

Abstract
Homocysteine, a metabolite of the methionine cycle has been reported to play a role in neurotoxicity through activation of NMDAR-mediated signaling pathway. The proposed mechanisms associated with homocysteine-NMDAR induced neurotoxicity involve a unique signaling pathway that triggers a crosstalk between ERK and p38 MAPKs, where activation of p38 MAPK is downstream of and dependent on ERK MAPK. However, the molecular basis of the ERK MAPK mediated p38 MAPK activation is not understood. The current study investigates whether AMPARs play a role in facilitating the ERK MAPK mediated p38 MAPK activation. Using surface biotinylation and immunoblotting approaches we show that treatment with homocysteine leads to a decrease in surface expression of GluA2-AMPAR subunit in neurons, but has no effect on the surface expression of GluA1-AMPAR subunit. Inhibition of NMDAR activation with D-AP5 or ERK MAPK phosphorylation with PD98059 attenuates homocysteine-induced decrease in surface expression of GluA2-AMPAR subunit. The decrease in surface expression of GluA2-AMPAR subunit is associated with p38 MAPK phosphorylation, which is inhibited by NASPM, a selective antagonist of GluA2-lacking Ca2+-permeable AMPARs. These results suggest that homocysteine-NMDAR mediated ERK MAPK phosphorylation leads to a decrease in surface expression of GluA2-AMPAR subunit resulting in Ca2+ influx through the GluA2-lacking Ca2+-permeable AMPARs and p38 MAPK phosphorylation. Cell death assays further show that inhibition of AMPAR activity with NBQX/CNQX or GluA2-lacking Ca2+-permeable AMPAR activity with NASPM attenuates homocysteine-induced neurotoxicity. We have identified an important mechanism involved in homocysteine-induced neurotoxicity that highlights the intermediary role of GluA2-lacking Ca2+-permeable AMPARs in the crosstalk between ERK and p38 MAPKs.
Keywords: Homocysteine, NMDA receptors, GluA2-AMPA receptors, ERK MAPK, p38 MAPK, neurotoxicity
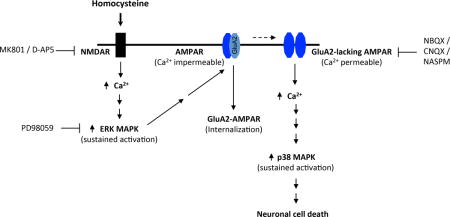
Graphical abstract
Homocysteine-induced neurotoxicity involves NMDAR-mediated activation of a unique crosstalk between ERK and p38 MAPKs. We report here that ERK MAPK activation leads to decrease in GluA2-AMPAR surface expression causing increased Ca2+ influx through GluA2-lacking Ca2+-permeable AMPARs resulting in p38 MAPK phosphorylation. The findings reveal an important intermediary role of GluA2-lacking AMPARs in homocysteine-NMDAR mediated crosstalk between ERK and p38 MAPKs.
INTRODUCTION
Hyperhomocysteinemia is a common metabolic disorder that is characterized by systemic elevation of homocysteine, a thiol containing amino acid formed as a metabolite of the methionine cycle. Hyperhomocysteinemia has been recognized as an independent risk factor for multiple neurodegenerative disorders (Obeid & Herrmann 2006, Seshadri et al. 2002, Sacco et al. 1998, Zoccolella et al. 2006, Miller 1999). It has been shown that elevated levels of homocysteine can induce neuronal injury and subsequent brain damage by stimulation of glutamate receptors (Duan et al. 2002, Kruman et al. 2002, Kruman et al. 2000). In vitro studies in cultured neurons further established the role of N-methyl-D-aspartate (NMDA) subtype of glutamate receptors in mediating homocysteine-induced neuronal cell death (Lipton et al. 1997, Kruman et al. 2000, Kruman et al. 2002, Mattson & Shea 2003, Jara-Prado et al. 2003, Poddar & Paul 2009, Poddar & Paul 2013). However, the intracellular signaling pathways associated with homocysteine-NMDA receptor (NMDAR) induced neuronal cell death is still not well understood.
Earlier studies have shown that Ca2+ influx following NMDAR stimulation with glutamate is biphasic in nature (Paul & Connor 2010, Hyrc et al. 1997, Cheng et al. 1999). A rapid low level initial increase through NR2A-containing NMDARs is followed by a delayed and larger increase that is NR2B-NMDAR dependent. Prior studies have also shown that Ca2+ influx following NMDAR stimulation with homocysteine is substantially smaller than that observed with glutamate (Kruman et al. 2000), which raises the possibility that low level Ca2+ influx in response to homocysteine treatment is mediated through NR2A-containing NMDARs. Consistent with this view, our earlier findings demonstrated that homocysteine-induced NMDAR stimulation leads to sustained phosphorylation of the extracellular signal-regulated kinase/mitogen activated protein kinase (ERK MAPK) and inhibition of NR2B-NMDARs with selective antagonists Ro-256981 or Ifenprodil failed to attenuate homocysteine-induced ERK MAPK phosphorylation. The study also showed that the active ERK MAPK plays a critical role homocysteine-NMDAR mediated neurotoxicity (Poddar & Paul 2009). These findings strongly indicate that homocysteine-NMDA receptor induced neurotoxicity involves a unique signaling pathway that requires stimulation of NR2A containing NMDARs. In a more recent study, we further demonstrated that homocysteine-mediated NMDAR stimulation leads to activation of the stress-activated kinase p38 MAPK, which is dependent on and downstream of ERK MAPK activity (Poddar & Paul 2013). Induction of p38 MAPK activation in turn leads to activation of caspase-3 resulting in neuronal apoptosis (Poddar & Paul 2013). The findings highlight a novel interplay between ERK and p38 MAPKs in response to homocysteine-mediated NMDAR stimulation and provide the first evidence that a crosstalk between the two MAPKs is essential for neuronal cell death.
The present study sought to determine the role of AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)-type glutamate receptors (AMPAR) in the crosstalk between ERK and p38 MAPKs. Our findings show that activation of ERK MAPK in response to homocysteine treatment leads to decrease in surface expression of the GluA2-AMPAR subunit. The removal of GluA2 subunit from cell surface is associated with increased influx of Ca2+ through the GluA2-lacking Ca2+-permeable AMPARs resulting in p38 MAPK phosphorylation that causes neuronal cell death.
EXPERIMENTAL PROCEDURES
Materials and reagents
All reagents required for primary cortical neuron cultures were obtained from Invitrogen (Carlsbad, CA, USA). L-homocysteine thiolactone, cytosine D-arabinofuranoside, glycine and Hoechst 33342 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-phospho-ERK1/2 (Thr202/Tyr204) monoclonal antibody (p-ERK, RRID:AB_331768), anti-phospho-p38 (Thr180/Tyr182) polyclonal antibody (p-p38, RRID:AB_331762), anti-p38 polyclonal antibody (RRID:AB_330731), anti-GluA2 antibody (RRID:AB_2650557), anti-rabbit (RRID:AB_2099233) and anti-mouse (RRID:AB_330924) horse-radish-peroxidase conjugated secondary antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-ERK2 polyclonal antibody (ERK, RRID:AB_2141292) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-GluA1 antibody (RRID:AB_2113602) and anti-phospho-GluA2-Ser880 (p-GluA2, RRID:AB_880227) were obtained from Abcam (Cambridge, MA, USA). Anti-rabbit-Alexa 488-conjugated antibody (RRID:AB_143165) was obtained from Molecular Probes (Eugene, OR, USA) and anti-mouse-Cy3 conjugated antibody (RRID:AB_2338690) was purchased from Jackson ImmunoResearch laboratories (West Grove, PA USA). West Pico supersignal chemiluminescence reagents, EZ-link NHS-PEG4-Biotin and NeutrAvidin agarose was obtained from Pierce (Rockland, IL, USA). Complete protease inhibitor cocktail was obtained from Roche (Nutley, NJ, USA). Selective pharmacological inhibitors were obtained as follows: MK801 hydrogen maleate and 6-cyano-7-nitroquinoxaline-2, 3, -dione (CNQX) from Sigma-Aldrich (St. Louis, MO, USA), 2,3-Dioxo-6-nitro-1,2,3,4,tetrahydrobenzoquinoxaline-7-sulfonamide (NBQX) and 1-Napthyl acetyl spermine trihydrochloride (NASPM) from Tocris, (Bristol, UK), D-(−)-2-Amino-5-phosphopentanoic acid (D-AP5) and PD98059 were obtained from EMD biosciences (Billerica, MA, USA). For establishing primary neuron cultures, Sprague-Dawley (RRID:RGD_737903) female pregnant rats (gestation day 15) were purchased from Harlan Laboratories (Livermore, CA, USA). Institutional Animal Care and Use Committee of University of New Mexico, Health Sciences Center approved all animal procedures used in the current study.
Neuron culture, L-Homocysteine preparation and stimulation
Primary cortical neuronal cultures were established from 16–17 day old rat embryos as described earlier (Poddar & Paul 2009, Poddar & Paul 2013), and the cells were maintained in culture for 12–14 days prior to experiment. L-homocysteine was prepared by alkali hydrolysis of L-homocysteine thiolactone hydrochloride followed by neutralization, and maintained in 0.02 mM N-Tris(hydroxymethyl)methyl-2-aminoethanesulfonicd acid (TES) buffer pH 7.4 (Poddar et al. 2001). For receptor stimulation neurons were treated with freshly prepared 50 μM of L-homocysteine in Hank’s balanced salt solution (Poddar & Paul 2009, Poddar & Paul 2013) containing 50 μM of glycine (Lipton et al. 1997). Cells were processed for immunoblotting and biotinylation studies after 4 h of homocysteine treatment or for cell death assay after 18 h of treatment. Initial studies evaluating the effect of tetrodotoxin (0.5 μM) showed that L-homocysteine induced changes in ERK and p38 MAPK phosphorylation were insensitive to tetrodotoxin treatment (data not shown). For some studies, selective antagonists D-AP5, MK801, NBQX, CNQX, NASPM or PD98059 were added either 30 min or 3.5 h after the onset of L-homocysteine treatment (Poddar & Paul 2013) to assess the effect of both long-term and short-term inhibition of NMDAR, AMPAR and GluA2-lacking Ca2+ permeable AMPAR activity as well as ERK MAPK phosphorylation on GluA2-AMPAR subunit surface expression as well as ERK and p38 MAPK phosphorylation. Cell lysates were analyzed by immunoblotting, biotinylation or cell death assay as specified in each experiment.
Subcellular fractionation
Subcellular fractionation of cultured neurons, treated with L-homocysteine (4 h) was performed as described previously (Dunah & Standaert 2001) with minor modifications. Briefly, neurons were washed twice with ice-cold TEVP buffer (10 mM Tris-HCl, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, and 1 mM EGTA, pH 7.4) following treatment with homocysteine and then homogenized on ice in TEVP buffer containing 320 mM sucrose. The cell homogenate was centrifuged at 4°C for 10 min at 800 × g to remove the nuclei and large debris. The supernatant (S1) was collected into a separate tube and further centrifuged at 4°C for 15 min at 9,200 × g to obtain a pellet that was comprised of the crude synaptosomal fraction (P2). The resulting supernatant (S2) was removed and centrifuged again at 4°C for 2 h at 1,65,000 × g to obtain the final cytosolic fraction (S3). The pellet obtained (P3) consisted of the light membrane/microsome-enriched fraction. At each step, small amounts of the supernatants (S1, S2 and S3) were removed for immunoblot analysis. To avoid possible crossover contamination, all the pellets (P1, P2 and P3) were rinsed once with cold TEVP buffer before immunoblot analysis.
Surface Biotinylation assay
Neurons were treated with L-homocysteine for 4 h in the presence or absence of pharmacological inhibitors as specified in each experiment. The inhibitors were added either 30 min or 3.5 h after the onset of homocysteine treatment. At the end of the treatment the cells were rinsed twice in ice-cold phosphate-buffered saline, pH 7.4 (PBS). Cell surface protein biotinylation and isolation of biotinylated proteins were performed using EZ-link NHS-PEG4-Biotin according to manufacturer’s protocol. Briefly, neurons were incubated on ice with PBS containing 500 μg/ml EZ-link NHS-PEG4-Biotin for 20 min. Following incubation, the unreacted biotinylation reagent was removed by washing the cells three times with an ice-cold buffer containing 20 mM Tris-HCl and 150 mM NaCl (pH 7.4). Cells were then harvested with lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Triton-X 100, 0.5% Sodium deoxycholate, 50 mM NaF; 10 mM Na4P2O7; 1 mM Na3VO4 and complete protease inhibitor cocktail. Homogenates were centrifuged at 14,000 × g for 15 min at 4°C. Equal amounts of protein from the resulting supernatants were incubated overnight with NeutrAvidin agarose (50 μL) to isolate the biotinylated proteins. The purified biotinylated proteins were analyzed by immunoblotting as specified in each experiment. The amount of total protein of interest present in each of the input lysate was determined by immunoblot analysis of equal proteins from the input lysates.
Immunoblotting
Equal protein from total cell lysates and subcellular fractions as well as the purified biotinylated proteins were resolved in 8% SDS-PAGE, and subjected to immunoblot analysis with antibodies as described in each experiment. All primary antibodies (anti-p-ERK, -ERK, -p-p38, -p38, -GluA1, -GluA2 and -p-GluA2-ser880) and secondary-horse radish peroxidase conjugated antibody concentrations were used according to recommendations made by the manufacturer. Signals from immune complexes were developed using West Pico supersignal chemiluminescence reagents and then captured on X-ray films. Densitometric analysis of the images was performed using the Image J software.
Cell death assay
Neuronal cultures grown on 2-well culture slides were treated with L-homocysteine in the presence or absence of selective inhibitors for 18 h. Cells were then fixed with 4% paraformaldehyde in PBS (pH 7.4) and stained with Hoechst 33342 dye as previously described (Poddar & Paul 2009). The percentage of pyknotic cells was assessed using fluorescent microscopy after counting a total of 1,500 nuclei for each set of experiments (n = 4).
Statistical analysis
Statistical analysis and comparison was performed using One-way analysis of variance (ANOVA, Bonferroni’s multiple comparison test) and differences were considered significant when p < 0.05.
RESULTS
Differential regulation of ERK and p38 MAPK by AMPAR
Our previous studies showed that homocysteine-NMDAR induced phosphorylation of both ERK and p38 MAPK in neurons follows a two-tier pattern (Poddar & Paul 2013). A rapid initial increase by 2.5 min is followed by a delayed larger increase by 4 h and the delayed increase in ERK and p38 MAPKs phosphorylation is primarily responsible for homocysteine-NMDAR dependent neurotoxicity. To examine the role of AMPARs in regulating the delayed increase in the phosphorylation of ERK and p38 MAPKs, neuronal cultures were treated with L-homocysteine (50 μM) for 4 h in the presence or absence of NMDA or AMPAR antagonists. The NMDAR antagonists, D-AP5 (100 μM) and MK801 (5 μM) or the AMPAR antagonists, NBQX (10 μM) and CNQX (50 μM) were added either 30 min (long-term inhibition) or 3.5 h (short-term inhibition) after the onset of homocysteine treatment. Immunoblot analysis was performed with anti-p-ERK or -p-p38 MAPK antibody. As previously reported (Poddar & Paul 2013), phosphorylation of both ERK and p38 MAPKs increased significantly following treatment with homocysteine (Fig. 1A–D). Application of D-AP5 or MK801, either 30 min or 3.5 h after the onset of homocysteine treatment blocked homocysteine-induced phosphorylation of both ERK MAPK (Fig. 1A) and p38 MAPK (Fig. 1B). However, treatment with NBQX or CNQX had no effect on ERK MAPK phosphorylation (Fig. 1C), regardless of the time of application of the inhibitors, but they effectively blocked p38 MAPK phosphorylation (Fig. 1D). This indicates a role of AMPARs in regulating homocysteine-induced p38 MAPK phosphorylation. The findings also raise the possibility that AMPAR activity play an intermediary role in regulating the cross talk between ERK MAPK and p38 MAPK.
FIGURE 1. AMPAR activity regulates homocysteine-induced p38 MAPK phosphorylation and neuronal cell death.

(A – D) Neuronal cultures were treated with 50 μM L-homocysteine (L-Hcy) for 4 h. (A, B) NMDAR antagonist D-AP5 (100 μM) and MK801 (5 μM) or (C, D) AMPAR antagonist NBQX (10 μM) and CNQX (50 μM) were added 30 min or 3.5 h after the onset of L-homocysteine treatment. Extracts with equal amounts of protein from each sample were analyzed with (A, C) anti-phospho-pERK1/2 (p-ERK) or anti-ERK antibody; and (B, D) anti-phospho-p38 (p-p38) or anti-p38 antibody. (A-D) Phosphorylated ERK and p38 MAPKs were quantified using computer-assisted densitometry and Image J analysis. Bar diagrams represent mean ± SEM (n = 4). *p < 0.001 from 0 min time point or control; #p < 0.001 from 4 h L-homocysteine treatment.
To examine the role of AMPARs in homocysteine-induced neuronal cell death, neurons were incubated with L-homocysteine (50 μM) in the absence or presence of NBQX (10 μM) or CNQX (50 μM) and cell viability was assessed 18 h later by Hoechst DNA staining (Fig. 2). As previously demonstrated (Poddar & Paul 2009, Poddar & Paul 2013), treatment with L-homocysteine alone significantly increased neuronal cell death (17.39% ± 1.35 for control vs. 44.65% ± 1.6 for homocysteine). Application of NBQX or CNQX during homocysteine treatment led to a significant reduction in neuronal cell death (20.82% ± 1.29 for NBQX and 18.98% ± 1.81 for CNQX), confirming a role of AMPARs in homocysteine-induced neuronal cell death.
FIGURE 2. Role of AMPAR activity in homocysteine-induced neuronal cell death.

Neuronal cultures were treated with L-homocysteine (50 μM, 18 h) in the absence or presence of NBQX (10 μM) or CNQX (50 μM) that were added 30 min after the onset of L-homocysteine treatment and then stained with Hoechst 33342, 18 h later. Representative photomicrographs show pyknotic DNA stained with Hoechst 33342 (indicated with arrows). Percentage of neurons with pyknotic nuclei is represented as mean ± SEM (n = 1500 cells/condition from 4 experiments). *p < 0.001 from control; #p < 0.05 from 18 h homocysteine treatment.
Homocysteine reduces cell surface expression of GluA2-AMPAR subunit
AMPARs at excitatory synapses in the forebrain consist predominantly of GluA1 and GluA2 subunits with low levels of GluA3 and GluA4, and alteration in surface expression of any subunit can dramatically alter AMPAR activity and function (Sprengel 2006, Malinow & Malenka 2002, Bredt & Nicoll 2003, Geiger et al. 1995, Sans et al. 2003, Tsuzuki et al. 2001). To explore the possible role of homocysteine in altering cell surface expression of GluA1 and GluA2 AMPAR subunits, neurons were treated with or without L-homocysteine (50 μM) for 4 h followed by biotin labeling of cell surface proteins and affinity purification of the biotinylated proteins. Immunoblot analysis of the purified biotinylated proteins, using GluA1 and GluA2 antibodies showed that L-homocysteine treatment had no effect on the cell surface expression of GluA1-AMPAR subunit (Fig. 3A, upper panel). However, the cell surface expression of GluA2-AMPAR subunit decreased significantly in the presence of L-homocysteine (Fig. 3B, upper panel). Immunoblot analysis of the total GluA1 and GluA2 protein levels in the input lysate showed no alteration in the expression of either GluA1 or GluA2 with homocysteine treatment (Fig. 3A, B, lower panel). Taken together, these findings indicate that L-homocysteine plays a role in the loss of surface expression GluA2-AMPAR subunit. Such decreases in GluA2 subunit are known to increase AMPAR Ca2+ permeability and has profound consequences on intracellular signaling (Hanley 2014).
FIGURE 3. Homocysteine regulates surface expression of GluA2-AMPAR subunit.

(A–C) Neuron cultures were treated with 50 μM of L-homocysteine (L-Hcy) for 4 h. (A, B) L-homocysteine treated cells were subjected to biotinylation of surface proteins followed by immunoblot analysis of the purified biotinylated proteins using (A) anti-GluA1 (upper panel) or (B) anti-GluA2 (upper panel) antibody. Equal amounts of protein from total cell extracts (input lysate) were also analyzed by immunoblotting using (A) anti-GluA1 (lower panel) or (B) anti-GluA2 (lower panel) antibody. (C) An equal amount of protein from total cell lysates was assessed for GluA2 phosphorylation at Ser880 by immunoblot analysis using anti-phospho-GluA2-S880 antibody (p-GluA2; upper panel). Total GluA2 was also analyzed to indicate equal protein loading (lower panel). (A–C) Quantitative analysis of GluA1 and GluA2 surface expression as well as GluA2 phosphorylation at Ser880 is represented as mean ± SEM (n = 4). *p < 0.001 from untreated control.
Earlier studies have provided strong evidence that loss of surface expression of GluA2-AMPAR subunit is associated with phosphorylation of serine880 residue (ser880) at the C-terminus of GluA2 (Perez et al. 2001, Matsuda et al. 1999, Chung et al. 2000, Chung et al. 2003, Iwakura et al. 2001, Kim et al. 2001, Xia et al. 2000, Lin & Huganir 2007). These findings also indicate that phosphorylation of ser880 facilitates the internalization of GluA2 as well as inhibits the recycling of GluA2, thus retaining GluA2 intracellularly. To evaluate whether the decrease in surface expression of GluA2 following exposure to L-homocysteine also involves phosphorylation of ser880, in subsequent experiments neurons treated with L-homocysteine (50 μM, 4 h) were processed for immunoblot analysis with anti-p-ser880 antibody. Consistent with the earlier findings, a significant increase in the phosphorylation of GluA2-AMPAR subunit at Ser880 was observed following treatment with L-homocysteine (Figure 3C), suggesting a causal link between the loss of surface expression of GluA2-AMPAR subunit and its phosphorylation.
Role of NMDARs in homocysteine-induced surface expression of GluA2-AMPAR subunit
To determine the role of NMDARs in the homocysteine-mediated decrease in surface expression of GluA2-AMPAR subunit, neurons were treated with L-homocysteine (50 μM, 4 h) in the absence or presence of D-AP5 (100 μM) added either 30 min or 3.5 h after the onset of homocysteine treatment. Surface expression of GluA2-AMPAR subunit was evaluated by immunoblot analysis following biotinylation and purification of cell surface proteins. Figure 4A (upper panel) shows that application of the inhibitor either 30 min or 3.5h after the onset of homocysteine treatment attenuated homocysteine-dependent loss in surface expression of GluA2-AMPAR subunit. Analysis of the GluA2 total protein levels in the input lysates shows that the expression level of GluA2 did not alter with NMDAR inhibition. To examine the effect of NMDAR inhibition on phosphorylation of GluA2-AMPAR subunit at ser880, neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of D-AP5, added either 30 min or 3.5h after the onset of L-homocysteine treatment. Immunoblot analysis (Fig. 4B) shows that the homocysteine-induced phosphorylation of GluA2 at ser880 was inhibited in the presence of D-AP5, irrespective of the time of its application. The findings indicate a role of NMDARs in homocysteine-induced loss of surface expression GluA2-AMPAR subunit and its phosphorylation at Ser880.
FIGURE 4. Homocysteine-induced alteration in GluA2-AMPAR surface expression is regulated by NMDAR.

(A, B) Neuron cultures were exposed to 50 μM of L-homocysteine (L-Hcy) for 4 h and D-AP5 was added either 30 min or 3.5 h after the onset of L-homocysteine treatment. (A) Biotinylated surface proteins were analyzed by immunoblot analysis using anti-GluA2 antibody (upper panel). Total GluA2 in the input lysates was also analyzed by immunoblotting with anti-GluA2 antibody (lower panel). (B) Neuronal lysates were analyzed by immunoblotting with anti-p-GluA2 antibody (upper panel) and blots were re-probed with anti-GluA2 antibody (lower panel). (A, B) Quantitative analysis of GluA2 surface expression and phosphorylation at Ser880 is represented as mean ± SEM (n=4). *p < 0.05 from 0 min time point or control; #p < 0.05 from 4 h homocysteine treatment.
Role of ERK MAPK in regulating homocysteine-induced surface expression of GluA2-AMPAR subunit
To test the possibility that homocysteine-NMDAR mediated phosphorylation of ERK MAPK plays a role in GluA2-AMPAR subunit internalization, initial studies examined the intra-cellular localization of phosphorylated ERK MAPK. Neurons were treated with L-homocysteine (50 μM) for 30 min or 4 h and then analyzed by immunocytochemistry using p-ERK (red) and ERK (green) antibodies. Figure 5A shows that within 30 min of homocysteine exposure, phosphorylated ERK MAPK was predominantly localized in the extranuclear region, where it remained sequestered for at least 4 h after the onset of treatment. To examine the subcellular localization of the extra-nuclear phosphorylated ERK MAPK, neurons treated with L-homocysteine (50 μM, 4 h) were subjected to biochemical fractionation (see Methods) and immunoblot analysis. Figure 5B (upper panel) shows that in the homocysteine treated samples, the supernatant (S1 fraction), obtained following the removal of nuclear material was highly enriched in phosphorylated ERK MAPK. Figure 5C shows that in the homocysteine treated samples, phosphorylated ERK MAPK was present in both the supernatant (S2 fraction) and the crude synaptosomal pellet (P2) obtained from the S1 fraction, although it was present at higher levels in the S2 fraction. High-speed centrifugation of the S2 fraction resulted in the separation of the cytoplasmic fraction (S3) and the light membrane/microsomal enriched fraction (P3). Immunoblot analysis of the S3 and P3 fractions showed that phosphorylated ERK MAPK was highly enriched in the cytoplasmic fraction (Fig. 5C, upper panel). Analysis of marker proteins for each sub-cellular compartment (Fig. 5C, panels 3 and 4) further confirmed that synaptophysin, a synaptic vesicle membrane protein was highly enriched in the crude synaptosomal fraction (P2), while calnexin, an integral membrane protein found in endoplasmic reticulum was highly enriched in the light membrane fraction (P3).
FIGURE 5. Subcellular localization of phosphorylated ERK MAPK in neurons treated with homocysteine.

(A) Immunocytochemical analysis illustrating the cytoplasmic and nuclear distribution of p-ERK (red) and ERK (green) MAPK in neurons following 50 μM of L-homocysteine (L-Hcy) treatment for 0 min, 30 min (0.5 h) or 4 h. The cell nuclei were counterstained with DAPI (blue). (B) Immunoblot analysis of the S1 fraction (supernatant following centrifugation of cell homogenate at 1,000 × g) obtained from neurons treated with L-homocysteine (50 μM) for 0 or 4 h using anti-p-ERK (upper panel) or anti-ERK (lower panel) antibody. (C) Sub-cellular distribution of phosphorylated ERK MAPK in neurons treated with L-homocysteine (50 μM, 4 h) was analyzed by immunoblot analysis of biochemical fractions with anti-p-ERK (panel 1) or anti-ERK (panel 2) antibody. Purity of the fractions was also evaluated by immunoblot analysis with anti-synaptophysin (panel 3) and anti-calnexin (panel 4) antibodies. The biochemical fractions evaluated are S2 (supernatant following centrifugation of S1 fraction at 10,000 × g), P2 (pellet comprising of crude synaptosomal membrane proteins following centrifugation of S1 fraction at 10,000 × g), S3 (final cytosolic fraction obtained following centrifugation of S2 fraction at 1,65,000 × g) and P3 (pellet comprising mainly of light membrane proteins obtained following centrifugation of S2 fraction at 1,65,000 × g).
The sub-cellular localization of phosphorylated ERK MAPK in the synaptosomal fraction (P2) suggests that ERK MAPK signaling could play a role in regulating homocysteine-dependent loss of surface expression of GluA2-AMPAR subunit. To test this possibility in subsequent studies, neurons were treated with L-homocysteine (50 μM, 4 h) in the absence or presence of PD98059 (15 μM), a pharmacological inhibitor of ERK MAPK. The inhibitor was added either 30 min or 3.5h after the onset of homocysteine treatment. Cells were then processed for biotinylation and purification of surface proteins followed by immunoblot analysis with anti-GluA2 antibody. As shown in Figure 6A (upper panel), PD98059 attenuated homocysteine-induced loss in surface expression of GluA2-AMPAR subunit, irrespective of the time of application of the inhibitor. Total level of GluA2 remained unaltered by this treatment (Fig. 6A, lower panel). Figure 6B shows that inhibition of ERK MAPK with PD98059, added either 30 min or 3.5h after the onset of homocysteine treatment, blocked the phosphorylation of ser880 in GluA2-AMPAR subunit. In addition, homocysteine-induced phosphorylation of p38 MAPK was also blocked following both long-term (3.5h) and short-term (30 min) inhibition of ERK MAPK (Fig. 6C), which is consistent with earlier findings (Poddar & Paul 2013). Thus, it appears that phosphorylated ERK MAPK regulates both the surface expression of GluA2-subunit of AMPARs and phosphorylation of p38 MAPK.
FIGURE 6. Homocysteine-NMDAR induced alteration in GluA2-AMPAR surface expression is regulated by ERK MAPK.

(A–C) Neuron cultures were exposed to 50 μM of L-homocysteine (L-Hcy) for 4 h and PD98059 was added either 30 min or 3.5 h after the onset of L-Hcy treatment. (A) Biotinylated surface proteins were processed for immunoblot analysis with anti-GluA2 antibody (upper panel). The total GluA2 in the input lysates was also analyzed using anti-GluA2 antibody (lower panel). (B, C) Equal amount of protein from total cell extracts was analyzed by immunoblotting using (B) anti-p-GluA2 or (C) anti-p-p38 antibody (upper panels). The blots were re-probed with (B) anti-GluA2 or (C) anti-p38 antibody (lower panels). (A, B) Quantitative analysis of GluA2 surface expression and phosphorylation at Ser880 is represented as mean ± SEM (n = 4). *p < 0.05 from 0 min time point or control; #p < 0.05 from 4 h homocysteine treatment.
Inhibition of GluA2-lacking AMPAR activity reduces homocysteine-induced p38 MAPK phosphorylation and neuronal cell death
Our earlier studies have shown that homocysteine-induced p38 MAPK phosphorylation and subsequent neuronal cell death are Ca2+ dependent (Poddar & Paul 2013). Since AMPARs lacking GluA2 subunit are Ca2+-permeable (Hollmann et al. 1991, Burnashev et al. 1992, Geiger et al. 1995, Swanson et al. 1997), it raises the possibility that Ca2+ influx through GluA2-lacking AMPARs may play a role in p38 MAPK phosphorylation. To examine the contribution of GluA2 lacking AMPAR activity in facilitating homocysteine-mediated p38 MAPK phosphorylation, neurons were treated with L-homocysteine (50 μM, 4h) in the absence or presence of NASPM (100 μM), a selective antagonist of GluA2-lacking Ca2+ permeable AMPARs (Koike et al. 1997, Russell et al. 2016, Gerace et al. 2014, Oguro et al. 1999, Dong et al. 2015, Conrad et al. 2008, Noh et al. 2005). The inhibitor was added either 30 min or 3.5 h after the onset of homocysteine treatment. Immunoblot analysis with anti-p-p38 antibody shows that both long-term (3.5h) and short-term (30 min) inhibition of GluA2-lacking Ca2+-permeable AMPAR activity with NASPM blocked homocysteine-induced phosphorylation of p38 MAPK (Fig 7A). However, homocysteine-mediated phosphorylation of ERK MAPK remained unaltered in the presence of NASPM, irrespective of the time of its application (Fig 7B). To further clarify the role of the GluA2 lacking Ca2+-permeable AMPARs in homocysteine-mediated neuronal cell death, neurons were treated with homocysteine in the absence or presence of NASPM for 18 h and analyzed by Hoechst DNA staining. Figure 7C shows that treatment with NASPM significantly reduced homocysteine-induced neuronal cell death (44.65% ± 1.6 for homocysteine vs. 23.07% ± 2.45 for NASPM). The findings establish the role of Ca2+ permeable AMPAR activity in homocysteine-induced p38 MAPK phosphorylation and subsequent cell death.
FIGURE 7. Role of GluA2-lacking AMPAR activity in homocysteine-mediated phosphorylation of p38 MAPK and subsequent neuronal death.

(A, B) Neuronal cultures were treated with 50 μM of L-homocysteine (L-Hcy) for 4 h and NASPM (100 μM) or NBQX (10 μM) was added 30 min or 3.5 h after the onset of homocysteine treatment. Phosphorylation of ERK MAPK and p38 MAPKs was analyzed using (A) anti-p-ERK antibody or (B) anti-p-p38 antibody (upper panels). Total pERK MAPK or p38 MAPK was analyzed by reprobing the blots with either (A) anti-ERK or (B) anti-p38 antibody (lower panels). Quantitative analysis of phosphorylated ERK and p38 MAPKs is represented as mean ± SEM (n = 4). *p < 0.001 from 0 min time point or control; #p < 0.001 from 4 h homocysteine treatment. (C) Neurons were treated with L-homocysteine (50 μM, 18 h) in the absence or presence of NASPM (100 μM, added 30 min after the onset of L-homocysteine treatment) and then stained with Hoechst 33342, 18 h later. Representative photomicrographs show pyknotic DNA stained with Hoechst 33342 (indicated with arrows). Percentage of neurons with pyknotic nuclei is represented as mean ± SEM (n = 1500 cells/condition from 4 experiments). *p < 0.001 from control; #p < 0.05 from 18 h homocysteine treatment.
DISCUSSION
The key finding of the current study is that active ERK MAPK, localized in the extranuclear compartments plays a role in homocysteine-NMDAR dependent decrease in surface expression of the GluA2-AMPAR subunit. Ca2+ influx through GluA2-lacking, Ca2+-permeable AMPARs lead to phosphorylation and activation of p38 MAPK resulting in neuronal cell death. The findings demonstrate for the first time that AMPAR activity plays a role in homocysteine-induced neurotoxicity by facilitating the crosstalk between ERK and p38 MAPKs.
AMPARs are the principal mediators of fast synaptic neurotransmission at majority of the excitatory synapses in the central nervous system (Dingledine et al. 1999, Traynelis et al. 2010). AMPARs are tetrameric assemblies of GluA1-GluA4 subunits, and the vast majority of the AMPARs at the adult forebrain synapses are heterotetramers of GluA2 and another subunit (Dingledine et al. 1999, Tsuzuki et al. 2001, Geiger et al. 1995, Loschmann et al. 2004). Majority of GluA2 subunit in the adult brain contains an arginine residue in the pore lining membrane domain at position 607 in place of the genomically encoded glutamine, owing to RNA editing, which renders functional AMPARs impermeable to Ca2+ (Sommer et al. 1991, Hume et al. 1991, Hollmann & Heinemann 1994, Man 2011). In the adult brain, both transient and prolonged alteration in the level of GluA2-AMPAR at the synapses in response to neuronal activity modulates Ca2+ entry into neurons and has profound implications on neuronal communication, long-term potentiation and homeostatic plasticity (Greger & Esteban 2007, Beattie et al. 2000, Hollmann et al. 1991, Sutton et al. 2006, Man 2011, Hou et al. 2008). AMPAR subunit profile also changes in some brain areas during the critical period of postnatal development with the recruitment of GluA2-lacking Ca2+-permeable AMPARs (Stubblefield & Benke 2010, Kumar et al. 2002, Henley & Wilkinson 2016, Pellegrini-Giampietro et al. 1992a), a process that is thought to be important for glutamatergic synapse maturation and experience-dependent strengthening of neural connectivity (Isaac et al. 2007, Liu & Savtchouk 2012, Liu & Cull-Candy 2000, Ho et al. 2007, Takahashi et al. 2003, Shi et al. 1999, Louros et al. 2014). However, such increase in GluA2-lacking Ca2+-permeable AMPARs during development may also increase the vulnerability of the immature brain to hypoxia-induced injury and seizure (Talos et al. 2006a, Talos et al. 2006b, Hagberg et al. 1994, Koh et al. 2004). As such, neurons tightly regulate the function of this subunit through a number of processes at the level of gene expression, RNA editing, recycling and trafficking to ensure normal brain function (Hou et al. 2008, Tanaka et al. 2000).
In addition to its role in synaptic plasticity and adaptation during development and in the adult brain GluA2-lacking, Ca2+ permeable AMPARs also appears to be involved in neurological disorders and diseases associated with excitotoxicity (Liu & Zukin 2007, Pellegrini-Giampietro et al. 1997). Considerable evidence indicates that delayed neurodegeneration following global ischemia and limbic seizures is triggered by enhanced AMPAR-elicited Ca2+ influx in vulnerable hippocampal neurons (Gorter et al. 1997, Opitz et al. 2000, Pellegrini-Giampietro et al. 1992b, Noh et al. 2005, Friedman et al. 1994, Huang et al. 2002, Pollard et al. 1993). The Ca2+ permeability of AMPAR under these pathological conditions involves down regulation of GluA2 mRNA and subunit expression resulting in a long lasting switch from GluA2-containing to GluA2-lacking functional AMPARs (Grooms et al. 2000, Park et al. 2009). Studies using cultured neurons exposed to glutamate or subjected to oxygen glucose deprivation further showed that internalization of GluA2 subunit along with increased trafficking of GluA2-lacking AMPAR to the plasma membrane could lead to enhanced Ca2+ accumulation resulting in excitotoxic cell death (Bose et al. 2012, Vikman et al. 2008). Additional studies in the hippocampus demonstrated that expression of long-term depression also involves internalization of GluA2-subunit of AMPARs, which is triggered by NMDAR activation (Beattie et al. 2000). NMDAR activation has also been associated with increased prevalence of GluA2-lacking AMPARs in the hypothalamus under hypertensive conditions and is involved in neuronal hyperactivity (Li et al. 2012). Persistent inflammation and nerve injury also increases GluA2-lacking AMPAR prevalence in the spinal dorsal horn and contributes to nociceptive hypersensitivity and neuropathic pain (Galan et al. 2004, Li et al. 2012, Vikman et al. 2008, Park et al. 2009). Such increased prevalence of GluA2-lacking AMPARs in the spinal cord is also associated with NMDAR activation. Consistent with these earlier findings, the present study now demonstrates that homocysteine-induced signaling through NMDAR is critical in the phenotypic switch from predominantly GluA2-containing to GluA2-lacking AMPARs in neurons and ERK MAPK plays an intermediary role in this phenomenon. The causal link between the decrease in surface expression of GluA2-AMPAR subunit and neuronal cell death suggests that Ca2+-influx through the GluA2-lacking, Ca2+-permeable AMPARs contributes to homocysteine-induced neurotoxicity. The ability of NASPM, which selectively inhibits GluA2-lacking AMPAR activity, to attenuate homocysteine-induced cell death further confirms the role of these receptors in mediating the neurotoxic effects of homocysteine.
The intracellular mechanisms associated with activity dependent modulation of AMPAR trafficking are still poorly understood. Multiple studies have examined the role of Ras and Ras-related protein (Rap), two members of the Ras superfamily of GTPases in AMPAR dependent synaptic plasticity. The findings showed that Ras mediates activity-induced synaptic enhancement by facilitating synaptic insertion of GluA1 or GluA4 subunits, while Rap mediates activity induced synaptic depression by removing synaptic GluA2 or GluA3 subunits (Zhu et al. 2002). The downstream molecule(s) that relay Ras and Rap signaling includes ERK MAPK and p38 MAPK. While ERK MAPK activation is involved in synaptic insertion of AMPAR subunits, p38 MAPK is thought to play a role in removal of AMPARs from the synapses (Zhu et al. 2002, Iwakura et al. 2001, Derkach et al. 2007). A key finding of our study is that homocysteine-NMDAR mediated sustained activation and extranuclear retention of the active ERK MAPK promotes removal of surface GluA2-AMPAR subunits, resulting in the prevalence of GluA2-lacking Ca2+-permeable AMPARs that facilitates Ca2+ influx and cell death. Such sequestration of active ERK MAPK in extranuclear compartments has also been reported to be a mediator of gamma-interferon induced cell death where cytosolic ERK promotes the pro-apoptotic function of the Death associated protein kinase by phosphorylation (Xia et al. 2000). The opposing role of ERK MAPK on AMPAR trafficking under physiological and pathological conditions suggest that depending on the nature of the upstream signaling, the activation of ERK MAPK could trigger a unique sequence of events causing differential regulation of AMPAR subunits. Identification of the downstream effectors of ERK MAPK in future studies may further our understanding of the function of ERK MAPK in regulating AMPAR activity in neurological disorders.
Although the importance of Ca2+-signaling through GluA2 lacking, Ca2+-permeable AMPARs is well recognized (Hollmann et al. 1991, Burnashev et al. 1992, Geiger et al. 1995, Swanson et al. 1997), relatively little is known about its downstream signaling targets. Our results now show that homocysteine dependent decrease in cell surface expression of GluA2-AMPAR subunit is associated with p38 MAPK phosphorylation, suggesting that Ca2+ entry through GluA2-lacking Ca2+-permeable AMPARs triggers the activation of a signaling cascade necessary for p38 MAPK phosphorylation. Consistent with this interpretation, selective inhibition of GluA2-lacking AMPAR activity with NASPM blocks the homocysteine-dependent p38 MAPK phosphorylation. Whether such phosphorylation of p38 MAPK in the presence of GluA2-lacking AMPAR, as observed in our study, is unique to the homocysteine signaling pathway or could be triggered by stimulating GluA2-lacking AMPARs through other excitatory signals remains to be investigated.
In conclusion, the present study provides mechanistic insight into the signaling cascade underlying homocysteine-NMDAR mediated neurotoxicity. The findings highlight the role of ERK MAPK in regulating homocysteine-NMDAR mediated trafficking of GluA2-AMPAR subunit. In addition, the study also provides evidence for a role of GluA2 lacking Ca2+-permeable AMPARs in the crosstalk between ERK and p38 MAPKs. These findings demonstrating the effect of homocysteine in an ex vivo neuron culture model provides the molecular basis for evaluating the mechanisms identified here in animal models of hyperhomocysteienemia. Based on the findings subsequent studies will assess whether this unique signaling pathway triggered by homocysteine acts in conjunction with other deleterious cascades to enhance the pathology of neurological disorders under hyperhomocysteinemic conditions.
Acknowledgments
This work was supported by the National Institutes of Health grants RO1 NS083914 (Poddar, R); R21 NS065343 (Poddar, R); RO1 NS059962 (Paul, S).
Abbreviations
- AMPAR
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- NMDAR
N-methyl-D-aspartate receptors
- ERK MAPK
Extracellular-signal regulated kinase/mitogen activated protein kinase
Footnotes
CONFLICT OF INTEREST DISCLOSURE: The authors have no conflict of interest.
ARRIVE guidelines have been followed:
Yes
=> if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert "All experiments were conducted in compliance with the ARRIVE guidelines." unless it is a Review or Editorial
References
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nature neuroscience. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Bose A, Tsai J, Li D. Early non-response in patients with severe depression: escitalopram up-titration versus switch to duloxetine. Clin Drug Investig. 2012;32:373–385. doi: 10.2165/11631890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Cheng C, Fass DM, Reynolds IJ. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca(2+)](i) increases and NMDA receptor mRNA levels. Brain research. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science (New York, NY. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nature reviews. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological reviews. 1999;51:7–61. [PubMed] [Google Scholar]
- Dong LD, Gao F, Wang XH, et al. GluA2 trafficking is involved in apoptosis of retinal ganglion cells induced by activation of EphB/EphrinB reverse signaling in a rat chronic ocular hypertension model. J Neurosci. 2015;35:5409–5421. doi: 10.1523/JNEUROSCI.4376-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. Journal of neurochemistry. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21:5546–5558. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Gerace E, Masi A, Resta F, Felici R, Landucci E, Mello T, Pellegrini-Giampietro DE, Mannaioni G, Moroni F. PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca(2+)-permeable AMPA receptors. Neurobiol Dis. 2014;70:43–52. doi: 10.1016/j.nbd.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Esteban JA. AMPA receptor biogenesis and trafficking. Current opinion in neurobiology. 2007;17:289–297. doi: 10.1016/j.conb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Grooms SY, Opitz T, Bennett MV, Zukin RS. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3631–3636. doi: 10.1073/pnas.050586497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Gilland E, Diemer NH, Andine P. Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol Neonate. 1994;66:205–213. doi: 10.1159/000244109. [DOI] [PubMed] [Google Scholar]
- Hanley JG. Subunit-specific trafficking mechanisms regulating the synaptic expression of Ca(2+)-permeable AMPA receptors. Semin Cell Dev Biol. 2014;27:14–22. doi: 10.1016/j.semcdb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA. Synaptic AMPA receptor composition in development, plasticity and disease. Nature reviews. 2016;17:337–350. doi: 10.1038/nrn.2016.37. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science (New York, NY. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual review of neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science (New York, NY. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17:6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Nagano T, Kawamura M, Horikawa H, Ibaraki K, Takei N, Nawa H. N-methyl-D-aspartate-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor down-regulation involves interaction of the carboxyl terminus of GluR2/3 with Pick1. Ligand-binding studies using Sindbis vectors carrying AMPA receptor decoys. The Journal of biological chemistry. 2001;276:40025–40032. doi: 10.1074/jbc.M103125200. [DOI] [PubMed] [Google Scholar]
- Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A. Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotoxicity research. 2003;5:237–243. doi: 10.1007/BF03033381. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Tibayan FD, Simpson JN, Jensen FE. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569–575. doi: 10.1111/j.0013-9580.2004.69103.x. [DOI] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neuroscience research. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- Kruman II, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, et al. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Byan HS, Pan HL. Switch to glutamate receptor 2-lacking AMPA receptors increases neuronal excitability in hypothalamus and sympathetic drive in hypertension. J Neurosci. 2012;32:372–380. doi: 10.1523/JNEUROSCI.3222-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Savtchouk I. Ca(2+) permeable AMPA receptors switch allegiances: mechanisms and consequences. The Journal of physiology. 2012;590:13–20. doi: 10.1113/jphysiol.2011.213926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in neurosciences. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Loschmann PA, De Groote C, Smith L, Wullner U, Fischer G, Kemp JA, Jenner P, Klockgether T. Antiparkinsonian activity of Ro 25-6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson’s disease. Experimental neurology. 2004;187:86–93. doi: 10.1016/j.expneurol.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Louros SR, Hooks BM, Litvina L, Carvalho AL, Chen C. A role for stargazin in experience-dependent plasticity. Cell Rep. 2014;7:1614–1625. doi: 10.1016/j.celrep.2014.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annual review of neuroscience. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY. GluA2-lacking, calcium-permeable AMPA receptors–inducers of plasticity? Current opinion in neurobiology. 2011;21:291–298. doi: 10.1016/j.conb.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. Journal of neurochemistry. 1999;73:1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends in neurosciences. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Miller JW. Homocysteine and Alzheimer’s disease. Nutr Rev. 1999;57:126–129. doi: 10.1111/j.1753-4887.1999.tb06936.x. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz T, Grooms SY, Bennett MV, Zukin RS. Remodeling of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit composition in hippocampal neurons after global ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13360–13365. doi: 10.1073/pnas.97.24.13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Voitenko N, Petralia RS, et al. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J Neurosci. 2009;29:3206–3219. doi: 10.1523/JNEUROSCI.4514-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Connor JA. NR2B-NMDA receptor-mediated increases in intracellular Ca2+ concentration regulate the tyrosine phosphatase, STEP, and ERK MAP kinase signaling. Journal of neurochemistry. 2010;114:1107–1118. doi: 10.1111/j.1471-4159.2010.06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Bennett MV, Zukin RS. Are Ca(2+)-permeable kainate/AMPA receptors more abundant in immature brain? Neuroscience letters. 1992a;144:65–69. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends in neurosciences. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proceedings of the National Academy of Sciences of the United States of America. 1992b;89:10499–10503. doi: 10.1073/pnas.89.21.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Paul S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. Journal of neurochemistry. 2009;110:1095–1106. doi: 10.1111/j.1471-4159.2009.06207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Paul S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. Journal of neurochemistry. 2013;124:558–570. doi: 10.1111/jnc.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- Pollard H, Heron A, Moreau J, Ben-Ari Y, Khrestchatisky M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-y. [DOI] [PubMed] [Google Scholar]
- Russell SE, Puttick DJ, Sawyer AM, Potter DN, Mague S, Carlezon WA, Jr, Chartoff EH. Nucleus Accumbens AMPA Receptors Are Necessary for Morphine-Withdrawal-Induced Negative-Affective States in Rats. J Neurosci. 2016;36:5748–5762. doi: 10.1523/JNEUROSCI.2875-12.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco RL, Roberts JK, Jacobs BS. Homocysteine as a risk factor for ischemic stroke: an epidemiological story in evolution. Neuroepidemiology. 1998;17:167–173. doi: 10.1159/000026169. [DOI] [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, et al. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci. 2003;23:9367–9373. doi: 10.1523/JNEUROSCI.23-28-09367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science (New York, NY. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Sommer B, Kohler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Sprengel R. Role of AMPA receptors in synaptic plasticity. Cell and tissue research. 2006;326:447–455. doi: 10.1007/s00441-006-0275-4. [DOI] [PubMed] [Google Scholar]
- Stubblefield EA, Benke TA. Distinct AMPA-type glutamatergic synapses in developing rat CA1 hippocampus. Journal of neurophysiology. 2010;104:1899–1912. doi: 10.1152/jn.00099.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science (New York, NY. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. The Journal of comparative neurology. 2006a;497:42–60. doi: 10.1002/cne.20972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. The Journal of comparative neurology. 2006b;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Grooms SY, Bennett MV, Zukin RS. The AMPAR subunit GluR2: still front and center-stage. Brain research. 2000;886:190–207. doi: 10.1016/s0006-8993(00)02951-6. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological reviews. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki K, Lambolez B, Rossier J, Ozawa S. Absolute quantification of AMPA receptor subunit mRNAs in single hippocampal neurons. Journal of neurochemistry. 2001;77:1650–1659. doi: 10.1046/j.1471-4159.2001.00388.x. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Rycroft BK, Christie MJ. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. The Journal of physiology. 2008;586:515–527. doi: 10.1113/jphysiol.2007.145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zoccolella S, Martino D, Defazio G, Lamberti P, Livrea P. Hyperhomocysteinemia in movement disorders: Current evidence and hypotheses. Curr Vasc Pharmacol. 2006;4:237–243. doi: 10.2174/157016106777698414. [DOI] [PubMed] [Google Scholar]