin, and Ki67 immunohistochemistry images of HBEGF-driven tumors from each strain of mice injected with viruses containing HBEGF and Cre. Scale bar represents 300 μm. The age of the mouse shown for HBEGF Ink4a/Arf lox/lox was 39d, for HBEGF Ptenlox/lox was 31d, and for HBEGF Ink4a/Arf lox/lox Ptenlox/lox was 31d. (b) Representative high power H&E and Ki67 images of HBEGF-driven tumors from each strain of mice injected with viruses containing HBEGF and Cre. Scale bar represents 300μm. (c) Percentage of each tumor grade observed in Nestin-TVA;Ink4a/Arf lox/lox, Nestin-TVA;Ptenlox/lox, and Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mice injected with viruses containing HBEGF alone or in combination with Cre. Low grade (LG) tumors were characterized by nuclear atypia and pleomorphism while high grade tumors (HG) were characterized by the presence of necrosis, vascular proliferation, and mitotic figures. (d) Quantification of Ki-67 proliferation index for HBEGF-driven tumors. For each tumor, the average of four high-power fields is shown. Data are represented as mean percentage ± S.E.M. Statistical significance is shown by the following key: ** P < 0.005 with not significant (ns) denoting P > 0.05.
EGFR and AXL mediate HBEGF signaling in mouse astrocytes
To determine if HBEGF signals through other RTKs, we performed an unbiased approach using the R&D Proteome Profiler, a membrane-based antibody array, which detects phosphorylation of 39 mouse receptor tyrosine kinase receptors simultaneously. Isogenic astrocytes from Nestin-TVA;Ink4a/Arf lox/lox mice were infected with either RCAS-Cre or RCAS-Cre and RCAS-HBEGF in combination. The cells were serum starved for 24 hours, lysed, and subjected to the Proteome Profiler immunoassay. As expected, Egfr was phosphorylated in the presence of HBEGF (Figure 4a and S3). Interestingly, the only other RTK that had a significant increase in phosphorylation in the presence of HBEGF was Axl (Figure 4a and S3). To ascertain the functional significance of Axl phosphorylation in this context, we used Clustered Regularly-Interspaced Short Palindromic Repeats (CRISPR)-based gene editing to delete Egfr and/or Axl in mouse astrocytes. Guide RNAs specific for mouse Egfr and Axl were designed and expressed in vectors containing resistance genes for blasticidin and puromycin, respectively. Single-cell clones were selected and expanded. Gene editing and loss of expression was confirmed through high-resolution melt analysis (HRMA), sequencing, and western blot (Figure 4b). Because the guide RNAs are specific for the mouse genome, we performed rescue experiments using the human orthologs of EGFR and AXL. Anchorage-independent growth was assessed by soft agar assay in this context. Cells lacking either Axl, Egfr, or both had significantly fewer colonies than the unmodified parental cells (P<0.0005; Figure 4c). Expression of human AXL or EGFR in the corresponding single knockout cells was able to partially rescue the phenotype (P<0.005; Figure 4c). In the single knockout cells, endogenous levels of Axl or Egfr are still present, respectively. Therefore, we also performed rescue experiments in cells lacking both Axl and Egfr. Interestingly, EGFR was able to rescue the double knockout cells to the same extent as the corresponding single knockout cells. However, Axl was not able to rescue the double knockout cells to the same extent as the corresponding single knockout cells suggesting that Egfr is required for activation of Axl in this context (P<0.0005; Figure 4c). Expression of both AXL and EGFR completely rescued the double knockout cells in the anchorage-independent growth assay (Figure 4c).
Figure 4. Both Egfr and Axl mediate HBEGF signaling.
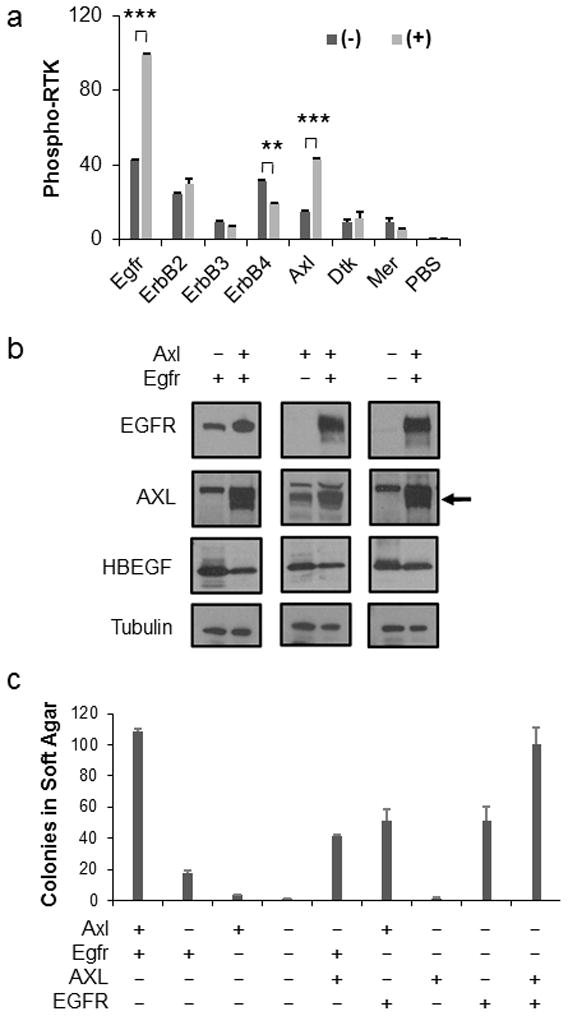
(a) Phosphorylation of multiple RTKs was simultaneously assayed in isogenic astrocytes in the presence (+) or absence (−) of HBEGF. A subset of RTKs analyzed representing the ErbB and TAM family members are shown. Densitometry of the array was used to determine the amount of RTK phosphorylation. Data are represented as mean ± S.E.M. Statistical significance is shown by the following key: ** P < 0.005, *** P < 0.0005. (b) Endogenous Axl and/or Egfr expression was eliminated using CRISPR-mediated gene editing (−) in Nestin-TVA;Ink4a/Arf lox/lox astrocytes. Human AXL and EGFR were expressed following RCAS-mediated infection for rescue (+). The arrow represents the AXL band. Blots were probed with tubulin antibody as a loading control. (c) Astrocytes from (b) were assayed for soft-agar colony formation to determine the functional significance of Axl and Egfr loss. Data are represented as mean±S.E.M. Cells lacking either Axl, Egfr, or both had significantly fewer colonies than the unmodified parental cells (P < 0.0005). Rescue with human AXL or EGFR in the corresponding single knockout cells was significant (P < 0.005). There was no significant difference between EGFR rescue in the single versus double knockout cells; however, a significant difference was observed between AXL rescue in the single versus double knockout cells (P < 0.0005). Expression of both AXL and EGFR completely rescued the double knockout cells as no significant difference was observed between the double rescue and the control parental cells.
HBEGF-driven mouse GBM most closely resembles the classical subtype of human GBM
Human GBM has been classified into four different subtypes: proneural, classical, mesenchymal, and neuronal.3 High grade HBEGF-driven tumors in mice resemble human GBM histologically (Figure 3a and 5a), but it is unclear which subtype, if any, they model. To determine if HBEGF-driven GBM resembled a particular subtype, we generated high-grade gliomas in Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mice by delivery of RCAS-Cre and RCAS-HBEGF. In addition, in the same mouse strain, gliomas were generated with RCAS-Cre in combination with RCAS-PDGFA or RCAS-BRAFV600E, other drivers previously found to form high-grade gliomas (Figure 5a).19,23 Previously, gliomas driven by PDGFA in the context of Ink4a/Arf and Pten loss were shown to resemble the proneural subtype using microarray bead chip technology.23 RNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissue that had been histologically confirmed by a board-certified Neuropathologist (J.A.S.). Three tumors from each cohort along with brain tissue from six control Nestin-TVA-negative Ink4a/Arf lox/lox;Ptenlox/lox mice were analyzed by RNA sequencing from a Ribo-Zero treated library (Figure 5a and S4a). The transcript coverage and exonic mapping rate were typical for FFPE tissue (Figure S3b and c). The sample cross correlation heat map of the Pearson correlations shows that each of the tumor replicates clustered with each other and the controls clustered separately from the tumors (Figure S4a). The transcriptome of each cohort was compared to the TCGA GBM subtype classifier genes after converting those genes to their mouse orthologs. The highest correlation for HBEGF was with the classical subtype (r=0.14), for PDGFA was the proneural subtype (r=0.30), for BRAF was the mesenchymal subtype (r=0.47) and for the control was the neural subtype (r=0.26) as shown on the heat map (Figure 5b) and in Table S3. We also performed Gene Set Enrichment Analysis (GSEA) to compare the expression pattern of these tumors with the datasets from the four GBM subgroups as previously described. 24 Based on GSEA, the transcriptome profile of the HBEGF driven tumors was a significant match to the signature of the Classical subtype (Figure S5a) and was in agreement with the Pearson correlations (Figure 5b). As expected, signature gene sets associated with mesenchymal and proneural GBM were significantly enriched in the gene expression profile of BRAF driven and PDGFA driven tumors, respectively (Figure S5b and c).
Figure 5. Molecular comparison between mouse and human GBM.
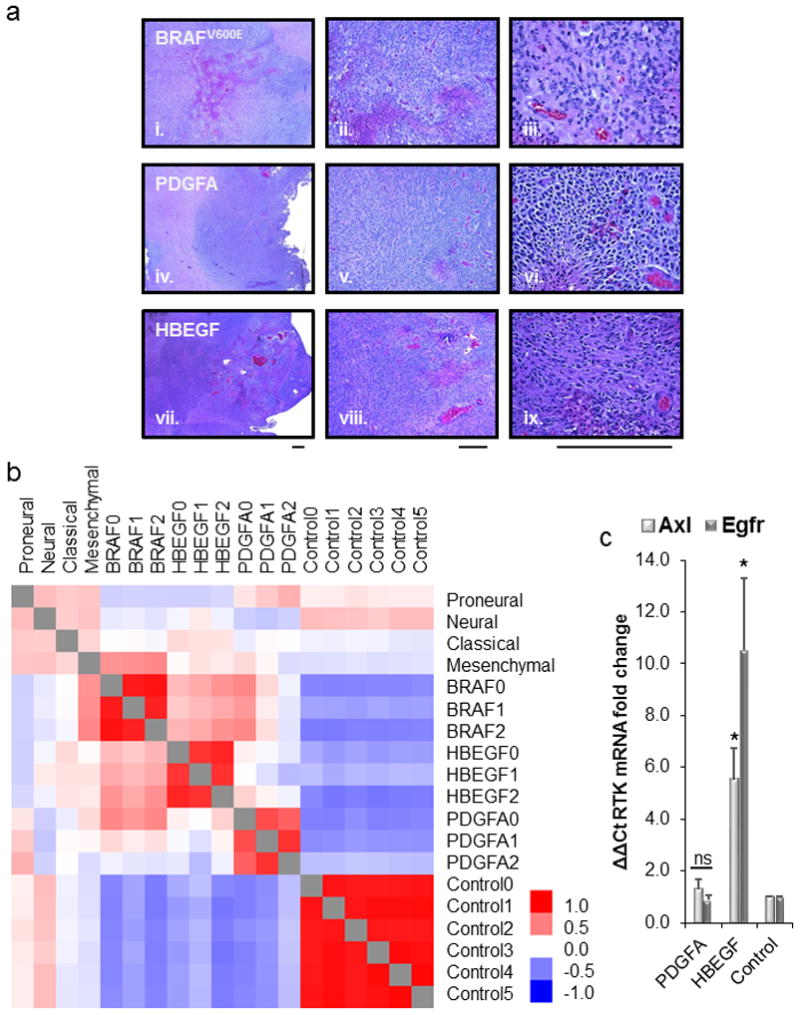
(a) Representative H&E sections for high-grade gliomas derived in the Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mouse model using RCAS-Cre and three different drivers: BRAFV600E(i., ii., iii.), PDGFA (iv., v., vi.), and HBEGF (vii., viii., ix.) showing 4x, 10x and 40x magnification, respectively. Scale bar represents 300μm. (b) Heatmap showing Pearson correlations between the TCGA subtypes (proneural, neural, classical, mesenchymal) and high-grade mouse gliomas driven by RCAS-BRAFV600E, PDGFA, and HBEGF in the absence of Ink4a/Arf and Pten. (c) Real-time PCR analysis of Axl and Egfr mRNA levels in either HBEGF or PDGFA tumors. Rn18s was used as an endogenous reference. Relative quantification was carried out using the comparative Ct method with TVA negative injected controls. Statistical significance is compared with control and is shown by the following key: * P < 0.05 with not significant (ns) denoting P > 0.05.
In both our in vitro and in vivo assays evaluating the oncogenic potential of HBEGF, we did not observe a significant difference with combined overexpression of EGFR (Figure 1b and 2b). This suggests that endogenous EGFR is sufficient when HEBGF is overexpressed or that an alternative signaling pathway could be possible. The Proteome Profiler and CRISPR knockout functional studies suggest a role for Axl in HBEGF signaling (Figure 4a and c). Differential expression analysis in the tumors from RNA sequencing showed specific upregulation of Egfr and Axl transcripts in HBEGF tumors (Figure S6). In PDGFA-driven tumors, there was specific upregulation of Pdgfra (Figure S6). These results were confirmed with real-time PCR for Axl and Egfr on the same tumor samples (Figure 5c). HBEGF-driven tumors had a 10-fold increase in Egfr transcript and 6-fold increase in Axl transcript compared to healthy controls and PDGFA-driven tumors (P<0.03). HBEGF induced expression of EGFR likely explains why endogenous EGFR was sufficient both in vitro and in vivo.
Suppression of HBEGF expression results in tumor regression and increased survival in vivo
In vitro neutralizing antibodies that recognize HBEGF decrease the proliferation of cultured glioma cells suggesting that HBEGF is important for tumor cell maintenance.5,7,25 Translating these findings in vivo is complicated by pharmacological limitations in drug kinetics and perfusion across the blood brain barrier. To bypass these limitations, we utilized a genetic approach to determine whether HBEGF is required for tumor maintenance in vivo. We have previously described the generation of a viral vector that allows expression of inserted genes to be regulated post-delivery using the tet-regulated system.26 A tet-responsive element (TRE) was inserted immediately downstream of the envelope gene in the replication-competent avian retroviral vector RCANBP(A) such that expression of any gene inserted downstream of the TRE was driven from this promoter and not the viral LTR. Expression from the TRE requires the presence of a tetracycline transcriptional activator (tTA) such as tet-off or a reverse tTA (rtTA) such as tet-on. In the context of tet-on, the tet-responsive gene is expressed only in the presence of doxycycline (Dox); in the context of tet-off, the tet-responsive gene is repressed in the presence of Dox.27 In the context of tet-off expression in vitro, we observed HBEGF expression that was effectively suppressed by the administration of Dox (Figure 6a).
Figure 6. Suppression of HBEGF expression in vitro and in vivo.
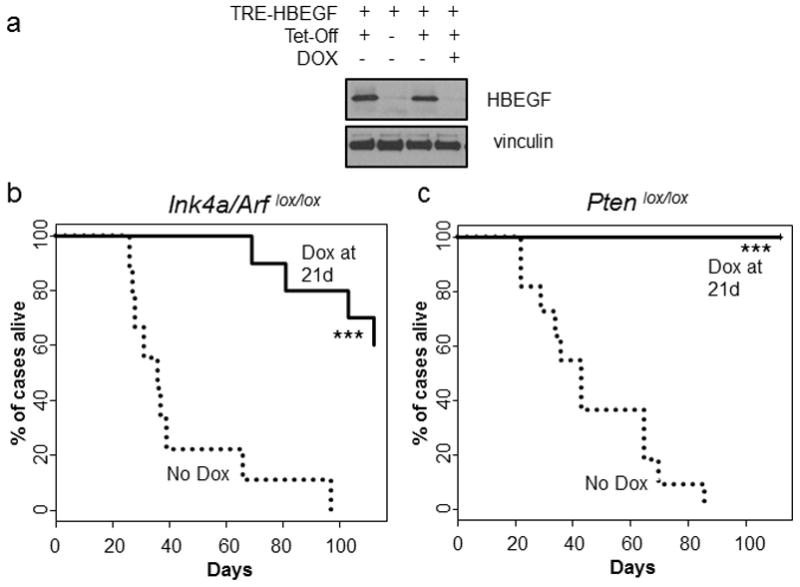
(a) Western blot of isogenic astrocytes infected with TRE-HBEGF and/or tet-off with or without 2 μg/ml of doxycycline for 72 h. (b) Kaplan-Meier survival analysis of tumor-bearing Nestin-TVA;Ink4a/Arf lox/lox injected with RCAN-TRE-HBEGF, RCAS-tet-off, and RCAS-Cre that were not treated (n = 9, round dash) and treated starting at 21 days of age (n = 21, solid line). Statistical significance is shown by the following key: * P < 0.05, ** P < 0.005, *** P < 0.0005. (c) Same as in B but for Nestin-TVA;Ptenlox/lox injected with RCAN-TRE-HBEGF, RCAS-tet-off, and RCAS-Cre (n = 11, round dash) (n=21, solid line). Statistical significance is shown by the following key: * P < 0.05, ** P < 0.005, *** P < 0.0005. For treatment studies, diet containing 625 mg/kg of doxycycline was used (Envigo TD.08434).
Tumors were generated in Nestin-TVA;Ink4a/Arf lox/lox and Nestin-TVA;Ptenlox/lox mice with retroviruses containing TRE-HBEGF, tet-off, and Cre and confirmed with MRI imaging at 21 days of age. In the Nestin-TVA;Ink4a/Arf lox/lox mice, tumor-bearing mice were split into two cohorts, one receiving standard feed (n=9) and one receiving Dox feed (n=10). While the mean survival in the untreated mice was 43.0±7.9 days, the mean survival in the mice treated was 103.7±4.9 days (P<0.0001) (Figure 6b). In the Nestin-TVA;Ptenlox/lox mice, tumor-bearing mice were divided into two cohorts, one receiving standard feed (n=11) and one receiving Dox feed (n=15). While the mean survival in the untreated mice was 46.8±6.5 days, all Dox treated mice survived to the experimental endpoint of 112 days (P<0.0001) (Figure 6c). We have previously demonstrated that Dox has no innate tumor suppressive properties; therefore, tumor regression is due to inhibition of HBEGF expression in this context.28
Intracranial injection of TRE-HBEGF, tet-off, and Cre containing retroviruses into newborn Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mice led to the formation of high-grade tumors in all injected mice (Figure 7a). To evaluate the kinetics of virally delivered HBEGF expression and activity following Dox treatment, we treated mice with Dox for 3 days. Expression of HBEGF as detected by IHC was visible without Dox treatment; however, following 3 days of Dox treatment very little HBEGF expression was visible by IHC (Figure 7a). A significant difference was observed in the mean number of cells staining positively for the proliferation marker Ki67 between the mice that received standard feed and the mice that received Dox feed. While 41.4±4.3 Ki67 positive cells were detected per high powered field (HPF) in the untreated tumor samples, 5.7±1.2 Ki67 positive cells were detected per HPF in the samples treated with Dox for 3 days (P=0.002) (Figure 7b).
Figure 7. Suppression of HBEGF expression prolongs survival in vivo.
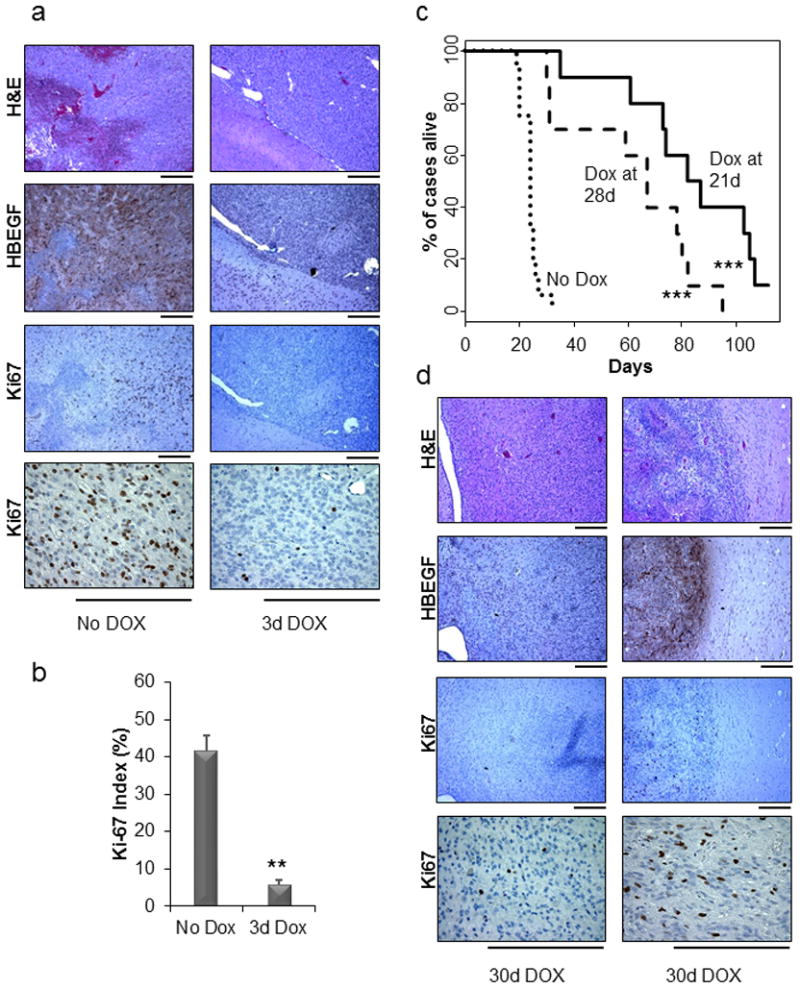
(a) Immunohistochemical analysis of HBEGF and Ki67 in brain tissue isolated from Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mice injected with RCAN-TRE-HBEGF, RCAS-tet-off, and RCAS-Cre that were either untreated or treated with doxycycline for 3 days after being weaned at 21 days of age. The two samples are from the same timepoint of 3 days. Scale bar represents 300 μm. (b) The Ki67 staining was quantified using two representative tumors for each cohort. For each tumor, the average of four high-power fields is shown. Data are represented as mean ± S.E.M (** P < 0.005). (c) Kaplan-Meier survival analysis of Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox injected with RCAN-TRE-HBEGF, RCAS-tet-off, and RCAS-Cre that were not treated (n = 16, round dash), treated starting at 21 days of age (n = 10, solid line), or treated starting at 28 days of age (n = 10, short dash). The Dox treated cohorts were compared with the untreated cohort (*** P < 0.0005). (d) Immunohistochemical analysis of regressed tumors using antibodies against HBEGF and Ki67 with corresponding H&E. Scale bar represents 300 μm. Both regressed tumors are from two different mice that were treated for 30 days with doxycycline.
MRI was performed at 21 days of age to identify tumor-bearing mice and to monitor tumor growth or regression at multiple time points throughout the study. One cohort of tumor-bearing mice received standard feed (n=16) and the other cohort received Dox feed continuously starting at 21 days of age (n=10). Survival rates were compared between untreated mice and mice given Dox to determine whether the suppression of HBEGF expression increased survival. While the mean survival in the untreated mice was 23.9±0.8 days, the mean survival in the mice treated starting at 21 days was 83.9±7.7 days (P<0.0001), with one mouse still alive at the experimental endpoint of 16 weeks (Figure 7c). This mouse remained tumor-free by MRI at 20 weeks of age (Figure S7a). To assess tumor maintenance in more advanced tumors, another cohort received Dox feed starting at 28 days of age (n=10) (Figure 7c). The mean survival in the mice treated starting at 28 days was 62.0±7.5 days, which was significantly different from the untreated mice (P<0.0001). However, the mice treated at 28 days had significantly reduced mean survival compared to the mice treated at 21 days (P=0.02), demonstrating that earlier treatment increases survival.
Recurrent tumors develop in the absence of HBEGF expression in the context of Ink4a/Arf and Pten loss
Tumors recurred in 19/20 responding Nestin-TVA;Ink4a/Arf lox/lox;Ptenlox/lox mice while on Dox treatment. The MRI data from a representative mouse that developed recurrent disease while on Dox treatment is shown in Figure S7b. A small tumor is visible pre-Dox treatment but no tumor was visible at 28, 42, or 64 days; however, tumor recurrence was visible starting at 84 days and a large recurrent tumor was detected at 104 days. Brain tissue was examined and H&E analysis confirmed GBM in this mouse. Of evaluable mice with recurrent tumors, 50% expressed HBEGF and 50% recurred in the absence of HBEGF expression (Figure 7d and S8). Expression of PDGFRA, AXL, EGFR, and MET was also evaluated by IHC in these tumors (Figure S9). Interestingly, EGFR expression was not detected in any of the recurrent tumors. In contrast, PDGFRA was detected in 9/10 recurrent tumors, AXL was detected in 7/10 recurrent tumors and MET was detected in 3/10 recurrent tumors. Future studies will evaluate which of these RTKs, if any, mediate resistance in this context.
Discussion
Using an established mouse model of glioma,19 we assessed the sufficiency of HBEGF in gliomagenesis. While not sufficient on its own, HBEGF cooperates with Ink4a/Arf or Pten loss to promote gliomagenesis. To our knowledge, this is the first report showing that HBEGF can lead to malignant glioma development in this context. Comparison of these HBEGF tumors to the human subtype signatures show that while they positively correlated with the neural, pronerual, and classical signatures, the strongest correlation was with the classical subtype.3 We also show that previously published mouse models for PDGFA and BRAFV600E resemble the proneural and mesenchymal subtypes, respectively.3,19,23
We have observed that in astrocytes in vitro, HBEGF activates not only Egfr but also Axl, a member of the TAM family of RTKs. AXL has been shown to be upregulated in human glioma cell lines and tissue, but activation of AXL in the presence of HBEGF has not previously been reported in gliomas or other cancers.17,18 Although unreported in the context of HBEGF, a role for AXL in mediating EGFR signaling has been cited as a mechanism of resistance against targeted EGFR therapies in lung and breast cancer.29,30 In breast cancer cells, the two receptors were found to physically interact. In addition, while activation of EGFR through the addition of EGF transactivated AXL in this context, the addition of an AXL activating antibody did not phosphorylate EGFR.31 It is therefore possible that HBEGF activates AXL via transactivation from an activated EGFR receptor.
While determination of the initial events in the genesis of a glioma are biologically important, it is perhaps more important clinically to ascertain the conditions for tumor maintenance to help guide clinical intervention. As an initial step toward developing targeted therapies, we used a tetracycline-regulated system to control HBEGF expression to assess the necessity of HBEGF in sustaining tumor growth. We demonstrated increased mean survival in all cohorts of mice after inhibition of HBEGF. While half of the recurrent tumors in the Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox mice expressed HBEGF, half recurred in the absence of HBEGF expression. While EGFR expression was not detected in these recurrent tumors, several other RTKs were expressed. Pathway crosstalk and signaling redundancies have been implicated in mediated drug resistance in several cancers, including gliomas. Future studies will evaluate the mechanism(s) of resistance in these tumors. Our study suggests that HBEGF promotes the formation of gliomas, is necessary for tumor maintenance, and therefore may be a novel therapeutic target.
Materials and methods
Mice and Genotyping
Nestin-TVA;Ink4a/Arflox/lox;Ptenlox/lox mice were generated from pre-existing strains. PCR for the TVA transgene and for the Ink4a/Arflox, wild-type Ink4a/Arf, Ptenlox, and wild-type Pten alleles was performed as previously described.32 All experiments were IACUC approved before experimentation.
Gene editing
Astrocytes were established as previously described.33 To disrupt Egfr and Axl via CRISPR-based gene editing, astrocytes were infected with VSV-G-pseudotyped plenti-CRISPRv2-Egfr (target: CCTCCATTAGACCATCCAGG) containing blasticidin resistance and/or plenti-CRISPRv2-Axl (target: CAGGTGCCAGAGGACTCACG) containing puromycin resistance.34 Clones were selected using 2.5 μg/ml puromycin and/or 80 μg/ml blasticidin, screened with high resolution melt analysis (HRMA), and confirmed with sanger sequencing followed by Western blot analysis.35
Viral constructs
The RCAS-CRE and RCAS-TETOFF avian retroviral vectors and RCAS-BRAFV600E have been described previously.19,20 Cloning details for RCAS-HBEGF and RCAN-TRE-HBEGF are available upon request. Virus was propagated and in vitro infections were done as previously described.33
Soft Agar assay
To assess anchorage-independent growth, cells were assayed in triplicate as described previously and visualized with 1μg/μl nitroblue tetrazolium stain (Invitrogen) added 200 μl overnight.19,33
Western blotting
Protein quantification and immunostaining were done as previously described.33 Antibodies used are available upon request.
R&D Proteome Profiler RTK-Array
Astrocytes were serum-starved overnight and then analyzed using the Proteome Profiler Mouse Phospho-RTK Array Kit (R&D) following the manufacturer’s instructions.
In vivo infection
Newborn mice were injected intracranially 2 mm ventral from bregma as described previously.19,36
Histological analysis
Brain tissue from injected mice was fixed, embedded, stained, and imaged as previously described.33
Bioinformatics and Genomics
TCGA glioblastoma data were accessed through cBioPortal.37,38 GBM classifier genes were converted to mouse orthologues using Biomart.3,23 RNA was isolated using the Recoverall Kit (Ambion), prepped with TruSeq Stranded Total RNA using Ribo-Zero Gold, and sequenced with HiSeq 50-Cycle Single-Read Sequencingv4 (Illumina). Reads were aligned to the mouse genome (mm10) and differential gene expression was determined with the open source USeq/DESeq2 package. Genes were selected using two thresholds, an FDR of <10% and absolute log2 ratio > 1. Tumor and control variance normalized expression data (rlog values) was converted to median centered log2 fold change and compared to the TCGA classifier genes using pairwise Pearson correlations. GSEA (v2.2.0) was used to analyze the enrichment of signature gene sets from different GBM subtypes as described.24
Immunohistochemistry
Tissue sections were immunostained as previously described.19,33 Antibodies used are available upon request.
Magnetic Resonance Imaging
Mice were anesthetized and imaged as previously described. 20,28
Statistical Analysis
Western blot and RTK arrays were performed in triplicate and duplicate, respectively, and density was analyzed using Image J. Survival data (n>9) was analyzed using a log-rank test of the Kaplan Meier estimate of survival. To compare means, two-tailed Student’s t test was used. P values below 0.05 were considered significant.
Supplementary Material
Acknowledgments
Financial Support: NIH R01NS073870 for S.L.H. and NIH F30CA203096 for C.H.S.
We thank the members of the VanBrocklin, McMahon, and Holmen labs as well as E. Holland, R. DePinho, and M. Bosenberg for providing mouse strains, reagents, and advice. We thank the Huntsman Cancer Institute (HCI) Vivarium staff for assistance. We thank Tim Parnell for his bioinformatics expertise. We acknowledge the use of the Mutation Generation and Detection Core, the DNA Synthesis Core, the DNA Sequencing Core, and the Small Animal Imaging Core supported by P30CA042014 awarded to HCI from the National Cancer Institute (NCI). We also acknowledge use of the HCI Shared Resources for high-throughput genomics and bioinformatics analysis, glass wash, and research histology. This work was supported by the National Institute of Neurological Disorders and Stroke [R01NS073870] and the NCI [F30CA203096].
Footnotes
Conflict of Interest: The authors disclose no potential conflicts of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishima K, Higashiyama S, Asai A, Yamaoka K, Nagashima Y, Taniguchi N, et al. Heparin-binding epidermal growth factor-like growth factor stimulates mitogenic signaling and is highly expressed in human malignant gliomas. Acta Neuropathol. 1998;96:322–8. doi: 10.1007/s004010050901. [DOI] [PubMed] [Google Scholar]
- 6.Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, et al. Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 2011;71:7198–206. doi: 10.1158/0008-5472.CAN-11-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramnarain DB, Park S, Lee DY, Hatanpaa KJ, Scoggin SO, Otu H, et al. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006;66:867–74. doi: 10.1158/0008-5472.CAN-05-2753. [DOI] [PubMed] [Google Scholar]
- 8.Ray KC, Moss ME, Franklin JL, Weaver CJ, Higginbotham JN, Song Y, et al. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene. 2013;33:1–9. doi: 10.1038/onc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yotsumoto F, Oki E, Tokunaga E, Maehara Y, Kuroki M, Miyamoto S. HB-EGF orchestrates the complex signals involved in triple-negative and trastuzumab-resistant breast cancer. Int J Cancer. 2010;127:2707–2717. doi: 10.1002/ijc.25472. [DOI] [PubMed] [Google Scholar]
- 10.He C, Lv X, Hua G, Lele SM, Remmenga S, Dong J, et al. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression. Oncogene. 2015:1–15. doi: 10.1038/onc.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornblum HI, Zurcher SD, Werb Z, Derynck R, Seroogy KB. Multiple trophic actions of heparin-binding epidermal growth factor (HB-EGF) in the central nervous system. Eur J Neurosci. 1999;11:3236–3246. doi: 10.1046/j.1460-9568.1999.00744.x. [DOI] [PubMed] [Google Scholar]
- 12.Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, et al. Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell. 2003;2:175–83. doi: 10.1046/j.1474-9728.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- 13.Puschmann TB, Zandén C, Lebkuechner I, Philippot C, De Pablo Y, Liu J, et al. HB-EGF affects astrocyte morphology, proliferation, differentiation, and the expression of intermediate filament proteins. J Neurochem. 2014;128:878–889. doi: 10.1111/jnc.12519. [DOI] [PubMed] [Google Scholar]
- 14.Ongusaha PP, Kwak JC, Zwible AJ, Macip S, Higashiyama S, Taniguchi N, et al. HB-EGF is a potent inducer of tumor growth and angiogenesis. Cancer Res. 2004;64:5283–90. doi: 10.1158/0008-5472.CAN-04-0925. [DOI] [PubMed] [Google Scholar]
- 15.Fu Sl, Bottoli I, Goller M, Vogt PK. Heparin-binding epidermal growth factor-like growth factor, a v-Jun target gene, induces oncogenic transformation. Proc Natl Acad Sci U S A. 1999;96:5716–21. doi: 10.1073/pnas.96.10.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Chakraborty S, Yang C-R, Hatanpaa KJ, Cipher DJ, Puliyappadamba VT, et al. An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII. Oncogene. 2014;33:4253–64. doi: 10.1038/onc.2013.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vajkoczy P, Knyazev P, Kunkel A, Capelle H, Behrndt S, von Tengg-Kobligk H, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006;103:5799–804. doi: 10.1073/pnas.0510923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutterer M, Knyazev P, Abate A, Reschke M, Maier H, Stefanova N, et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:130–138. doi: 10.1158/1078-0432.CCR-07-0862. [DOI] [PubMed] [Google Scholar]
- 19.Robinson JP, VanBrocklin MW, Guilbeault aR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–44. doi: 10.1038/onc.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Brocklin MW, Robinson JP, Lastwika KJ, McKinney AJ, Gach HM, Holmen SL. Ink4a/Arf loss promotes tumor recurrence following Ras inhibition. Neuro Oncol. 2012;14:34–42. doi: 10.1093/neuonc/nor184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson Da, Horner J, et al. Activated Kras and Ink4a / Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen A-J, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozawa T, Riester M, Cheng YK, Huse J, Squatrito M, Helmy K, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu F, Chen Y, Zhao C, Wang H, He D, Xu L, et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell. 2016;29:669–683. doi: 10.1016/j.ccell.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato S, Drake AW, Tsuji I, Fan J. A potent anti-HB-EGF monoclonal antibody inhibits cancer cell proliferation and multiple angiogenic activities of HB-EGF. PLoS One. 2012;7:e51964. doi: 10.1371/journal.pone.0051964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005;65:8250–5. doi: 10.1158/0008-5472.CAN-05-1173. [DOI] [PubMed] [Google Scholar]
- 27.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lübbert H, et al. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:10933–8. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JP, Vanbrocklin MW, McKinney AJ, Gach HM, Holmen SL. Akt signaling is required for glioblastoma maintenance in vivo. Am J Cancer Res. 2011;1:155–167. [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 31.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, et al. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep. 2015;13:898–905. doi: 10.1016/j.celrep.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin CH, Grossmann AH, Holmen SL, Robinson JP. The BRAF kinase domain promotes the development of gliomas in vivo. Genes Cancer. 2015;6:9–18. doi: 10.18632/genesandcancer.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu R, Wallace J, Dahlem TJ, Grunwald DJ, O’Connell RM. Targeting human microRNA genes using engineered Tal-effector nucleases (TALENs) PLoS One. 2013;8:e63074. doi: 10.1371/journal.pone.0063074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson JP, Vanbrocklin MW, Lastwika KJ, McKinney aJ, Brandner S, Holmen SL. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene. 2011;30:1341–50. doi: 10.1038/onc.2010.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.