

Abstract
Dysregulation of the oncogenic transcription factor MYC induces B cell transformation and is a driver for B cell non-Hodgkin lymphoma (B-NHL). MYC overexpression in B-NHL is associated with more aggressive phenotypes and poor prognosis. Although genomic studies suggest a link between MYC overexpression and B cell receptor (BCR) signaling molecules in B-NHL, signaling pathways essential to Myc-mediated B-cell transformation have not been fully elucidated. We utilized intracellular phospho-flow cytometry to investigate the relationship between Myc and BCR signaling in pre-malignant B cells. Utilizing the Eμ-myc mouse model, where Myc is overexpressed specifically in B cells, both basal and stimulated BCR signaling were increased in precancerous B lymphocytes from Eμ-myc mice compared to wild-type littermates. B cells overexpressing Myc displayed constitutively higher levels of activated CD79α, Btk, Plcγ2, and Erk1/2. Notably, Myc overexpressing B cells maintained elevated BCR signaling despite treatment with ibrutinib, a Bruton’s tyrosine kinase inhibitor. Furthermore, PI3K/Akt pathway signaling was also increased in Eμ-myc B cells, and this increase was partially suppressed with ibrutinib. Additionally, experiments with Btk-null B cells revealed off-target effects of ibrutinib on BCR signaling. Our data show that in pre-malignant B cells, Myc overexpression is sufficient to activate BCR and PI3K/Akt signaling pathways and further enhances signaling following BCR ligation. Therefore, our results indicate precancerous B cells have already acquired enhanced survival and growth capabilities prior to transformation, and that elevated MYC levels confer resistance to pharmacologic inhibitors of BCR signaling, which has significant implications for B-NHL treatment.
Keywords: Myc, B cell receptor signaling, Btk, ibrutinib
Introduction
Non-Hodgkin lymphoma (NHL) represents a spectrum of morphologically, phenotypically, and genetically distinct lymphomas with ~85% of B lymphocyte lineage. Overexpression of the oncogenic transcription factor MYC is associated with an aggressive phenotype and poor overall survival, regardless of the B-NHL subtype (1–4). Dysregulated Myc expression in primary murine B cells drives cellular transformation and lymphomagenesis and is necessary for both human and murine lymphoma maintenance (5–7).
Signaling through the B cell receptor (BCR) in the absence of antigenic stimulation is important for normal B cell differentiation and survival (8, 9). Additionally, ligation of the BCR induces a cascade of phosphorylation events and activation of multiple kinases, including the spleen tyrosine kinase (SYK) and Bruton’s tyrosine kinase (BTK), leading to MAPK and NF-κB activation and a gene expression signature enriched for MYC-induced genes (10–12). Independently of BTK, SYK can activate an alternative pro-survival pathway through PI3K/AKT (13). Aberrant or chronic BCR signaling is commonly observed in B-NHL and contributes to lymphoma growth and survival, but the mechanisms for this remain incompletely understood (14–16). Targeting the BCR pathway by silencing signaling proteins has deleterious effects on lymphoma cells with chronic BCR activation (14), and pharmacologic inhibition of BTK by ibrutinib has shown encouraging results in the treatment of relapsed/refractory B-NHL (17–19).
Recent studies in human lymphoma cell lines identified components of the BCR pathway as possible MYC transcriptional targets (20). Additionally, the MYC-regulated miRNA cistron miR-17~92 was reported to influence BCR signaling in lymphoma cell lines (21). Although these studies suggest a link between MYC overexpression and BCR signaling in lymphoma, the relationship between MYC and BCR signaling in non-transformed B lymphocytes was unknown. Therefore, we investigated BCR signaling in pre-malignant B cells. We determined the levels of activated forms of multiple components of the BCR signaling pathway were increased both at rest and following ligation of the BCR in primary B cells overexpressing Myc. Myc overexpressing B cells were less sensitive to Btk inhibition by ibrutinib, and off-target effects of ibrutinib were also identified. These studies highlight the relationship between Myc overexpression and pro-survival signaling, identify a means by which Myc drives lymphomagenesis, and provide insight into treatment strategies for patients with lymphoma expressing high levels of MYC protein.
Results
Myc overexpression increases basal and stimulated BCR signaling in primary B cells
To investigate the influence of Myc on BCR signaling prior to cellular transformation, spleens were harvested from Eμ-myc transgenic mice, which overexpress Myc specifically in B cells ((5) and Supplemental Fig S1), before the development of lymphoma. Spleens from wild-type littermates served as controls. Basal activity of BCR signaling proteins was interrogated in IgM+, CD19+ splenic B cells by intracellular phospho-flow cytometry (schematic Fig 1A). Unstimulated B lymphocytes from Eμ-myc mice demonstrated significantly increased levels of phospho-Btk (36% elevated, p=0.0179), phospho-Plcγ2 (48% and 40% elevated at Y759 and Y1217, p=0.0013 and 0.0050, respectively), and phospho-Erk1/2 (56% elevated, p=0.0007) compared to wild-type B cells (Fig 1B). Levels of phospho-CD79α and phospho-Syk were also increased in unstimulated Eμ-myc splenic B cells (28% and 9% elevated, respectively; Fig 1B), but differences did not reach statistical significance (p=0.07 and p=0.12, respectively). Therefore, Myc overexpression alone increased basal signaling of several proteins in the BCR pathway in primary, non-transformed B cells.
Figure 1. Myc overexpressing non-transformed B cells have increased BCR signaling.
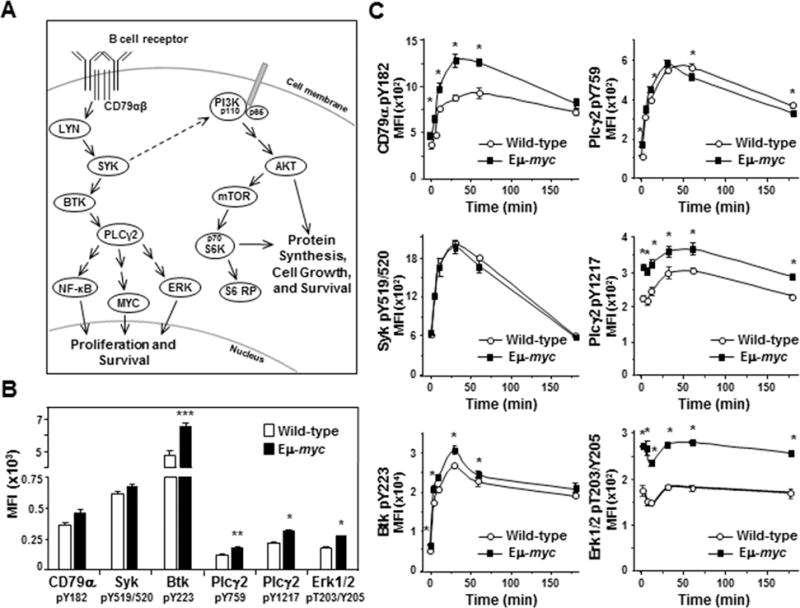
A) Schematic of the BCR signaling cascade. The BCR and its coreceptor CD79αβ are embedded in the plasma membrane. Following ligation of the BCR, the coreceptor becomes phosphorylated and initiates signaling cascades that result in phosphorylation of multiple kinases and phospholipase C. This leads to activation of proteins such as NF-κB, MYC, ERK, and S6 ribosomal protein and ultimately to cellular proliferation and/or survival. B, C) Levels of activated/phosphorylated proteins in the BCR signaling pathway were determined by intracellular phospho-flow cytometry in splenic B cells from Eμ-myc mice and wild-type littermates either unstimulated (not IgM ligated) (B) or at intervals following IgM ligation (C). Each protein was measured in at least three independent experiments with 2–4 mice of each genotype per experiment. Mean fluorescence intensities (MFI) from a representative experiment are shown. Error bars indicate SEM; p-values compare the levels of phospho-protein in Eμ-myc B cells to the levels in wild-type littermates. In B, *p<0.0015, **p≤0.005, and ***p=0.0179; in C, *p≤0.0115 CD79α pY182, *p≤0.0385 Plcγ2 pY759 and pY1217, *p≤0.0496 Btk pY223, and *p≤0.0013 Erk pT203/Y205.
Ligation of the BCR activates signaling of the pathway above basal levels (22). To determine whether Myc expression affects activated BCR signaling, we ligated the BCR with anti-IgM F(ab’)2. At intervals after BCR ligation, proteins in the BCR pathway were evaluated by intracellular phospho-flow cytometry. We detected robust activation of proteins that are activated early following IgM ligation (e.g., CD79α, Syk, Btk, and Plcγ2) in both Eμ-myc and wild-type splenic B cells (Fig 1C). Although the activation curves were similar in Eμ-myc and wild-type cells, with 2–4 fold increases in each phospho-protein following ligation of the BCR, there were notable differences. Specifically, although basal levels of activated CD79α were statistically equivalent in Eμ-myc and wild-type B cells, there was a sharp increase in phospho-CD79α in Eμ-myc cells that significantly exceeded that of wild-type cells at 5 (p=0.0041), 10 (p=0.0115), 30 (p=0.0065), and 60 minutes (p=0.0055) following BCR ligation (upper left, Fig 1C). Phospho-CD79α peaked within 30 minutes in Eμ-myc B cells at a level 2.8-fold above the baseline. In contrast, phospho-CD79α peaked later in wild-type B cells, achieving a level 2.6-fold above baseline 60 minutes after BCR ligation (upper left, Fig 1C). Additionally, although activation of Syk in Eμ-myc B cells paralleled that of wild-type B cells (middle left, Fig 1C), the levels of activated downstream proteins phospho-Btk (bottom left, Fig 1C) and phospho-Plcγ2 (Y1217) (middle right, Fig 1C) started and remained significantly higher in Eμ-myc B cells over 60 minutes after BCR ligation. Levels of phospho-Plcγ2 (Y759) were slightly higher in Eμ-myc cells until 30 minutes following BCR ligation and then decreased at a faster rate than wild-type cells (upper right, Fig 1C). Together these data indicate Myc overexpression altered the activation of critical BCR signaling proteins in non-transformed B cells following BCR ligation resulting in augmentation of BCR signaling.
Although phospho-Erk1/2 levels in Eμ-myc B cells were constitutively higher (52–81% higher) than in wild-type cells, we did not detect an expected increase in phospho-Erk levels following BCR ligation in either genotype (bottom right, Fig 1C). Other known BCR downstream effector proteins, including NF-κB and p38MAPK, exhibited similar results (Supplemental Fig S2). Because multiple signals converge on downstream effector proteins, we postulated their activation is likely to be tightly regulated by phosphatases and thus, more difficult to detect. To further investigate Erk phosphorylation, we added hydrogen peroxide (H2O2) after BCR ligation to transiently inactivate protein tyrosine phosphatases and allow accumulation of phosphorylated Erk. Phospho-Erk1/2 levels increased nearly 5-fold in both Eμ-myc and wild-type splenic B cells 10 minutes after BCR ligation and addition of H2O2 (top left, Fig 2A). Phosphatases typically recover from H2O2 inactivation resulting in reduced phospho-protein levels by 30 minutes (23). However, we observed in repeated experiments phospho-Erk1/2 levels in Eμ-myc B cells declined more slowly compared to wild-type B cells in the presence of H2O2, remaining above baseline and nearly 50% higher than wild-type levels 30 minutes after BCR ligation. Therefore, Erk1/2 activity is differentially regulated in B cells that overexpress Myc, resulting in constitutively higher phospho-Erk1/2 levels and sustained signaling following BCR ligation.
Figure 2. Inhibition of phosphatases further reveal increased BCR signaling in precancerous Eμ-myc B cells.
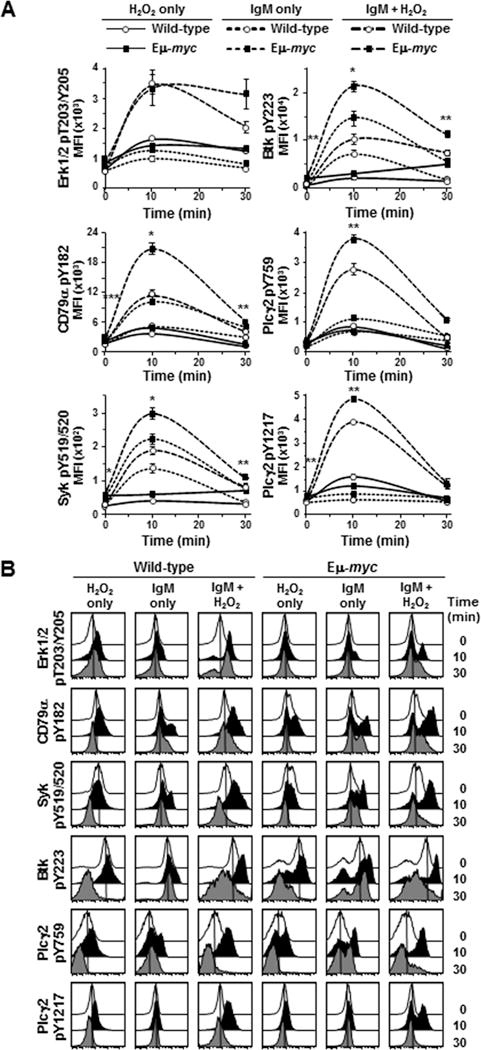
BCR signaling was evaluated by intracellular phospho-flow cytometry at intervals after IgM ligation of splenic B cells and addition of H2O2. A) Mean fluorescence intensities (MFI) from a representative experiment are shown. Each protein was measured in at least three experiments with 2–3 mice of each genotype per experiment. Error bars are SEM; p-values compare phospho-protein levels in Eμ-myc B cells to the levels in wild-type littermates with anti-IgM+H2O2 (large dashed lines); *p≤0.002, **p≤0.03 and ***p≤0.05 (p values for other comparisons not denoted). B) Histograms of fluorescence of the indicated phospho-proteins from B cells from a representative mouse of each genotype from one experiment at intervals after IgM ligation and/or H2O2 addition.
We also evaluated other proteins in the BCR signaling pathway in the presence of H2O2. Addition of H2O2 alone caused minimal accumulation of phosphorylated proteins in either genotype. However, following BCR ligation, H2O2 augmented activation of CD79α, Syk, Btk, and Plcγ2, which peaked by 10 minutes and returned to near baseline levels by 30 minutes in both genotypes, and again levels were significantly higher in Eμ-myc B cells. For example, following BCR ligation and addition of H2O2, phospho-CD79α and phospho-Plcγ2 (Y759) levels in Eμ-myc B cells increased 10- and 11-fold, respectively, as compared to 7- and 9- fold in wild-type B cells and resulted in significantly higher peak phospho-protein levels at 10 minutes [p=0.0013 for CD79α and p=0.0150 for Plcγ2 (Y759)] (middle, Fig 2A). Although phospho-Syk and phospho-Plcγ2 (Y1217) levels increased 5- and 7-fold, respectively in B cells of both genotypes 10 minutes after BCR ligation and addition of H2O2 (bottom, Fig 2A), peak phospho-protein levels were significantly higher in Eμ-myc B cells compared to wild-type [p=0.0012 for Syk and p=0.0154 for Plcγ2 (Y1217)]. Additionally, the phospho-Btk levels were significantly higher in Eμ-myc cells (p=0.0001) 10 minutes after BCR ligation and H2O2 addition (upper right panel, Fig 2A), even though phospho-Btk increased nearly 25-fold in wild-type B cells compared to 11-fold in Eμ-myc cells. Histograms of fluorescence in B cells from a representative experiment illustrate increased phospho-protein levels 10 minutes after BCR ligation and/or H2O2 addition by a shift of the curves to the right and return toward baseline by 30 minutes (Fig 2B). These data provide additional evidence that untransformed Myc overexpressing B cells have increased basal BCR signaling and that Myc further increases BCR signaling upon receptor ligation.
Eμ-myc splenic B cells exhibit resistance to Btk inhibition
Following BCR ligation, Syk phosphorylates tyrosine-551 of Btk, which prompts autophosphorylation of tyrosine-223 in Btk (24). Ibrutinib covalently binds Btk near the ATP binding domain to irreversibly inhibit Btk activity (25). Given the robust activation of Btk in Eμ-myc B lymphocytes following BCR ligation, we postulated ibrutinib would be a potent inhibitor of Btk activation in B cells that overexpressed Myc. We first evaluated basal levels of phospho-Btk (Y223) in splenic B cells treated with increasing concentrations of ibrutinib. Treatment with ibrutinib induced equivalent dose-dependent decreases in basal levels of phospho-Btk in wild-type and Eμ-myc B cells (Fig 3A), with a 38% decrease using 12.5 μM ibrutinib and a 58% decrease with 62.5 μM ibrutinib. After BCR stimulation, there was a greater reduction in phospho-Btk levels in Eμ-myc B cells compared to wild-type cells, with 12.5 μM ibrutinib achieving 42% reduction in phospho-Btk in Eμ-myc B cells, but only 26% reduction in wild-type cells (Fig. 3B). Nevertheless, absolute levels of phospho-Btk remained higher in Eμ-myc B cells than in wild-type cells over the range of ibrutinib concentrations (Fig. 3B). Notably, the level of phospho-Btk in 12.5 μM ibrutinib-treated Eμ-myc B lymphocytes was analogous to the level of phospho-Btk in untreated wild-type cells 10 minutes after BCR ligation (p = 0.54; Fig. 3B).
Figure 3. Pre-cancerous Eμ-myc B cells exhibit relative resistance to Btk inhibition by ibrutinib.
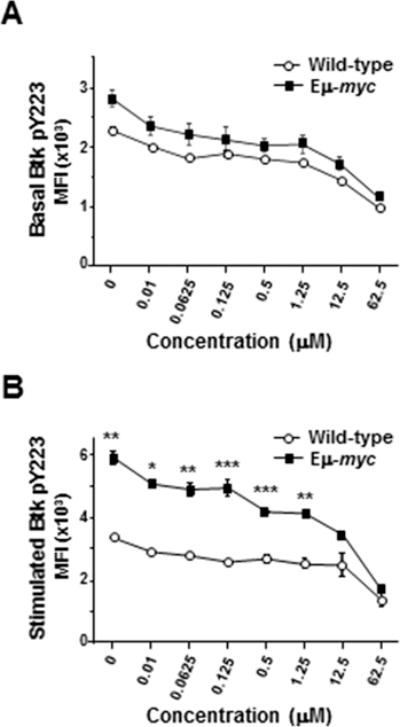
CD19+, IgM+ splenic B cells from Eμ-myc or wild-type mice at rest (A) or 10 minutes after IgM ligation (B) were pretreated with escalating concentrations of ibrutinib, and Btk phosphorylation was measured by intracellular phospho-flow cytometry. Mean fluorescence intensities (MFI) of representative experiments containing 3 mice of each genotype are shown. Error bars represent SEM; *p=0.007, **p≤0.025, ***p≤0.04.
We also investigated downstream of Btk by assessing basal and BCR stimulated levels of phospho-Plcγ2 (Y759) and phospho-Erk1/2 in the presence of ibrutinib. Ibrutinib treatment led to dose-dependent decreases in phospho-Plcγ2 (Y759) in unstimulated B cells and B cells after BCR ligation (Supplemental Fig S3A and B). Ibrutinib did not affect basal levels of phospho-Erk (T203/Y205) even in the presence of H2O2 (Supplemental Fig S3C), but potently reduced stimulated phospho-Erk levels (Supplemental Fig S3D). Importantly, at any concentration of ibrutinib, levels of phospho-Btk, phospho-Plcγ2, and phospho-Erk remained higher in Eμ-myc B cells than in wild-type. Therefore, Myc overexpression renders B cells relatively resistant to Btk inhibition by ibrutinib.
Based on its mechanism of action and reported specificity for Btk inhibition (25), we expected ibrutinib would inhibit Btk phosphorylation and consequently, phosphorylation of downstream components of BCR signaling (e.g., Plcγ2 and Erk1/2; Fig 1A). While treatment with ibrutinib (12.5 μM) reduced phospho-Btk levels in IgM-ligated Eμ-myc B cells by 47%, these levels were equivalent to the IgM-stimulated phospho-Btk levels in wild-type B cells that did not receive ibrutinib (MFI 255 versus 194, respectively, p=0.11) (Fig 4A, 3rd bar versus last bar). Ibrutinib (12.5 μM) had little effect on basal levels of phospho-Plcγ2 (Y759) in both genotypes (Fig 4B, 0 min DMSO versus 0 min ibrutinib). However, the levels of phospho-Plcγ2 (Y759) after BCR ligation remained ~50% higher in ibrutinib-treated Eμ-myc B cells than ibrutinib-treated wild-type cells (MFI 93 versus 60, respectively, p=0.0016, Fig 4B last two bars), and this level in ibrutinib-treated Eμ-myc B cells was analogous to the level of phospho-Plcγ2 (Y759) in IgM-stimulated wild-type B cells that had not been treated with ibrutinib (MFI 95, p=0.42; Fig 4B, 3rd bar versus last bar). Absolute levels of phospho-Erk and phospho-Plcγ2 (Y1217) were significantly higher in Eμ-myc B cells than in wild-type B cells, but we did not detect an increase in activation following BCR ligation or an effect of ibrutinib in the absence of H2O2 (Fig 4C and Supplemental Fig S4A, respectively).
Figure 4. BCR downstream signaling in Eμ-myc B cells is relatively resistant to ibrutinib.
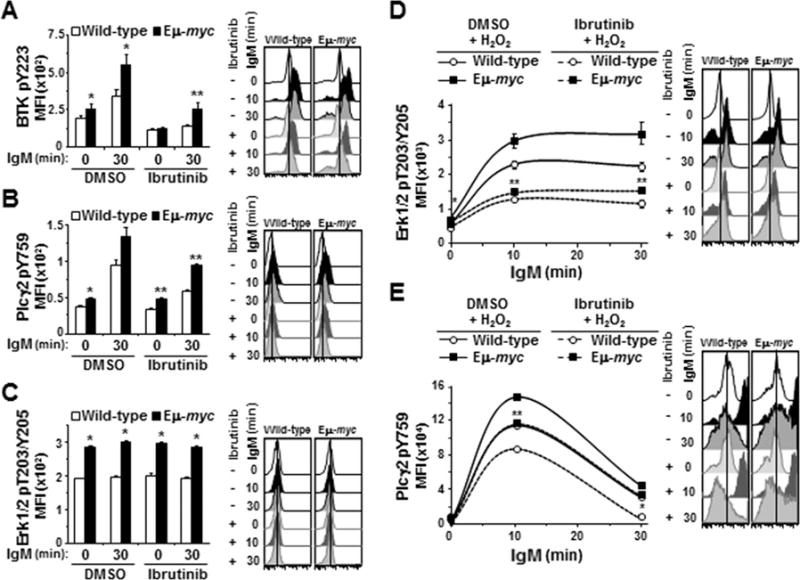
BCR signaling was measured by intracellular phospho-flow cytometry in splenic B cells incubated with ibrutinib prior to IgM ligation without (A-C) or with (D and E) H2O2. A-C) Mean fluorescence intensities (MFI) prior to and at intervals after IgM ligation from one representative experiment (left) and histograms showing one representative mouse per genotype (right) for Btk pY223 (A), Plcγ2 pY759 (B), and Erk1/2 pT203/Y205 (C). D, E) MFI without and intervals after BCR ligation and H2O2 addition for Erk1/2 pT203/Y205 (D) and Plcγ2 pY759 (E). Each protein was measured in at least three experiments with 3–4 mice of each genotype per experiment. Error bars are SEM; A, *p≤0.025 and **p≤0.005; B, *p≤0.050 and **p≤0.005; C, *p≤0.005; D, p*≤0.001 and **p≤0.05; E, p*≤0.004 and **p=0.02. In A-C, p-values compare phospho-protein levels between Eμ-myc and wild-type littermates, and in D and E compare phospho-protein levels in Eμ-myc and wild-type littermate B cell only treated with ibrutinib (p values for other comparisons not denoted).
We again utilized H2O2 to better visualize the effects of ibrutinib on phospho-Erk1/2. As we observed previously with addition of H2O2, Erk1/2 activation was detectable 10 minutes after BCR ligation and persisted in Eμ-myc cells beyond that in wild-type cells (Fig 4D). Treatment with ibrutinib attenuated activation of Erk1/2 in both genotypes, but phospho-Erk1/2 levels remained higher in the Eμ-myc B cells at every time evaluated (Fig 4D, p=0.0008, 0.0522, and 0.0413 at 0, 10, and 30 minutes, respectively). H2O2 also accentuated the differences between Eμ-myc and wild-type B cells upon ibrutinib treatment when evaluating Plcγ2. Phospho-Plcγ2 (Y759) in ibrutinib-treated Eμ-myc B cells was 1.3-fold and 4.2-fold greater than in ibrutinib-treated wild-type cells at 10 and 30 minutes, respectively, after BCR ligation and addition of H2O2 (p=0.0201 and p=0.0013, respectively, Fig 4E). The absolute levels of phospho-Plcγ2 (Y759) in ibrutinib-treated Eμ-myc B cells were statistically equivalent to levels in wild-type cells that were not treated with ibrutinib (MFI 116000 and 113250, respectively, at 10 min, p=0.39; MFI 34402 and 31520, respectively, at 30 minutes, p=0.28) (Fig 4E, dashed line with square symbol versus solid line with circle symbol). Levels of phospho-Plcγ2 (Y1217) also remained higher in Eμ-myc cells than in wild-type B cells despite ibrutinib treatment (Supplemental Fig S4B). Thus, as predicted, ibrutinib blunted the phosphorylation of Plcγ2 and Erk1/2; however, more complete inhibition was evident in wild-type B cells.
Ibrutinib has off-target effects on other kinases
We also evaluated the effect of ibrutinib on the BCR pathway upstream of Btk (Fig 1A), originally as a negative control, as ibrutinib is a Btk-specific inhibitor and should not alter signaling upstream of Btk (25). Unexpectedly, in both wild-type and Eμ-myc B cells, CD79α and Syk phosphorylation was also inhibited by ibrutinib (Fig 5A and 5B). There was little effect on basal levels of phospho-CD79α with ibrutinib treatment (Fig 5A, first two bars versus 5th and 6th bars), but a pronounced reduction was evident in BCR stimulated cells (~30% reduction in both genotypes) (Fig 5A, 2nd and 3rd bars versus last two bars). As with Btk, BCR stimulated phospho-CD79α levels were similar in ibrutinib-treated Eμ-myc B cells and wild-type B cells that were not ibrutinib-treated (Fig 5A, 3rd bar versus last bar, p=0.506). Treatment with ibrutinib (12.5 μM) reduced basal and BCR-stimulated phospho-Syk levels in both genotypes, with a 48% reduction in phospho-Syk levels in wild-type cells and a 34% decrease in Eμ-myc cells 30 minutes after BCR ligation (Fig 5B, 3rd bar versus 7th bar and 4th bar versus last bar, respectively). Dose titration experiments indicated that even 0.0025 μM ibrutinib caused significant reductions in basal phospho-Syk in wild-type (9.3% reduction, p=0.0263) and Eμ-myc B cells (10.3% reduction, p=0.0188) (Supplemental Fig S5A). Higher concentrations of ibrutinib (0.25 μM for wild-type, p=0.1367 and 0.03125 μM for Eμ-myc, p=0.0044) were required to achieve 10% reductions in phospho-Syk after BCR ligation (Supplemental Fig S5B). These data indicate ibrutinib treatment inhibits more than just Btk activation in B cells.
Figure 5. Ibrutinib has off-target effects in the BCR signaling pathway.
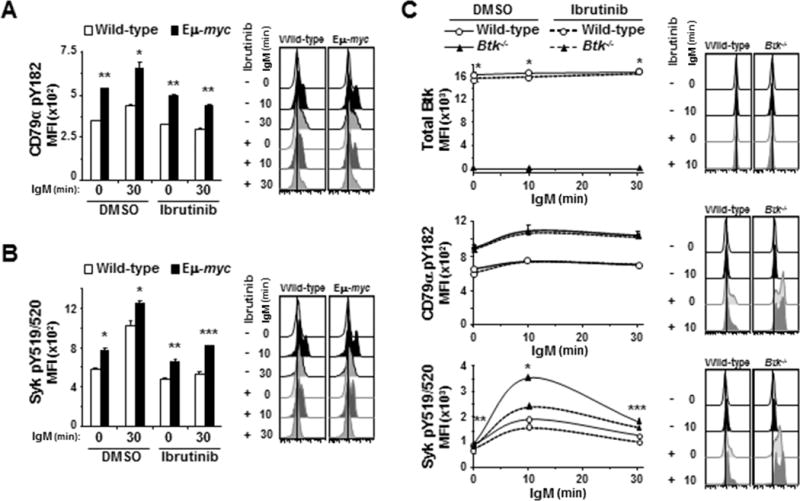
Activation of BCR signaling pathway proteins after IgM ligation was evaluated by intracellular phospho-flow cytometry in splenic B cells pre-treated with ibrutinib. A, B) Mean fluorescence intensities (MFI) from one representative experiment (left) and histograms (right) from one representative mouse are shown for CD79α pY182 (A) and Syk pY519/520 (B). Each protein was measured in at least three experiments with 3–4 2mice of each genotype. Error bars are SEM; A, *p≤0.006 and **p≤5 × 10−4; B, *p≤0.05, **p≤0.01, ***p≤0.005. C) Splenic B cells from Btk−/− or wild-type mice were incubated with ibrutinib for 1 hour prior to ligation of IgM. MFI (left) from one representative experiment of 4 total experiments and histogram (right) of fluorescence of cells from one representative mouse of each genotype are shown for the indicated phospho-proteins. Error bars are SEM; p-values compare total Btk levels between wild-type and Btk−/− B cells (top) and phospho-Syk between DMSO and ibrutinib-treated Btk−/− cells (bottom); *p=7.834 × 10−4, **p≤2.53 × 10−3, ***p≤0.0173 (p values for other comparisons not denoted).
To determine whether ibrutinib inhibits phosphorylation of CD79α and Syk directly or by a feedback loop with Btk or downstream effector molecules, we first evaluated total Btk levels in wild-type B cells after ibrutinib treatment. Ibrutinib treatment did not alter total Btk levels in B cells (top panel, Fig 5C). We also evaluated B cells from Btk-null mice, which do not express Btk protein ((26, 27) and Fig 5C, top panel). In BCR ligated Btk−/− B cells, ibrutinib treatment did not affect phosphorylation of CD79α (middle panel, Fig 5C), whereas there was significantly reduced phosphorylation of Syk (bottom panel, Fig 5C). Interestingly, levels of activated CD79α and Syk following BCR ligation were significantly higher in Btk−/− B cells than wild-type B cell controls. In addition, Btk−/− B cells were more sensitive to Syk inhibition by ibrutinib than wild-type B cells, as evidenced by a 26% reduction in phospho-Syk 10 minutes after ligation of the BCR in ibrutinib-treated Btk−/− B cells but only 11% reduction in phospho-Syk levels in wild-type B cells (p=0.0015). Additionally, dose-dependent decreases in phospho-Syk levels in Btk−/− B cells were similar to wild-type and Eμ-myc B cells (Supplemental Fig S6A). Finally, Myc levels were evaluated in wild-type and Eμ-myc B cells and shown to not be affected by ibrutinib treatment in either genotype (Supplemental Fig S6B), indicating the inhibition of signaling upstream of Btk by ibrutinib was not due to a reduction in Myc. Together these results imply that direct off-target effects are likely responsible for the reduction in phospho-Syk detected in B cells treated with ibrutinib.
Activation of BCR signaling by Myc overexpression induces Akt and ribosomal S6 kinases
The PI3K/Akt pathway is activated following ligation of the BCR by cross-talk with kinases such as Syk (Fig 1A and (13)). Thus, we evaluated the effect of Myc overexpression on activation of Akt and ribosomal S6 kinases by evaluating phosphorylation of threonine-308 and serine-473 of Akt and ribosomal protein S6 (S6RP), respectively. In both Eμ-myc and wild-type B cells, small increases in phospho-Akt (T308) occurred quickly following BCR ligation (left panel, Fig 6A), and returned to baseline within 30 minutes, whereas no significant changes were detected in phospho-Akt (S473) (middle panel, Fig 6A). Notably, levels of phospho-Akt (T308 and S473) were constitutively higher in Eμ-myc B cells compared to wild-type B cells (Fig 6A). Following ligation of the BCR, phosphorylation of S6RP increased similarly in Eμ-myc and wild-type B cells over 60 minutes (right panel, Fig 6A), but diverged by 180 minutes, when higher levels of phospho-S6RP were detected in Eμ-myc B cells compared to wild-type cells (p=0.0562). Although H2O2 has little direct effect on serine/threonine phosphatases, it can indirectly impact them through upstream tyrosine kinases (28); consequently, we evaluated both phospho-Akt sites following addition of H2O2. Addition of H2O2 immediately after BCR ligation further accentuated the early increase in phospho-Akt (T308) (left panel, Fig 6B) and revealed a BCR ligation-induced increase in phospho-Akt (S473) that was not evident in absence of H2O2 (middle panel, Fig 6B). H2O2 had minimal effect on detection of phospho-S6RP in wild-type B cells, but allowed a slight accumulation of phospho-S6RP in Eμ-myc cells (right panel, Fig 6B), suggesting altered regulation of S6-kinases in Myc overexpressing cells.
Figure 6. Signaling through PI3k/Akt is enhanced in Eμ-myc B cells and inhibited by ibrutinib.
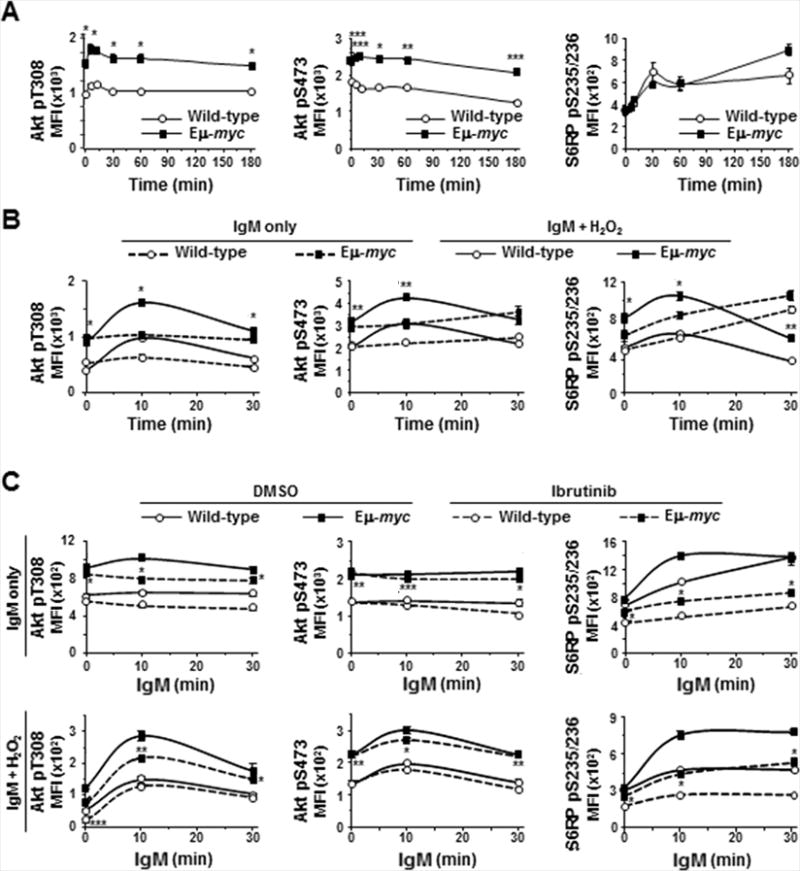
Phosphorylation of Akt and S6 ribosomal protein (S6RP) in splenic B cells was measured by intracellular phospho-flow cytometry at intervals after IgM ligation without (A, upper panels C) or with (B, lower panels C) addition of H2O2 in the absence (A, B) or presence (C) of ibrutinib. Each protein was measured in at least three experiments with 2–3 mice of each genotype. Mean fluorescence intensities (MFI) from one representative experiment are shown; p-values compare phospho-protein levels in Eμ-myc and wild-type B cells in IgM-stimulated (A), IgM-stimulated + H2O2 (B, solid lines), ibrutinib-treated (C, dashed lines); *p≤0.01, **p≤0.03, and ***p≤0.05 (p values for other comparisons not denoted).
We also assessed whether ibrutinib affected Akt phosphorylation. Treatment with ibrutinib reduced basal and BCR-stimulated phospho-Akt (T308) in Eμ-myc and wild-type B cells, but absolute levels were higher in ibrutinib-treated Eμ-myc cells than in untreated wild-type cells (left upper panel, Fig 6C). Upon exposure to H2O2, proportionally larger reductions in phospho-Akt (T308) occurred in ibrutinib-treated Eμ-myc B cells, but again the levels remained higher in ibrutinib-treated Eμ-myc cells compared to wild-type cells that were not treated with ibrutinib (left lower panel, Fig 6C). Phosphorylation of Akt (S473) was not affected by ibrutinib treatment in either genotype in the absence or presence of H2O2 (upper and lower middle panels, Fig 6C). Ibrutinib treatment significantly inhibited basal and BCR-stimulated phosphorylation of S6RP in both Eμ-myc and wild-type B cells in the absence (upper right panel, Fig 6C) and presence (lower right panel, Fig 6C) of H2O2. Again, phospho-S6RP levels remained higher in ibrutinib-treated Eμ-myc cells than wild-type B cells and were similar to levels in untreated wild-type B cells. Together these data indicate Myc overexpression activates PI3K/Akt signaling and that ibrutinib has previously unappreciated effects beyond Btk inhibition.
Discussion
Myc is required for the growth and survival of B cell lymphomas, and its overexpression is frequently selected for in these malignancies (7). Genetically engineered mice showed Myc overexpression induces B cell transformation, solidifying Myc as a driver of B cell lymphoma (5). However, the cellular mechanisms by which Myc dysregulation induces B cell transformation had not been fully explored. Our study focused on cell signaling and revealed that prior to cellular transformation, Myc overexpression alone led to increased signaling through pro-growth and survival pathways, including the BCR and PI3K/Akt pathways. There was significantly increased basal activation of Btk, Plcγ2, Erk1/2, and Akt in non-transformed, primary splenic B cells from Eμ-myc mice compared to wild-type littermates. Moreover, our data show these Myc-mediated increases in signaling cannot be fully inhibited with ibrutinib, which has clinical implications.
Ligation of the BCR prompts phosphorylation of downstream kinases and effector molecules, such as Syk, Btk, Plcγ2, and Erk1/2, as well as components of the PI3K/Akt pathway in normal B lymphocytes (Fig 1A). Following BCR ligation in B cells from Eμ-myc mice, we detected more robust activation of CD79α, Btk, and Plcγ2. Additionally, levels of phospho-Erk1/2 and phospho-Akt were constitutively higher in B cells from Eμ-myc mice. Increased BCR signaling is reported to contribute to B cell lymphoma development (29). Finally, as evidenced by increased levels of phospho-S6RP 180 minutes after BCR ligation, Myc overexpression led to sustained activity of ribosomal S6-kinases, which are important regulators of cell growth and metabolism.
Although we detected high levels of activated Erk1/2 and p38MAPK in B cells from Eμ-myc mice, we were unable to detect further activation in either genotype following BCR ligation. Consequently, we utilized H2O2, which allowed phosphorylated proteins to accumulate, and as such, we were able to detect BCR-induced increases in phospho-Erk and phospho-p38MAPK, implying these kinases are tightly regulated. Although phosphatases that regulate upstream proteins in the BCR pathway had rebounded by 30 minutes after H2O2 addition, phospho-Erk1/2 levels remained elevated, suggesting phosphatases that regulate Erk activation are more sensitive to H2O2 inhibition. Furthermore, altered regulation of Erk1/2 activation in Eμ-myc B cells allowed for higher basal and more sustained BCR-stimulated Erk signaling. Levels of phospho-p38MAPK were also higher in Eμ-myc B cells compared to wild-type. These results are consistent with our previous report that Myc overexpression was sufficient to induce Ras/MAPK signaling (30). Together these data indicate Erk1/2 and p38MAPK are involved in BCR signaling and are regulated by mechanisms that are different than upstream kinases.
Activation of BCR signaling leads to a transcriptional program that is enriched for MYC and MYC-induced genes (10, 11). Additionally, genomic studies in Burkitt lymphoma cell lines identified CD79β and SYK as potential transcriptional targets of MYC (20), and the MYC-regulated miR-17~92 cluster augments signaling through the BCR in transformed lymphoma cells (21). These reports together with our data suggest a feed-forward loop, whereby MYC expression increases BCR signaling, which further amplifies MYC activation, and so on. Importantly, our data indicate that such a feed-forward mechanism is not dependent on oncogenic transformation, as Myc overexpression alone augmented both basal and stimulated signaling by multiple proteins in the BCR signaling cascade in non-transformed B cells.
Aberrant activation of BCR and PI3K/Akt signaling has been demonstrated in B-NHL. Prolonged or continuous signaling via the BCR pathway and reduced sensitivity to negative regulation have been identified in various lymphoid malignancies (14, 16, 31, 32). Our data indicate that elevated Myc levels likely contribute to this increased signaling. Moreover, since inhibition of BCR activation in vitro by genetic or pharmacologic means has proven deleterious to lymphoma growth and survival (14, 16, 33, 34), pharmacologic inhibition has been an attractive prospect for drug development. Indeed, ibrutinib is efficacious in the treatment of indolent B-NHL (17–19); however, a subset is refractory to ibrutinib. We determined that hyperactivation of BCR signaling by Myc confers relative resistance to Btk inhibition by ibrutinib. While ibrutinib did reduce phosphorylation and activation of Btk in Myc overexpressing B cells, absolute levels of activated Btk and downstream proteins Plcγ2 and Erk1/2 remained higher following ibrutinib treatment than the levels in untreated wild-type B cells. Our studies predict that B-NHL with increased MYC expression may be less likely to respond to ibrutinib specifically and possibly BCR inhibition more generally. While MYC expression in indolent lymphomas is not routinely evaluated, it may be worthwhile if ibrutinib treatment is being considered. Furthermore, although aberrant BCR signaling has been demonstrated in aggressive large cell lymphomas, ibrutinib therapy has not been as successful (17). We postulate that differential responses to ibrutinib treatment may correlate with MYC expression, recognizing that MYC overexpression is associated with more aggressive lymphoma phenotypes.
We observed treatment with ibrutinib impaired activation of Btk and downstream signaling molecules, but also reduced activation of upstream signaling proteins CD79α, Syk, and Akt in both Myc overexpressing and wild-type B cells. These results were unexpected, since ibrutinib is reported to be highly specific for Btk (25, 35). Ibrutinib did not affect SYK phosphorylation in a previous report, but only tyrosine-352 was interrogated (34), whereas SYK activation is complicated by phosphorylation of multiple tyrosine residues, the roles of which have not been fully elucidated (36, 37). Nevertheless, tyrosine-352 is believed to lie in a regulatory domain, whereas tyrosine-525/526 are in the activation loop and are autophosphorylated upon ligation of the BCR (37), and reportedly are better predictors of SYK activity in diffuse large B cell lymphoma (38). Therefore, we evaluated phosphorylation of the murine homologous residues, tyrosine-519/520. Data showed inhibition of Syk phosphorylation by ibrutinib occurred in Myc overexpressing and wild-type B cells in a dose-dependent manner. This inhibition was also observed in Btk−/− B cells, indicating it is an off-target effect rather than the result of a regulatory feedback loop. Therefore, the ibrutinib-mediated reduction in phospho-Akt and phospho-S6RP we detected may be secondary to Syk inhibition, as Syk activates PI3K/Akt signaling following BCR ligation (13). These unexpected findings could provide an alternative explanation for the increased bleeding risk in patients treated with ibrutinib, which has been previously linked to ineffective collagen-evoked signaling in platelets (39, 40). Phosphorylation of SYK (Y525) is associated with platelet activation and thrombus formation in the setting of sheer stress (41). Therefore, inhibition of SYK by ibrutinib may lead to ineffective thrombosis and bleeding.
Our results expand understanding of the role of Myc in cell signaling and indicate Myc overexpression alone is sufficient to cause aberrant activation of pro-growth and survival signaling that likely contributes to lymphomagenesis. Our data have significant clinical implications for targeting the BCR pathway in human lymphomas that highly express MYC. Additionally, we showed off-target effects of ibrutinib on Syk that may explain some side effects of ibrutinib therapy. Furthermore, the relative resistance to Btk inhibition by ibrutinib in cells that overexpress Myc suggest MYC expression may serve as a therapeutic biomarker.
Materials and Methods
Mice
Male C57Bl/6 Eμ-myc mice (5) were mated to C57Bl/6 females to generate transgenic and nontransgenic littermates. C57Bl/6 Btk−/− mice (26) and C57Bl/6 wild-type controls were provided by Dr. Peggy Kendall (Vanderbilt University Medical Center). Spleens were harvested from mice 4–6 weeks of age, prior to lymphoma development. All mouse studies were in accordance with state and federal guidelines and were approved by the Vanderbilt Institutional Animal Care and Use Committee.
Intracellular flow cytometry
The protocol was modified from Irish, et al (42). Briefly, following disaggregation and red blood cells lysis, 1×107 splenocytes/ml in culture medium were incubated at 37C for 60 minutes. For assessment of upstream BCR signaling proteins, culture medium was DMEM + 10% fetal bovine serum (FBS). For Erk, p38MAPK, Akt, and S6RP assessment, FBS was reduced to 2.5% for the 3 hour kinetic experiments or 0% for shorter experiments. BCR signaling was stimulated by ligation of IgM with 10 μg/ml anti-mouse IgM F(ab’)2 fragments (Jackson ImmunoResearch). For some experiments, splenocytes were pretreated for 1 hour with ibrutinib (Selleckchem) or DMSO (control). In indicated experiments, H2O2 (550 μM) was added immediately after BCR ligation. Cells were fixed with 1.5% paraformaldehyde (10 minutes, room temperature) at one time after intervals following IgM ligation. Cells were incubated with anti-IgM and anti-CD19 antibodies, permeabilized with 0.1% Triton-X (5 minutes, 4C), and then incubated with phospho-specific antibodies in PBS, 3% FBS, 15 mM sodium azide. In some cases unconjugated phospho-specific antibodies were used, followed by AlexaFluor 647-conjugated goat anti-rabbit IgG (Cell Signaling Technology) for 30 minutes at room temperature. For a list of antibodies used see Supplemental Table S1. Fluorescence was measured on a BD Fortessa flow cytometer, and data was analyzed with FlowJo software.
Statistics
Unless otherwise stated, values represent mean ± SEM, and paired t-test was used for comparisons.
Supplementary Material
Acknowledgments
We thank Peggy Kendall for providing Btk−/− mice and members of the Eischen laboratory for their advice.
Funding: This work was supported by NIH/NCI R21CA184352 (CME), the Hematology Helping Hands Research Award (TKM), and the NCI Cancer Center support grant P30CA68485 that supports the Vanderbilt Flow Cytometry Shared Resource and the NCI Cancer Center support grant P30CA056036.
Footnotes
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Xu X, Zhang L, Wang Y, Zhang Q, Zhang L, Sun B, et al. Double-hit and triple-hit lymphomas arising from follicular lymphoma following acquisition of MYC: report of two cases and literature review. Int J Clin Exp Pathol. 2013;6(4):788–94. [PMC free article] [PubMed] [Google Scholar]
- 2.Young KH, Xie Q, Zhou G, Eickhoff JC, Sanger WG, Aoun P, et al. Transformation of follicular lymphoma to precursor B-cell lymphoblastic lymphoma with c-myc gene rearrangement as a critical event. Am J Clin Pathol. 2008;129(1):157–66. doi: 10.1309/NKK3FEX2BE5L7EKB. [DOI] [PubMed] [Google Scholar]
- 3.Christie L, Kernohan N, Levison D, Sales M, Cunningham J, Gillespie K, et al. C-MYC translocation in t(14;18) positive follicular lymphoma at presentation: An adverse prognostic indicator? Leuk Lymphoma. 2008;49(3):470–6. doi: 10.1080/10428190701836845. [DOI] [PubMed] [Google Scholar]
- 4.Huh YO, Lin KI, Vega F, Schlette E, Yin CC, Keating MJ, et al. MYC translocation in chronic lymphocytic leukaemia is associated with increased prolymphocytes and a poor prognosis. Br J Haematol. 2008;142(1):36–44. doi: 10.1111/j.1365-2141.2008.07152.x. [DOI] [PubMed] [Google Scholar]
- 5.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318(6046):533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 6.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67(20):9762–70. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 7.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 8.Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117(6):787–800. doi: 10.1016/j.cell.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90(6):1073–83. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 10.Pede V, Rombout A, Vermeire J, Naessens E, Mestdagh P, Robberecht N, et al. CLL cells respond to B-Cell receptor stimulation with a microRNA/mRNA signature associated with MYC activation and cell cycle progression. PLoS One. 2013;8(4):e60275. doi: 10.1371/journal.pone.0060275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeomans A, Thirdborough SM, Valle-Argos B, Linley A, Krysov S, Hidalgo MS, et al. Engagement of the B-cell receptor of chronic lymphocytic leukemia cells drives global and MYC-specific mRNA translation. Blood. 2016;127(4):449–57. doi: 10.1182/blood-2015-07-660969. [DOI] [PubMed] [Google Scholar]
- 12.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120(6):1175–84. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beitz LO, Fruman DA, Kurosaki T, Cantley LC, Scharenberg AM. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J Biol Chem. 1999;274(46):32662–6. doi: 10.1074/jbc.274.46.32662. [DOI] [PubMed] [Google Scholar]
- 14.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaffer AL, 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. doi: 10.1146/annurev-immunol-020711-075027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–20. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100(14):8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Psathas JN, Doonan PJ, Raman P, Freedman BD, Minn AJ, Thomas-Tikhonenko A. The Myc-miR-17-92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: a novel lymphomagenic feed-forward loop. Blood. 2013;122(26):4220–9. doi: 10.1182/blood-2012-12-473090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41(6–7):599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 23.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9(2):387–99. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 24.Baba Y, Hashimoto S, Matsushita M, Watanabe D, Kishimoto T, Kurosaki T, et al. BLNK mediates Syk-dependent Btk activation. Proc Natl Acad Sci U S A. 2001;98(5):2582–6. doi: 10.1073/pnas.051626198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1):58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 26.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3(3):283–99. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 27.Satterthwaite AB, Cheroutre H, Khan WN, Sideras P, Witte ON. Btk dosage determines sensitivity to B cell antigen receptor cross-linking. Proc Natl Acad Sci U S A. 1997;94(24):13152–7. doi: 10.1073/pnas.94.24.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding J, Takano T, Gao S, Han W, Noda C, Yanagi S, et al. Syk is required for the activation of Akt survival pathway in B cells exposed to oxidative stress. J Biol Chem. 2000;275(40):30873–7. doi: 10.1074/jbc.M004813200. [DOI] [PubMed] [Google Scholar]
- 29.Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B cell antigen receptor and overexpression of MYC can cooperate in the genesis of B cell lymphomas. PLoS Biol. 2008;6(6):e152. doi: 10.1371/journal.pbio.0060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gramling MW, Eischen CM. Suppression of Ras/Mapk pathway signaling inhibits Myc-induced lymphomagenesis. Cell Death Differ. 2012;19(7):1220–7. doi: 10.1038/cdd.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44(12):1321–5. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44(12):1316–20. doi: 10.1038/ng.2469. [DOI] [PubMed] [Google Scholar]
- 33.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–80. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374(4):323–32. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurosaki T, Johnson SA, Pao L, Sada K, Yamamura H, Cambier JC. Role of the Syk autophosphorylation site and SH2 domains in B cell antigen receptor signaling. J Exp Med. 1995;182(6):1815–23. doi: 10.1084/jem.182.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geahlen RL. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009;1793(7):1115–27. doi: 10.1016/j.bbamcr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111(4):2230–7. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib Inhibits Platelet Integrin alphaIIbbeta3 Outside-In Signaling and Thrombus Stability But Not Adhesion to Collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326–35. doi: 10.1161/ATVBAHA.115.306130. [DOI] [PubMed] [Google Scholar]
- 40.Manne BK, Badolia R, Dangelmaier C, Eble JA, Ellmeier W, Kahn M, et al. Distinct pathways regulate Syk protein activation downstream of immune tyrosine activation motif (ITAM) and hemITAM receptors in platelets. J Biol Chem. 2015;290(18):11557–68. doi: 10.1074/jbc.M114.629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Speich HE, Grgurevich S, Kueter TJ, Earhart AD, Slack SM, Jennings LK. Platelets undergo phosphorylation of Syk at Y525/526 and Y352 in response to pathophysiological shear stress. Am J Physiol Cell Physiol. 2008;295(4):C1045–54. doi: 10.1152/ajpcell.90644.2007. [DOI] [PubMed] [Google Scholar]
- 42.Irish JM, Czerwinski DK, Nolan GP, Levy R. Kinetics of B cell receptor signaling in human B cell subsets mapped by phosphospecific flow cytometry. J Immunol. 2006;177(3):1581–9. doi: 10.4049/jimmunol.177.3.1581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.