

Abstract
Enterohemorrhagic Escherichia coli (EHEC) produces Shiga toxin 1 (Stx1) and Shiga toxin 2 (Stx2). Nitric oxide (NO), which acts as an antimicrobial defense molecule, was found to enhance the production of Stx1 and Stx2 in EHEC under anaerobic conditions. Although EHEC O157 has two types of anaerobic NO reductase genes, an intact norV and a deleted norV, in the deleted norV‐type EHEC, a high concentration of NO (12–29 μmol/L, maximum steady‐state concentration) is required for enhanced Stx1 production and a low concentration of NO (~12 μmol/L, maximum steady‐state concentration) is sufficient for enhanced Stx2 production under anaerobic conditions. These results suggested that different concentration thresholds of NO elicit a discrete set of Stx1 and Stx2 production pathways. Moreover, the enhancement of Shiga toxin production in the intact norV‐type EHEC required treatment with a higher concentration of NO than was required for enhancement of Shiga toxin production in the deleted norV‐type EHEC, suggesting that the specific NorV type plays an important role in the level of enhancement of Shiga toxin production in response to NO. Finally, Fur derepression and RecA activation in EHEC were shown to participate in the NO‐enhanced Stx1 and Stx2 production, respectively.
Keywords: enterohemorrhagic Escherichia coli, Fur, nitric oxide, NO reductase, RecA, Shiga toxin
1. Introduction
Enterohemorrhagic Escherichia coli (EHEC) O157 is a causative agent of intestinal disorders ranging from mild infection to severe, bloody diarrhea (hemorrhagic colitis) (Hofmann, 1993; Keusch & Acheson, 1997; Lansbury & Ludlam, 1997). EHEC O157 associated with hemolytic‐uremic syndrome (HUS) has been shown to produce Shiga toxin 1 (Stx1) and Shiga toxin 2 (Stx2). Stx1, which is identical to the Shiga toxin produced by Shigella dysenteriae type 1 (Tesh & O'Brien, 1991), and Stx2 share 54% amino acids sequence homology (Jackson, Neill, O'Brien, Holmes, & Newland, 1987). Both Stx1 and Stx2 are members of the AB5 toxin family (Calderwood, Auclair, Donohue‐Rolfe, Keusch, & Mekalanos, 1987; De Grandis et al., 1987; Fraser et al., 2004; Jackson, Newland, Holmes, & O'Brien, 1987; Yutsudo, Nakabayashi, Hirayama, & Takeda, 1987). The A subunit is an RNA‐N‐glycosidase that plays a role in protein synthesis inhibition in eukaryotic cells (Endo et al., 1988). The pentamer of B subunits binds to the surface receptor, glycolipid receptor Gb3, on the target cells (Jacewicz, Clausen, Nudelman, Donohue‐Rolfe, & Keusch, 1986).
Both Shiga toxin genes in EHEC are located within Stx‐encoding phages that are related to the λ phage, which is well‐characterized with respect to both its genome arrangement and transcription patterns (Shimizu, Ohta, & Noda, 2009; Waldor & Friedman, 2005). However, the conditions under which they are highly expressed are different (Calderwood & Mekalanos, 1987; Calderwood et al., 1987; De Grandis et al., 1987; Hull, Acheson, Echeverria, Donohue‐Rolfe, & Keusch, 1993). Stx2 production is activated by phage‐inducing agents, such as mitomycin C, indicating that the stx2 gene is transcribed from the phage late promoter (P R') of the Stx2‐encoding phage. Since the phage lysis genes of Stx2‐encoding phage as well as stx2 are transcribed by the P R' during Stx2‐encoding phage induction, the Stx2‐encoding phage and Stx2 are released from bacterial cells at the same time (Shimizu et al., 2007, 2009). In contrast, the Stx1‐encoding phage in EHEC is less sensitive to phage‐inducing agents than the Stx2‐encoding phage, with the result that the level of Stx1 production induced by mitomycin C is lower. The Stx1‐encoding phage carrying the stx1 contains the stx1 promoter (P Stx1) region, which represents the functional operator‐binding site (Fur box) for the Fur (Calderwood & Mekalanos, 1987; Porcheron & Dozois, 2015). Fur is an iron‐responsive repressor of iron‐transport systems in E. coli (Lee & Helmann, 2007). Under low‐iron conditions, Stx1 production is increased (Calderwood & Mekalanos, 1987; Shimizu et al., 2009). Thus, there are two types of promoters for Stx1 expression in EHEC. Stx1 was mainly regulated by the P Stx1 and remained in bacterial cells (Shimizu et al., 2007, 2009).
Nitric oxide (NO) is a crucial cellular signaling molecule involved in many physiological and pathological events, e.g., cell cycle regulation, vascular relaxation, apoptosis, hypoxia, nutrient deficiency, and antimicrobial defense (Fang, 2004; Thomas et al., 2008). Because of the enormous variety of chemical reactions and biological properties associated with NO, the responses to this molecule are highly diverse. The maintenance of steady‐state NO concentrations has emerged as a key determinant of the biological function of NO. Precise cell responses are differentially regulated by the specific NO concentration. Though the synthesis and diffusion of NO are partial determinants of its concentration, consumption of NO is also important for determining its biological function. The rate of NO consumption by cells is directly dependent on the oxygen concentration (Thomas, Liu, Kantrow, & Lancaster, 2001), suggesting an important regulatory relationship between NO signaling and oxygen concentration in different environments. Whereas increased oxygen levels will increase NO consumption, NO consumption is decreased under anaerobic conditions.
In humans, NO is produced by the inducible isoform of NO‐synthase (iNOS) in several cell types, including macrophages, as part of the immune response to counteract microbial infection. NO induces a bacterial SOS response, which plays a central role in the E. coli response to a wide variety of genotoxic agents (Lobysheva, Stupakova, Mikoyan, Vasilieva, & Vanin, 1999; Schapiro, Libby, & Fang, 2003; Spek et al., 2001; Stupakova, Lobysheva, Mikoyan, Vanin, & Vasilieva, 2000). The SOS regulatory system, which is involved in the induction of more than 40 genes upon blockage of ongoing DNA replication, controls the response to DNA damage or the inhibition of DNA replication (Walker, 1984). DNA damage causes RecA polymerization around a single‐stranded DNA (Kowalczykowski, 2000). This active filament form can direct homologous recombination (Cox, 1999). Activated RecA in turn mediates the cleavage of LexA (Little, 1991), inactivating it and resulting in derepression of the SOS regulon (Fernandez De Henestrosa et al., 2000). Moreover, to perform specific roles in SOS mutagenesis, RecA also promotes UmuD cleavage (Pham et al., 2002). In addition, RecA promotes cleavage of the phage CI repressor, triggering induction of the lytic cycle (Little, 1984, 1993), and late gene expression by the Stx‐encoding prophage results from the Stx‐encoding prophage induction in EHEC (Wagner et al., 2002). Conversely, NO decreases stx2 expression in EHEC O157 to repress the SOS response by the NO sensor nitrite‐sensitive repressor NsrR (Vareille, de Sablet, Hindre, Martin, & Gobert, 2007). Moreover, NO also inhibits the expression of the locus of enterocyte effacement (LEE) genes in EHEC (Branchu et al., 2014). NO is thus a signaling mediator with many diverse and often opposing biological activities in bacterial cells.
As a strategy to evade the host immune attack, pathogenic bacteria have evolved a biochemical pathway to degrade NO. E. coli has evolved several mechanisms for NO detoxification (Poole, 2005; Spiro, 2006, 2007). It expresses a flavorubredoxin (NorV), which reduces NO to N2O under anaerobic conditions (Gardner, Costantino, & Salzman, 1998; Gardner & Gardner, 2002; Gardner, Helmick, & Gardner, 2002; Spiro, 2012). It also expresses a flavohemoglobin (HmpA), and then utilizes O2 to convert NO to nitrate under aerobic conditions and reduces NO to N2O under anaerobic conditions (Householder, Fozo, Cardinale, & Clark, 2000; Kim, Orii, Lloyd, Hughes, & Poole, 1999; Poole, 2005; Watmough et al., 1999). However, the rate of NO reduction by HmpA is very low (Gardner & Gardner, 2002). Since NorV is sensitive to O2, NO reductase activity detoxifies NO under anaerobic conditions. Recent study suggested that the E. coli Hcp may be a high affinity NO reductase that is one of the enzymes used to reduce NO to N2O under anaerobic conditions (Wang et al., 2016). Since NorV, HmpA and Hcp in E. coli can detoxify NO throughout the physiological O2 range, E. coli is able to resist large amounts of NO. However, in EHEC O157, there are two types of norV genes, an intact norV and a 204 bp‐deleted norV (Gardner et al., 2002). The products of deleted norV did not exhibit NO reductase activity under anaerobic conditions (Shimizu, Tsutsuki, Matsumoto, Nakaya, & Noda, 2012). The intact norV‐type EHEC induced lower NO production and higher Stx2 production within macrophages than deleted norV‐type EHEC O157 (Shimizu et al., 2012). Therefore, intact norV‐type EHEC O157 showed a better level of survival than deleted norV‐type EHEC, suggesting that the intact norV was a direct virulence determinant of EHEC O157 under anaerobic conditions. Furthermore, an evolutionary analysis revealed that intact norV in EHEC O157 was strictly correlated with subgroup C cluster 1, and deleted norV was correlated with subgroup C clusters 2 and 3 (Shimizu, Hirai, Yokoyama, Ichimura, & Noda, 2015).
In this study, we examined the effect of NO on Shiga toxin production in EHEC O157 under anaerobic conditions. We elucidated the role of intact NorV in NO‐promoted Shiga toxin production. We also found that enhanced Stx1 and Stx2 production in response to NO was involved in Fur derepression and RecA activation in EHEC O157, respectively.
2. Materials and Methods
2.1. EHEC strains, plasmids, and oligonucleotides
The EHEC strains and plasmids used in this study are listed in Table 1. The oligonucleotides used for this study are shown in Table S1.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristics | Reference |
|---|---|---|
| EDL933 | EHEC O157, stx1, stx2, deleted norV | Perna et al. (2001) |
| E1‐E2S | EDL933, insertion of luxE in downstream of stxB1 | Shimizu et al. (2011) |
| E(SR)2‐E1S | EDL933, insertion of luxE in downstream of stxB2, deletion of S‐R in Stx2‐phage | Shimizu et al. (2011) |
| ERSA | EDL933, insertion of luxE in downstream of recA | This study |
| EVm | EDL933, replacement of intact norV | Shimizu et al. (2012) |
| K2 | EHEC O157, stx1, stx2, deleted norV | Shimizu et al. (2012) |
| K16 | EHEC O157, stx1, stx2, intact norV | Shimizu et al. (2012) |
| K24 | EHEC O157, stx1, deleted norV | Shimizu et al. (2012) |
| K42 | EHEC O157, stx2, deleted norV | Shimizu et al. (2012) |
| K43 | EHEC O157, stx2, intact norV | Shimizu et al. (2012) |
| K58 | E. coli O157, intact norV | Shimizu et al. (2012) |
| K15 | EHEC O157, stx1, stx2, intact norV | Shimizu et al. (2012) |
| K15(‐V) | K15, deletion of intact norV | Shimizu et al. (2012) |
| ERA1‐1 | EDL933, deletion of recA | This study |
| ERG22Y | EDL933, recA (G22Y) | This study |
| ERG24Y | EDL933, recA (G24Y) | This study |
| ERE123A | EDL933, recA (E123A) | This study |
| ERG204S | EDL933, recA (G204S) | This study |
| ERD224A | EDL933, recA (D224A) | This study |
| ERG288Y | EDL933, recA (G288Y) | This study |
| EP | EDL933, deletion of stx1 promoter | This study |
| EH | EDL933, deletion of hmpA | Shimizu et al. (2012) |
| EDLf | EDL933, deletion of fur | This study |
| E1Q1 | EDL933, deletion of Q of Stx1‐encoding phage | This study |
| DH5α (λpir) | recA1, endA1, gyrA96, thi‐1, hsdR17, supE44, relA1, deoR, (lacZYA‐argF)U169, ⌊pir + | Elliott and Kaper (1997) |
| pRed/ET (amp) | Red/ET expression plasmid, Ampr | Gene Bridges GmbH |
| placlux8 | LuxCDABE expression plasmid, Ampr, P lac | Shimizu et al. (2011) |
| pRPL3 | NO reporter plasmid, norR‐norV promoter‐luxCDABE fusion, Ampr | Shimizu et al. (2012) |
| pLCE19 | Suicide plasmid, luxE and loxP‐flanked Cmr cassette | Shimizu et al. (2011) |
| pluxCDAB3 | LuxCDAB expression plasmid, Ampr, P lac | Shimizu et al. (2011) |
| pluxStx1P2 | stx1 reporter plasmid, stx1 promoter‐luxCDABE fusion, Ampr | This study |
| pluxStx1PGG6 | Mutated stx1 reporter plasmid, stx1 promoter‐luxCDABE fusion, mutation of Fur box sequence, Ampr | This study |
| pFRT‐Kan | Suicide plasmid, FRT‐flanked PGK‐gb2 neo cassette | Gene Bridges GmbH |
| pFRT‐recA2 | Suicide plasmid, recA and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAG22Y1 | Suicide plasmid, recA (G22Y) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAG24Y1 | Suicide plasmid, recA (G24Y) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAE123A3 | Suicide plasmid, recA (E123A) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAG204S3 | Suicide plasmid, recA (G204S) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAD224A1 | Suicide plasmid, recA (D224S) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| precAG288Y1 | Suicide plasmid, recA (G288Y) and FRT‐flanked PGK‐gb2 neo cassette | This study |
| pFT‐A | Thermonsensitive FLP expression plasmid, Ampr | National BioResource Project (NIG, Japan) |
| pCreA1 | Thermonsensitive Cre expression plasmid, Ampr | Shimizu et al. (2011) |
| pTrcHis2A | Expression plasmid, Ampr, Ptrc | Invitrogen |
| pTrcHis2A‐fur | Fur‐His expression plasmid, Ampr, Ptrc | This study |
Ampr, Ampicillin‐resistant; Cmr, Chloramphenicol‐resistant.
2.2. Reagents and media
NO was generated by four NO donors, namely DETA‐NONOate (DETA/NO) (Cayman Chemical Company, MI, USA), NOC12 (Dojindo Laboratories, Kumamoto, Japan), Spermine‐NONOate (Sper/NO) (Cayman Chemical Company), and PROLI‐NONOate (PROLI/NO) (Cayman Chemical Company). When a heat‐inactivated DETA/NO was prepared, it was dissolved in hydrochloric acid solution (0.1 mol/L) and then incubated for 4 hr at 60°C. The NOS inhibitor N G‐Monomethyl‐L‐arginine (L‐NMMA) (Dojindo Laboratries) and an iron‐chelating agent, deferoxamine (Sigma‐Aldrich, MO, USA) were used. NaNO2 and NaNO3 were purchased from Wako (Tokyo, Japan). LB broth was dissolved in 10 g of Tryptone (Nacalai Tesque, Japan), 5 g of yeast extract (Nacalai Tesque), and 10 g of sodium chloride (Wako) in 1 L of DW, adjusted to pH 7.2 and autoclaved. Polyclonal antisera for Stx1 and Stx2 were prepared as described previously (Noda, Yutsudo, Nakabayashi, Hirayama, & Takeda, 1987; Yutsudo et al., 1987). The anti‐Stx1 and anti‐Stx2 antisera primarily reacted with the A subunit of Stx1 and Stx2, respectively. Anti‐RNA α and anti‐RecA antibodies were obtained from NeoClone Biotechnology International (Madison, WI, USA) and Bio Academia (Osaka, Japan).
2.3. Plasmid construction
1 stx1 promoter plasmids: stx1 promoter plasmids were constructed by using PCR to amplify a 214‐bp stx1 promoter DNA fragment from the genomic DNA of EHEC EDL933 using the primer set P1049 ‐ P1050. The DNA fragment was digested with NcoI–PvuII (Takara, Tokyo, Japan), and was then cloned into placlux8 (Shimizu, Ohta, Tsutsuki, & Noda, 2011) to yield the plasmid pluxStx1P2. In pluxStx1P2, the stx1 promoter fragment is placed upstream of the promoter‐less P. luminescens luxCDABE gene. To construct the mutated stx1 promoter plasmid, a PrimeSTAR Mutagenesis kit (Takara) was used with the plasmid DNA of pluxStx1P2 as a template and the primer set P1215 ‐ P1216 to yield the plasmid pluxStx1PGG6 according to the manufacturer's instructions. The construct was confirmed using restriction digestion and DNA sequencing.
2 Template plasmids for homologous recombination: To construct a template plasmid using a Red/ET recombination system for recA mutation, the recA gene was amplified by PCR from genomic DNA of the EDL933 strain using the primers P1077 and P1078. This fragment was cloned by ligation of a NotI fragment into pFRT‐Kan to generate plasmids pFRT‐recA2. Further, to construct template recA‐mutated plasmids, a PrimeSTAR Mutagenesis kit (TAKARA) was used with plasmid DNA of pFRT‐recA2 as a template and primer set (Table S1 and S2) to yield template recA‐mutated plasmids (Table 1). These constructs were confirmed using restriction digestion and DNA sequencing.
3 Fur expression plasmid: To construct plasmids expressing Fur, fur was amplified by PCR from the genomic DNA of the EHEC EDL933 strain using the primer set (P10011 and P715). This DNA fragment was cleaved with NcoI and SalI (Takara), and then was inserted into these sites in pTrcHis 2A (Invitrogen, NY, USA) to yield pTrcHis 2A‐fur expressing His‐tagged Fur. The construct was confirmed using restriction digestion and DNA sequencing.
2.4. Strain construction
The mutant EHEC strains were derivatives of the EDL933 strain and were obtained by using a Red/ET recombination system (Gene Bridges GmbH, Heidelberg, Germany). Briefly, PCR primers containing 50 bp immediately upstream and downstream, respectively, of the target sequence were used to amplify a DNA fragment containing the FRT‐flanked PGK‐gb2‐neo cassette (Kanr) found in appropriate plasmids (Table S3). The linear PCR product was electroporated into the appropriate parent strain which had earlier been transformed with plasmid pRed/ET (ampr).
To construct the stx1 promoter‐deficient EHEC strain EP, fur‐deficient EHEC strain EDLf, Q‐deficient EHEC strain E1Q1 and recA‐deficient EHEC strain ERA1‐1 (Table 1), a DNA fragment containing the Kanr cassette was obtained by PCR from pFRT‐Kan plasmid DNA using the primer sets P613‐P628, P724‐P725, P1064‐P600, and P10002‐P845, respectively. The PCR product was electroporated with the EDL933 previously transformed with plasmid pRed/ET.
To construct recA mutants (ERG22Y, ERG24Y, ERE123A, ERG204S, ERD224A and ERG288Y) (Table 1), the respective DNA fragments containing the mutated recA gene and Kanr cassette were obtained by PCR from the plasmid DNA of each template recA‐mutated plasmid using the primer set P1100 and P845. Each PCR product was electroporated with the ERA1‐1 previously transformed with plasmid pRed/ET. Recombinants containing Kanr in place of the target sequence were selected on Kan plates and confirmed by PCR. Deletion of Kanr elements was completed by transformation with plasmid pFT‐A. Kanamycin‐sensitive mutants were confirmed by PCR. The plasmids pRed/ET and pFT‐A are temperature‐sensitive and were removed by overnight growth at 37°C.
2.5. Culture conditions
For anaerobic culture, EHEC strains grown overnight [the optical density at 600 nm (OD600) = 1.0 ~ 1.5 × 109 cfu/ml] were diluted 1:100 with LB broth containing 10 mmol/L HEPES (pH 7.0) (LB‐pH 7.0) and grown statically at 37°C using Anaero Pack‐Anaero 5% (Mitsubishi Gas Chemical Company, Tokyo, Japan). For aerobic culture, EHEC strains grown overnight (OD600 = 1.0 ~ 1.5 × 109 cfu/ml) were diluted 1:100 with LB‐pH 7.0 and grown statically at 37°C under aerobic conditions.
2.6. Cell fractionation
Cells were pelleted by centrifugation at 20,630g for 5 min, and the supernatant obtained was used as the culture supernatant fraction. The pellet was suspended in an equal volume of ice‐cold PBS (pH 7.4) and sonicated for 15 s on ice. After sonication, the cell homogenate was centrifuged at 20,630g for 5 min, and the supernatant obtained was used as the cell‐associated fraction.
2.7. Kinetic analysis of NO release from NO donors under anaerobic conditions
Five microliters of various concentrations of NO donor solution was added to 5 ml of LB‐pH 7.0. To estimate NO release from NO donors, the NO concentration in LB‐pH 7.0 was measured using an amiNO‐2000 NO electrode (Innovative Instruments Inc., Tampa, FL, USA) at 37°C under anaerobic conditions in an anaerobic chamber. The anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI, USA) maintains a strict anaerobic (0–5 ppm) environment through a hydrogen gas mix reacting with a palladium catalyst to remove oxygen by forming a water molecule.
2.8. NO growth inhibition assay
EHEC strains grown overnight (OD600 = 1.0 ~ 1.5 × 109cfu/ml) were diluted 1:100 with LB‐pH 7.0 containing various concentrations of NO donor and grown statically for 18 hr at 37°C under anaerobic conditions. OD600 in culture was measured by a spectrophotometer (Ultrospec 3100 pro, GE Healthcare, USA).
2.9. Reporter assay
Reporter strains grown overnight (OD600 = 1.0 ~ 1.5 × 109 cfu/ml) were diluted 1:100 with LB‐pH 7.0 containing various concentrations of NO donor and grown statically for 18 hr at 37°C under anaerobic conditions. Relative light units (RLU) and the number of bacteria were measured by a GLOMAX 20/20 luminometer (Promega, Madison, USA) and bacteria plate counts (cfu), respectively.
2.10. Infection assay
RAW264.7 cells were obtained from the Riken Cell Bank (Tsukuba, Japan) and maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma‐Aldrich) supplemented with 10% fetal bovine serum (FBS) and a 1% antibiotic plus antimycotic solution (Sigma‐Aldrich). The RAW264.7 cells were seeded at 5 × 105 cells per well and then 5 × 106 bacteria of EHEC reporter strains were added to the monolayer per well. The plate was centrifuged briefly to synchronize the infection and then incubated for 20 min (0 hr) at 37°C under an atmosphere of 5% CO2. The medium was collected for the measurement of NO2 − derived from NO under aerobic conditions. The cells were washed and fresh DMEM‐10% FBS containing 100 μg/ml of gentamicin was added to kill the extracellular bacteria. After 2 hr at 37°C under an atmosphere of 5% CO2, the medium was collected for measurement of NO2 −, the cells were washed, and the medium was changed to include 12 μg/ml of gentamicin with or without a NOS inhibitor, L‐NMMA (4 mmol/L). The infected monolayers were either lysed from the tissue culture dishes by addition of PBS containing 0.1% deoxycholic acid or further incubated at 37°C under 5% CO2. The number of surviving bacteria and RLU were determined by cfu and luminometer, respectively. The concentrations of NO metabolite NO2 − in medium were determined by Griess assay.
2.11. SDS‐PAGE and Immunoblot analysis
Samples were subjected to SDS‐PAGE and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, USA). After blocking with 5% skim milk in TBS containing 0.1% Tween 20, the membranes were incubated with the indicated antibody, followed by incubation with HRP‐conjugated antirabbit IgG or HRP‐conjugated antimouse IgG (R&D Systems, USA). Antibody‐antigen complexes were detected using an ECL detection kit (SuperSignal® West Pico Chemiluminescent Substrate kit, Thermo Scientific) and an LAS‐1,000 luminescent image analyzer (Fujifilm, Tokyo, Japan). Densitometric analysis was performed by Image Gauge software (Fujifilm), and protein levels were normalized to the α subunit of RNA polymerase (RNA α).
2.12. Gb3 receptor ELISA
Porcine erythrocyte Gb3 (Nacalai Tesque) was coated on microtiter plate wells (C96 Maxisorp, Nunc‐immuno plate; Nalge Nunc International, Rochester, NY) by evaporation from an ethanolic solution. A 100‐μl aliquot of ethanolic Gb3 (2 μg/ml for Stx1 and 8 μg/ml for Stx2) was added per microtiter plate well in triplicate, and the ethanol was allowed to evaporate at room temperature for 6 hr. Each well was blocked with 200 μl of 0.2% (w/v) BSA in PBS (BSA‐PBS) for 6 hr and washed twice with 200 μl/well BSA‐PBS. Dilutions of standard Stxs were prepared in BSA‐PBS (Shimizu et al., 2007). The solution was dispensed into the wells and incubated overnight at 4°C. The wells were emptied and washed three times (1 min each) with 200 μl of BSA‐PBS. Then, 100 μl of diluted rabbit antiserum against Stx1 or antiserum against Stx2 in BSA‐PBS was added to the wells for 1 hr at room temperature. The wells were washed as before, then diluted HRP‐conjugate anti‐rabbit IgG in BSA‐PBS was added to the wells and left to sit for 1 hr at room temperature. The substrate, 2,2′‐azino‐bis‐(3‐ethylbenzthiazoline‐6‐sulphonic) diammonium salt (ABTS), was dissolved in 0.1 mol/L citric acid (pH 4.35) at 0.3 mg/ml. A volume of 8.75 μl of 30% H2O2 was added per 10 ml of solution. The wells were emptied and washed three times with 200 μl of BSA‐PBS for 3 min each. Finally, the wells were washed once with PBS, and 100 μl/well of ABTS solution was added to the plate, which was then shaken gently and placed in the dark. After sufficient color had developed (usually 40–60 min), the absorbance of each well at 415 nm was determined using an ELISA plate reader.
2.13. Bacterial mRNA analysis
Total RNA was isolated by ISOGEN II (Wako), and the concentration was determined by measuring the A260 value. Subsequently, 0.5 μg of RNA from each sample was reverse‐transcribed by using PrimeScript™ II 1st strand cDNA Synthesis Kit (Takara) according to the manufacturer's protocol. To obtain standard curves for the hmpA, fur, and tufA genes, genomic DNA from EHEC EDL933 was 10‐fold serially diluted from 1.0 × 105 to 0.1 cfu/2 μl and amplified in the 7,300 Real‐Time PCR System (Applied Biosystems, CA, USA) with the primers (P16 ‐ P17), (P1243 – P1244) or (P890 ‐ P891) using the Power SYBER Green PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol, respectively. Serial dilutions of cDNA were amplified in the 7300 Real‐Time PCR System under the same conditions as for the standard curves with the primers (P16 ‐ P17), (P1243 – P1244), or (P890 ‐ P891). The levels of hmpA, fur, and tufA mRNA were quantified by noting the fluorescence crossing point of the samples on the corresponding standard curve, and the results are presented as the ratio among the expression levels of hmpA mRNA, fur mRNA, and tufA mRNA.
2.14. stx1 promoter assay
Reporter strains grown overnight (OD600 = 1.0 ~ 1.5 × 109 cfu/ml) were diluted 1:100 with LB‐pH 7.0 and grown for 2 hr at 37°C, and then 100 μmol/L PROLI/NO or 200 μmol/L deferoxamine was added. Further growth was allowed statically for 20 min at 37°C under anaerobic conditions. RLU and the number of bacteria were measured by a luminometer and bacteria plate counts (cfu).
2.15. Statistics
Student's t‐test was used to determine significant differences when only two treatment groups were being compared. One‐way ANOVA with Student‐Newman‐Keuls multiple comparisons test was used to analyze significant differences among multiple groups.
3. Results
3.1. NO inhibits anaerobic growth of the deleted norV‐type EHEC
The steady‐state concentration of NO depends on both the rate of NO production and rate of NO consumption. NO release from NO donors is solely dictated by temperature and pH level (Thomas et al., 2002), whereas NO consumption depends on the NO reaction with oxygen. To quantitate the precise NO production levels within our assay system, the concentration of NO in LB broth (pH 7.0) treated with different amounts of NO donor was measured using an NO electrode over time at 37°C under anaerobic conditions. The results showed that, under treatment with 200 μmol/L DETA‐NONOate (DETA/NO), the maximum steady‐state concentration of NO in LB medium was 12 μmol/L after 4 hr (Figure 1a). However, the maximum steady‐state concentration of NO was 29 μmol/L after 5 hr of treatment with 400 μmol/L DETA/NO (Figure 1a). Similarly, the maximum steady‐state concentrations of NO in LB medium under the treatments with 100 μmol/L PROLI‐NONOate (PROLI/NO), 200 μmol/L NOC12, and 400 μmol/L Spermine‐NONOate (Sper/NO) were 117 μmol/L after 1.8 min, 25 μmol/L after 1.7 hr, and 140 μmol/L after 0.8 hr, respectively (Figure 1a). Table 2 provides a summary of the peak NO concentration and the time in treatment with various NO donors.
Figure 1.
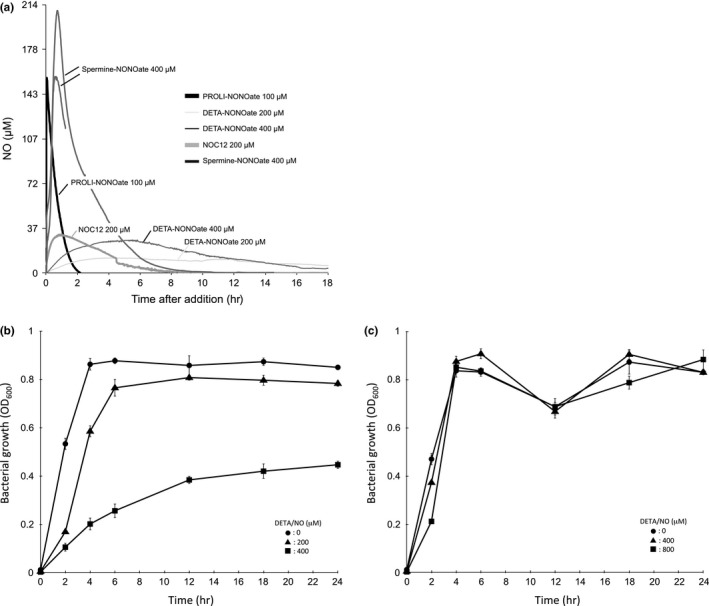
Real‐time quantification of NO concentration and growth of EHEC in the presence of various NO donors under anaerobic conditions. (a) The NO steady‐state levels in LB broth (pH 7.0) in the presence of various NO donors were measured using an amiNO‐2000 NO electrode at 37°C under anaerobic conditions. Representative NO electrode data are shown (n = 3). (b, c) EHEC strains grown overnight were diluted with LB broth containing various concentrations of NO donor and grown statically for 24 hr at 37°C under anaerobic conditions. The OD 600 in culture was measured by spectrophotometer at the indicated times. Data are the means ± standard deviations of values from three experiments. Results are shown for the deleted norV‐type wild EHEC EDL933 (b) and the intact norV‐type wild EHEC K15 (c).
Table 2.
Peak NO concentration and its time in treatment with various NO donors
| Donor (μmol/L) | Peak NO (μmol/L) | Peak time (hr) | |
|---|---|---|---|
| DETA‐NONOate | 200 | 12 | 4.1 |
| 400 | 29 | 4.9 | |
| NOC12 | 200 | 25 | 1.7 |
| 400 | 53 | 2.2 | |
| Spermine‐NONOate | 200 | 29 | 1.2 |
| 400 | 140 | 0.8 | |
| PROLI‐NONOate | 100 | 117 | 0.03 |
Representative data are shown as the mean (n = 3).
The intact norV in EHEC O157 played an important role in protecting the anaerobic growth from NO‐mediated growth inhibition (Shimizu et al., 2012). Therefore, to confirm the growth inhibition of EHEC O157 by NO under anaerobic conditions, the deleted norV‐type wild EHEC EDL933 and the intact norV‐type wild EHEC K15 were cultured in LB broth with various concentrations of DETA/NO at 37°C. In the deleted norV‐type EHEC EDL933, the bacterial growth (OD600) was significantly decreased in the presence of 400 μmol/L DETA/NO from 2 hr to 24 hr of treatment compared to that of untreated cells (Figure 1b). When we used 200 μmol/L DETA/NO, the growth of EHEC EDL933 was not inhibited from 12 hr to 24 hr (Figure 1b). In contrast, the bacterial growth of the intact norV‐type EHEC K15 was not decreased in the presence of 800 μmol/L DETA/NO from 4 hr to 24 hr under anaerobic conditions (Figure 1c). The deleted norV‐type and the intact norV‐type EHEC demonstrated different threshold sensitivities of anaerobic growth inhibition to NO.
3.2. NO increases Shiga toxin production in EHEC under anaerobic conditions
Next, we investigated the effect of NO on Shiga toxin production in the deleted norV‐type EHEC O157 by Immunoblot analysis. Stx1 has been shown to be predominantly located in the cell‐associated fraction, while Stx2 is mainly found in the extracellular fraction (Shimizu et al., 2009). In the presence of 400 μmol/L DETA/NO, Stx1 production in the cell‐associated fraction was increased as compared to that of the control during 2–24 hr incubation (Figure 2a). However, treatment with 200 μmol/L DETA/NO did not enhance Stx1 production (Figure 2b). On the other hand, addition of 200 μmol/L DETA/NO to the bacterial culture enhanced Stx2 production in the culture supernatant from 6 hr to 24 hr (Figure 2c). However, when 20 μmol/L DETA/NO was added, Stx2 productions were not enhanced (Figure 2d). The enhancements of Stx1 and Stx2 production in the deleted norV‐type EHEC at the late‐stationary phase were sufficient for exposure to NO at the log phases (Fig.S1). When Gb3‐ELISA was used for quantitative analysis of Stx1 and Stx2, Stx1 production in the cell‐associated fraction of the deleted norV‐type EHEC was found to be increased by two–threefold and Stx2 production in the culture supernatant of the deleted norV‐type EHEC was increased by ~fivefold as compared to that of the control (Fig. S2). Moreover, transcriptional analysis using C‐P reporter strains revealed that the transcriptional levels of stx1 and stx2 in the deleted norV‐type EHEC were also enhanced in the treatment with 400 and 200 μmol/L DETA/NO at 18 hr under anaerobic conditions, respectively (Fig. S3). These findings indicated that a high concentration of NO (12–29 μmol/L, maximum steady‐state concentration) is required for enhanced Stx1 production and a low concentration of NO (~12 μmol/L, maximum steady‐state concentration) is sufficient for enhanced Stx2 production in the deleted norV‐type EHEC under anaerobic conditions.
Figure 2.
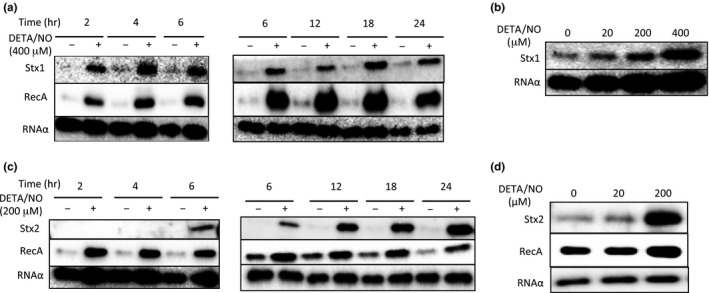
NO enhances Shiga toxin production and RecA expression in the deleted norV‐type EHEC under anaerobic conditions. EHEC EDL933 grown overnight was diluted with LB broth containing DETA‐NONOate (DETA/NO) and then grown statically at 37°C under anaerobic conditions. (a) EHEC strains were fractionated into the cell‐associated fractions at the indicated times. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐Stx1 antiserum, anti‐RecA antibody, and anti‐RNA α antibody as an internal control. (b) EHEC strains were fractionated into the cell‐associated fractions at 18 hr. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐Stx1 antiserum and anti‐RNA α antibody as an internal control. (c) EHEC strains were fractionated into the cell‐associated fractions and the culture supernatant fractions at the indicated times. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐RecA antibody and anti‐RNA α antibody as an internal control. Each volume, which corresponds to 0.2 μg of protein of the cell‐associated fraction, of the supernatant fraction was analyzed by Immunoblot analysis using anti‐Stx2 antiserum. (d) EHEC strains were fractionated into the cell‐associated fraction and the culture supernatant fractions at 18 hr. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐RecA antibody and anti‐RNA α antibody as an internal control. Each volume, which corresponds to 0.2 μg of protein of the cell‐associated fraction, of the supernatant fraction was analyzed by Immunoblot analysis using anti‐Stx2 antiserum.
The expression of Shiga toxin in EHEC is regulated through induction of the integrated bacteriophage that encodes the Shiga toxin genes. The inductions of Stx‐encoded phages and productions of Shiga toxin are linked to induction of the SOS response, a ubiquitous response to DNA damage (Kimmitt, Harwood, & Barer, 2000). DNA damage activates the bacterial SOS response, leading to activation and upregulation of RecA (Walker, 1984). Thus, we investigated the effect of NO on the expression level of RecA in the deleted norV‐type EHEC under anaerobic conditions. When we incubated the deleted norV‐type EHEC in a culture containing 200 μmol/L DETA/NO, the expression level of RecA was increased (Figure 2d). However, in the treatment with 20 μmol/L DETA/NO, the amounts of RecA were not enhanced (Figure 2d). Moreover, transcriptional analysis using C‐P reporter strains revealed that the transcriptional levels of recA were enhanced in the treatment with 200 and 400 μmol/L DETA/NO at 18 hr under anaerobic conditions (Fig. S3).
To further confirm the ability of NO in the deleted norV‐type EHEC to enhance Stx1 and Stx2 production, we examined the effects of other NO donors and an inactive NO donor on Shiga toxin production under anaerobic conditions. In the presence of two kinds of NO donors, NOC12 or Spermine‐NONOate (Sper/NO), both Stx1 and Stx2 production in the cell‐associated fractions and the culture supernatant fractions of the deleted norV‐type EHEC were increased in a dose‐dependent manner (Figure 3a). Moreover, a heat‐inactivated NO donor did not enhance Stx1 and Stx2 production in the deleted norV‐type EHEC (Figure 3b). We considered the possibility that other intermediates of denitrification might induce Shiga toxin production in addition to NO under anaerobic conditions. The nitrate and nitrite of NO metabolites did not enhance Shiga toxin production (Figure 3c, 3d). These results indicated that NO, not intermediates, acts to enhance Stx1 and Stx2 production in the deleted norV‐type EHEC under anaerobic conditions.
Figure 3.
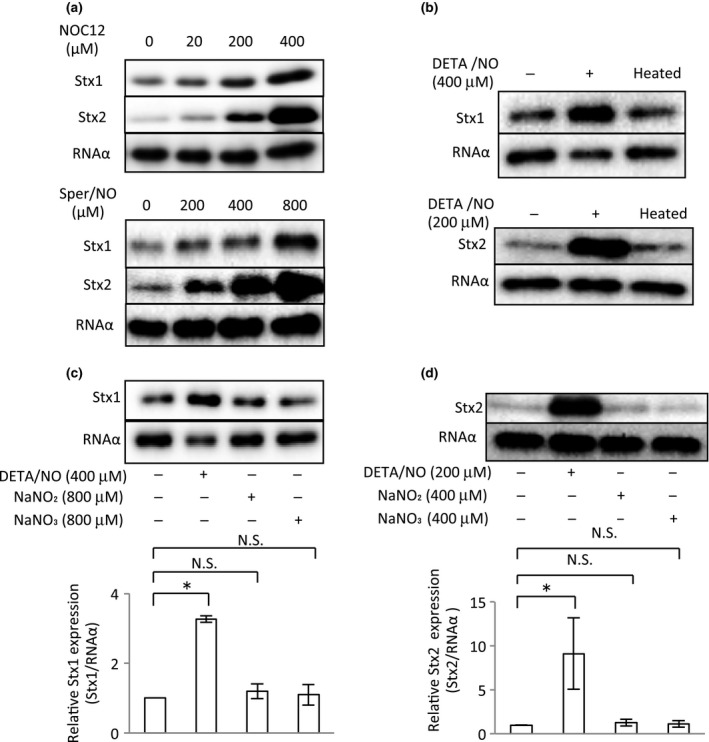
Effect of two kinds of NO donors, inactivated NO donor and NO metabolite, on Shiga toxin production in the deleted norV‐type EHEC under anaerobic conditions. EHEC EDL933 grown overnight were diluted with LB broth containing various concentrations of NO donors [NOC12 (a) or Spermine‐NONOate (Sper/NO) (a)], inactivated NO donor (b) or NO metabolite (c, d) and then grown statically for 18 hr at 37°C under anaerobic conditions. The culture supernatant fractions and cell‐associated fractions from the culture of EHEC strains were collected. The cell‐associated fractions were analyzed by Immunoblot analysis using anti‐Stx1 antiserum and anti‐RNA α antibody as an internal control. The culture supernatant fractions were analyzed by Immunoblot analysis using anti‐Stx2 antiserum. The relative amounts of Stx1 and Stx2 were quantified by densitometry and normalized to internal control RNA α . Data are the means ± standard deviations of values from four experiments. *p < 0.01; N. S., not significant.
In previous studies, NO has been shown to inhibit Stx2 production in the deleted norV‐type EHEC under aerobic conditions (Vareille et al., 2007). Next, we examined the effect of growth of the deleted norV‐type wild EHEC EDL933 in treatment with NO under aerobic conditions. The growth was inhibited by around 40% with 800 μmol/L DETA/NO (Figure 4a), whereas the EHEC growth was inhibited by 50% with 400 μmol/L DETA/NO under anaerobic conditions (Figure 1b). This result suggested that the NO level in bacterial cells treated with 400 μmol/L DETA/NO under anaerobic conditions was higher than that in bacterial cells treated with 800 μmol/L DETA/NO under aerobic conditions. To confirm this, we determined the NO level in bacterial cells treated with an NO donor under aerobic and anaerobic conditions using a novel NO reporter plasmid, pRPL3 (Shimizu et al., 2012). In the deleted norV‐type EDL933 (pRPL3), the specific luminescence (RLU/cfu) of the reporter under the treatment with 800 μmol/L DETA/NO after 18 hr was 0.83 ± 0.07 under aerobic conditions (Figure 4a). In contrast, the specific luminescence (RLU/cfu) of the reporter by the treatment with 400 μmol/L DETA/NO was 1.15 ± 0.04 under anaerobic conditions (Figure 5c). These results indicated that the level of NO in the deleted norV‐type EHEC EDL933 by treatment with 800 μmol/L DETA/NO under aerobic conditions was lower than that by treatment with 400 μmol/L DETA/NO under anaerobic conditions. Next, we examined the effect of Stx2 production in response to NO under aerobic conditions. In the presence of 800 μmol/L DETA/NO, the production of Stx2 in the deleted norV‐type wild EHEC was increased (Figure 4b). When the deleted norV‐type wild EHEC was used under aerobic conditions, NO did not inhibit Stx2 production in the presence of 100 μmol/L DETA/NO (Figure 4b). NorV and Hcp in E. coli have NO reductase activity under anaerobic conditions (Shimizu et al., 2012; Wang et al., 2016). Under aerobic conditions, HmpA in E. coli has been shown to degrade NO to reduce the NO concentration in the aerobic environment (Gardner & Gardner, 2002; Kim et al., 1999). We next analyzed the effect of NO treatment on the aerobic growth and Stx2 production in the deleted norV‐type hmpA‐deficient mutant EH. The expression levels of hmpA mRNA in the deleted norV‐type wild EHEC were increased twofold by the treatment with 100 and 800 μmol/L DETA/NO (Figure 4b). Although aerobic growth was inhibited in the treatment with more than 100 μmol/L DETA/NO, Stx2 production was increased in the treatment with 800 μmol/L DETA/NO (Figure 4b). In the treatment with 100 μmol/L DETA/NO, Stx2 production was repressed in the deleted norV‐type hmpA‐deficient mutant EH (Figure 4b). Therefore, when EHEC were exposed to NO, at lower NO concentrations this exposure promoted the repression of Stx2 production, whereas at higher levels it tended to enhance Stx2 production.
Figure 4.
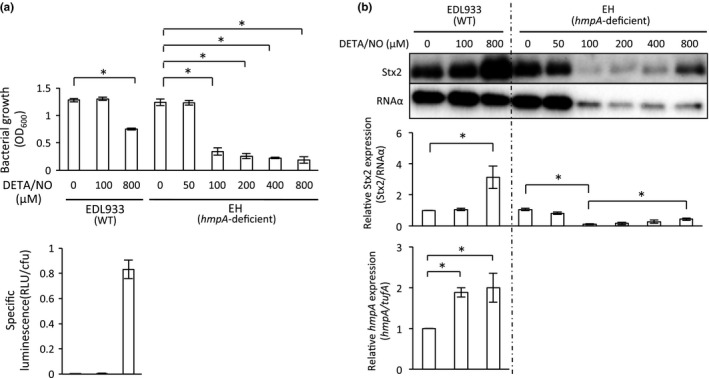
Effect of NO on Stx2 production in wild and hmpA‐deficient mutant EHEC strains under aerobic conditions. The deleted norV‐type wild EDL933 and the deleted norV‐type hmpA‐deficient EH grown overnight were diluted with LB broth containing various concentrations of DETA‐NONOate (DETA/NO) and grown statically for 18 hr at 37°C under aerobic conditions. (a) The optical density at 600 nm (OD 600) was determined. NO level in the deleted norV‐type wild EDL933 in medium. EHEC strains harboring the NO reporter plasmid pRPL3 were cultured in LB medium containing a various concentrations of DETA/NO for 18 hr at 37°C under aerobic conditions. Relative light units (RLU) and the number of bacteria were measured by a luminometer and bacteria plate counts (cfu), respectively. Data are the means ± standard deviations of values from three experiments. *p < 0.01. (b) EHEC strains were fractionated into the supernatant fraction and cell‐associated fractions. They were then analyzed by Immunoblot analysis using anti‐Stx2 antiserum and anti‐RNA α antibody, respectively. The relative amounts of Stx2 were quantified by densitometry and normalized to internal control RNA α. The inductions of hmpA in wild‐type EHEC were analyzed by real‐time qRT‐PCR. Data are the means ± standard deviations of values from five experiments. *p < 0.01
Figure 5.
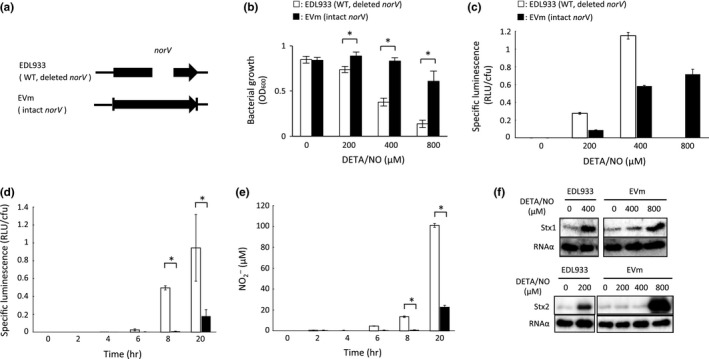
Role of intact NorV in NO‐mediated anaerobic growth inhibition, NO level in bacterial cells and Shiga toxin production in EHEC under anaerobic conditions. (a) Gene structure of the norV in the deleted norV‐type wild EHEC EDL933 and the intact norV‐replacement mutant EHEC EVm. (b) Comparison of NO‐mediated anaerobic growth inhibition between the deleted norV‐type EDL933 and the intact norV‐type EVm. EHEC strains were cultured in LB medium containing various concentrations of DETA‐NONOate (DETA/NO) for 18 hr at 37°C under anaerobic conditions. The optical density at 600 nm (OD 600) was measured. Data are the means ± standard deviations of values from five experiments. *p < 0.01. (c) Comparison of the NO level in bacterial cells in medium between the deleted norV‐type EDL933 and the intact norV‐type EVm. EHEC strains harboring the NO reporter plasmid pRPL3 were cultured in LB medium containing various concentrations (200 or 400 μmol/L for EDL933; 200, 400 or 800 μmol/L for EVm) of DETA/NO for 18 hr at 37°C under anaerobic conditions. Relative light units (RLU) and the number of bacteria were measured by a luminometer and bacteria plate counts (cfu), respectively. Data are the means ± standard deviations of values from three experiments. (d, e) NO level in the deleted norV‐type EDL933 within macrophages and the concentration of the NO metabolite NO − 2 in the culture medium of infected macrophages. The EHEC strain harboring the NO reporter plasmid pRPL3 was added to the monolayer of RAW264.7 cells, and incubated for 20 min (0 hr). The medium was changed to include 100 μg/ml gentamicin. After 2 hr, the cells were washed, and the medium was changed to include 12 μg/ml gentamicin with (black) or without (white) 4 mM NOS inhibitor, L‐NMMA. The infected monolayers were either lysed or further incubated. The RLU and number of surviving bacteria were determined by luminometry and bacteria plate counts (cfu) (d). The concentrations of NO metabolite NO − 2 in medium were determined by Griess assay (e). Data are the means ± standard deviations of values from three experiments. *p < 0.01. (f) Comparison of NO‐enhanced Shiga toxin production between the deleted norV‐type EDL933 and the intact norV‐type EVm. EHEC strains were cultured in LB medium containing various concentrations of DETA/NO for 18 hr at 37°C under anaerobic conditions. EHEC strains were fractionated into culture supernatant fractions and cell‐associated fractions. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐Stx1 antiserum and anti‐RNA α antibody as an internal control. Each volume, which corresponds to 0.2 μg of protein of the cell‐associated fraction, of the supernatant fraction was analyzed by Immunoblot analysis using anti‐Stx2 antiserum.
3.3. Shiga toxin production in EHEC is influenced by NO reductase in the presence of NO under anaerobic conditions
To clarify the function of NorV in EHEC O157 under anaerobic conditions, we investigated the effect of DETA/NO on anaerobic growth in the deleted norV‐type wild EHEC and intact norV‐replacement mutant EHEC (Figure 5a). The deleted norV‐type wild EDL933 displayed a reduction of anaerobic growth in response to 400 μmol/L DETA/NO (Figure 5b). In contrast, the growth of the intact norV‐type EVm was not inhibited by 800 μmol/L DETA/NO (Figure 5b). We next determined the NO level in bacterial cells treated with an NO donor under anaerobic conditions. In the deleted norV‐type EDL933 (pRPL3), the specific luminescence (RLU/cfu) of the reporter in the treatments with 200 and 400 μmol/L DETA/NO after 18 hr were 0.28 ± 0.01 and 1.15 ± 0.04, respectively, under anaerobic conditions (Figure 5c). In the intact norV‐type EVm (pRPL3), in contrast, the specific luminescence (RLU/cfu) of the reporter in the treatments with 200, 400, and 800 μmol/L DETA/NO were 0.087 ± 0.003, 0.58 ± 0.01, and 0.72 ± 0.06, respectively, under anaerobic conditions (Figure 5c). These results indicated that the deleted norV‐type EHEC EDL933 produced a higher level of NO within bacterial cells compared with the intact norV‐type EHEC EVm during treatment with the same concentrations of NO donor. We also determined the NO level within macrophages of the deleted norV‐type EHEC EDL933 using this NO reporter system. The results showed that both the specific luminescence (RLU/cfu) of the reporter strain within RAW264.7 cells and the concentration of the NO metabolite NO2 − in the culture medium of infected RAW264.7 cells began to increase after 6 hr postinfection and reached a high level of specific luminescence (0.95 ± 0.37 RLU/cfu) and a high concentration of NO (101 ± 2 μmol/L) at 20 hr postinfection (Figure 5d, 5e). To investigate that the specific luminescence (RLU/cfu) of the reporter strain corresponded to the NO level within macrophages, we used N G‐Monomethyl‐L‐arginine (L‐NMMA), which is a specific inhibitor for the NO production of NO‐synthase (NOS) in macrophages. Treatment of 4 mmol/L L‐NMMA did not significantly increase either the specific luminescence (RLU/cfu) of the reporter strain or the concentration of NO2 − in the culture medium at 20 hr postinfection (Figure 5d, 5e). The specific luminescence (RLU/cfu) of the deleted norV‐type NO reporter EHEC EDL933 within RAW264.7 cells at 20 hr postinfection was 0.95 ± 0.37, similar to the level (1.15 ± 0.04) observed with 400 μmol/L DETA/NO after 18 hr of incubation under anaerobic conditions, suggesting that the NO level in the deleted norV‐type EHEC EDL933 within RAW264.7 cells was equivalent to that in treatment with 400 μmol/L DETA/NO under anaerobic conditions. These findings suggest that the enhancements of Shiga toxin production in EHEC by NO might occur under physiological conditions, such as within activated macrophages during infection.
Next, to investigate that NO reductase activity suppressed the enhancement of Shiga toxin production by NO under anaerobic conditions, we examined the effect of intact NorV on the NO‐induced Shiga toxin production. The level of production of Stx1 in the deleted norV‐type wild EDL933 was increased in the culture using 400 μmol/L DETA/NO (Figure 5f). In the intact norV‐type EVm, Stx1 production was not increased in the culture with 400 μmol/L DETA/NO (Figure 5f). In the same way, Stx2 production was also enhanced by 200 μmol/L DETA/NO in the deleted norV‐type EHEC EDL933 (Figure 5f). However, NO‐enhanced Stx2 production in the intact norV‐type EVm was required for the high concentration of NO donor (800 μmol/L DETA/NO) (Figure 5f).
NO reductase activity of NorV under anaerobic conditions is stronger than that under aerobic conditions. We next examined whether NO reductase activity would affect the NO‐induced enhancement of Shiga toxin production under aerobic conditions. The results showed that the roles played by NO and NO reductase in EHEC for the productions of Stx1 and Stx2 were generally consistent with those under anaerobic conditions, although the effective concentrations of NO donor were much higher than those under aerobic conditions (Fig. S4).
In this study, EDL933, a deleted norV‐type strain of EHEC, was used as the parent EHEC, and also we used the intact norV‐type wild EHEC K15 as a parent strain (Figure 6a). We next investigated the effect of NO reductase in EHEC on Stx1 and Stx2 productions under anaerobic conditions using wild type K15 and the norV‐deficient K15(‐V) strains. Growth in the intact norV‐type EHEC K15 was not inhibited even in the presence of 1200 μmol/L DETA/NO (Figure 6b). However, the norV‐deficient K15(‐V) dramatically decreased anaerobic growth in response to 400 μmol/L DETA/NO (Figure 6b). The intact norV‐type EHEC K15 produced a lower level of NO within bacterial cells compared with the norV‐deficient K15(‐V) following treatment with the same concentrations of DETA/NO (200 or 400 μmol/L) (Figure 6c).
Figure 6.
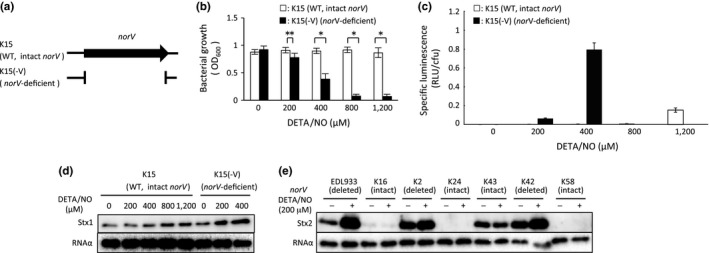
Comparison of NO‐mediated anaerobic growth inhibition, the NO level in bacterial cells and NO‐induced Shiga toxin production between intact norV‐type EHEC and norV‐deficient EHEC under anaerobic conditions. (a) Gene structure of the norV in the intact norV‐type wild EHEC K15 and norV‐deficient mutant EHEC K15(‐V). (b) Comparison of NO‐mediated anaerobic growth inhibition between the intact norV‐type K15 and norV‐deficient K15(‐V). EHEC strains were grown with LB medium containing various concentrations of DETA‐NONOate (DETA/NO) for 18 hr at 37°C under anaerobic conditions. The optical density at 600 nm (OD 600) was monitored. Data are the means ± standard deviations of values from five experiments. *p < 0.01; **p < 0.05. (c) Comparison of the NO level in bacterial cells in medium between the intact norV‐type K15 and the norV‐deficient K15(‐V). EHEC strains harboring the NO reporter plasmid pRPL3 were cultured in LB medium containing various concentrations [200, 400, 800, or 1200 μmol/L for K15; 200 or 400 μmol/L for K15(‐V)] of DETA/NO for 18 hr at 37°C under anaerobic conditions. Relative light units (RLU) and the number of bacteria were measured by a luminometer and bacteria plate counts (cfu), respectively. (d) Comparison of NO‐enhanced Shiga toxin production between the intact norV‐type K15 and norV‐deficient K15(‐V). EHEC strains grown containing various concentrations of DETA/NO for 18 hr at 37°C under anaerobic conditions. EHEC strains were fractionated into culture supernatant fractions and cell‐associated fractions. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐Stx1 antiserum and anti‐RNA α antibody as an internal control. (e) E. coli strains were cultured with LB broth containing 200 μmol/L DETA/NO for 18 hr at 37°C under anaerobic conditions. EHEC strains were fractionated into culture supernatant fractions and cell‐associated fractions. Each volume, which corresponds to 0.2 μg of protein of the cell‐associated fraction, of the supernatant fraction was analyzed by Immunoblot analysis using anti‐Stx2 antiserum. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐RNA α antibody as an internal control.
Stx1 production in intact norV‐type EHEC K15 was only increased in the presence of 1200 μmol/L DETA/NO (Figure 6d). In the norV‐deficient K15(‐V), Stx1 production was increased by culture with 400 μmol/L DETA/NO (Figure 6d). Next, to confirm that the enhancement of Stx2 production in EHEC by the addition of an NO donor was suppressed by intact NorV under anaerobic conditions, we examined the enhancement of Stx2 production in four additional Stx2‐producing EHEC strains by Immunoblot analysis. When EHEC were incubated with 200 μmol/L DETA/NO, the deleted norV–type wild EHEC strains (K2, K42) exhibited the enhancement of Stx2 production (Figure 6e). The intact norV‐type wild EHEC strains (K16, K43) did not show enhanced Stx2 production in the culture containing 200 μmol/L DETA/NO (Figure 6e).
3.4. Mechanism of the enhancement of Stx1 production by NO under anaerobic conditions
With respect to the Stx1 expression in EHEC, there are two types of promoters in the Stx1‐encoding phage of EHEC (Shimizu et al., 2009). The first is a P Stx1, which is adjacent to the stx1 (not the stx2) and regulated by the environmental iron concentration in EHEC (Calderwood & Mekalanos, 1987). A functional Fur consensus box exists in the P Stx1 region (Calderwood & Mekalanos, 1987, 1988). Fur is a global repressor that is regulated by iron (Hantke, 2001). The second type of promoter is transcribed from the P R' of the Stx1‐encoding phage, which is important for Stx1 production (Wagner et al., 2002). Therefore, to examine whether the P Stx1 or the P R' of the Stx1‐encoding phage regulates NO‐induced Stx1 production under anaerobic conditions, we constructed Q of the Stx1‐encoding phage‐deficient mutant E1Q1 and the P Stx1‐deficient mutant EP. Since Q of the Stx1‐encoding phage is a late antiterminator gene (Wagner & Waldor, 2002; Wagner et al., 2002), induction of the Stx1‐encoding phage in E1Q1 could not occur. In the Q‐deficient E1Q1, the expression of Stx1 was increased by the NO‐donor treatment to the same degree as in the wild‐type strain (Figure 7a). In the P Stx1‐deficient mutant, however, the level of Stx1 production was much lower than that in the wild‐type EHEC (Figure 7a). These results indicated that the enhancement of Stx1 production was related with the P Stx1, which was regulated by the iron regulator Fur. To determine whether NO treatment affected the fur expression in EHEC, we analyzed the expression levels of fur mRNA. The expression level of fur in EHEC at log phase in treatment with 400 μmol/L DETA/NO was equivalent to that of the control under anaerobic conditions (Figure S5). Next, to investigate that derepression of Fur contributes to the enhancement of Stx1 production in EHEC treated with an NO donor, we constructed the stx1 reporter plasmid pluxStx1P2 (Figure 7b). The P Stx1 activity (RLU/cfu) of EDL933 harboring the stx1 reporter plasmid pluxStx1P2, EDL933 (pluxStx1P2), was increased in treatment with either 100 μmol/L PROLI‐NONOate (PROLI/NO) or 200 μmol/L of an iron chelator, deferoxamine (Figure 7c). The P Stx1 activity (RLU/cfu) of the fur‐deficient mutant EDLf (pluxStx1P2) was increased at the basal level. But, it was not enhanced following treatment with either 100 μmol/L PROLI/NO or 200 μmol/L deferoxamine alone (Figure 7c). When we used the Fur box‐mutated reporter plasmid pluxStx1PGG6, the level of the P Stx1 activity (RLU/cfu) of EDL933 (pluxStx1PGG6) was similar to those of EDL933 (pluxStx1PGG6) in treatment with 100 μmol/L PROLI/NO or 200 μmol/L deferoxamine, respectively (Figure 7b, c). These results indicated that the inhibition of Fur binding to the Fur box in the P Stx1 region functions to enhance the P Stx1 activity in response to NO.
Figure 7.
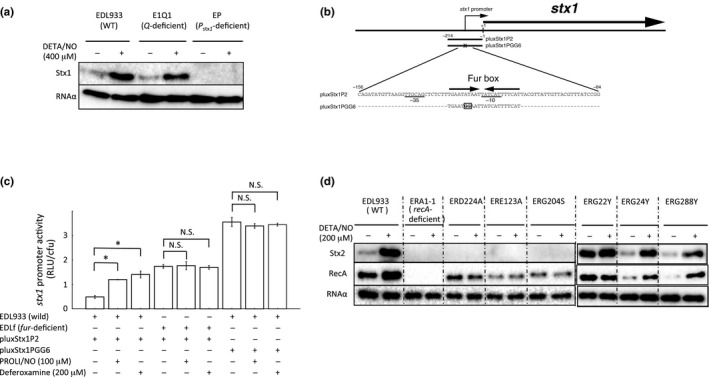
Effects of NO on Stx1 and Stx2 production in various mutant EHEC under anaerobic conditions. (a) Effects of NO on Stx1 production in wild EHEC EDL933, Q‐deficient mutant EHEC E1Q1 and P stx1‐deficient mutant EHEC EP under anaerobic conditions. EHEC were cultured in medium containing 400 μmol/L DETA‐NONOate (DETA/NO) for 18 hr at 37°C under anaerobic conditions. EHEC were fractionated into cell‐associated fractions. A 0.2 μg sample of each protein of the cell‐associated fraction was analyzed by Immunoblot analysis using anti‐Stx1 antiserum and anti‐RNA α antibody as an internal control. (b) Promoter sequence of stx1 and schematic representation of various stx1 promoter‐luxCDABE fusion genes utilized in the mutation analysis. The arrows indicate a Fur box, and the boxed region within the Fur box is a mutated site. The −35 and −10 regions of the proposed promoter are underlined. The number indicates the nucleotide position with base pairs upstream of the start codon (+1) of stx1. (c) Comparison of the specific luminescence of EDL933 harboring an stx1 reporter plasmid, pluxStx1P2, fur‐deficient EDLf harboring pluxStx1P2 and EDL933 harboring a mutated stx1 reporter plasmid, pluxStx1PGG6. Reporters were cultured in LB broth supplemented with or without 100 μmol/L PROLI‐NONOate (PROLI/NO) or 200 μmol/L deferoxamine at 37°C and then were collected after 20 min for the estimation of specific luminescence. Data are the means ± standard deviations of values from three experiments. *p < 0.01; N.S., not significant. (d) Effects of NO on Stx2 production in the EHEC EDL933, recA‐deficient mutant EHEC ERA1‐1 and recA point‐mutant EHEC strains under anaerobic conditions. EHEC strains were grown with LB broth containing 200 μmol/L DETA‐NONOate (DETA/NO) for 18 hr at 37°C under anaerobic conditions. EHEC strains were fractionated into culture supernatant fractions and cell‐associated fractions. Each volume, which corresponds to 0.2 μg of protein of the cell‐associated fraction, of the supernatant fraction was analyzed by Immunoblot analysis using anti‐Stx2 antiserum. The cell‐associated fraction protein was analyzed by Immunoblot analysis using anti‐RecA antibody and anti‐RNA α antibody as an internal control.
3.5. Mechanism of the enhancement of Stx2 production by NO under anaerobic conditions
Because, in the Stx2‐encoding phage, there was no promoter for Stx2 production adjacent to stx2, the recA‐deficient mutant EHEC ERA1‐1 did not produce Stx2 (Figure 7d). Therefore, we could not investigate the effect of NO on RecA‐dependent Stx2 expression using the recA‐deficient mutant. A previous study (Adikesavan et al., 2011) reported that the introduction of a point mutation of recA (e.g., E123A, G204S, or D224A) leads to the loss of upregulation of RecA in each recA‐deficient E. coli strain in the presence of nalidixic acid (100 μg/ml), while the point mutations G22Y, G24Y, and G288Y of recA do not impair the upregulation of RecA (Table S4). The mutations of G24Y and G288Y of recA continued to cleave LexA in the presence of nalidixic acid (100 μg/ml), but that of G22Y of recA lead to upregulation of RecA without LexA cleavage upon DNA damage (Table S4) (Adikesavan et al., 2011). These results suggested that one of the functions of RecA in E. coli allows E. coli to separate the activation of RecA and induction of SOS response. To investigate whether RecA affects the enhancement of Stx2 production in the presence of an NO donor under anaerobic conditions, we constructed the same isogenic recA point‐mutated strains using EHEC EDL933 as a parent strain (Table 1 and Table S4). Wild or recA point‐mutated EHEC strains (e.g., ERG123A, ERG204S, ERD224A) were cultured with LB broth containing 200 μmol/L DETA/NO for 18 hr under anaerobic conditions (Figure 7d). Stx2 production was not detected in the ERE123A, ERG204S, or ERD224A strains in the presence or absence of an NO donor, which was similar to the findings for the recA‐deficient ERA1‐1. The levels of RecA were increased by exposure to NO after 18 hr of incubation in the wild‐type, ERG24Y and ERG288Y strains. In the recA‐mutant ERG22Y strain, the level of RecA was similar to that of the wild‐type strain in the absence of NO‐donor treatment, and RecA was not increased by addition of the NO donor. Moreover, in recA‐mutant ERG22Y, the basal levels of Stx2 production were increased in comparison with those of the wild‐type strain. However, the NO‐enhanced Stx2 production was not observed in recA‐mutant ERG22Y. On the other hand, in the recA‐mutant strains, ERG24Y and ERG288Y, Stx2 production in the culture with an NO donor was enhanced to a degree similar to that in the wild‐type strain. These results indicated that the increase of RecA in EHEC was related to the NO‐enhanced Stx2 production under anaerobic conditions.
4. Discussion
The steady‐state NO concentration has emerged as a key determinant of bacterial response. Our data have revealed the approximate threshold concentration limits for growth inhibition, activation of Stx1 production and activation of Stx2 production in EHEC under anaerobic conditions. The results indicated that there were three concentration levels of NO activity in the deleted norV‐type EHEC EDL933 under anaerobic conditions: a growth inhibition range (12–29 μmol/L, maximum steady‐state concentration), an enhancement of Stx1 production range (12–29 μmol/L, maximum steady‐state concentration), and an enhancement of Stx2 production range (~12 μmol/L, maximum steady‐state concentration). This suggested that the bacterial responses were differentially influenced by specific NO concentrations. In this study, we used four kinds of NO donors (DETA/NO, PROLI/NO, NOC12, and Sper/NO) as the source of NO. Under anaerobic conditions, our results were consistent with previous reports under aerobic conditions (Thomas et al., 2004, 2008), namely, the steady‐state concentrations of NO in medium were much lower than those of the NO donors, except in the case of PROLI/NO.
Macrophages are an important component of the innate immune response. One of the antimicrobial systems of macrophages is the iNOS pathway, which is responsible for the generation of NO (Bogdan, 2001; MacMicking, Xie, & Nathan, 1997; Nathan & Hibbs, 1991). We observed that the specific luminescences (RLU/cfu) of the deleted norV‐type NO reporter within macrophages were 0.50 ± 0.02, which corresponds to the enhancement of Stx2 production, and 0.95 ± 0.37, which corresponds to the enhancement of Stx1 production, at 8 hr and 20 hr postinfection, respectively (Figure 5d). These results suggested that the NO generated in macrophages might induce Stx2 production in EHEC after 8 hr postinfection, and enhancement of Stx1 production in EHEC by NO within macrophages might occur at 20 hr postinfection or later. However, it was previously reported that NO suppressed the activation of RecA and then inhibited Stx2 synthesis in EHEC (Vareille et al., 2007). These results appear to be similar to our results under aerobic conditions using an hmpA‐deficient mutant strain (Figure 4b). The transcriptional repressor NsrR has a low threshold for sensing NO relative to its lesser sensitivity for SOS response (Karlinsey et al., 2012; Spek et al., 2001). The inhibition of stx2 expression in EHEC was essential for a long‐term exposure to a significant level of NO (Vareille et al., 2007). Under our experimental conditions, Stx2 production alone might have been inhibited by treatment with 100 μmol/L DETA/NO at 18 hr of incubation under aerobic conditions when we used the deleted norV‐type hmpA‐deficient EHEC. At lower concentrations of NO, RecA activation and Stx2 production might be inhibited by NO via the NsrR (Vareille et al., 2007). On the other hand, at higher concentrations of NO, RecA in EHEC might be activated by NO to induce an SOS response.
As an iron‐containing transcriptional regulator, Fur has been shown to directly respond to NO in E. coli (Fleischhacker & Kiley, 2011; Mukhopadhyay, Zheng, Bedzyk, LaRossa, & Storz, 2004; Spiro, 2006). As a transcriptional repressor, Fur regulates Stx1 production via P Stx1 in EHEC and is inactivated by exposure to NO, and then the derepression of Fur‐regulated P Stx1 is observed in EHEC. In contrast, the induction of the SOS response is caused by NO, resulting in the indirect activation of RecA (Lobysheva et al., 1999; Schapiro et al., 2003). We revealed that a higher concentration of NO is required for enhanced Stx1 production than for enhanced Stx2 production in EHEC. These results indicated that the concentration of NO required to depress Fur was higher than that required to activate RecA in EHEC, suggesting that Fur has a high threshold for sensing NO (Karlinsey et al., 2012). When Fur was overexpressed in EHEC, there was no significant inhibition of the enhancement of Stx1 production by NO‐donor treatment (Fig. S6). Fur is also a repressor of hmpA transcription in E. coli (Hernandez‐Urzua et al., 2007). Thus, not only inhibition of NO‐enhanced Stx1 production but also repression of hmpA expression might occur through the overexpression of Fur in EHEC.
It was reported that an recA‐deficient E. coli strain harboring a mutant recA expression plasmid (G288Y, G22Y, or G24Y) exhibited upregulation of RecA levels after DNA damage, while an recA‐deficient E. coli strain harboring a mutant recA expression plasmid (E123A, D224A, or G204S) did not (Adikesavan et al., 2011) (Table S4). Our results suggest that, in order for Stx2 to be expressed in EHEC, higher level expression of RecA or upregulation of RecA in response to DNA damage might be necessary. Moreover, E. coli (recA G24Y) exhibited both upregulation of RecA and LexA cleavage activity after DNA damage, but E. coli (recA G22Y) exhibited upregulation of RecA even in the absence of LexA cleavage activity (Adikesavan et al., 2011) (Table S4). Both E. coli (recA G24Y) and E. coli (recA G22Y) showed the UmuD cleavage activity (Adikesavan et al., 2011) (Table S4). Although the interaction of LexA with activated RecA triggers the cleavage reaction, activated RecA can also mediate the cleavage of two other groups of proteins. The first is a group of temperate phage repressors, exemplified by the λ CI repressor, which is cleaved in lysogens upon SOS‐induction treatment. The second set of substrates is a set of mutagenesis proteins, exemplified by the host UmuD protein, which is activated by specific cleavage to perform specific roles in SOS mutagenesis. Since the ERG22Y mutant strain did not exhibit an enhancement of Stx2 production when cultured in the presence of an NO donor, the LexA cleavage activity, not UmuD cleavage activity, of activated RecA in EHEC was related to NO‐enhanced Stx2 production, and the λ CI repressor of Stx2‐encoding phage in EHEC might be cleaved by the LexA cleavage activity of RecA to enhance Stx2 production (Table S4).
Conflict of Interest
None declared.
Supporting information
Acknowledgments
We thank Ms. Kanako Hirano for technical assistance. This research was supported by Grants‐in‐Aid for Scientific Research (26670208 and 24390104) from Japan Society for the Promotion of Science (JSPS).
Ichimura K, Shimizu T, Matsumoto A, et al. Nitric oxide‐enhanced Shiga toxin production was regulated by Fur and RecA in enterohemorrhagic Escherichia coli O157. MicrobiologyOpen. 2017;6:e461 https://doi.org/10.1002/mbo3.461
References
- Adikesavan, A. K. , Katsonis, P. , Marciano, D. C. , Lua, R. , Herman, C. , & Lichtarge, O. (2011). Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genetics, 7, e1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, C. (2001). Nitric oxide and the immune response. Nature Immunology, 2, 907–916. [DOI] [PubMed] [Google Scholar]
- Branchu, P. , Matrat, S. , Vareille, M. , Garrivier, A. , Durand, A. , Crepin, S. , … Gobert, A. P. (2014). NsrR, GadE, and GadX interplay in repressing expression of the Escherichia coli O157:H7 LEE pathogenicity island in response to nitric oxide. PLoS Pathogens, 10, e1003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood, S. B. , Auclair, F. , Donohue‐Rolfe, A. , Keusch, G. T. , & Mekalanos, J. J. (1987). Nucleotide sequence of the Shiga‐like toxin genes of Escherichia coli . Proceedings of the National Academy of Sciences USA, 84, 4364–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood, S. B. , & Mekalanos, J. J. (1987). Iron regulation of Shiga‐like toxin expression in Escherichia coli is mediated by the fur locus. Journal of Bacteriology, 169, 4759–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood, S. B. , & Mekalanos, J. J. (1988). Confirmation of the Fur operator site by insertion of a synthetic oligonucleotide into an operon fusion plasmid. Journal of Bacteriology, 170, 1015–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, M. M. (1999). Recombinational DNA repair in bacteria and the RecA protein. Progress in Nucleic Acid Research and Molecular Biology, 63, 311–366. [DOI] [PubMed] [Google Scholar]
- De Grandis, S. , Ginsberg, J. , Toone, M. , Climie, S. , Friesen, J. , & Brunton, J. (1987). Nucleotide sequence and promoter mapping of the Escherichia coli Shiga‐like toxin operon of bacteriophage H‐19B. Journal of Bacteriology, 169, 4313–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, S. J. , & Kaper, J. B. (1997). Role of type 1 fimbriae in EPEC infections. Microbial Pathogenesis, 23, 113–118. [DOI] [PubMed] [Google Scholar]
- Endo, Y. , Tsurugi, K. , Yutsudo, T. , Takeda, Y. , Ogasawara, T. , & Igarashi, K. (1988). Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N‐glycosidase activity of the toxins. European Journal of Biochemistry, 171, 45–50. [DOI] [PubMed] [Google Scholar]
- Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nature Reviews Microbiology, 2, 820–832. [DOI] [PubMed] [Google Scholar]
- Fernandez De Henestrosa, A. R. , Ogi, T. , Aoyagi, S. , Chafin, D. , Hayes, J. J. , Ohmori, H. , & Woodgate, R. (2000). Identification of additional genes belonging to the LexA regulon in Escherichia coli . Molecular Microbiology, 35, 1560–1572. [DOI] [PubMed] [Google Scholar]
- Fleischhacker, A. S. , & Kiley, P. J. (2011). Iron‐containing transcription factors and their roles as sensors. Current Opinion in Chemical Biology, 15, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, M. E. , Fujinaga, M. , Cherney, M. M. , Melton‐Celsa, A. R. , Twiddy, E. M. , O'Brien, A. D. , & James, M. N. (2004). Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. Journal of Biological Chemistry, 279, 27511–27517. [DOI] [PubMed] [Google Scholar]
- Gardner, P. R. , Costantino, G. , & Salzman, A. L. (1998). Constitutive and adaptive detoxification of nitric oxide in Escherichia coli. Role of nitric‐oxide dioxygenase in the protection of aconitase. Journal of Biological Chemistry, 273, 26528–26533. [DOI] [PubMed] [Google Scholar]
- Gardner, A. M. , & Gardner, P. R. (2002). Flavohemoglobin detoxifies nitric oxide in aerobic, but not anaerobic, Escherichia coli. Evidence for a novel inducible anaerobic nitric oxide‐scavenging activity. Journal of Biological Chemistry, 277, 8166–8171. [DOI] [PubMed] [Google Scholar]
- Gardner, A. M. , Helmick, R. A. , & Gardner, P. R. (2002). Flavorubredoxin, an inducible catalyst for nitric oxide reduction and detoxification in Escherichia coli . Journal of Biological Chemistry, 277, 8172–8177. [DOI] [PubMed] [Google Scholar]
- Hantke, K. (2001). Iron and metal regulation in bacteria. Current Opinion in Microbiology, 4, 172–177. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Urzua, E. , Zamorano‐Sanchez, D. S. , Ponce‐Coria, J. , Morett, E. , Grogan, S. , Poole, R. K. , & Membrillo‐Hernandez, J. (2007). Multiple regulators of the Flavohaemoglobin (hmp) gene of Salmonella enterica serovar Typhimurium include RamA, a transcriptional regulator conferring the multidrug resistance phenotype. Archives of Microbiology, 187, 67–77. [DOI] [PubMed] [Google Scholar]
- Hofmann, S. L. (1993). Southwestern Internal Medicine Conference: Shiga‐like toxins in hemolytic‐uremic syndrome and thrombotic thrombocytopenic purpura. American Journal of the Medical Sciences, 306, 398–406. [DOI] [PubMed] [Google Scholar]
- Householder, T. C. , Fozo, E. M. , Cardinale, J. A. , & Clark, V. L. (2000). Gonococcal nitric oxide reductase is encoded by a single gene, norB, which is required for anaerobic growth and is induced by nitric oxide. Infection and Immunity, 68, 5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, A. E. , Acheson, D. W. , Echeverria, P. , Donohue‐Rolfe, A. , & Keusch, G. T. (1993). Mitomycin immunoblot colony assay for detection of Shiga‐like toxin‐producing Escherichia coli in fecal samples: Comparison with DNA probes. Journal of Clinical Microbiology, 31, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz, M. , Clausen, H. , Nudelman, E. , Donohue‐Rolfe, A. , & Keusch, G. T. (1986). Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin‐binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. Journal of Experimental Medicine, 163, 1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M. P. , Neill, R. J. , O'Brien, A. D. , Holmes, R. K. , & Newland, J. W. (1987). Nucleotide sequence analysis and comparison of the structural genes for Shiga‐like toxin I and Shiga‐like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiology Letters, 44, 109–114. [Google Scholar]
- Jackson, M. P. , Newland, J. W. , Holmes, R. K. , & O'Brien, A. D. (1987). Nucleotide sequence analysis of the structural genes for Shiga‐like toxin I encoded by bacteriophage 933J from Escherichia coli . Microbial Pathogenesis, 2, 147–153. [DOI] [PubMed] [Google Scholar]
- Karlinsey, J. E. , Bang, I. S. , Becker, L. A. , Frawley, E. R. , Porwollik, S. , Robbins, H. F. , … Fang, F. C. (2012). The NsrR regulon in nitrosative stress resistance of Salmonella enterica serovar Typhimurium. Molecular Microbiology, 85, 1179–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keusch, G. T. , & Acheson, D. W. (1997). Thrombotic thrombocytopenic purpura associated with Shiga toxins. Seminars in Hematology, 34, 106–116. [PubMed] [Google Scholar]
- Kim, S. O. , Orii, Y. , Lloyd, D. , Hughes, M. N. , & Poole, R. K. (1999). Anoxic function for the Escherichia coli flavohaemoglobin (Hmp): Reversible binding of nitric oxide and reduction to nitrous oxide. FEBS Letters, 445, 389–394. [DOI] [PubMed] [Google Scholar]
- Kimmitt, P. T. , Harwood, C. R. , & Barer, M. R. (2000). Toxin gene expression by shiga toxin‐producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerging Infectious Diseases, 6, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski, S. C. (2000). Initiation of genetic recombination and recombination‐dependent replication. Trends in Biochemical Sciences, 25, 156–165. [DOI] [PubMed] [Google Scholar]
- Lansbury, L. E. , & Ludlam, H. (1997). Escherichia coli O157: Lessons from the past 15 years. Journal of Infection, 34, 189–193. [DOI] [PubMed] [Google Scholar]
- Lee, J. W. , & Helmann, J. D. (2007). Functional specialization within the Fur family of metalloregulators. BioMetals, 20, 485–499. [DOI] [PubMed] [Google Scholar]
- Little, J. W. (1984). Autodigestion of lexA and phage lambda repressors. Proceedings of the National Academy of Sciences USA, 81, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, J. W. (1991). Mechanism of specific LexA cleavage: Autodigestion and the role of RecA coprotease. Biochimie, 73, 411–421. [DOI] [PubMed] [Google Scholar]
- Little, J. W. (1993). LexA cleavage and other self‐processing reactions. Journal of Bacteriology, 175, 4943–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobysheva, I. I. , Stupakova, M. V. , Mikoyan, V. D. , Vasilieva, S. V. , & Vanin, A. F. (1999). Induction of the SOS DNA repair response in Escherichia coli by nitric oxide donating agents: Dinitrosyl iron complexes with thiol‐containing ligands and S‐nitrosothiols. FEBS Letters, 454, 177–180. [DOI] [PubMed] [Google Scholar]
- MacMicking, J. , Xie, Q. W. , & Nathan, C. (1997). Nitric oxide and macrophage function. Annual Review of Immunology, 15, 323–350. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, P. , Zheng, M. , Bedzyk, L. A. , LaRossa, R. A. , & Storz, G. (2004). Prominent roles of the NorR and Fur regulators in the Escherichia coli transcriptional response to reactive nitrogen species. Proceedings of the National Academy of Sciences USA, 101, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, C. F. , & Hibbs, J. B. Jr (1991). Role of nitric oxide synthesis in macrophage antimicrobial activity. Current Opinion in Immunology, 3, 65–70. [DOI] [PubMed] [Google Scholar]
- Noda, M. , Yutsudo, T. , Nakabayashi, N. , Hirayama, T. , & Takeda, Y. (1987). Purification and some properties of Shiga‐like toxin from Escherichia coli 0157:H7 that is immunologically identical to Shiga toxin. Microbial Pathogenesis, 2, 339–349. [DOI] [PubMed] [Google Scholar]
- Perna, N. T. , Plunkett, G. 3rd , Burland, V. , Mau, B. , Glasner, J. D. , Rose, D. J. , … Blattner, F. R. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature, 409, 529–533. [DOI] [PubMed] [Google Scholar]
- Pham, P. , Seitz, E. M. , Saveliev, S. , Shen, X. , Woodgate, R. , Cox, M. M. , & Goodman, M. F. (2002). Two distinct modes of RecA action are required for DNA polymerase V‐catalyzed translesion synthesis. Proceedings of the National Academy of Sciences USA, 99, 11061–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole, R. K. (2005). Nitric oxide and nitrosative stress tolerance in bacteria. Biochemical Society Transactions, 33, 176–180. [DOI] [PubMed] [Google Scholar]
- Porcheron, G. , & Dozois, C. M. (2015). Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Veterinary Microbiology, 179, 2–14. [DOI] [PubMed] [Google Scholar]
- Schapiro, J. M. , Libby, S. J. , & Fang, F. C. (2003). Inhibition of bacterial DNA replication by zinc mobilization during nitrosative stress. Proceedings of the National Academy of Sciences USA, 100, 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, T. , Hirai, S. , Yokoyama, E. , Ichimura, K. , & Noda, M. (2015). An evolutionary analysis of nitric oxide reductase gene norV in enterohemorrhagic Escherichia coli O157. Infect Genet Evol, 33, 176–181. [DOI] [PubMed] [Google Scholar]
- Shimizu, T. , Kawakami, S. , Sato, T. , Sasaki, T. , Higashide, M. , Hamabata, T. , … Noda, M. (2007). The serine 31 residue of the B subunit of Shiga toxin 2 is essential for secretion in enterohemorrhagic Escherichia coli . Infection and Immunity, 75, 2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, T. , Ohta, Y. , & Noda, M. (2009). Shiga toxin 2 is specifically released from bacterial cells by two different mechanisms. Infection and Immunity, 77, 2813–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, T. , Ohta, Y. , Tsutsuki, H. , & Noda, M. (2011). Construction of a novel bioluminescent reporter system for investigating Shiga toxin expression of enterohemorrhagic Escherichia coli . Gene, 478, 1–10. [DOI] [PubMed] [Google Scholar]
- Shimizu, T. , Tsutsuki, H. , Matsumoto, A. , Nakaya, H. , & Noda, M. (2012). The nitric oxide reductase of enterohaemorrhagic Escherichia coli plays an important role for the survival within macrophages. Molecular Microbiology, 85, 492–512. [DOI] [PubMed] [Google Scholar]
- Spek, E. J. , Wright, T. L. , Stitt, M. S. , Taghizadeh, N. R. , Tannenbaum, S. R. , Marinus, M. G. , & Engelward, B. P. (2001). Recombinational repair is critical for survival of Escherichia coli exposed to nitric oxide. Journal of Bacteriology, 183, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro, S. (2006). Nitric oxide‐sensing mechanisms in Escherichia coli . Biochemical Society Transactions, 34, 200–202. [DOI] [PubMed] [Google Scholar]
- Spiro, S. (2007). Regulators of bacterial responses to nitric oxide. FEMS Microbiology Reviews, 31, 193–211. [DOI] [PubMed] [Google Scholar]
- Spiro, S. (2012). Nitrous oxide production and consumption: Regulation of gene expression by gas‐sensitive transcription factors. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 367, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupakova, M. V. , Lobysheva, I. I. , Mikoyan, V. D. , Vanin, A. F. , & Vasilieva, S. V. (2000). A role of iron ions in the SOS DNA repair response induced by nitric oxide in Escherichia coli . Biochemistry (Mosc), 65, 690–695. [PubMed] [Google Scholar]
- Tesh, V. L. , & O'Brien, A. D. (1991). The pathogenic mechanisms of Shiga toxin and the Shiga‐like toxins. Molecular Microbiology, 5, 1817–1822. [DOI] [PubMed] [Google Scholar]
- Thomas, D. D. , Espey, M. G. , Ridnour, L. A. , Hofseth, L. J. , Mancardi, D. , Harris, C. C. , & Wink, D. A. (2004). Hypoxic inducible factor 1α, extracellular signal‐regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proceedings of the National Academy of Sciences USA, 101, 8894–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D. D. , Liu, X. , Kantrow, S. P. , & Lancaster, J. R. Jr (2001). The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proceedings of the National Academy of Sciences USA, 98, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D. D. , Miranda, K. M. , Espey, M. G. , Citrin, D. , Jourd'heuil, D. , & Paolocci, N. , … Wink, D. A. (2002). Guide for the use of nitric oxide (NO) donors as probes of the chemistry of NO and related redox species in biological systems. Methods in Enzymology, 359, 84–105. [DOI] [PubMed] [Google Scholar]
- Thomas, D. D. , Ridnour, L. A. , Isenberg, J. S. , Flores‐Santana, W. , Switzer, C. H. , Donzelli, S. , … Wink, D. A. (2008). The chemical biology of nitric oxide: Implications in cellular signaling. Free Radical Biology and Medicine, 45, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vareille, M. , de Sablet, T. , Hindre, T. , Martin, C. , & Gobert, A. P. (2007). Nitric oxide inhibits Shiga‐toxin synthesis by enterohemorrhagic Escherichia coli . Proceedings of the National Academy of Sciences USA, 104, 10199–10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, P. L. , Livny, J. , Neely, M. N. , Acheson, D. W. , Friedman, D. I. , & Waldor, M. K. (2002). Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli . Molecular Microbiology, 44, 957–970. [DOI] [PubMed] [Google Scholar]
- Wagner, P. L. , & Waldor, M. K. (2002). Bacteriophage control of bacterial virulence. Infection and Immunity, 70, 3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor, M. K. , & Friedman, D. I. (2005). Phage regulatory circuits and virulence gene expression. Current Opinion in Microbiology, 8, 459–465. [DOI] [PubMed] [Google Scholar]
- Walker, G. C. (1984). Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli . Microbiological Reviews, 48, 60–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Vine, C. E. , Balasiny, B. K. , Rizk, J. , Bradley, C. L. , Tinajero‐Trejo, M. , … Cole, J. A. (2016). The roles of the hybrid cluster protein, Hcp and its reductase, Hcr, in high affinity nitric oxide reduction that protects anaerobic cultures of Escherichia coli against nitrosative stress. Molecular Microbiology, 100, 877–892. [DOI] [PubMed] [Google Scholar]
- Watmough, N. J. , Butland, G. , Cheesman, M. R. , Moir, J. W. , Richardson, D. J. , & Spiro, S. (1999). Nitric oxide in bacteria: Synthesis and consumption. Biochimica et Biophysica Acta, 1411, 456–474. [DOI] [PubMed] [Google Scholar]
- Yutsudo, T. , Nakabayashi, N. , Hirayama, T. , & Takeda, Y. (1987). Purification and some properties of a Vero toxin from Escherichia coli O157:H7 that is immunologically unrelated to Shiga toxin. Microbial Pathogenesis, 3, 21–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials