

ABSTRACT
The term ‘prion-like’ is used to define some misfolded protein species that propagate intercellularly, triggering protein aggregation in recipient cells. For cell binding, both direct plasma membrane interaction and membrane receptors have been described for particular amyloids. In this respect, emerging evidence demonstrates that several β-sheet enriched proteins can bind to the cellular prion protein (PrPC). Among other interactions, the physiological relevance of the binding between β-amyloid and PrPC has been a relevant focus of numerous studies. At the molecular level, published data point to the second charged cluster domain of the PrPC molecule as the relevant binding domain of the β-amyloid/PrPC interaction. In addition to β-amyloid, participation of PrPC in binding α-synuclein, responsible for neurodegenerative synucleopathies, has been reported. Although results indicate relevant participation of PrPC in the spreading of α-synuclein in living mice, the physiological relevance of the interaction remains elusive. In this comment, we focus our attention on summarizing current knowledge of PrPC as a receptor for amyloid proteins and its physiological significance, with particular focus on α-synuclein.
KEYWORDS: α-synuclein, charged cluster domain, interneuronal transport, LAG3, neurodegeneration, PrPC, Parkinson disease
Parkinson´s disease (PD) is the second most common neurodegenerative disease worldwide. Motor disabilities, globally called parkinsonism, are characteristic of the disorder; these may be preceeded by neurovegetative symptoms, sleep disorders, and loss of olfaction; neurocognitive deficits may also appear with disease progression leading to dementia (i.e., Parkinson disease dementia, PDD). Histologically, PD is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta, which is causative of parkinsonism, and the presence of intraneuronal insoluble inclusions called Lewy Bodies (LB) and Lewy Neurites (LN) in several brain regions.1 LB/LN aggregates contain various misfolded proteins such us ubiquitin,2 tau,3 and lipids4 but their major component is hyperphosphorylated α-synuclein.5 Abnormal α-synuclein aggregates appear, in addition to PD, in various α-synucleinopathies such as Dementia with Lewy Bodies (DLB) and multiple system atrophy (MSA).6 In these disorders, aggregates are deposited in the brain in a filamentous form displaying a β-sheet structure7 which is abnormally phosphorylated at Serine129 and is also ubiquitinated.8 In addition, PDD, and particularly, DLB post-mortem brains often display accumulations of β-amyloid forming diffuse and neuritic plaques, and neurofibrillary tangles composed of abnormally hyperphosphorylated tau. In addition, the potential contribution of α-synuclein to Alzheimer disease (AD) pathology is considered important today, with ≈ 40% of AD cases presenting LB/LN, often as amygdala-predominant Lewy body disease (LBD).9
‘PRION-LIKE’ PROTEINS AND NEURONAL SPREADING
Most of the above-mentioned misfolded proteins (i.e., β-amyloid, hyperphosphorylated tau, and abnormal α-synuclein) are able to ‘propagate’ or ‘spread’ between cells in the same manner as infectious prions, a mechanism termed ‘prion-like’, ‘prionoid’, or simply ‘prions’ in several studies.10,11 However, not all the molecular properties of infective prions observed in transmissible spongiform encephalopathies (TSEs) have been demonstrated in these proteinaceous species.11 The infectious spreading of pathogenic prions occurs between cells, tissues and, more relevantly, organisms. This is not described for all ‘prion-like’ molecules. As recently indicated by Harbi and Harrison in Prion,12 ‘prion-like activity’ or ‘prion-like propagation’ may also refer to self-propagating protein aggregates that may not yet meet a stricter ‘prion’ definition. The term ‘prion-like’ for the ‘propagative’ activities of the misfolded proteins will be used in the present article. Moreover, in our review misfolded α-synuclein protein becomes a ‘prion-like’ protein.
Although there is some disagreement, it is well established that the disease progression of some neurodegenerative disorders, such as AD and PD, seems to correlate with brain propagation of the misfolded protein between predictable anatomical pathways.13-17 In PD and DLB, the sequential accumulation of α-synuclein starts in the olfactory bulb, medulla oblongata, midbrain and forebrain, and neocortex.18 Indeed, the spreading of pathological α-synuclein is closely correlated with disease progression and is considered to be the underlying mechanism of progression of the disease.19
Extensive research has demonstrated that spreading of α-synuclein can also be reproduced in vivo and in vitro. In vivo, peripheral (i.e,20) or intracerebral injections of recombinant α-synuclein protofibrils or insoluble fractions of α-synuclein derived from affected brains trigger conversion of natural endogenous α-synuclein into the abnormal misfolded form, and this misfolded α-synuclein propagates in the brains of wild-type mice,6,21 α-synuclein transgenic mice,22,23 monkeys,24 and marmosets.25 In vitro experiments, and more relevantly, the emerging development of ‘lab-on-a-chip’ device cultures based on microfluidics, have been of great value in helping to determine the cell-to-cell transport of several amyloids including α-synuclein (i.e.,26-29). Thus, recombinant α-synuclein fibrils27,28 as well as LB extracts26 can spread between neurons and astroglial cells growing in these devices.26 Several mechanisms have been reported for α-synuclein spreading using these or other in vitro methods, such as extracellular vesicles and tunneling nanotubes (TNTs).13,30-35 In addition to these descriptions, several groups have started to measure the presence of membrane receptors for α-synuclein and/or other amyloids.
A CHARGED DOMAIN OF PRPC IS A COMMON ‘DOCKING-DOMAIN’ FOR SEVERAL ‘PRION-LIKE’ PROTEINACEOUS SPECIES
Several studies have reported that β-sheet rich amyloid proteins (including α-synuclein) can interact with plasma membrane (e.g.,36). Although this interaction might be involved in amyloid internalization leading to cytotoxicity, ‘docking’ and receptor-mediated interaction activities at plasma membrane might support most of the physiological activities of oligomeric proteinaceous species.37 PrPC can bind with numerous membrane-associated molecules including adhesion molecules, growth-factor receptors, and neurotransmitter receptors, among others. More relevantly, PrPC has been described as a high-affinity binding partner of oligomeric β-amyloid (Aβo)—a relevant finding in determining the early trigger in AD.38-43 However, this interaction (Aβo-PrPC) is not exclusive since other studies have determined that the N-terminal domain of PrPC can bind to several β-rich peptides, including Aβ.44,45 Indeed, mapping studies point to the ≈ 90–110 amino acids located in the second charged cluster of PrPC as the main residues responsible for Aβo binding40,46 (see47 for a recent description of PrPC domains). Although interaction between PrPC and Aβo has been demonstrated in several studies,38-43 some observations indicate that PrPC is not a mediator of the neurotoxic effects of Aβo (i.e.,48-51). Starting from these pioneering PrPC-Aβo binding descriptions, several laboratories have started to analyze whether PrPC may also be a ‘cellular partner’ for other proteinaceous species displaying ‘propagative’ properties, and to determine whether PrPC participates in or regulates the spreading of these ‘prion-like’ proteins and their associated neuropathology. Results published recently by our group point out that, in addition to Aβo, membrane-anchored PrPC can also bind to α-synuclein fibers.27
Several receptors have been described as binding α-synuclein. It has been shown that α-synuclein binds to Na+/K+-ATPase subunit α3,52 lymphocyte-activation gene 3 (LAG3),53 neurexin,52,53 amyloid β precursor-like protein 1 (APLP1),53 and PrPC.27 Although α-synuclein is implicated in the binding, uptake, and/or trafficking of α-synuclein protofibrils, some details of the process are missing, and for most of them except LAG3 and PrPC, their putative participation in the spreading of α-synuclein and interneuronal transport has not been fully investigated. For LAG3 and PrPC, their binding and participation in α-synuclein expansion has been analyzed in vitro and in vivo.27,53 In both cases, the absence of the protein receptor (LAG3 or PrPC) largely decreases but does not fully impair α-synuclein spreading in vivo.27,53 Recent unpublished from our group noted that this decrease in α-synuclein spreading in the absence of PrPC may occur in different strains of mice lacking functional PrPC, by avoiding an indirect effect of the ‘Prnp-flanking genes’ observed in the Zurich I (ZH1)-derived mice47,54,55 (Fig. 1). Conversely, the overexpression of Prnp enhances α-synuclein spreading and the generation of the phosphorylated form of α-synuclein (p-α-synuclein) in anatomically connected regions (i.e., striatum → motor cortex27) (Fig. 1c). These results were obtained using different Prnp genotypes, Prnp+/+ and Prnp0/0 (B6.129 (ZH1) Prnp0/0 27 and Zurich 3 (ZH3) Prnp0/0 mice54), and Tga20 (Prnp-overexpressing) mice under a B6.129 background27 (Fig. 1).
FIGURE 1.
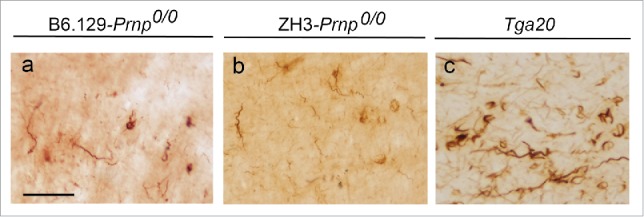
Increased p-α-synuclein aggregate labeling in the motor neocortex in Tga20 mice compared with B6.129 (ZH1 Prnp0/0) and ZH3 Prnp0/0 mice. Examples of p-α-synuclein aggregates in the cortex of B6.129 (ZH1) Prnp0/0 (a); ZH3 Prnp0/0 (b) and Tga20 mice (c) injected with recombinant mouse α-synuclein fibrils in the post-commissural striatum. Note the relevant accumulation of p- α-synuclein in intracellular deposits of retrograde labeled neurons in cortical layer V of Tga20 mice. Scale bar: a = 100 µm pertains to b and c.
TOWARD UNCOVERING OF THE PHYSIOLOGICAL RELEVANCE OF α-SYNUCLEIN-PRPC INTERACTION: LAST-MINUTE QUESTIONS
Although the interaction between α-synuclein and PrPC has been described, as has a correlation between Prnp expression and p-α-synuclein spreading in vitro and in vivo, new, challenging questions have emerged. Several open questions at both the cellular level and that of neurodegeneration warrant further study:
α-Synuclein can be transported intercellularly through several mechanisms. However, the participation of PrPC in α-synuclein seeding properties and particular transport mechanisms needs further research.
Astroglial cells participate in α-synuclein spreading.26 Since PrPC is expressed in neurons and glial cells,56 PrPC might also play a role in α-synuclein glia-to-neuron transmission.
The putative participation of PrPC in the neurotoxic effects of α-synuclein calls for further attention. Conversely, the actions of this binding in PrPC biology and physiology, both healthy and unhealthy, are still unkown.
The vast majority of PD patients are sporadic, but mutations in the SNCA gene encoding α-synuclein A53T, A30P, E46K, A53E, H50Q, and G51D cause autosomal-dominant forms of PD.57 As the α-synuclein/PrPC interaction takes place in the second charged cluster of the PrPC, the question of whether these point mutations may alter α-synuclein/PrPC interaction needs to be addressed.
α-synuclein protofibrils bind to PrPC in their second charged cluster domain, sharing this binding motif with Aβo. This suggests that the effort to block PrPC/Aβo interaction with molecules (i.e., antibodies58) or chemical compounds could also be a potential therapeutic intervention for α-synuclein spreading and, likely, for synucleopathies.
As Santiago Ramón y Cajal said “Ideas do not last long. We must do something with them.” We hope that the ideas discussed above will evolve very quickly through the use of newly emerging techniques (i.e, lab-on-a-chip, organoids, etc.) under the umbrella of new mutant mice to more fully reveal the role of PrPC in neurodegeneration.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGEMENTS
The authors thank Tom Yohannan for editorial advice. We would also like to thank Prof. Adriano Aguzzi for the gift of the Zurich 3 (ZH3) Prnp0/0 mice and Dr. Mario Nuvolone for his comments.
FUNDING
This research was supported by grants from the Spanish Ministry of Economy, Industry and Competitiveness (MEICO) (BFU2015–67777-R), the Spanish Prion Network (Prionet Spain, AGL2015–71764-REDT), the Generalitat de Catalunya (SGR2014–1218), CIBERNED (PRY-2016–2, MFDEND), La Caixa Obra Social Foundation, and La Marató de TV3 to JADR. IF was funded by the Ministerio de Economía, Industria y Competitividad (MEICO), Instituto de Salud Carlos III - Fondos FEDER, a Way to Build Europe, FIS grant PI14/00757 and CIBERNED.
REFERENCES
- [1].Dauer W, Przedborski S. Parkinson's disease: Mechanisms and models. Neuron 2003; 39:889-909; PMID:12971891 [DOI] [PubMed] [Google Scholar]
- [2].Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol 1988; 155:9-15; PMID:2837558; https://doi.org/10.1002/path.1711550105 [DOI] [PubMed] [Google Scholar]
- [3].Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 2003; 62:389-97; PMID:12722831 [DOI] [PubMed] [Google Scholar]
- [4].Noda K, Kitami T, Gai WP, Chegini F, Jensen PH, Fujimura T, Murayama K, Tanaka K, Mizuno Y, Hattori N. Phosphorylated IkappaBalpha is a component of Lewy body of Parkinson's disease. Biochem Biophys Res Commun 2005; 331:309-17; PMID:15845394; https://doi.org/10.1016/j.bbrc.2005.03.167 [DOI] [PubMed] [Google Scholar]
- [5].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997; 388:839-40; PMID:9278044; https://doi.org/10.1038/42166 [DOI] [PubMed] [Google Scholar]
- [6].Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun 2014; 2:88; PMID:25095794; https://doi.org/10.1186/PREACCEPT-1296467154135944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Nati Acad Sci U S A 2000; 97:4897-902; PMID:10781096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002; 4:160-4; PMID:11813001; https://doi.org/10.1038/ncb748 [DOI] [PubMed] [Google Scholar]
- [9].Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesné SE. Soluble alpha-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci 2012; 32:10253-66; PMID:22836259; https://doi.org/10.1523/JNEUROSCI.0581-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [10].Ashe KH, Aguzzi A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 2013; 7:55-9; PMID:23208281; https://doi.org/10.4161/pri.23061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Erana H, Venegas V, Moreno J, Castilla J. Prion-like disorders and transmissible spongiform encephalopathies: An overview of the mechanistic features that are shared by the various disease-related misfolded proteins. Biochem Biophys Res Commun 2017; 483:1125-36; PMID:27590581; https://doi.org/10.1016/j.bbrc.2016.08.166 [DOI] [PubMed] [Google Scholar]
- [12].Harbi D, Harrison PM. Classifying prion and prion-like phenomena. Prion 2014; 8:161-5; PMID:24549098; https://doi.org/10.4161/pri.27960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Costanzo M, Zurzolo C. The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem J 2013; 452:1-17; PMID:23614720; https://doi.org/10.1042/BJ20121898 [DOI] [PubMed] [Google Scholar]
- [14].Saper CB, Wainer BH, German DC. Axonal and transneuronal transport in the transmission of neurological disease: Potential role in system degenerations, including Alzheimer's disease. Neuroscience 1987; 23:389-98; PMID:2449630; https://doi.org/10.1016/0306-4522(87)90063-7 [DOI] [PubMed] [Google Scholar]
- [15].Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T. Limbic neuropathology in idiopathic Parkinson's disease with concomitant dementia. Folia Neuropathol 2004; 42:141-50; PMID:15535032 [PubMed] [Google Scholar]
- [16].Braak H, Del Tredici K. Neuroanatomy and pathology of sporadic Parkinson's disease. Adv Anat Embryol Cell Biol 2009; 201:1-119; PMID:19230552 [PubMed] [Google Scholar]
- [17].Goedert M, Masuda-Suzukake M, Falcon B. Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 2017; 140:266-78; PMID:27658420; https://doi.org/10.1093/brain/aww230 [DOI] [PubMed] [Google Scholar]
- [18].Goedert M. Neurodegeneration. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015; 349:1255555; PMID:26250687; https://doi.org/10.1126/science.1255555 [DOI] [PubMed] [Google Scholar]
- [19].Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24:197-211; PMID:12498954; https://doi.org/10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- [20].Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamguney G. Neuroinvasion of alpha-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 2016; 90:9182-93; PMID:27489279; https://doi.org/10.1128/JVI.01399-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949-53; PMID:23161999; https://doi.org/10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bernis ME, Babila JT, Breid S, Wusten KA, Wullner U, Tamguney G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 2015; 3:75; PMID:26612754; https://doi.org/10.1186/s40478-015-0254-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mougenot AL, Bencsik A, Nicot S, Vulin J, Morignat E, Verchere J, Bétemps D, Lakhdar L, Legastelois S, Baron TG. Transmission of prion strains in a transgenic mouse model overexpressing human A53T mutated alpha-synuclein. J Neuropathol Exp Neurol 2011; 70:377-85; PMID:21487306; https://doi.org/10.1097/NEN.0b013e318217d95f [DOI] [PubMed] [Google Scholar]
- [24].Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014; 75:351-62; PMID:24243558; https://doi.org/10.1002/ana.24066 [DOI] [PubMed] [Google Scholar]
- [25].Hasegawa M, Nonaka T, Masuda-Suzukake M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther 2017; 172:22-33; PMID:27916654; https://doi.org/10.1016/j.pharmthera.2016.11.010 [DOI] [PubMed] [Google Scholar]
- [26].Cavaliere F, Cerf L, Dehay B, Ramos-Gonzalez P, De Giorgi F, Bourdenx M, Bessede A, Obeso JA, Matute C, Ichas F, et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol Dis 2017; 103:101-12; PMID:28411117; https://doi.org/10.1016/j.nbd.2017.04.011 [DOI] [PubMed] [Google Scholar]
- [27].Urrea L, Segura-Feliu M, Masuda-Suzukake M, Hervera A, Pedraz L, Aznar JM, et al. Involvement of cellular prion protein in alpha-synuclein transport in neurons. Mol Neurobiol 2017; Epub ahead of print; https://doi.org/10.1007/s12035-017-0451-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol 2012; 72:517-24; PMID:23109146; https://doi.org/10.1002/ana.23747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Song HL, Shim S, Kim DH, Won SH, Joo S, Kim S, Jeon NL, Yoon SY. beta-amyloid is transmitted via neuronal connections along axonal membranes. Ann Neurol 2014; 75:88-97; PMID:24114864; https://doi.org/10.1002/ana.24029 [DOI] [PubMed] [Google Scholar]
- [30].Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016; 10:344-51; PMID:27715442; https://doi.org/10.1080/19336896.2016.1223003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol 2005; 15:102-11; PMID:15695097; https://doi.org/10.1016/j.tcb.2004.12.002 [DOI] [PubMed] [Google Scholar]
- [32].Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R, Buée L, Colin M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 2016; 4:117; PMID:27809932; https://doi.org/10.1186/s40478-016-0386-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zeinabad HA, Zarrabian A, Saboury AA, Alizadeh AM, Falahati M. Interaction of single and multi wall carbon nanotubes with the biological systems: Tau protein and PC12 cells as targets. Sci Rep 2016; 6:26508; PMID:27216374; https://doi.org/10.1038/srep26508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dieriks BV, Park TI, Fourie C, Faull RL, Dragunow M, Curtis MA. alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci Rep 2017; 7:42984; PMID:28230073; https://doi.org/10.1038/srep42984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Okuda S, Uemura N, Takahashi R. Alpha-synuclein fibrils propagate through tunneling nanotubes. Mov Disord 2017; 32:394; PMID:28218419; https://doi.org/10.1002/mds.26909 [DOI] [PubMed] [Google Scholar]
- [36].Monsellier E, Bousset L, Melki R. alpha-synuclein and huntingtin exon 1 amyloid fibrils bind laterally to the cellular membrane. Sci Rep 2016; 6:19180; PMID:26757959; https://doi.org/10.1038/srep19180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Linden R. The biological function of the prion protein: A cell surface scaffold of signaling modules. Front Mol Neurosci 2017; 10:77; PMID:28373833; https://doi.org/10.3389/fnmol.2017.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen H, Schluter H, Hildebrand D, Zerr I, Matschke J, Glatzel M. High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer's disease. Brain 2014; 137:873-86; PMID:24519981; https://doi.org/10.1093/brain/awt375 [DOI] [PubMed] [Google Scholar]
- [39].Freir DB, Nicoll AJ, Klyubin I, Panico S, McDonald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011; 2:336; PMID:21654636; https://doi.org/10.1038/ncomms1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128-32; PMID:19242475; https://doi.org/10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, et al. Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem 2011; 286:15095-105; PMID:21393248; https://doi.org/10.1074/jbc.M110.199356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ganzinger KA, Narayan P, Qamar SS, Weimann L, Ranasinghe RT, Aguzzi A, Dobson CM, McColl J, St George-Hyslop P, Klenerman D. Single-molecule imaging reveals that small amyloid-beta1-42 oligomers interact with the cellular prion protein (PrP(C)). Chembiochem 2014; 15:2515-21; PMID:25294384; https://doi.org/10.1002/cbic.201402377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, et al. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013; 288:7857-66; PMID:23362282; https://doi.org/10.1074/jbc.M112.423954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057-70; PMID:21441896; https://doi.org/10.1038/emboj.2011.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Resenberger UK, Winklhofer KF, Tatzelt J. Cellular prion protein mediates toxic signaling of amyloid beta. Neurodegener Dis 2012; 10:298-300; PMID:22156337; https://doi.org/10.1159/000332596 [DOI] [PubMed] [Google Scholar]
- [46].Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role of N-terminal residues. J Biol Chem 2010; 285:26377-83; PMID:20576610; https://doi.org/10.1074/jbc.M110.145516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].del Rio JA, Gavin R. Functions of the cellular prion protein, the end of Moore's law, and Ockham's razor theory. Prion 2016; 10:25-40; PMID:26890218; https://doi.org/10.1080/19336896.2015.1126038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306-14; PMID:20665634; https://doi.org/10.1002/emmm.201000082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:E3-4; discussion E-5; PMID:20703260; https://doi.org/10.1038/nature09217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, Strittmatter SM. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain 2016; 139:526-46; PMID:26667279; https://doi.org/10.1093/brain/awv356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, Strittmatter SM. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J Biol Chem 2015; 290:17415-38; PMID:26018073; https://doi.org/10.1074/jbc.M115.643577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Léna C, et al. alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 2015; 34:2408-23; PMID:26323479; https://doi.org/10.15252/embj.201591397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016; 353:aah3374, 1-12; https://doi.org/10.1126/science.aah3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nuvolone M, Hermann M, Sorce S, Russo G, Tiberi C, Schwarz P, Minikel E, Sanoudou D, Pelczar P, Aguzzi A, et al. Strictly co-isogenic C57BL/6J-Prnp-/- mice: A rigorous resource for prion science. J Exp Med 2016; 213:313-27; PMID:26926995; https://doi.org/10.1084/jem.20151610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nuvolone M, Sorce S, Paolucci M, Aguzzi A. Extended characterization of the novel co-isogenic C57BL/6J Prnp-/- mouse line. Amyloid 2017; 24:36-7; PMID:28434290; https://doi.org/10.1080/13506129.2017.1289913 [DOI] [PubMed] [Google Scholar]
- [56].Vilches S, Vergara C, Nicolas O, Sanclimens G, Merino S, Varon S, Acosta GA, Albericio F, Royo M, Del Río JA, et al. Neurotoxicity of prion peptides mimicking the central domain of the cellular prion protein. PloS One 2013; 8:e70881; PMID:23940658; https://doi.org/10.1371/journal.pone.0070881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Burre J, Sharma M, Sudhof TC. Cell biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb Perspect Med 2017; Epub ahead of print;PMID:28108534; https://doi.org/10.1101/cshperspect.a024091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci 2010; 11:130; PMID:20946660; https://doi.org/10.1186/1471-2202-11-130 [DOI] [PMC free article] [PubMed] [Google Scholar]