

Abstract
Protein O-mannosylation is crucial for the biology of Mycobacterium tuberculosis but the key mannosylated protein(s) involved and its(their) underlying function(s) remain unknown. Here, we demonstrated that the M. tuberculosis mutant (Δpmt) deficient for protein O-mannosylation exhibits enhanced release of lipoarabinomannan (LAM) in a complex with LprG, a lipoprotein required for LAM translocation to the cell surface. We determined that LprG is O-mannosylated at a unique threonine position by mass spectrometry analyses of the purified protein. However, although replacement of this amino acid by an alanine residue completely abolished LprG O-mannosylation, the increased release of the LAM/LprG complex was preserved. We found that the increased secretion of this complex is due to enhanced LAM production in the Δpmt M. tuberculosis and M. smegmatis mutants relative to their wild-type counterparts. This abnormal release of LAM/LprG has functional consequences on the induction of inflammatory responses and provides a possible explanation for the reduced virulence of the M. tuberculosis Δpmt mutant.
Introduction
Glycosylation is one of the most common post-translational modifications of proteins that considerably affects protein properties/functions and biological activities in eukaryotes. Until recently, protein glycosylation was thought to occur only rarely in prokaryotes with limited impact on their biology1. However, the analysis of protein reactivity with the lectin Concanavalin A and the development of mass spectrometry (MS) for protein analysis have revealed that several proteins in actinomycetes are O-mannosylated, especially in the human major pathogen, Mycobacterium tuberculosis. The first identified glycoproteins were the secreted antigens PstS1 (38 kDa), the alanine-proline rich Apa protein (45–47 kDa) and the lipoprotein LpqH (19 kDa)2–5. These results garnered immediate widespread attention because this post-translational modification may play an important role in the biology of M. tuberculosis and the host/pathogen interaction. Indeed, M. tuberculosis is the causative agent of human tuberculosis (TB), a disease which still remains a major public health concern with 10.4 million new cases and 1.8 million deaths in 20156. A better understanding of the strategies used by this pathogen to circumvent the host immune defense, and thus to persist and to cause disease, may reveal weak spots in the TB bacilli that can be exploited to develop innovative approaches to control this important pathogen and the associated disease.
O-mannosylated proteins purified from mycobacteria bind to host innate-immune receptors, such as lung surfactant protein A, DC-SIGN (dendritic-cell-specific intercellular molecule-3-grabbing non-integrin), and the mannose receptor and contribute to phagocyte invasion7–9. In addition, T cell antigenicity of secreted M. tuberculosis glycoproteins is modulated by the pattern of O-mannosylation. The best documented example is the Apa antigen, which contains up to nine mannose residues when purified from M. tuberculosis or the model mycobacterial strain Mycobacterium smegmatis, but no sugar residue when expressed in, and purified from, Escherichia coli 10, 11. The glycosylated forms of Apa induce strong delayed-type hypersensitivity reactions (DTH) in vivo and exhibit a high capacity to stimulate T lymphocyte responses of guinea pigs immunized with BCG. In contrast, non-glycosylated Apa, obtained either following deglycosylation of the native form from M. tuberculosis or purified from E. coli, is more than 10-fold less potent in inducing these responses10, 11. Similar results have been obtained in mice and humans showing that O-mannosylation of Apa enhances its T cell antigenicity12. However, glycosylation of this antigen is dispensable for the induction of a protective immune response in mice12.
The key finding demonstrating the major role played by O-mannosylation in the biology of TB bacilli came from the construction and the phenotypic characterization of a defined M. tuberculosis mutant deficient for this post-translational modification13. VanderVen et al.14 identified a protein, encoded by the Rv1002c gene in M. tuberculosis, that exhibits sequence similarities with membrane-associated protein O-mannosyl transferase from Saccharomyces cerevisiae. They demonstrated that Rv1002c (pmt) encodes an O-mannosyl transferase that catalyzes the initial step of protein glycosylation in M. tuberculosis and that this process is coupled to Sec-translocation14. Knock-out of Rv1002c, and its ortholog Msmeg_5447, in M. tuberculosis and M. smegmatis respectively demonstrated that these genes are dispensable for in vitro mycobacterial growth, in contrast to eukaryotes where protein O-mannosylation is required for viability13. Mass-spectrometry (MS) analyses showed that the mutant strains were indeed deficient in protein O-mannosylation consistent with the hypothesis that Rv1002c and MSMEG_5447 have no paralogs in M. tuberculosis and M. smegmatis respectively. The M. tuberculosis ΔRv1002c::km (Δpmt) mutant displayed several phenotypes in vitro and in vivo demonstrating that protein O-mannosylation is a major post-translational modification for the biology of this pathogen. First, the ability of the Δpmt mutant to form colonies on solid medium was profoundly impaired and it exhibited delayed growth in liquid medium containing dextrose as the sole carbon source. Second, this mutant was strongly attenuated in several cellular and animal infection models: its growth in mouse alveolar macrophages or primary human blood monocyte-derived macrophages and its capacity to kill immunocompromised mice were severely impaired13. These results established Rv1002c as a membrane-associated protein O-mannosyl transferase of crucial importance for the biology of M. tuberculosis and especially for its interaction with the host. However, these studies did not identify the Rv1002c-target proteins responsible for the in vitro growth defect and attenuated phenotype when not glycosylated.
Two main strategies have been used to identify glycosylated proteins in M. tuberculosis. Zamorano et al.15 performed glycoproteins enrichment using the lectin Concanavalin A, followed by 2D gel electrophoresis and conventional mass spectrometry analysis of the individual spots. They identified 41 proteins, but did not provide direct evidence that they were indeed glycosylated. More recently, advances in MS technologies have allowed the development of analytical approaches to directly search for glycopeptides in complex protein extracts. For example, Smith et al.16 used liquid chromatography (LC) coupled to tandem MS analyses to identify 14 glycopeptides, and to localize their glycosylsation sites, in M. tuberculosis culture filtrate corresponding to 13 glycosylated proteins. These results largely extended the repertoire of M. tuberculosis glycoproteins but did not address the question of the impact of this post-translational modification on the functions of these proteins.
Here, we aimed to address this question and to examine how the O-mannosylation of M. tuberculosis proteins may contribute to host/pathogen interactions. We identified the lipoprotein, LprG, as a new O-mannosylated protein and characterized its glycosylation site. We demonstrated that the lack of protein O-mannosylation is associated with increased production of lipoarabinomannnan (LAM), a critical mycobacterial virulence factor17, and increased release of this lypoglycan complexed with LprG. We found that this perturbation is associated with enhanced stimulation of Toll-Like Receptor 2 (TLR2), a key host receptor to control M. tuberculosis infection18, and a stronger induction of inflammatory responses by infected macrophages.
Results
Phenotypes of M. tuberculosis mutants deficient in selected mannoprotein production
We applied the recently developed glycopeptidomics approach13 for the analysis of the M. tuberculosis secretome to further enrich the mannoprotein repertoire of M. tuberculosis. The combination of high efficiency SDS-PAGE based shotgun MS proteomics and automated post-analytical data mining led to the unambiguous characterization of several tryptic glycopeptides allowing the identification of the parent glycoproteins. Five of them, i.e. Rv0175, Rv0315, Rv0838 (LpqR), Rv1411c (LprG) and Rv3491, were then selected on the basis of the confidence of their glycopeptide identification (Supplementary Figure S1), their novelty with regards to previously characterized O-mannosylated M. tuberculosis proteins and their putative role in pathogenicity (Table 1). We reasoned that if O-mannosylation affects the function of a protein in a defined condition, then a knock-out mutation of the corresponding gene should also affect the phenotype in the same condition. Therefore, we individually disrupted each of the five target genes in the M. tuberculosis reference strain H37Rv and introduced a kanamycin resistant marker (km) together with an oligonucleotide specific tag by homologous recombination (Supplementary Figure S2). None of the mutants exhibited growth defects in standard ADC-enriched 7H9 liquid medium (Fig. 1A). However, the colony forming capacity of ΔlprG was impaired on 7H11 supplemented with OADC (Fig. 1B). This observation is similar to the strong growth defect on solid medium described for the H37Rv Δpmt 13, although the phenotype was not as severe for the ΔlprG strain. None of the other mutants displayed growth alterations on solid medium (Supplementary Figure S3). Complementation of the ΔlprG mutant with the lprG-Rv1410c operon restored the capacity to form colonies on agar plates (Fig. 1B and Supplementary Figure S3).
Table 1.
Selected M. tuberculosis mannoproteins with high confidence glycopeptidomics identification.
| Peptide sequence | Experimental Mass (Da) | Calculated Mass (Da) | Delta (Da) | Hexose number | Peptide score | Protein Access # | Rv gene | Gene Name | Protein description | required for | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| in-vitro growthd | in-vivo growthe | ||||||||||
| D100C*VAAT aQAPDAGAMSASMQK119 b | 2170,9 | 2008,9 | 162,1 | 1 | 121,2 | O07419 | Rv0175 | mce1F | Mce associated membrane protein | − | + |
| P39AGPTPAPAAPAAATGGLLFHDEFDGPAGSVPDPSK74 | 3544,7 | 3382,6 | 162,1 | 1 | 28,62 | O07242 | Rv0315c | β-1,3-glucanase precursor | − | + | |
| T 47 TTPSGPVPPVSEAAR62 | 1889,9 | 1565,8 | 324,1 | 2 | 55,3 | O53850 | Rv0838 | lpqR | D-alanyl-D-alanine dipeptidase | − | nr |
| V228QVTKPPVS 236 | 1115,6 | 953,6 | 162,1 | 1 | 24,94 | P9WK45 | Rv1411cc | lprG | Lipoarabinomannan carrier protein LprG | − | + |
| Q164PFSLQLIGPPPSPVQR180 b | 2022,1 | 1860,0 | 162,1 | 1 | 49,06 | I6XHD1 | Rv3491c | Predicted secreted protein | − | nr | |
Figure 1.
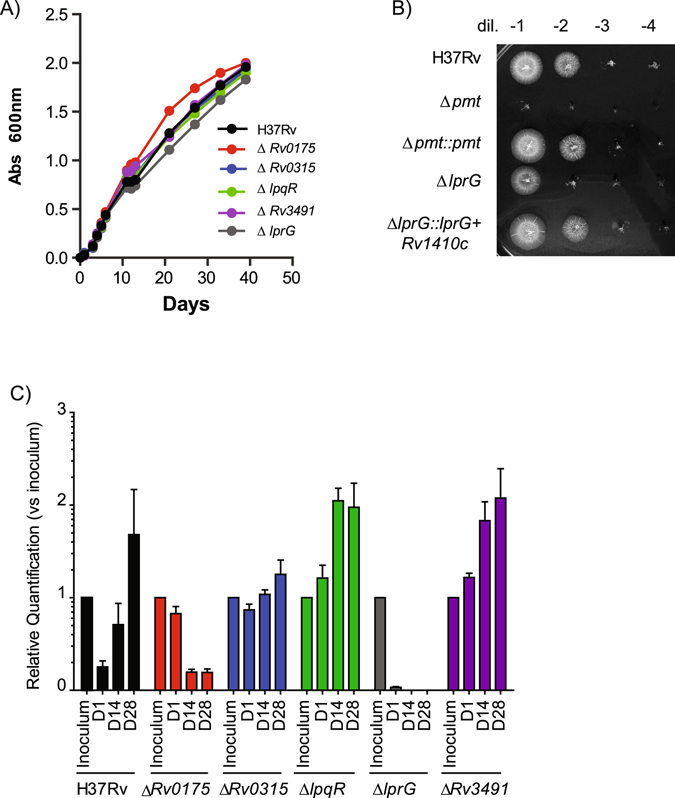
Contribution of selected mannoproteins to M. tuberculosis growth in vitro and in vivo. (A) Growth of WT M. tuberculosis and mutants in liquid medium. Mycobacterial strains were inoculated at 106cfu/ml. Absorbance at 600 nm was followed over time for the various cultures. (B) Growth of WT M. tuberculosis and mutants on 7H11 Petri plates. Serial dilutions of adjusted liquid cultures of the various mutants were spotted on solid medium and incubated at 37 °C. (C) Growth of tagged-WT M. tuberculosis and tagged-mutants in BALB/c mice. Mice (5 for each time points) were infected with 6.102 cfu of a population containing similar concentration of each strains. At 1, 14 or 28 days post-infection, the bacterial population was recovered from the lung of the infected mice and grown for 7 days in liquid medium. Total DNA was extracted and the quantity of each mutant evaluated by qPCR using primers specific for the tags. The amount of each mutant relative to the inoculum is plotted.
One of the more striking feature of the M. tuberculosis Δpmt mutant is its strong attenuation in cellular and animal infection models. We therefore evaluated the in vivo growth of the five mutants in BALB/c mice. We monitored the in vivo replication using qPCR in a competition experiment as described by Blumenthal et al.,19 to avoid any mis-interpretation associated with the altered capacity to form colonies on solid medium. Each mutant and the wild type strain was tagged using a specific oligonucleotide sequence. The tagged strains were grown independently in liquid medium and an inoculum was prepared by mixing similar quantities of each clone (Supplementary Figure S4). Mice were then infected intranasally using 6 × 102 cfu of the mixed population (Supplementary Figure S4). At each time point, the bacterial load in lung and spleen was quantified by plating serial dilution of lung and spleen homogenates. As expected, we observed a five-log increase during the four-week-infection period (Figure S4). In parallel, the organ homogenates were used to grow a short-term culture used to extract total DNA and to evaluate the relative quantity of each strain by qPCR. The relative quantity of ΔRv0175 and ΔlprG was similar to that of the other mutants and wild type control in the inoculum, but their proportional representation decreased substantially during the infection time-course (Fig. 2C and Supplementary Figure S4). Conversely, the quantity of the other strains increased. These results show that the ΔRv0175 and ΔlprG mutants were attenuated in this competition experiment.
Figure 2.
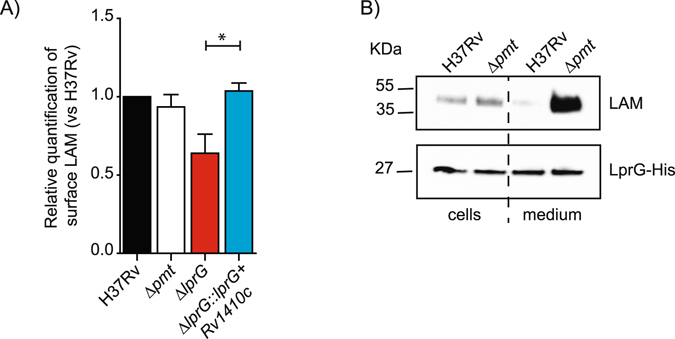
Impact of protein O-mannosylation on LAM exposition to the bacterial cell surface and release of the LprG/LAM complex. (A) FACS analyses of LAM staining on the surface of the various M. tuberculosis strains. The bar graph values correspond to the median fluorescence value after normalization to H37Rv. Results are representative of two independent experiments. *P < 0.05. (B) Western-blot analyses of LAM associated with LprG in WT M. tuberculosis or Δpmt mutant. Similar amounts of LprG-His protein purified from the bacteria or culture medium were separated by SDS-PAGE. After transfer, the LAM or LprG-His molecules were revealed using the anti-LAM antibody CS-35 or an anti-His antibody. Blots are representative of at least three independent experiments.
Based on these results, we focused our study on the LprG protein. Indeed, the phenotype of the H37Rv ΔlprG strain exhibited striking similarities with those of the Δpmt mutant, suggesting that the latter may be due, at least in part, to lack of LprG mannosylation and subsequent alteration of LprG functions.
Impact of O-mannosylation on LprG-associated localization of LAM
LprG belongs to the M. tuberculosis lipoprotein family and appears to act in concert with the efflux pump P55, encoded by the gene Rv1410c, which forms an operon together with lprG on the M. tuberculosis chromosome20. Several biological roles of LprG were recently described which provide tractable activities on which the impact of O-mannosylation can be evaluated. LprG together with P55 provides protection against toxic compounds such as ethidium bromide and malachite green21, 22. LprG is able to bind to various glycolipids such as lipoarabinomannan (LAM), lipomannan and phosphatidylinositol mannoside (three groups of molecules which share a similar core structure)23 and triglycerides24. LprG mutants exhibit impaired surface exposition of LAM25, 26, suggesting that the biological role of this lipoprotein is to transfer LAM from the plasma membrane to the bacterial cell surface.
We examined whether the lack of O-mannosylation alters one or more functions of LprG. We first evaluated the presence of LAM at the surface of the WT H37Rv strain and the Δpmt and ΔlprG isogenic mutants using FACS (Fig. 2A). As expected, disruption of lprG decreased the surface display of LAM, a phenotype reversed by complementation with the lprG-Rv1410c operon. However, the Δpmt strain exhibited a level of LAM exposition similar to that of the WT strain, suggesting that the lack of LprG glycosylation does not prevent the translocation of LAM. We further explored the impact of glycosylation on LprG function by testing whether co-purification of LAM with LprG was affected in a mycobacterial strain deficient for O-mannosylation. We expressed LprG fused to a Hexahistidine (His)-tag in the WT H37Rv or the Δpmt isogenic mutant. We then affinity purified the LprG-His protein from either the culture filtrate or the bacterial cells and measured the amount of LAM co-purified with LprG-His by Western blot using an anti-LAM antibody. Similar amounts of LAM were associated with the LprG-His protein purified from the pellet of the Δpmt mutant and the WT strain (Fig. 2B). In sharp contrast, the amount of LAM associated with LprG-His recovered from the culture filtrates was much higher for the Δpmt mutant than for the WT strain. This suggests that the O-mannosylation defect favors the release of the LAM associated with LprG in the bacterial culture filtrate. We tested whether this effect was restricted to M. tuberculosis by expressing the LprG-His protein in M. smegmatis mutated for the MSMEG_5447 (the ortholog of Rv1002c) gene (MSMEG-5447::res or Δpmt mutant) or the isogenic strain complemented with Rv1002c from M. tuberculosis. Similar amounts of LAM were co-purified with LprG-His from the pellets of the M. smegmatis Δpmt mutant and the Rv1002c-complemented strain, whereas much higher LAM were associated with the protein purified from the Δpmt mutant culture supernatant, as observed in the M. tuberculosis background (Supplementary Figure S5).
Altogether, these results suggest that the lack of O-mannosylation increased the amount of LAM released in the supernatant in association with LprG.
Identification of the LprG glycosylation site
We tested whether the lack of LprG O-mannosylation was responsible for the higher association of LAM with LprG observed with the Δpmt mutant, by targeting specifically the LprG glycosylation. We first further characterized the LprG mannosylation status and precisely determined the glycosylation site(s) by high resolution mass spectrometry (HR-MS) and top-down fragmentation experiments of the intact protein. Deconvolution of the ESI-HRMS spectrum of the purified recombinant LprG-His expressed in M. tuberculosis revealed two molecular species of molecular mass of 22,379 Da and 22,541 Da (Fig. 3A and B). The 162 Da difference in mass between these two peaks is consistent with the mass of an hexose moiety, suggesting that these two molecular species correspond to the unmodified and monoglycosylated proteoforms of a truncated form of LprG (Fig. 3A and B) missing the N-terminal pentapeptide (expected average mass of 22,379 Da and 22,541 Da). Top-down fragmentation of the intact protein precursors supported these assignments and further allowed the identification of T231 as the unique glycosylation site of the glycoform 22,541 Da protein (Figs 3B and S6). We produced a lprG A691G point mutant (lprG*), encoding a LprG protein containing a T231A mutation, fused with a His-tagged coding sequence, to evaluate the impact of the mannosylation on the activities of the protein. This construct was expressed in the M. smegmatis Δpmt mutant complemented with Rv1002c and the LprG-His T231A protein was purified and analyzed by MS in a same way. As expected, we detected a unique molecular species with a molecular mass of 22,436 Da (Fig. 3C and D) consistent with a mutated T231A form of LprG, confirming that the mutated protein was no longer O-mannosylated. However, although the molecular mass of this molecular species is consistent with the T231A mutation, it is noteworthy that it corresponds to a slightly longer truncated form of LprG starting from the upstream N terminal serine residue (Fig. 3C and D). This alternative sequence truncation was confirmed by top-down MS experiments and suggests that the absence of glycosylation may affect the proteolytic cleavage of the protein N terminus as previously reported5.
Figure 3.
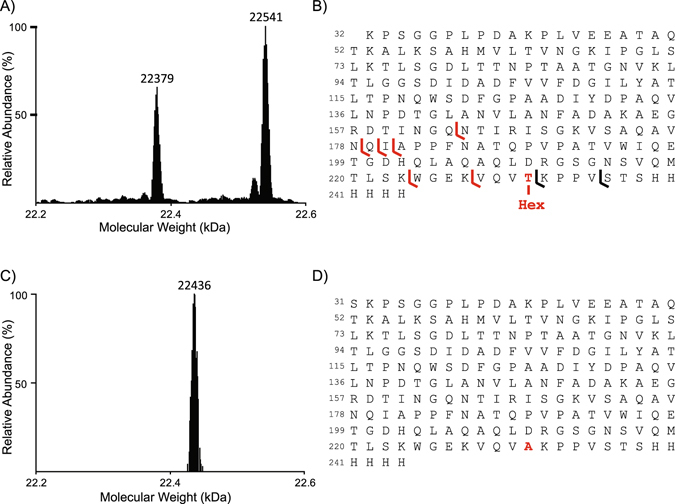
MS analyses of LprG and LprG T231A glycosylation. (A) Deconvoluted ESI-HRMS spectrum of the purified recombinant LprG-His expressed in M. tuberculosis. (B) Peptide sequence of the molecular species observed in A reporting the fragment ions detected in the top-down electron transfer dissociation spectrum of the fragmentation of the 22,541 Da molecular mass ion precursor allowing the localization of the unique hexose on the T231. (C) Deconvoluted ESI-HRMS spectrum of purified recombinant LprG-His T231A expressed in M. smegmatis demonstrating the absence of glycosylation of the mutated protein. (D) Peptide sequence of the molecular species observed in C.
Impact of LprG O-mannosylation on mycobacterial growth and association with LAM
We next addressed whether the lack of LprG glycosylation directly affects its functions. We generated a complementation plasmid carrying the lprG* mutated gene in an operon with Rv1410c and transferred this construct into the H37Rv ΔlprG mutant. We verified whether the production of LprG T231A complements the ΔlprG mutation. We first evaluated their colony-forming capacity on 7H11 OADC solid medium and found no difference between the strains producing the wild type LprG or the LprG T231A mutated protein, indicating that the LprG T231A is functional (Supplementary Figure S7). We next tested whether the T231A mutation affects the capacity of LprG to co-purify with LAM. The LprG-His and LprG-His T231A proteins were purified either from the M. smegmatis Δpmt mutant or from the isogenic strain complemented with Rv1002c. Western-blot analyses revealed that similar amount of LAM co-purified with LprG-His and LprG-His T231A recovered from the same M. smegmatis background. However, the LAM signal was much higher when the proteins were purified from the supernatant of the Δpmt mutant than complemented M. smegmatis (Fig. 4). In contrast, the amount of LAM in the cell extracts was slightly higher for the complemented strain than for the Δpmt mutant suggesting that the LAM/LprG complex is more efficiently retained in the cells when protein-O-mannosylation is preserved (Fig. 4).
Figure 4.
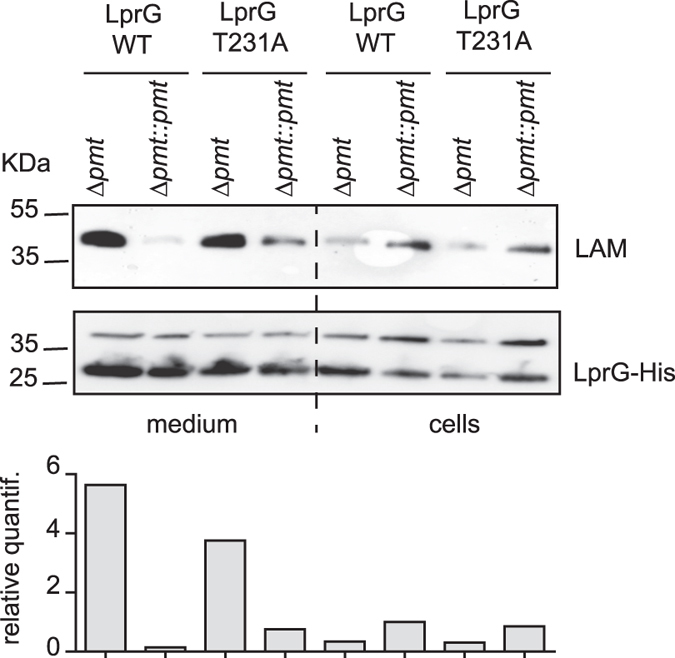
Impact of LprG O-mannosylation on the release of the LprG/LAM complex in M. smegmatis. Similar amounts of LprG-His or LprG-His T231A purified from either culture medium or bacterial cells were separated by SDS-PAGE, transferred onto membranes and revealed using anti-LAM or anti-His antibodies. The genetic backgrounds in which the protein was produced are indicated. The upper band revealed by the anti-His antibody is a non-specific cross-reacting protein. The signal intensity for LAM was quantified and plotted in the graph below the Western blot image. This blot is representative of three independent experiments.
Altogether these results establish that the enhanced capacity of LAM to co-purify with secreted LprG when produced in M. smegmatis Δpmt mutants is not due to LprG mannosylation but rather to an indirect effect.
Impact of protein O-mannosylation on steady state level of LAM
The higher amount of LAM found associated with LprG in the supernatant of the Δpmt mutants might be due to a higher steady state level of these compounds. We evaluated the total amount of LAM found in the M. tuberculosis WT, Δpmt and ΔlprG mutants and complemented strains. We prepared crude extracts from bacterial pellets or culture supernatants, separated the compounds by SDS-PAGE, and then performed Western blot analysis using either anti-LAM, or anti-hsp65 and anti-Ag85A antibodies as controls for the cell-associated or extracellular proteins, respectively (Fig. 5). As expected, the bacterial pellets contained similar amounts of LAM, but the culture supernatant of the Δpmt mutant exhibited approximately 10 fold more LAM than that of the WT cells. This phenotype was reverted by complementation with pmt from either M. tuberculosis or M. smegmatis (data not shown). In contrast, the amount of LAM was similar in both fractions for the other mutant and complemented strains, including the strain expressing the LprG-His T231A. This finding was not restricted to M. tuberculosis because similar results were obtained for crude extracts from M. smegmatis WT and the Δpmt mutant (data not shown). These results demonstrate that the production of LAM and its release into the culture medium is enhanced in the Δpmt mutants, independently of the LprG mannosylation.
Figure 5.
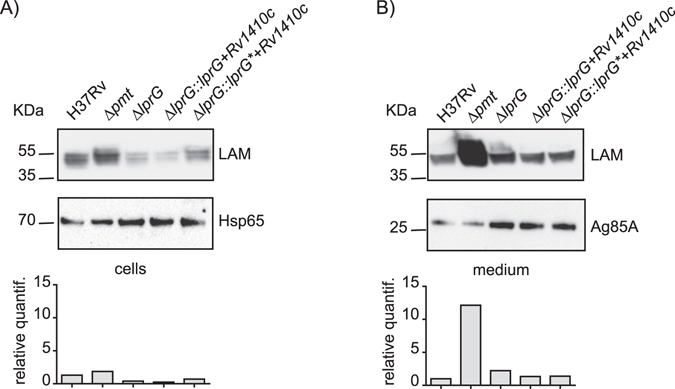
Impact of protein O-mannosylation on LAM production. Crude extracts obtained from the culture medium (B) or bacterial cells (A) were separated by SDS-PAGE, transferred onto membranes, and analyzed using antibodies raised against LAM and either hsp65 (for cellular extracts) or Ag85A (for medium supernatants). The signal intensity for LAM was quantified and plotted in the graph below the western blot image. This blot is representative of two independent experiments.
Protein O-mannosylation and induction of inflammatory response
LprG plays a key role in the cross-talk between M. tuberculosis and the host by interacting with the host receptor, TLR2, and by modulating the inflammatory response23, 27. This TLR2 agonist activity is strongly enhanced by LAM association with LprG23. The higher release of LAM/LprG complex may be associated with modification of the interaction of M. tuberculosis Δpmt mutant with macrophages. We first infected human monocyte-derived macrophages (hMDM) at an MOI of 1:1 for 1 h with the Δpmt or ΔlprG mutants, the WT control or the complemented strains and monitored the percentage of infected cells and the number of bacteria per cell for seven days. For this purpose, the strains were transformed with a plasmid expressing the fluorescent protein GFP. As we previously observed, the Δpmt mutant showed impaired invasion and growth in hMDM13. The ΔlprG mutant exhibited a similar defect of invasion capacity (lower percentage of infected cells and lower number of bacteria/cell) after seven days of infection. Complementation with either the WT lprG or mutated lprG A691G in operon with Rv1410c restored full virulence in this cellular model (Fig. 6A and Supplementary Figure S8). These results demonstrate that the lack of LprG O-mannosylation was not responsible for the attenuation observed with either the ΔlprG or Δpmt mutant.
Figure 6.
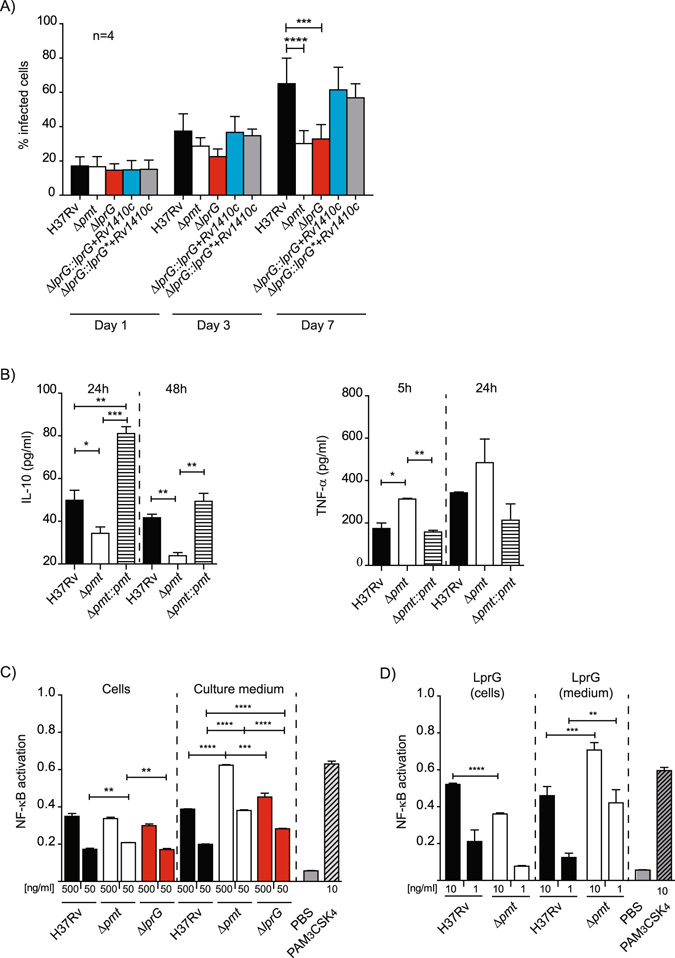
Protein O-mannosylation affects the inflammatory response of infected cells. (A) Growth of WT, mutants and complemented strains in hMDM. hMDM were infected at an MOI 1 for 1 h with the indicated strains. Data are presented as the mean +/− SD of four independent experiments performed in duplicate. (B) Production of TNF-α and IL-10 by MHS cells infected by WT, Δpmt and complemented strains. MHS cells were infected at MOI 1:1 for 1 h and TNF-α and IL-10 were evaluated after 5 h or 24 h and 24 or 48 h respectively. Data are representative of two independent experiments performed in triplicate. (C) NK-κB activation in HEK-TLR2 cells incubated with crude extracts from WT and Δpmt M. tuberculosis strains. Activation of NF-κB, evaluated using SEAP activity, following incubation with 500 ng/ml or 50 ng/ml of crude extracts recovered from either bacterial cells or culture medium from the indicated M. tuberculosis strains. PBS 1× or PAM3CSK4 (10 ng/ml) were used as negative and positive controls respectively. Data are presented as the mean +/− SD of three independent experiments. (D) NF-κB activation in HEK-TLR2 cells incubated with LprG/LAM complex purified from bacterial cells or culture medium of WT or Δpmt M. tuberculosis strains. HEK-TLR2 cells were incubated with 10 ng/ml or 1 ng/ml of LprG-His and co-purified LAM. SEAP activity was quantified after 24 h. PBS 1× or PAM3CSK4 (10 ng/ml) were used as negative and positive controls respectively. Data are presented as the mean +/− SD of at least five independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next evaluated whether the inflammatory response of infected cells was affected by the Δpmt mutation. We used cell lines to limit the variability associated with primary cells. We infected human (THP-1) or mice macrophages (MHS) at MOI 1:1 for 1 h with either the wild type H37Rv, the Δpmt mutant, and the Rv1002c-complemented strain and assayed inflammatory (TNF-α) or anti-inflammatory (IL-10) cytokines at 5, 24 and 48 h post-infection. We detected a significantly greater TNF-α release by MHS cells after infection with the Δpmt mutant than with the WT or complemented strains and a similar trend was observed in THP-1 cells (Fig. 6B and Supplementary Figure S8). In contrast, the secretion of the anti-inflammatory cytokine IL-10 decreased markedly in both cell types. We next addressed whether this higher inflammatory response may be due to greater stimulation of TLR2 using a HEK293 cell line stably transfected with human TLR2 and CD14 genes and an NF-κB-inducible reporter gene. We prepared crude extracts from the bacterial pellets and culture supernatants of Δpmt, ΔlprG mutants, and controls and incubated them with HEK-TLR2 cells. The culture supernatants from the Δpmt mutant activated NF-κB more than the similar extract from the WT strain (1.6-fold increase, p < 0.001) (Fig. 6C). In contrast, similar TLR2 activation was observed for the ΔlprG mutant and strains producing either the WT LprG or the non mannosylated LprG T231A (data not shown). The difference observed for the Δpmt sample was restricted to the culture supernatant because the crude extracts obtained from the various bacterial pellets gave similar NF-κB activation. These results are consistent with the higher secretion of the LprG/LAM complex associated with the Δpmt mutation.
We next tested the LprG-His protein purified from the culture medium or bacterial pellets from the WT M. tuberculosis or the Δpmt mutant for their TLR2 agonist activity (Fig. 6D). NF-κB activation was slightly higher for the proteins purified from the WT bacterial pellets than the Δpmt cells when equal amounts of LprG-His were incubated with HEK-TLR2 cells. In contrast, we measured much higher NF-κB activation with LprG-His purified from the supernatant of the H37Rv Δpmt mutant than from the supernatant of the WT strain (1.5-fold increase, p < 0.001 for 10ng/ml purified proteins and 3.4-fold increase, p < 0.005 for 1 ng/ml). We obtained similar results with LprG-His purified from either the bacterial pellet or culture supernatant from the M. smegmatis Δpmt mutant or the isogenic strain complemented with Rv1002c (Supplementary Figure S9). The T231A mutation, and as a consequence the glycosylation status of LprG, did not affect the induction of NF-κB (data not shown).
Finally, we confirmed that LprG/LAM signals primarily through TLR2 using bone marrow-derived macrophages (BMDM) from WT, TLR2 −/−, or MyD88−/− mice. Incubation of WT BMDM with the LprG/LAM complex induced the production of TNF-α. Consistent with results obtained with HEK-TLR2 cells, we measured higher TNF-α release with LprG/LAM purified from culture medium from the M. smegmatis Δpmt than from the Rv1002c-complemented strain in WT macrophages and the opposite for samples obtained from the bacterial pellets. The TNF-α release was abrogated in TLR2 −/− BMDM as in macrophages deficient for MyD88, the common adaptor protein for most TLR (Supplementary Figure S10).
Altogether, these results clearly establish that the lack of protein glycosylation is associated with a greater induction of the TLR2-mediated inflammatory response during host-cell infection. This effect is explained, at least in part, by the higher release of the LAM/LprG complex.
Discussion
The molecular mechanisms explaining the various phenotypes associated with the lack of protein O-mannosylation in mycobacteria are unknown. In M. tuberculosis, these phenotypes include a marked deficiency to form colony and severe attenuation in mouse and cellular models of infection13. Here, we demonstrated that the M. tuberculosis Δpmt mutant induces a stronger inflammatory response by infected phagocytes than the WT strain. This pro-inflammatory phenotype may be explained by higher secretion of the LAM/LprG complex which stimulates the TLR2 signaling pathway leading to NF-κB activation. Although LprG is O-mannosylated, this modification does not influence release of the LAM/LprG complex. Rather, disruption of mycobacterial pmt leads to a higher steady-state level of LAM and release in the supernatant of mycobacterial cultures. The additional LAM in the medium, at least in part bound to LprG, is likely the molecular basis underlying the greater induction of the inflammatory response through potentiation of the TLR2 agonist activity of LprG.
This conclusion is supported by several lines of evidence. First the greater inflammatory response induced in human and mouse macrophages by the M. tuberculosis Δpmt mutant was reproduced in a simpler system using either non phagocytic HEK-TLR2 cells or BMDM from WT or TLR2 −/− mice and either the mycobacterial culture supernatant or the purified LprG/lipoglycan complex. Second, the T231A point mutation abolished LprG O-mannosylation but had no effect on NF-κB activation and accumulation of LprG/LAM complex. Finally, although the LprG lipoprotein was found to exhibit TLR2 agonist activity23, 27, association with mycobacterial lipoglycans (including LAM) strongly enhanced this activity23. However, further studies are required to determine whether this stronger induction of the TLR2 mediated response accounts for the attenuation observed with the M. tuberculosis Δpmt mutant in the mouse model. In the last decade, TLR2 has emerged as a major host receptor for host/pathogen cross talk and immune response against M. tuberculosis (for review28, 29). TLR2 signaling mediates the control of M. tuberculosis through a wide range of host immune responses including macrophage activation, pro-inflammatory cytokine release, Th1 response and apoptosis28. Accordingly, TLR2-deficient mice exhibit higher susceptibility to M. tuberculosis infection than WT mice18, 30. However, M. tuberculosis may also use TLR2 signaling to escape the host immune response. TLR2 agonists from M. tuberculosis were found to suppress dendritic cell activation, type I interferon induction and antigen processing and presentation31, 32. The fine tuning of the inflammatory response, in particular through TLR2 signaling, is thought to be a strategy developed by the tuberculosis bacillus to achieve the various steps of its infectious cycle, from survival of the initial encounter with the host, to persistence in granulomas and finally to the induction of the central necrosis and rupture of the granuloma favoring bacterial release in the airways and transmission33. Perturbation of this fine tuning may be deleterious to the pathogen through increased production of toxic compounds, but also to the host through uncontrolled inflammation34, 35.
A surprising observation from this study was that several phenotypes associated with the Δpmt mutation are also found for the ΔlprG mutant, including the defect in colony formation on solid media and a similar level of attenuation in hMDM. However, our analyses with the strain producing LprG T231A, which is no longer glycosylable by Pmt, strongly argues against a direct role of LprG glycosylation in the corresponding phenotype. It is possible that the activity of a protein acting in the same pathway as LprG is strongly influenced by O-mannosylation. An obvious candidate is the efflux pump encoded by the gene Rv1410c in M. tuberculosis H37Rv. This gene forms an operon together with lprG and most of the phenotypes described for the ΔlprG mutant were only complemented upon expression of both Rv1410c and lprG (this study21, 22). We examined our mass spectrometry data in depth but found no evidence that the Rv1410c-encoded protein is O-mannosylated (data not shown). The impact of glycosylation on LprG activity thus remains an open question.
Another unexpected result from this study was the increased LAM production in the Δpmt mutants. This finding is not restricted to M. tuberculosis H37Rv because we observed the same for the M. smegmatis Δpmt mutant. Most of the additional lypoglycans were found in the culture supernatant. As discussed before, this enhanced secretion has a major impact on the inflammatory response of the host cell but the molecular basis for such abnormal production and secretion remains unclear. None of the enzymes known to be involved in the LAM biosynthetic pathway were identified among the O-mannosylated proteins (data not shown). One possible explanation may reside in the mannose flux. Indeed, Pmt is thought to transfer mannose residues from the same decaprenyl-phospho-mannose donor used for the elongation of PIM during the formation of LM and LAM36. The lack of Pmt may increase the amount of decaprenyl-phospho-mannose donor available for these other pathways. Another possibility is that genes of the LAM biosynthesis pathways are upregulated in the Δpmt strains. This is still speculative and additional work would be required to establish this point. However, such side effect was previously observed in mycobacteria for other mutants. For example, mutations that abolish the synthesis or transport of glycopeptidolipid in the opportunistic pathogen Mycobacterium abscessus enhance the production of lipoproteins which, through their TLR2 agonist activity, stimulate an inflammatory response during host cell infection37.
In conclusion, protein O-mannosylation appears to modulate the production and secretion of inflammatory molecules such as LAM, but the underlying molecular mechanism remains to be established. The exposure of these lypoglycans, which act as strong TLR2 ligands, especially when associated with the lipoprotein LprG leads to modulation of the innate immune response, providing a possible explanation for the strong attenuation of the Δpmt M. tuberculosis strain in cellular and animal infection models.
Materials and Methods
Bacterial growth conditions
M. smegmatis mc2155 wild-type (WT) and M. smegmatis derivatives were cultured in Luria Bertani (LB) broth containing 0.05% Tween-80 or LB agar at 37 °C. M. tuberculosis WT and M. tuberculosis mutants were grown at 37 °C in liquid Middlebrook 7H9 broth (Difco) containing ADC (Becton Dickinson) and supplemented with 0.05% Tween-80 or in solid Middlebrook 7H11 broth supplemented with OADC (ADC with 0.005% oleic acid) (Becton Dickinson). Albumin free culture conditions for the M. tuberculosis strains were performed in 7H9 broth supplemented with dextrose (1 g/L) and catalase (1.5 mg/L). For analysis of LAM, bacteria were grown without Tween-80. When necessary, media were supplemented with Hygromycin (Hyg) and Kanamycin (Km) at 50 µg/ml and 40 µg/ml respectively. Liquid cultures were grown to logarithmic or stationary phases depending of the needs.
E. coli DH5α was used for cloning experiments. Strains were grown at 37 °C in LB broth or LB agar. When necessary, Hyg (100 µg/ml) and Km (40 µg/ml) were added for selection of the appropriate strains.
In order to characterize the growth rate of M. tuberculosis WT and M. tuberculosis mutants, growth curves were monitored in 50 mL of 7H9 ADC 0.05% Tween-80. M. tuberculosis strains were grown until log-phase and then inoculated at 106 cfu/mL. OD 600 nm was measured at different time points for one month. The capacity of the different mutants to grow on solid media was evaluated on 7H11 OADC. M. tuberculosis cultures were grown until exponential phase, OD was measured and bacterial concentration was adjusted to 106 cfu/mL. Then, 10 μL of serial dilutions were plated. Morphology and size of spots were evaluated at 2–4 weeks of growth.
Every experiment involving WT or recombinant M. tuberculosis strains were performed in BSL3 laboratories until the samples were inactivated. Experiments with E. coli or M. smegmatis were realized in BSL1 laboratories.
Construction of M. smegmatis and M. tuberculosis mutants and complementation
M. tuberculosis knockout (KO) strains were constructed by homologous recombination using the recombineering system38. First, the allelic exchange substrate (AES) for each gene of interest: Rv0175c, Rv0315, Rv0838, Rv1411c and Rv3491 were produced. Primers were designed with restriction sites to amplify upstream and downstream regions of the targeted genes (Supplementary Table S1) and PCR fragments were ligated to a Km resistance cassette. Then, M. tuberculosis H37Rv carrying a pJV53 derivative containing the hyg resistance gene was grown to mid-log phase, induced with acetamide and competent cells were prepared. This strain was transformed with each AES. Km-resistant colonies were obtained and replacement of the targeted gene by the AES was verified by PCR analysis (Supplementary Figure S1, Supplementary Table S1). As a result, M. tuberculosis ΔRv0175, ΔRv0315, ΔRv0838 (ΔlpqR), ΔRv1411c (ΔlprG) and ΔRv3491 mutants were obtained.
M. tuberculosis ΔlprG was complemented with Rv1410c, the lprG gene (Rv1411c) or both genes. Briefly, PCR products were amplified from M. tuberculosis H37Rv total DNA using primers indicated in Table S1 and inserted between PvuII and HindIII restriction sites of the integrative plasmid pMV361H39. In this construct, the gene expression is under the control of its own promoter.
M. tuberculosis H37Rv, ΔlprG and Δpmt gfp strains were made by insertion of the pMV361H GFP integrative plasmid. The various complemented strains expressing GFP were made using pMV361H GFP integrative plasmid carrying the PCR fragment of interest inserted between PvuII and HindIII sites (Supplementary Table S2).
Integrative plasmid pMV361H Rv1410c-lprG A691G gfp was constructed using the pMV361H Rv1410c-lprG gfp as template. Briefly, the whole plasmid was amplified using CGAGAAGGTCCAGGTCGCGAAGCCCCCGGTGAGCTGATC and GATCAGCTCACCGGGGGCTTCGCGACCTGGACCTTCTCG primers. Then, the PCR product was transformed in M. tuberculosis ΔlprG to obtain M. tuberculosis ΔlprG:: Rv1410c-lprG A691G gfp strain.
M. smegmatis_5447::res-Ωkm-res was complemented with pWM158 integrative plasmid which contains the Rv1002c gene from M. tuberculosis 13.
Mouse Infections and qPCR analysis
Female BALB/c mice (7–8 week old) were purchased from the Centre d’Elevage Janvier and housed in the Institut de Pharmacologie et de Biologie Structurale ASB3 animal facilities. The M. tuberculosis WT and mutant strains were grown independently in standard liquid medium and an inoculum was prepared by mixing similar quantity of each clones. Mice, five per group, were infected intranasally with 40 μL PBS containing 6 × 102 cfu of the mixed population. At 1, 14 and 28 days post-infection, 5 mice were euthanized and lungs and spleens were homogenized in 5 mL of PBS 0.05% Tween-80. The course of the global infection was followed by plating serial dilutions of 100 μL of lung and spleen homogenates on 7H11 OADC solid medium. The remains of organ homogenates were centifugated at 800 rpm to remove clumps. The supernatants were then centifugated at 14000 rpm and bacilli were resuspended in 5 mL of 7H9 ADC Km. After one week of culture, mycobacterial DNA were extracted using DNeasy Blood and tissue extraction kit (Qiagen) according to the recommendations of the manufacturer.
Quantitative PCR analysis was performed in 25 μL reactions using the suitable set of primers (Supplementary Table S3), 5 μL of DNA as template and the maxima syber green qPCR master mix (thermo scientific). Reactions were performed on the Applied Biosystems 7500. The amplification program for qPCR was: 10 min at 95 °C, followed by 40 cycles each consisting of 95 °C for 15 sec, 60 °C for 30 sec and 72 °C for 30 sec. Two PCR reactions were used for each mutant. The expression of the common part was used as internal control (qKmFint and qKmRint primers). Specific values were first normalized with the common region and relative quantification was then calculated.
Cell Culture and infections
Human monocyte-derived macrophages (hMDMs) were prepared as previously described40. Briefly, human blood samples, obtained from the Etablissement Français du Sang of Toulouse (France), were collected from anonymous nontuberculous donors. Peripheral blood monocytes were cultured to differentiation for 7 days on glass coverslips in 24-well tissue culture plates (5.105 cells/well) containing RPMI 1640 (Gibco, Cergy Pontoise, France) supplemented with 2 mM glutamine (Gibco) and 7% of heat-inactivated human AB serum. Before infections, hMDMs were washed twice with fresh RPMI medium. Bone marrow-derived macrophages from C57Bl6 (WT), TLR2 −/−, or MyD88−/− mice were differentiated in DMEM (Invitrogen) supplemented with 10% FCS (Fisher Scientific), 20% Macrophage Colony Stimulating Factor (M-CSF from L929 cell supernatant) and 10 mM HEPES (Invitrogen). The day before stimulation with LprG/LAM complex, macrophages were seeded into 96-well plates at a density of 5.104 cells/well.
Mycobacterial infections using hMDMs were performed as previously described40. Briefly, macrophages were infected with exponentially growing GFP-expressing mycobacteria at MOI 1 :1 in RPMI medium. Infection was allowed to proceed at 37 °C in 5% CO2 for 1 h, and the extracellular bacteria were removed by three successive washes with 1 mL fresh medium. At the end of the infection period, the cells were collected at 0, 3 and 7 days post-infection and fixed for 1 h at RT with 4% PFA in PBS supplemented with 15 mM sucrose, aldehyde groups were neutralized with 50 mM NH4Cl. Extracellular mycobacteria were labelled with rabbit anti-mycobacteria Ab, which was detected by a Rhodamine Red-conjugated goat anti-rabbit secondary Ab. Preparations were visualized under a Leica DMLA fluorescence microscope to count and calculate (i) the number of bacteria per macrophages and (ii) the number of macrophages containing at least one particle (percent internalization or phagocytosis). Data are expressed as relative values, calculated as the percentage of internalization or phagocytosis divided by control values. For each set of conditions, triplicate experiments were performed and at least 50 cells per slide were counted.
Quantification of NF-κB activity and cytokine secretion (TNF-α and IL-10)
The level of NF-κB activation was studied using HEK-BlueTM hTLR2 reporter cell lines (InvivoGen). This cell line is stably transfected with a reporter plasmid expressing a secreted embryonic alkaline phosphatase (SEAP) gene under the control of a NF-κB-dependent promoter. In all cases, cells were used in 96-well plates according to manufacturer instructions. Cells were treated with the purified proteins at the specified concentrations. Unstimulated cells were used as negative controls, whereas Pam3CSk4 10 ng/ml was used as positive control. After 16 h of incubation, QUANTI-BlueTM detection medium (InvivoGen) was used following manufacturer specifications and SEAP activity, corresponding to NF-κB activation, was determined by reading the absorbance at 630 nm. For infection experiments, HEK-BlueTM hTLR2 cells were infected with corresponding M. tuberculosis strains in 96-well plates at MOI 1:1 for 24 h. At the tested infection time, supernatants were harvested, filtered, and NF-κB activity was subsequently evaluated as described before.
To assess TNF-α or IL-10 secretion levels, THP-1 or MHS cells were infected at MOI 1:1 for 1 h and cytokines were evaluated after 5 h and 24 h for TNF-α and 24 and 48 h for IL-10. Cytokine secretions were quantified in supernatants of the THP-1 or MHS cells using the BD OptEIATM human TNF ELISA Set and BD OptEIATM human IL-10 ELISA set or the PeproTech murine TNF-α and IL-10 ABTS ELISA development kits respectively, according to the recommendations of the manufacturer. For BMDM, cells were incubated for 24 h with 100 ng/ml LprG/LAM complexes purified from bacterial cells or culture medium of M. smegmatis Δpmt or M. smegmatis Δpmt complemented with pmt from M. tuberculosis. Unstimulated cells were used as negative controls (PBS 1X), whereas Pam3CSK4 (100 ng/ml) and LPS (100 ng/ml) was used as positive control. TNF-α secretion was quantified in supernatants of the BMDM using the mouse TNF-α ELISA Set kit (eBioscience), according to the recommendations of the manufacturer. Data are expressed as picograms of cytokine per milliter of supernatant.
Cloning, expression and purification of His-tagged proteins
Cloning, expression and purification of LprG and LprG T231A His-tagged proteins were performed as previously described with minor modification23. The lprG gene was amplified from M. tuberculosis H37Rv reference strain by PCR using the 5′-TTTTTTCATATGCGGACCCCCAGACGCCACTG-3′ and 5′-TTTTTTACTAGTGCTCACCGGGGGCTTCG-3′ primers. The lprG A691G (lprG*) was amplified from the complementation plasmid using the same primers (underlined the NdeI and SpeI restriction enzyme sites). Both DNA fragments were inserted between the NdeI and SpeI sites of pWM218 mycobacterial replicative plasmid13 to obtain pLprG-His and pLprG T231A-His plasmids. The lprG gene expression is under the control of the pBlaF* promoter.
LprG and LprG T231A were expressed either in M. tuberculosis or in M. smegmatis. Cultures were centrifuged and pellet and medium were separated. Supernatants were filtered twice and concentrated to 2 mL by ultrafiltration with Vivaspin 10 kDa system (Sartorius). Cells were resuspended in 2 mL of PBS 1x pH 7.4 and disrupted. Insoluble material was removed from the lysate by centrifugation at 12000 rpm for 10 min at 4 °C and supernatant was incubated directly with Ni2+ beads (Qiagen) for 2–4 h at 4 °C in continuous agitation. The mix was transferred to polypropylene columns, washed with 5 volumes of wash buffer (PBS, 20 mM imidazole, pH7.4), and bound proteins were dissociated with aliquots of 100 μl of elution buffer (PBS, Imidazole from 100 to 500 mM). After purification, proteins were dialyzed 3 times in PBS pH 7.4. Protein purity was verified by SDS-PAGE with blue-Coomassie stain and Western-blot using anti-His antibody.
LAM detection in crude extracts
M. tuberculosis H37Rv, Δpmt, ΔlprG, ΔlprG:: lprG-Rv1410c and ΔlprG:: lprG* -Rv1410c were grown in 7H9 supplemented with ADC during one month. Cultures were centrifuged and pellets and medium were separated. Supernatants were filtered and concentrated to 2 mL by ultrafiltration with Vivaspin 10 kDa system (Sartorius). Bacterial pellets were resuspended in 2 mL of PBS 1x pH7.4 and disrupted. Insoluble material was removed from the lysate by centrifugation at 12000 rpm for 10 min at 4 °C. The total amount of LAM was analyzed by Western blot using the CS-35 anti-LAM antibody. Anti-hsp65 or anti-Ag85A antibodies were used as controls.
Western blot analysis
Western blot analysis was carried out using standard procedures. Equivalent quantities of protein extracts or purified proteins were separated by electrophoresis through a 4–20% precast protein gel (BioRad). Proteins were transferred to PVDF membranes for 50–60 min. After 1 h blocking with 5% milk in PBS supplemented with 0.05% Tween-20 (PBST), proteins were analyzed using specific antibodies: mouse monoclonal anti-LAM (CS-35, BEI Resources, 1:50, overnight at 4 °C), mouse monoclonal anti-His6 (Affymetrix, 1:3000, overnight at 4 °C), mouse monoclonal anti-Ag85 (Santa Cruz Biotechnology, 1 :1000, overnight at 4 °C), rabbit polyclonal anti-Hsp65 (gift from M. Daffé, 1:2000000, overnight at 4 °C) goat anti-mouse (BioRad, 1 :5000, 1 h RT) or goat anti-rabbit (Sigma-Aldrich, 1:20000, 1 h RT) secondary antibodies conjugated with horseradish peroxidase (HRP).
Surface LAM staining
Ten days cultures were centrifuged at 800 rpm for 10 min to remove clumps. Bacilli were washed with PBS and fixed in 4% PFA. After centrifugation, pellets were washed with PBS and incubated at RT in 5% BSA for 1 h. Bacilli were sedimented and suspended in 3% BSA with mouse anti-LAM CS-35 at 1/150 and incubated overnight at 4 °C. Bacilli were washed with PBS-0.05% Tween-20 and resuspended in PBS with goat anti-mouse IgG CMA-Rho red at 1/1000 for 2 h at RT. Bacilli were washed and analyzed by flow-cytometry. Relative quantification of LAM was calculated using H37Rv as reference level.
High-Resolution Mass Spectrometry
LC-ESI-MS and MS² analyses of intact recombinant LprG-His and LprG-His T231A were performed on a nanoRS UHPLC system (Dionex) coupled to an ETD-enabled LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific), with fluoranthene as reagent anion. 5 µL of sample was loaded on a C4-precolumn (300 µm ID × 5 mm, Thermo Fisher Scientific) at 20 µL/min in 5% acetonitrile, 0.05% TFA. After 5 min desalting, the precolumn was switched online with the analytical C4 nanocolumn (75 µm ID × 15 cm, in-house packed with C4 Reprosil) equilibrated in 95% solvent A (5% acetonitrile, 0.2% formic acid) and 5% solvent B (0.2% formic acid in acetonitrile). Proteins were eluted using the following gradient of solvent B at 300 nL/min flow rate: 5 to 40% during 5 min; 40 to 100% during 33 min. The LTQ-Orbitrap Velos was operated either in single MS or in data-dependent acquisition mode with the XCalibur software. MS scans were acquired in the 500–2000 m/z range with the resolution set to a value of 60,000. For top-down experiments, survey scan MS were acquired in the same way, with a resolution at 30,000. Precursor ions were selected from an inclusion list established thanks to previous MS analyses and were fragmented in ETD and the resulting fragment ions were analyzed in the Orbitrap, at a resolution of 60,000. Isolation width was set at 5 m/z and activation time was fixed to 20 ms for ETD.
Ethics Statement
Animal studies were performed in accordance with the CNRS guidelines for housing and care of laboratory animals in compliance with the European Community council directive 86/609/EEC guidelines and its implementation in France. The procedures involving mice were reviewed and approved by the relevant Ethics Committee (Comité d’éthique en experimentation animale N°01) and received approval from the French Ministry for High Education and Research after ethical evaluation (number 201508271122464 v2 (APAFIS#1535)). The Etablissement Français du Sang (Toulouse, France) provided human peripheral blood samples under contract 21/PLER/TOU/IPBS01/2013-0042. Experiments with human blood were performed in accordance with the relevant French guidelines and regulations (articles L1243-3, L1243-4 and L1245-2 of the French Public Health Code) and protocols were approved by the French Ministry for High Education and Research (agreement number DC 2012-1715). Informed consents were obtained from the donors before sample collection.
Electronic supplementary material
Acknowledgements
We thank Flavie Moreau and Pierre-Jean Bordignon for the help with animal experimentation and bone marrow-derived macrophages respectively. We are grateful to Dr Agathe Subtil (Institut Pasteur) for the generous gift of knock-out mice. The authors acknowledge support from the Centre National de la Recherche Scientifique, University of Toulouse, the National Research Agency (ANR-11-BSVB-016 and ANR-11-EQUIPEX-003), the Fondation pour la Recherche Médicale (ING20140129051 and DEQ20160334879), the French Ministry for Research (Investissement d’avenir, Proteomics French Infrastructure, ANR-10-INBS-08), The Bettencourt-Schueller Foundation, the Fonds Européens de Développement Régional, Toulouse Métropole, and the Région Midi-Pyrénées.
Author Contributions
H.A., J.P., D.P., O.B.-S., E.M., M.R. and C.G. designed the experiments. H.A., J.P., W.M., D.P., C.-F.L., C.B., C.R. performed the experiments. H.A., J.P., W.M., D.P. C.-F.L., E.M., O.B.-S., M.R. and C.G. analyzed and interpreted the results. H.A., J.P., O.B.-S., M.R. and C.G. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08489-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Michel Rivière, Email: Michel.Riviere@ipbs.fr.
Christophe Guilhot, Email: Christophe.Guilhot@ipbs.fr.
References
- 1.Cain JA, Solis N, Cordwell SJ. Beyond gene expression: the impact of protein post-translational modifications in bacteria. J. Proteomics. 2014;97:265–286. doi: 10.1016/j.jprot.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Garbe T, et al. Expression of the Mycobacterium tuberculosis 19-kilodalton antigen in Mycobacterium smegmatis: immunological analysis and evidence of glycosylation. Infect. Immun. 1993;61:260–267. doi: 10.1128/iai.61.1.260-267.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Espitia C, Mancilla R. Identification, isolation and partial characterization of Mycobacterium tuberculosis glycoprotein antigens. Clin. Exp. Immunol. 1989;77:378–383. [PMC free article] [PubMed] [Google Scholar]
- 4.Dobos KM, Swiderek K, Khoo K-H, Brennan PJ, Belisle JT. Evidence for glyosylation sites on the 45-kilodalton glycoprotein of Mycobacterium tuberculosis. Infect. Immun. 1995;63:2846–2853. doi: 10.1128/iai.63.8.2846-2853.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrmann JL, O’Gaora P, Gallagher A, Thole JER, Young DB. Bacterial glycoproteins: a link between glycosylation and proteolytic cleavage of a 19kDa antigen from Mycobacterium tuberculosis. EMBO J. 1996;15:3547–3554. [PMC free article] [PubMed] [Google Scholar]
- 6.WHO. Global Tuberculosis report 2016 (World Heatlh Organization, 2016).
- 7.Ragas A, Roussel L, Puzo G, Rivière M. The Mycobacterium tuberculosis cell-surface glycoprotein apa as a potential adhesin to colonize target cells via the innate immune system pulmonary C-type lectin surfactant protein A. J. Biol. Chem. 2007;282:5133–5142. doi: 10.1074/jbc.M610183200. [DOI] [PubMed] [Google Scholar]
- 8.Pitarque S, et al. Deciphering the molecular bases of Mycobacterium tuberculosis binding to the lectin DC-SIGN reveals an underestimated complexity. Biochem. J. 2005;392:615–624. doi: 10.1042/BJ20050709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diaz-Silvestre H, et al. The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb. Pathog. 2005;39:97–107. doi: 10.1016/j.micpath.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Horn C, et al. Decreased Capacity of Recombinant 45/47-kDa Molecules (Apa) of Mycobacterium tuberculosis to Stimulate T Lymphocyte Responses Related to Changes in Their Mannosylation Pattern. J. Biol. Chem. 1999;274:32023–32030. doi: 10.1074/jbc.274.45.32023. [DOI] [PubMed] [Google Scholar]
- 11.Romain F, et al. Deglycosylation of the 45/47-Kilodalton antigen complex of Mycobacterium tuberculosis decreases its capacity to elicit in vivo or in vitro cellular immune responses. Infect. Immun. 1999;67:5567–5572. doi: 10.1128/iai.67.11.5567-5572.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandakumar S, et al. O-mannosylation of the Mycobacterium tuberculosis adhesin Apa is crucial for T cell antigenicity during infection but is expendable for protection. PloS Pathogens. 2013;9:e1003705. doi: 10.1371/journal.ppat.1003705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CF, et al. Bacterial protein O-mannosylatiob enzyme is crucial for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA. 2014;110:6560–6565. doi: 10.1073/pnas.1219704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanderVen BC, Harder JD, Crick DC, Belisle JT. Export-mediated assembly of mycobacterial glycoproteins parallels eukaryotic pathways. Science. 2005;309:941–943. doi: 10.1126/science.1114347. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Zamorano M, et al. Mycobacterium tuberculosis glycoproteomics based on ConA-lectin affinity capture of mannosylated proteins. J. Proteome Res. 2009;8:721–733. doi: 10.1021/pr800756a. [DOI] [PubMed] [Google Scholar]
- 16.Smith GT, Sweredoski MJ, Hess S. O-linked glycosylation sites profiling in Mycobacterium tuberculosis culture filtrate proteins. J. Proteomics. 2014;97:296–306. doi: 10.1016/j.jprot.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergne I, Gilleron M, Nigou J. Manipulation of the endocytic pathway and phagocyte functions by Mycobacterium tuberculosis lipoarabinomannan. Front. Cell. Infect. Microbiol. 2014;4:187. doi: 10.3389/fcimb.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiling N, et al. Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J. Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 19.Blumenthal A, Trujillo C, Ehrt S, Schnappinger D. Simultaneous analysis of multiple Mycobacterium tuberculosis knockdown mutants in vitro and in vivo. PLoS One. 2010;5:e15667. doi: 10.1371/journal.pone.0015667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bigi F, et al. The gene encoding P27 lipoprotein and a putative antibiotic-resistance gene form an operon in Mycobacterium tuberculosis and Mycobacterium bovis. Microbiology. 2000;146:1011–1018. doi: 10.1099/00221287-146-4-1011. [DOI] [PubMed] [Google Scholar]
- 21.Farrow MF, Rubin EJ. Function of a mycobacterial major facilitator superfamily pump requires a membrane-associated lipoprotein. J. Bacteriol. 2008;190:1783–1791. doi: 10.1128/JB.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianco MV, et al. Role of P27–P55 operon from Mycobacterium tuberculosis in the resistance to toxic compounds. BMC Infect. Dis. 2011;11:195. doi: 10.1186/1471-2334-11-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drage MG, et al. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonist of Toll-like receptor 2. Nature structural & molecular biology. 2010;17:1088–1095. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinot AJ, et al. Mycobacterial metabolic syndrome: LprG and Rv1410 regulate triacylglyceride levels, growth rate and virulence in Mycobacterium tuberculosis. PloS Pathogens. 2016;12:e1005351. doi: 10.1371/journal.ppat.1005351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaur RL, et al. LprG-mediated surface expression of lipoarabinomannan is essential for virulence of Mycobacterium tuberculosis. PloS Pathogens. 2014;10:e1004376. doi: 10.1371/journal.ppat.1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shukla S, et al. Mycobacterium tuberculosis lipoprotein LprG binds lipoarabinomannan and determines its cell envelope localization to control phagolysosomal fusion. PloS Pathogens. 2014;10:e1004471. doi: 10.1371/journal.ppat.1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J. Immunol. 2004;173:2660–2668. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Zeng J, Xie J. Navigating through the maze of TLR2 mediated sigaling network for better mycobacterium infection control. Biochem. 2014;102:1–8. doi: 10.1016/j.biochi.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida A, Inagawa H, Kohchi C, Nishizawa T, Soma G. The role of Toll-like receptor 2 in survival strategies of Mycobacterium tuberculosis in macrophage phagosomes. Anticancer Res. 2009;29:907–910. [PubMed] [Google Scholar]
- 30.Drennan MB, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta D, et al. Suppression of TLR2-induced IL-12, reactive oxygen species, and inducible nitric oxide synthase expression by Mycobacterium tuberculosis antigens expressed inside macrophages during the course of infection. J. Immunol. 2010;184:5444–5455. doi: 10.4049/jimmunol.0903283. [DOI] [PubMed] [Google Scholar]
- 32.Simmons DP, et al. Mycobacterium tuberculosis and TLR2 agonist inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J. Immunol. 2010;185:2405–24015. doi: 10.4049/jimmunol.0904005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cambier CJ, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–222. doi: 10.1038/nature12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–294. doi: 10.1016/j.immuni.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tobin DM, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148:434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaur, et al. Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 2009;69:23–78. doi: 10.1016/S0065-2164(09)69002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roux AL, et al. Overexpression of proinflammatory TLR-2-signalling lipoproteins in hypervirulent mycobacterial variants. Cell. Microbiol. 2011;13:692–704. doi: 10.1111/j.1462-5822.2010.01565.x. [DOI] [PubMed] [Google Scholar]
- 38.van Kessel JC, Hatfull GF. Mycobacterial recombineering. Methods Mol. Biol. 2008;435:203–215. doi: 10.1007/978-1-59745-232-8_15. [DOI] [PubMed] [Google Scholar]
- 39.Burbaud S, et al. Trehalose polyphleates are produced by a glycolipid biosynthetic pathway conserved across plylogenetically distant mycobacteria. Cell Chem. Biol. 2016;23:1–12. doi: 10.1016/j.chembiol.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Astarie-Dequeker C, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in the invasion of human macrophages by inducing changes in the lipid organization of the plasma membrane. PloS Pathogens. 2009;5:e1000289. doi: 10.1371/journal.ppat.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 42.Griffin J, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PloS Pathogens. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.