

Abstract
Target-specific scoring methods are more commonly used to identify small-molecule inhibitors among compounds docked to a target of interest. Top candidates that emerge from these methods have rarely been tested for activity and specificity across a family of proteins. Here, we dock a chemical library to CaMKIIδ, a member of the Ca2+/calmodulin (CaM)-dependent protein kinase (CaMK) family, and rescore the resulting protein-compound structures using SVMSP, a target-specific method that we previously developed. Among the 35 selected candidates, three hits were identified, such as quinazoline compound 1 (KIN-1), which inhibited CaMKIIδ kinase activity with single-digit micromolar IC50. Activity across the kinome was assessed by profiling analogs of 1, namely 6 (KIN-236), and an analog of hit compound 2 (KIN-15), namely 14 (KIN-332), against 337 kinases. Interestingly, for 6 (KIN-236), CaMKIIδ and homolog CaMKIIγ were among the top 10 targets. Among the top 25 targets of 6 (KIN-236), IC50 values ranged from 5 to 22 μM. Compound 14 (KIN-332) was not specific towards CaMKII kinases, but the compound inhibited two kinases with sub-micromolar IC50s among the top 25. Derivatives of 1 were tested against several kinases including several members of the CaMK family. The data afforded a limited structure-activity relationship study. Molecular dynamics simulations with explicit-solvent followed by end-point MM-GBSA free energy calculations revealed strong engagement of specific residues within the ATP-binding pocket, and also changes in the dynamics as a result of binding. This work suggests that target-specific scoring approaches like SVMSP may hold promise for the identification of small-molecule kinase inhibitors that exhibit some level of specificity towards the target of interest across a large number of proteins.
Graphical Abstract
INTRODUCTION
Despite the large number of kinase inhibitors that have been discovered to date, a large portion of the human kinome remains unexplored.1 One example is the Ca2+/calmodulin (CaM)-dependent protein kinase (CaMK) superfamily of kinases.2, 3 Despite its nomenclature, the majority of kinases in the CaMK family do not possess the characteristic Ca2+/CaM-binding regulatory domain. Over the years, several members of the CaMK family have been studied in cancer, including members of the AMP-activated protein kinase (AMPK)-related kinase family such as maternal embryonic leucine zipper kinase (MELK) or Novel (nua) kinase family 1 (NUAK1).4–8 MELK has been found to be overexpressed in basal-like breast cancer and several studies have implicated the enzyme with tumor growth and metastasis.9, 10 NUAK1 a member of the AMPK-like family whose function has yet to be elucidated despite strong data associating it with promoting tumor growth and metastasis.11–13 CaMKIIγ is another CaMK family member that has been studied in cancer.14–17 CaMKIIγ was recently discovered to be the target of the potent anti-leukemia drug berbamine.18 The compound has been previously shown to be a potent inhibitor of MDA-MB-231 cancer cell proliferation.19 Additional examples of non-calmodulin dependent members of the CaMK are the PIM group of kinases, which have been explored for their role in cancer.20
Structure-based computational methods such as virtual screening have been extensively used to identify small-molecule inhibitors of kinases and other targets. These methods consist of docking a chemical library to the target of interest followed by re-scoring protein-compound complexes and rank-ordering compounds for experimental validation. Several docking methods have been implemented in various computer programs such as AutoDock,21, 22 Glide,23, 24 and Gold.25 Methods to predict the binding mode of small molecules have matured significantly, but there is a need for better scoring methods to rank-order protein-compound structures.26 Scoring methods for the rank-ordering of protein-compound complexes range from methods that attempt to predict the Gibbs free energy of binding to those that make use of physico-chemical or structural information. The methods that use free energy include molecular dynamics-based approaches, such as free energy perturbation, thermodynamic integration, and end-point methods (e.g. MM-PBSA and LIE). These methods use molecular dynamics simulations that require substantial computational resources. Scoring methods are less demanding, but they are generally used for library enrichment in virtual screening. Several scoring approaches have been developed ranging from empirical,24, 27, 28 force field,25, 29 to knowledge-based.30–32 Increasingly, scoring methods use machine learning techniques to improve database enrichment and rank-ordering.33–37
The enrichment power of scoring methods is generally dependent on the target. As a potential solution to this problem, we introduced a target-specific scoring approach (SVMSP, Support Vector Machine SPecific) that uses Support Vector Machine (SVM) trained on pair potentials from statistical energy functions.33, 38 SVMSP is trained using a positive and negative set. The positive set consists of crystal structures of the target in complex with small molecules. The negative set is composed of decoy molecules docked to the target protein. Since SVMSP is developed using the structure of the target of interest, we stipulated that the re-scoring method may favor small molecules that inhibit and potentially show specificity towards the target. Our previous work showed that SVMSP generally performed as well as or better than existing methods, and that the unique strength of the method is its ability to show uniform performance across a set of targets.19 SVMSP was compared to several other scoring methods including Glide. In particular, we found that SVMSP performed well in the enrichment of chemical libraries docked to kinases. The method was applied on several occasions to identify small-molecule inhibitors of kinases and more recently aldehyde dehydrogenases.33, 39
Here, we use virtual screening with SVMSP re-scoring to identify small-molecule inhibitors of CaMKIIδ, a representative member of the CaMKII sub-group of the CaMK family implicated in both cancer progression40 and cardiovascular disease.41 To that end, we docked 149,479 compounds to its ATP-binding pocket. SVMSP was used to rank-order these compounds and select top candidates for validation. We acquired the top 35 candidates and screened them for activity in-house using a FRET-based assay. A quinazoline compound 1 (KIN-1) inhibitor was identified and, along with an analog 6 (KIN-236), further evaluated for activity and activity across the kinome using a radiometric assay at Reaction Biology Corp (http://www.reactionbiology.com, Malvern, PA). We explored the activity profile of the derivative compound using the KinomeScan service and tested it for activity against 337 kinases. The top 25 targets from the kinome profiling were selected for a concentration-dependent study. To further explore the activity of 1, we acquired an additional 8 derivatives and tested them for activity against 11 kinases that includes three members of the CaMK family. Explicit-solvent molecular dynamics simulations followed by end-point free energy calculations were used to gain further insight into the activity and binding profile of the compound.
RESULTS
Virtual Screening Identifies Active Compounds
In an effort to identify small molecules that inhibit members of the CaMK family, we resorted to virtual screening using SVMSP, a target-specific re-scoring method. The workflow is summarized in Fig. 1a. A library of 149,979 small molecules was docked to the ATP-binding pocket of CaMKIIδ using AutoDock Vina.22 The resulting protein-compound complexes were re-scored and rank-ordered using SVMSP, which uses machine learning trained on a large number of kinase three-dimensional structures from the PDB.33, 34, 38, 39 The Support Vector Machine (SVM) algorithm requires a positive and negative training set. In our case, the positive set consisted of 389 co-crystal structures of kinase-compound complexes obtained from the PDB (Table S1), and the negative set consisted of randomly selected decoy compounds from ZINC42 docked to the target (in this case CaMKIIδ) (Table S2).
Figure 1.
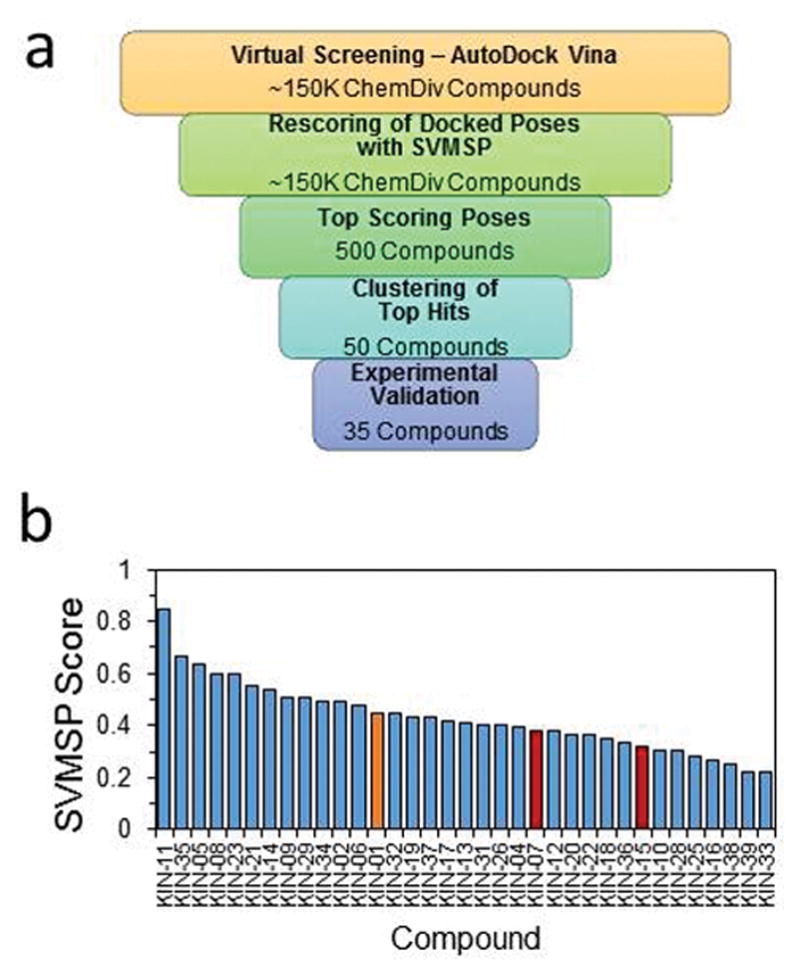
(a) Virtual screening workflow used to identify compounds that inhibited CaMKIIδ. Approximately 150,000 compounds were docked in AutoDock Vina and rescored using SVMSP. The top 500 compounds were identified and clustered to 50. Of the 50 compounds, 35 were experimentally validated. (b) SVMSP scores of the 35 experimentally validated compounds identified from virtual screening against CaMKIIδ. Three hits, 1 (orange), 2 (red), and 3 (red) were discovered from the initial virtual screening. 2 and 3 were later abandoned for possessing moieties commonly found in frequent hitters of pan-assay interference compounds.
We compare the performance of the SVMSP model to the Glide scoring function using Vina-docked and Glide-docked poses. We identified a set of 33 active and 58 inactive compounds against CaMKIIδ from ChEMBL (Table S3).43 We assessed the ability of the four different docking and scoring combinations in their ability to enrich for active compounds against inactive ones (Fig. S1a). Performance was assessed using the area under the ROC curve (ROC-AUC). A ROC-AUC of 1 corresponds to a perfect scoring function that ranks all active compounds before inactive compounds, while a ROC-AUC of 0.5 represents one that ranks compounds randomly. We find that the Vina-docked and Glide-docked poses rescored with SVMSP perform the best (ROC-AUC of 0.72 and 0.64, respective). Glide-docked poses scored with GlideScore and Vina-docked poses rescored with GlideScore had ROC-AUCs of 0.61 and 0.56, respectively. In virtual screening, only the top candidates are selected for validation. At low false discovery rates (FDR ≤ 5%), we see that Glide/SVMSP shows better performance than Vina/SVMSP. At a FDR of 5%, the true positive rate of Glide/SVMSP is 18% versus 9% in Vina/SVMSP. We then compared the performance of docked poses with and without energy minimization. Docked poses from Vina and Glide were refined using a local optimization algorithm. Minimization was limited to residues within 5 Å of the docked compound. We again assessed performance using ROC-AUC. We find that the ROC-AUCs of minimized and unminimized poses are generally similar (Fig. S1b). Again, Glide-docked and Vina-docked poses scored with SVMSP resulted in the best performance, with both having ROC-AUCs of 0.68. Similarly, Glide-docked and Vina-docked poses scored with GlideScore have ROC-AUCs of 0.56 and 0.57, respectively. At low FDRs, we again see the Glide/SVMSP outperforming Vina/SVMSP. We next examined how the difference in the Vina-docked and Glide-docked poses affected the subsequent SVMSP and Glide scores (Fig. S1c). We find that an increase in the absolute difference between the GlideScore of Vina-docked and Glide-docked poses does not correlate with a change in the absolute difference on the SVMSP score. Additionally, there is only a weak correlation between the RMSD of the Vina-docked and Glide-docked poses and the absolute difference in GlideScore (Fig. S1d) and SVMSP (Fig. S1e).
SVMSP was used to rank-order the 149,979 compounds. The top 500 candidates were selected and clustered into 50 sets. We selected a representative structure from each of the clusters. Among them, 35 compounds were available for purchase. The SVMSP scores for these compounds are provided in Fig. 1b.
The top 35 compounds selected from virtual screening were acquired and tested for inhibition of CaMKIIδ kinase activity using a FRET assay in-house. The physiochemical properties of these 35 compounds are provided in Table S4. We found three compounds inhibited CaMKIIδ enzyme activity by 50% or more at 50 μM, namely 1, 2 (KIN-7), and 3 (KIN-15) (Scheme 1). It is worth mentioning the fact that 2 contains an acylhydrazone moiety, which is known to be unstable at low pH, so this compound would not be suitable in future hit-to-lead and lead optimization efforts unless the acylhydrazone moiety is replaced with a more stable group. We profiled an analog of 2 to compare the inhibition profile across the kinome of the active compounds that emerged from this work. We did not test 3 since it was flagged as a pan-assay interference (PAINS) compound.44
Scheme 1.
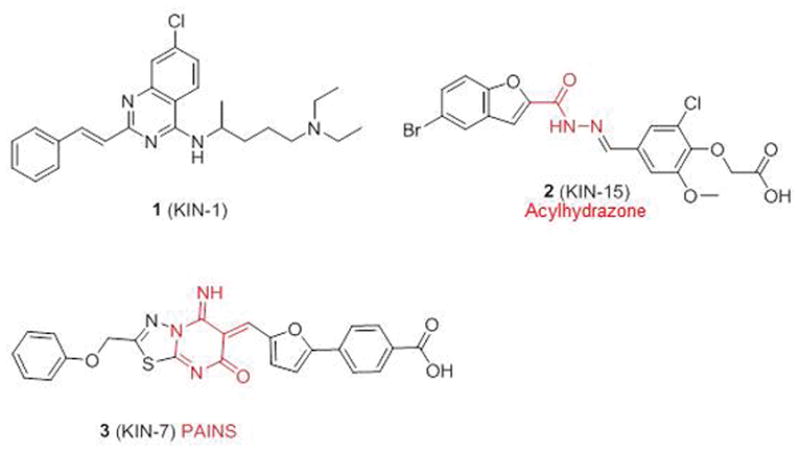
Chemical structure of active compounds that emerged from virtual screening. Problematic moieties are colored in red.
We compared the structure of compound 1 to the approximately 2,300 compounds tested against CaMKIIδ in the literature using ChEMBL.43 The most chemically similar compound is an inhibitor of CaMKIIδ that was discovered using pharmacophore search, which features a similar quinazoline core as compound 1 (ECFP4 fingerprints, Tanimoto coefficient of 0.154).45 In their compound, the alkene linker of the benzene substituent is shortened with a secondary amine, and the alkylamine tail of 1 is replaced with a methylpiperidine group. Additionally, we compared the structure of 1 against approximately 55,000 compounds in ChEMBL’s Kinase SARfari database. The most similar compound, chloroquine, has a Tanimoto coefficient of 0.256 to 1. Chloroquine is used to treat malaria, and features a similar core and alkylamine tail as 1. The quinazoline core is replaced by a quinoline ring while the bezene substituent and alkene linker of 1 are omitted in the compound.
A concentration-dependent study for prepared 1 (Scheme 2) and one of its analogs, 6 (Scheme 3), using a radiometric assay done at Reaction Biology Corp showed that the compounds inhibited enzyme activity with an IC50 of 7.8 ± 0.4 μM (Fig. 2). The ADMET of 1 and 6 were computationally predicted using QikProp46 and included in Table S5.
Scheme 2.
Reagents and conditions: (a) (CH3CO)2O, 110 °C, 5 h; (b) 28.0 – 30 % NH3 (aq), 50–60 °C, 10 h, 70% (two steps); (c) PheCHO, NaOAc, HOAc, 110 °C, 30 h; (d) POCl3, N,N-dimethylaniline, toluene, 110 °C, 8 h; (e) 2-amino-5-diethylaminopentane, DMAP, Et3N, toluene, 110 °C, 15 h.
Scheme 3.
Chemical structure of derivatives of 1.
Figure 2.
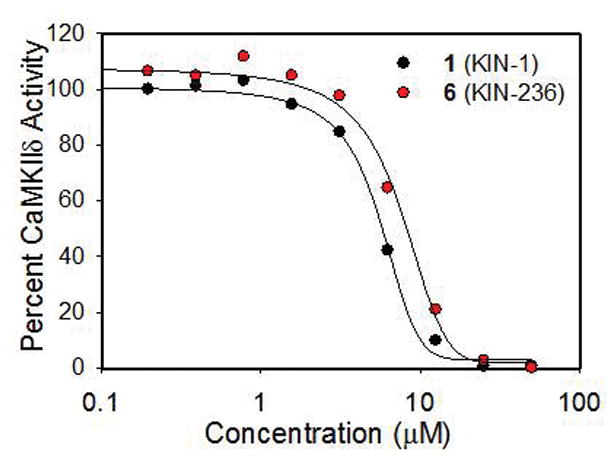
Concentration-dependent study for 1 and one of its analogs, 6, using a radiometric assay at Reaction Biology Corp showed that the compounds inhibited enzyme activity by an IC50 of about 5 μM.
Kinome-Wide Profiling
To explore the activity of 1 across the human kinome, we resorted to kinome profiling using the KinomeScan facility at Reaction Biology Corp, which included 337 unique kinases. We selected 6 for the profiling study. Considering the similarity in structure between 1 and 6 and their nearly identical activity for inhibition of CaMKIIδ (Fig. 2), it was expected that both compounds will share similar targets and inhibition profile. 6 was profiled against 337 kinases at 20 μM (Fig. 3a). The data show that the compound inhibited 16 kinases by 50% or more at 20 μM. To confirm these results, the top 25 targets of 6 were selected for a concentration-dependent study (Fig. 3b). There were 10 kinases that were inhibited by 6 with an IC50 of 10 μM or less, further highlighting the selective nature of the compound series. Interestingly, there were only 4 serine/threonine kinases among the top 25, and all belong to the CaMK superfamily, namely CaMKIIγ, MELK, CaMKIIδ and CaMK1α. Two (CaMKIIγ and CaMKIIδ) belong to the CaMKII sub-group, one belongs to the CaMK1 sub-group (CaMK1α), and another belongs to the CaMKL sub-group (MELK). The remaining targets among the top 25 were all tyrosine kinases.
Figure 3.

(a) Kinome-wide profiling of 6 against 337 kinases at 20 μM. 6 inhibited 16 kinases by 50% or more at 20 μM. (b) Concentration-dependent study of 6 against the top 25 targets.
The inhibition profile of 6 was compared to that of a derivative of 2 (compound 14) (Fig. S2a). We profiled compound 14 against the same 337 kinases using the KinomeScan facility at Reaction Biology Corp. 14 was profiled against the 337 kinases at 20 μM (Fig. S2b). The data show that the compound inhibited 46 kinases by 50% or more at 20 μM. This is in contrast to 16 kinases that were inhibited by 50% or more by 6. The greater number of targets inhibited 14 was reflected in follow-up concentration-dependent studies for the top 25 kinases (Fig. S2c). At 20 μM, the compound inhibited CaMKIIδ by 42%, suggesting an IC50 that is lower than that of 1 and 6. CaMKIIδ was at position 46 among the 337 kinases when rank-ordered by percent inhibition, in contrast to 6 where CaMKIIδ was among the top 10.
Binding Mode
An important question is whether the binding mode of 1 that emerged from virtual screening (Fig. 4a) could be adopted by its derivatives in multiple members of the CaMK subfamily. To explore this question, we first re-docked the compound with another program (Glide) to determine if the binding mode could be reproduced by the re-docking. We re-docked 1 and its derivatives against CaMKIIδ, CaMKIIγ, and MELK. We stipulated that a binding mode that is found in all three kinases will provide additional confidence for the predicted pose. Interestingly, in all three cases, we identified a binding mode for the Glide binding pose that adopted a similar arrangement in three-dimensional space to the Vina binding mode (Fig. 4b). However, the binding mode in Fig. 4a shows a shift in the position of the compound to better occupy the two hydrophobic pockets of the ATP binding domain. The RMSD between the binding pose from virtual screening and this new binding pose is 4 Å. It is well-known that among co-crystallized inhibitors bound to the ATP binding site, the majority use one or both of these hydrophobic pockets.47 The predicted binding mode of 1 was superimposed with the binding mode of ATP (Fig. 4c). The quinazoline core of 1 occupies the same hydrophobic pocket of the adenine of ATP, while the N-akylamine tail of 1 overlaps with the ribose and triphosphate tail of ATP.
Figure 4.

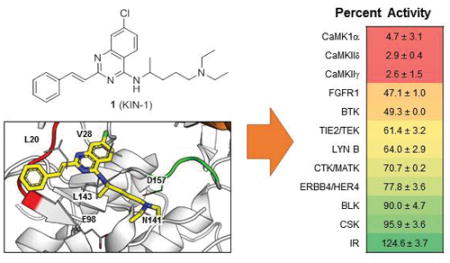
Characterization of the binding mode of 1 to CaMKIIδ. (a) Stereoview of the binding mode of 1 from the original virtual screening against CaMKIIδ in the R configuration. (b) Stereoview of the predicted binding mode of 1 after analysis of the compound series against CaMKIIδ, CaMKIIγ, and MELK. (c) Stereoview of ATP (PDB ID: 1ATP, 2.20 Å) is superimposed on the predicted binding mode of 1 (yellow). (d) Stereoview of an experimentally crystallized inhibitor featuring a quinazoline ring structure in a similar conformation (PDB ID: 3QKM, 2.20 Å, magenta) as the predicted binding mode of 1 (yellow). In (a–d), a crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) is shown as cartoon (gray) with the hinge loop (red), DFG-motif (green), and αC helix (orange) highlighted. The protein is shown with the glycine-rich loop removed (Residues Glu-19 to Arg-29) to expose the ATP binding pocket. Key residues between 1 and the protein kinase are shown in stick format. Two-dimensional representation of the (e) virtual screening and (f) Glide-predicted binding modes of 1 against CaMKIIδ, showing the key interactions between the atoms of the ligand and the ATP binding pocket of the receptor. Generated by PoseView (http://poseview.zbh.uni-hamburg.de/poseview). (g) A multiple sequence alignment of the top candidates of 6 was generated in Cluster Omega and the residues of CaMKIIδ within 4 Å of the original virtual screening binding mode of 1 were identified (in color). These residues were annotated based on the hydrophobicity of the residue, ranging from red (most hydrophobic) to blue (most hydrophilic). Residues in the hinge loop and activation loop are highlighted red and green, respectively, on the residue numbers.
To further confirm the binding pose, we searched the PDB for small-molecule kinase inhibitors that share substituents with 1. Since there is no crystal structure of 1 or any of its analogs bound to a kinase in the PDB, we resorted to crystal structures of compounds that possessed similar moieties. The quinazoline moiety of 1 is commonly found on kinase small-molecule inhibitors. We compared the orientation of the quinazoline scaffold of 1 to existing co-crystal structures of kinase inhibitors that also featured quinazoline rings in their scaffold. Generally, the quinazoline ring that forms the core scaffold of these compounds are positioned into one of three general orientations (Fig. S3). The first orientation features compounds that mimic the nucleobase of ATP with a benzene-substituent at the imidazole position (PDBID: 4OBO, 2.10 Å; 4OBQ, 2.19 Å) (Fig. S3a). The second group features a quinazoline orientation that mirrors the one found in our compound, whereby the benzene and pyrimidine rings of the quinazoline core occupies the other’s position (PDBID: 1M17, 2.60 Å; 3PRZ, 2.60 Å; 4AA5, 2.38 Å) (Fig. S3b). The third group includes four compounds (PDB ID: 3QKM, 2.20 Å; 4BB4, 1.65 Å; 4PP7, 3.40 Å; 4F7S, 2.20 Å) possessing a quinazoline moiety in a similar orientation to the one found in our predicted binding mode of 1 (Fig. S3c). One example is a spriocyclic sulfonamide inhibitor of AKT1 (Fig. 4d), whose quinazoline superimposes nearly perfectly with the predicted binding pose (Fig. 4b). This suggests that the position and orientation of the quinazoline of 1 is likely in the correct position in the predicted binding modes.
A comparison of the two-dimensional representations of the virtual screening and Glide-predicted binding modes reveal similarities in the binding interactions between them. From the two-dimensional representation of the virtual screening (Fig. 4e) and Glide-predicted (Fig. 4f) binding modes, the quinazoline ring occupies the hydrophobic pocket normally occupied by the adenine of ATP, while the benzene substituent of 1 occupies the hydrophobic pocket formed by residues in the hinge loop. In both binding modes, the alkylamine substituent of the compound forms a salt-bridge interaction with the aspartic acid on the DFG motif of the activation loop. In the original pose, the quinazoline ring of the compound forms a π-π interaction with Phe-90 of the hinge loop that is not seen in the predicted pose.
A multiple sequence alignment using Clustal Omega48 of the kinase domains of the top candidates was carried out. We identified residues in the ATP binding pocket of CaMKIIδ that formed interactions with the compound and color-coded them based on hydrophobicity,49 ranging from the most hydrophobic residues in red to the most hydrophilic in blue (Fig. 4g). Across the majority of these residues, either the residue was conserved across all the kinases (e.g. Lys-43 and Asn-141) or the alternate residue at the position did not result in a large difference in the hydrophobicity (e.g. Leu-20 to Ile and Val-74 to Ile). Leu-92 and Val-93 are replaced by less hydrophobic residues in the majority of the top ranked tyrosine kinases of 6. At residue 92, leucine is replaced by tyrosine or histidine in 7 of the 9 tyrosine kinases, and at residue 93, valine is replaced by methionine or alanine in 8 of 9 tyrosine kinases. These residues, along with Leu-20, border the benzene ring substituent site that is attached to the core quinazoline of 1.
The quinazoline core in compound 1 does not form hydrogen bonds to the hinge region in either the Vina or Glide pose. This hydrogen bonding pattern to the hinge region is frequently seen in existing kinase ATP inhibitors.50 The residues on the hinge loop that commonly form hydrogen bonds with inhibitors corresponds to the residues from position 90 to 94 in CaMKIIδ (amino acids FDLVT) in Fig. 4g. The quinazoline core of 1 forms hydrophobic contacts with Phe-90, Leu-92, and Val-93 of the hinge loop on CaMKIIδ. The sidechains of these residues do not have moieties that are amenable for hydrogen bonding. The most common sites for hydrogen bonding are at residues that correspond with positions 91 and 93 on CaMKIIδ, which are referred to as the hinge acceptor and hinge donor, respectively. On CaMKIIδ, these two residues are Asp-91 and Val-93. However, they also do not form any hydrogen bonding interaction with our compounds. While Asp-91 has a moiety amendable for hydrogen bonding, the sidechain in the crystal structure points away from the binding pocket, and is unlikely to form hydrogen bonds to the core of the compound in the crystal structure.
Structure-Activity Relationships
Compound 1 and 8 of its derivatives were tested for activity against 11 kinases selected from the list of the top targets of 6 (Fig. 5). These studies were done at 20 μM concentration of compound. Measurements at single concentrations can be prone to error, so these data are used qualitatively. First, an overall inspection of the data shown in Fig. 5 shows a striking preference for CaMK by the compounds as evidenced by the color-coding used to represent percent activity of the enzyme. Overall, 1 and several derivatives [4 (KIN-234), 5 (KIN-235), 6 (KIN-236), 7 (KIN-281), and 8 (KIN-284)] showed strong preference for the three CaMK members considered in the list. 9 (KIN-286), 10 (KIN-288), and 11 (KIN-289), however, showed much weaker activity towards the CaMKs. This is explained by the fact that 9, 10, and 11 lack the N-alkylamine moiety in the quinazoline ring. Instead, these compounds possess piperidine, azepane, and methyl-substituted piperidine moieties, respectively. 9 and 10 did not inhibit any of the other kinases with a few exceptions: 10 inhibited FGFR1, BTK, TIE2/TEK, and BLK by 60, 35, 25 and 50% respectively.
Figure 5.

Profiling of 1 and 8 derivatives for activity against a panel of 11 kinases at 20 μM concentration of compound. Numbers represent percent activity ± standard deviation.
Molecular Dynamics Simulations and Free Energy Calculations
To further gain insight into the interaction between 1 and its derivatives with CaMKIIδ, we ran explicit-solvent molecular dynamics simulations followed by free energy calculations. Using the Glide-predicted binding mode of 1, we generated binding modes for each of the derivatives of 1. We then subjected these protein-compound structures to 100-nanosecond (ns) explicit-solvent molecular dynamics simulations. Although the original docked compound of 1 is in its R configuration, the subsequent synthesis produces a racemic mixture of the compound that was not further separated. Given the presence of a chiral center in the compounds with the N-alkylamine moiety (1, 5, 6, 7, and 8), we considered both the R and S configurations for these 5 compounds. The components of the MM-GBSA free energy are listed in Table 1 and the RMSD measurements of each simulation is listed in Table S6. It is important to note that MM-GBSA calculations are not meant to reproduce the absolute value of the free energy of binding. Instead, these calculations are meant to rank-order compounds by their binding affinity. In addition, the multiple components of the MM-GBSA free energy provide deeper understanding of the contributions of the components of the free energy such as the non-polar, electrostatics and entropy. Pearson correlation coefficients between the experimental percent activity and the individual MM-GBSA components are similar for both R and S configurations (Table 2). Considering the bi-modal nature of the data, Spearman and Kendall coefficients may be more appropriate. Spearman’s ρ is 0.68 and 0.85 for the correlation between the MM-GBSA free energy and the percent activity of the compounds for the R and S enantiomers, respectively. The higher correlation for the S enantiomer suggests that this configuration is likely the one that binds to the kinase.
Table 1.
Calculated Free Energies (±95% Cl) of Compounds Against CaMKIIδ (kcal·mol−1)
| Compound | ΔEVDW | ΔEELE | ΔEGB | ΔESURF | ΔEGBTOT | ΔGMM-GBSA |
|---|---|---|---|---|---|---|
| R Conformation | ||||||
| (R)-1 (KIN-1) | −42.8 ± 0.3 | −160.7 ± 1.4 | 176.9 ± 1.4 | −5.7 ± 0.0 | −32.3 ± 0.3 | −14.2 ± 0.5 |
| (R)- 5 (KIN-235) | −50.2 ± 0.4 | −148.4 ± 1.0 | 168.5 ± 1.2 | −6.5 ± 0.0 | −36.7 ± 0.3 | −18.1 ± 0.6 |
| (R)-6 (KIN-236) | −52.1 ±0.3 | −148.1 ± 1.3 | 164.8 ± 1.3 | −6.6 ± 0.0 | −42.1 ±0.4 | −21.6 ± 0.6 |
| (R)-7 (KIN-281) | −58.8 ± 0.2 | −158.7 ± 1.2 | 178.3 ± 1.1 | −7.5 ±0.0 | −46.6 ± 0.3 | −24.5 ± 0.6 |
| (R)-8 (KIN-284) | −51.6 ±0.3 | −150.2 ± 1.3 | 165.2 ± 1.3 | −6.4 ± 0.0 | −42.9 ±0.3 | −23.5 ± 0.6 |
| S Conformation | ||||||
| (S)-1 (KIN-1) | −44.0 ± 0.3 | −163.5 ± 1.5 | 179.5 ± 1.4 | −5.9 ± 0.0 | −33.8 ± 0.3 | −14.8 ± 0.5 |
| (S)-5 (KIN-235) | −51.4 ± 0.3 | −146.4 ± 1.1 | 166.1 ± 1.1 | −6.8 ± 0.0 | −38.4 ± 0.3 | −19.4 ± 0.6 |
| (S)-6 (KIN-236) | −45.2 ± 0.3 | −170.2 ± 1.7 | 186.4 ± 1.6 | −5.8 ± 0.0 | −34.7 ± 0.3 | −15.4 ± 0.6 |
| (S)-7 (KIN-281) | −51.0 ± 0.3 | −180.8 ± 0.9 | 194.6 ± 0.9 | −7.0 ± 0.0 | −44.3 ± 0.3 | −23.1 ± 0.5 |
| (S)-8 (KIN-284) | −51.1 ± 0.3 | −146.4 ± 1.4 | 163.5 ± 1.3 | −6.4 ± 0.0 | −40.5 ± 0.3 | −20.3 ± 0.6 |
| Achiral | ||||||
| 4 (KIN-234) | −45.0 ± 0.3 | −186.1 ± 1.0 | 197.2 ± 1.0 | −6.4 ± 0.0 | −40.3 ±0.3 | −18.8 ± 0.6 |
| 9 (KIN-286) | −38.7 ± 0.2 | −1.5 ± 0.2 | 18.4 ± 0.2 | −4.7 ± 0.0 | −26.4 ± 0.2 | −11.0 ± 0.4 |
| 10 (KIN-288) | −39.2 ± 0.2 | −5.8 ± 0.2 | 21.5 ± 0.2 | −4.9 ± 0.0 | −28.4 ± 0.2 | −12.5 ± 0.5 |
| 11 (KIN-289) | −39.0 ± 0.2 | −1.1 ± 0.2 | 18.0 ± 0.2 | −4.8 ± 0.0 | −27.0 ± 0.2 | −11.4 ± 0.5 |
Table 2.
Correlation Coefficients of Computational Free Energies and Experimental Percent Inhibition for Compound 1 and Derivatives Against CaMKIIδ
| Component | r | ρ | T |
|---|---|---|---|
| R Conformation and Achiral | |||
| ΔEVDW | 0.77 | 0.65 | 0.48 |
| ΔEELE | 0.98 | 0.69 | 0.48 |
| ΔEGB | −0.99 | −0.73 | −0.59 |
| ΔESURF | 0.88 | 0.64 | 0.48 |
| ΔEGBTOT | 0.84 | 0.68 | 0.48 |
| ΔGMM-GBSA | 0.80 | 0.68 | 0.48 |
| S Conformation and Achiral | |||
| ΔEVDW | 0.85 | 0.89 | 0.76 |
| ΔEELE | 0.98 | 0.54 | 0.37 |
| ΔEGB | −0.98 | −0.56 | −0.42 |
| ΔESURF | 0.91 | 0.85 | 0.70 |
| ΔEGBTOT | 0.89 | 0.80 | 0.59 |
| ΔGMM-GBSA | 0.83 | 0.85 | 0.65 |
Analysis of the MM-GBSA components in Table 1 shows that compounds with the N-alkylamine moiety exhibit substantially more favorable electrostatic potential energies (ΔEELE) than compounds that lack the moiety. This suggests that the critical component of the N-alkylamine moiety is the charged tertiary amine that likely forms favorable electrostatic interactions with amino acid residues on the kinases. These interactions likely occur with the charged sidechains of Asp-157 (Fig. 4d). Overall the non-polar interaction van der Waals energy (ΔEVDW) is also more favorable among compounds with the N-alkylamine group (Table 1) consistent with the larger number of atoms for compounds with the N-alkylamine moiety. The electrostatic component of the solvation energy (ΔEGB), however, was dramatically more favorable for the compounds lacking the N-alkylamine, consistent with the fact that desolvation of a charged group results in significant penalty in the free energy. Finally, the flexible nature of the N-alkylamine moiety is likely to result in an entropy penalty for binding. We further explored the MM-GBSA data to determine whether the components of the MM-GBSA free energy correlate with compound activity data from Fig. 5. Using Spearman’s ρ, activity is highly correlated to the van der Waals energy (ΔEVDW), electrostatic energy (ΔEELE), non-polar solvation energy (ΔESURF), and combined internal and solvation terms (ΔEGBTOT). Correlations ranging from 0.64 to 0.69 in the R configuration and 0.54 to 0.89 in the S configuration. The polar solvation term (ΔEGB) is anti-correlated with compound activity (ρ = −0.73 and −0.56 for R and S, respectively).
To gain insight into the intermolecular interactions between compound and residues within the ATP-binding site of CaMKIIδ, we calculated MM-GBSA decomposition energies of each compound (Fig. 6a). Decomposition energies correspond to intermolecular energies between the bound compound and individual residues within the target protein. The decomposition energies are calculated in the same manner as the MM-GBSA energy except that they do not include the entropy term. Generally, both enantiomers share similar decomposition energies at individual residues. Highly favorable interaction energies were identified between compound and three hydrophobic residues, namely Leu-20, Val-28, and Leu-143 (Fig. 6b). Leu-20 forms hydrophobic interactions with the benzene ring substituent that is attached to the core quinazoline, while Val-28 and Leu-143 form hydrophobic interactions with the quinazoline core of the compound. There is a weak interaction near Asn-141 and Ala-156, which interacts with the N-alkylamine moiety of 1. These interactions disappear in 9, 10, and 11, which lack the N-alkylamine moiety. It is worth noting that for 9, 10 and 11, there is greater interaction with Leu-92, Val-93, and Thr-94 of the hinge loop.
Figure 6.

(a) Decomposition energies of key residues of binding for 1 and derivatives against CaMKIIδ. For compounds with a chiral center in the N-akylamine moiety, only the S configuration shown. Each row is a compound while each column is an individual residue. (b) A snapshot from the simulation of (S)-1 bound to CaMKIIδ, showing the sidechains of key residues from the interaction in line format. The crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) is shown as cartoon (gray) with the hinge loop (red), DFG-motif (green), and αC helix (orange) highlighted. The protein is shown with part of the glycine-rich loop removed (Residues Gly-21 to Ser-26) to expose the ATP binding pocket. (c) Plot of the RMSD of the compound (blue) and the distance from the positively charged amine hydrogen on the N-akylamine moiety of the compound from the negatively charged epsilon and delta oxygen atoms of Glu-97 (orange) and Asp-157 (gray), respectively, across 1000 snapshots for the R and S configurations of 1 and 7. (d) A snapshot from the simulation of (S)-7 (KIN-281) bound to CaMKIIδ, showing the additional hydrogen bond created by the amine group on the quinazoline core of the compound. The crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) is shown as cartoon (gray) with the hinge loop (red), DFG-motif (green), and αC helix (orange) highlighted. The protein is shown with the glycine-rich loop removed (residues Glu-19 to Arg-29) to expose the ATP binding pocket. Asp-157 and Glu-97 are also shown as element-colored stick models, with carbons colored as green and cyan, respectively. (e) Dynamic cross-correlation matrix (DCCM) cross section of 1 and 8 derivatives against the protein kinase domain of CaMKIIδ.
From the decomposition energies, there are unfavorable interactions between the compound and residues Glu-97 and Asp-157. Although the initial models feature a salt bridge between the positively charged protonated amine on the N-alkylamine substituent and the negatively charged aspartic acid of the DFG motif. We observe that this interaction is not stable in our simulations. Visualization of the individual simulations reveal that the nearby Glu-97 sidechain attracts the flexible N-akylamine and causes it to adopt multiple conformations over the course of the simulation (Fig. 6c). In (R)-1, the salt bridge with Asp-157 is broken in all but one of the individual trajectories as the N-alkylamine interacts with Glu-97. The dissociation of the salt bridge and subsequent flexibility is also observed in (S)-1, although in only approximately half of the trajectories. In 7, which features an amine substituent on the quinazoline ring instead of a chlorine, we see stabilization of both the salt bridge and compound as the additional amine strengthens the interaction with Asp-157 (Fig. 6d). This is consistent with the higher inhibitory activity of 7 compared with the parent compound (Fig. 5).
The relative flexibility of the different binding poses of compound 1 is explored in Fig. S4. Three binding poses of compound 1 emerged from this study: the Vina-docked pose from the original virtual screen of 1 in its R configuration, and the Glide-docked poses of 1 in both its R and S configurations. We compared the atomic fluctuations on a per-residue basis for each of these three binding poses against the apo protein. This is measured as a ratio between the B-factors of a set of atoms on a residue in the bound and unbound states. A normalized flexibility of 1 corresponds to similar levels of dynamics between the bound and unbound protein. A measure above 1 represents greater fluctuation in the bound state than the unbound state, while a measure below 1 represents a more stable bound state compared to the unbound state. This measure was compared for all atoms of a residue (Fig. S4a), only the backbone C, Cα, and N atoms (Fig. S4b), and only the sidechain atoms (Fig. S4c). We find that generally, the relative flexibility of the three binding modes are similar. Compound binding generally stabilizes residues that line the ATP binding pocket. However, there are exceptions where residues become more flexibility upon ligand binding. In the Vina-docked pose, Phe-90 of the hinge loop and His-126 show the largest difference in flexibility compared to the Glide-docked pose. While His-126 is not near the ATP binding pocket, Phe-90 forms a π-π interaction with the compound in the Vina-docked pose. The majority of the difference in flexibility at these two residues is attributed to dynamics in the backbone of these residues. Ala-175 is among the most flexible residue in the protein, but this change in flexibility is only prevalent in the R configuration of the Vina-docked and Glide-docked poses.
We next explored the dynamics of the compounds bound to CaMKIIδ using dynamic cross-correlation matrices (DCCM). The correlation of the motion between each of the compounds and CaMKIIδ is shown in Fig. 6e. Visual inspection reveals similar motions for 1 and its derivatives. Generally, there are positive correlations between the residues of the compounds and the residues of the binding pocket. This is reflected most prominently in the residues near the three substituents of 1. The alkylamine moiety that forms a salt-bridge with the DFG motif in 1 near Asp-157 features a largely positively correlated region in each of the compounds. However, this region is noticeably smaller in the residues following Asp-157 in 4, 9, and 11, which feature shorter moieties at this position. The benzene ring substituent site that is attached to the core quinazoline of 1 occupies a region following Val-93 of the hinge loop and a region near Leu-20. The core quinazoline not only occupies the region near Ala-41 and Lys-43, but also Phe-90 of the hinge loop. In most cases, both regions are positively correlated, except in (S)-8, which follows an opposite trend in the Leu-20 region. Overall, the dynamics of the compounds reveal similar patterns of correlation in CaMKIIδ.
Molecular Dynamics Simulations to Explore Inhibition Profile Among Kinases
To gain deeper insight into the basis for the inhibition of compounds, we again resorted to explicit-solvent molecular dynamics simulations. We carried out simulations of (S)-6 bound to 6 of the kinases identified from the kinome profiling of the top 25 targets of 6 in Fig. 3b. We picked the S enantiomer as it featured both higher correlation coefficients in the SAR and increased stability with the aspartic acid of the DFG motif. We selected kinases with the highest (CaMKIIγ, CaMKIIδ, BTK, and CSK) and lowest (FGFR4 and EPHB1) affinities to 6. The differences in the IC50 values between highest and lowest affinity kinases is 5-fold (5 μM in CamKIIγ versus 22 μM in EPHB1).
We docked (S)-6 against each of the 6 kinase structures and selected the pose that best resembled the predicted binding pose against CaMKIIδ using Glide. We ran 100-ns explicit-solvent molecular dynamics simulations and free energy calculations for these complexes (Table S7). These extensive simulations will allow the compound to adopt its most favorable binding pose within the ATP-binding pocket. We next explored the interaction of (S)-6 with individual residues on the protein kinases by using decomposition energy calculations. We focused on the interaction energies of these key residues in the binding pocket (Fig. 7a). Like the SAR, three residues were strongly engaged by the compound across the 6 protein kinases, namely Leu-20, Val-28, and Leu-143. When comparing the enzymes that are strongly inhibited by (S)-6 (i.e. CaMKIIδ, CaMKIIγ, BTK, and CSK), with those that are more weakly inhibited by the compound (i.e. EPHB1 and FGFR4), we see little differences in the decomposition energies at specific residues.
Figure 7.

(a) Total decomposition energy (ΔEGBTOT) of key residues of binding for (S)-6 against 6 kinase targets. Each row is a kinase target while each column is an aligned residue. (b) Summation of the van der Waals (ΔEVDW) and electrostatic (ΔEELE) terms from the total decomposition energy of key residues of binding for (S)-6 against the same 6 kinase targets. (c) Dynamic cross-correlation matrix (DCCM) cross section of (S)-6 against the protein kinase domain of 6 kinase targets. Gaps in the multiple sequence alignment are shown in gray. Cross sections were converted into vectors and hierarchically clustered using Euclidean distance and average linkage. (d) The correlation of motion of 6S bound to CaMKIIγ and EPHB1 were mapped onto the structures of the protein kinase domains. Correlation coefficients were colored according to the scale in panel c. (e) Per-residue DCCM of the protein kinase domain for (S)-6 bound to CaMKIIγ and EPHB1.
In CaMKIIδ and CaMKIIγ, the interaction energies of the compound with two negatively charged glutamic acid residues at positions 97 and 140 are favorable. However, the polar and non-polar solvation terms are unfavorable. At position 97, this residue is replaced by an aliphatic alanine and an uncharged polar asparagine in EPHB1 and FGFR4, respectively. Similarly, the negatively charged glutamic acid is replaced by positively charged arginine residues in the 4 tyrosine kinases at position 140. Two residues appear to interact with (S)-6 among these two groups: Glu-97 and Asp-157. These two negatively charged residues interact with the positively charged amine group of the N-alkylamine moiety to form hydrogen bonds with the compound depending on the orientation of the flexible N-alkylamine moiety. In the CaMKs, the interaction energy appears to be highly unfavorable with Glu-97 and slightly unfavorable with Asp-157. This can be attributed to the penalty of desolvation of the N-alkylamine. In fact, when the solvation energies are not considered, the interaction energy is highly favorable between the compound and these two amino acids (Fig. 7b).
In addition to interaction energies, we explored the effect of (S)-6 on the dynamics of the 6 kinases considered in the calculations. This was accomplished by measuring the degree of correlation in the motion of (S)-6 with the motion of individual residues on the kinases to which the compound is bound (Fig. 7c). Positive correlations suggest that the compound and residue tend to move in the same direction over the course of the trajectory. Overall inspection of the map in Fig. 7c reveals similar patterns of interaction between the enzymes that are strongly inhibited by 6 (i.e. CaMKIIδ, CaMKIIγ, BTK, and CSK) and those that are less strongly inhibited (i.e. EPHB1 and FGFR4). The map reveals strongly correlated motions between compound (S)-6 and residues in the protein kinases; these regions of strong correlated motions occur between (i) residues 14 and 30; (ii) 40 and 80; and (iii) 110 and 170. Strong anti-correlated motion with (S)-6 occurs in the other regions, such as those from residues (i) 9 to 14; (ii) 30 to 40; (iii) 95 to 110; and (iv) 170 to 275.
Since the patterns of motion are similar by visual inspection, we wondered if we could differentiate the two groups of kinases by clustering the protein-compound complexes in a quantitative manner. We stipulated that kinases with similar dynamics as a result of (S)-6 were more likely to also have similar inhibition profiles. For each kinase, we defined a fingerprint by taking a cross-section of the DCCM map and mapping the correlations to the multiple sequence alignment. This fingerprint consists of correlation coefficients that correspond to the correlation of motion between compound and each residue on the protein. To compare these fingerprints, we calculated the Euclidean distance between each pair of kinases and hierarchically cluster these fingerprints using average linkage (Fig. 7c). Interestingly, CaMKIIγ exhibited dynamics that was most dissimilar to the other kinases. Among the other CaMK, CaMKIIδ is clustered with BTK. The two kinases with the highest IC50, EPHB1 and FGFR4, are clustered together. We visualized the dynamics of (S)-6 on CaMKIIγ and EPHB1 by applying the correlation coefficients from the DCCM cross-sections on structures of their kinase domains (Fig. 7d). Generally, the patterns of motion are similar between the two kinase targets, with the strongest correlations occurring adjacent to the binding pocket and strongest anti-correlated motion occurring distal to the binding pocket. In EPHB1, there is less correlation inside the binding pocket than in CaMKIIγ. The DCCMs of the CaMKIIγ and EPHB1 protein in Fig. 7e reveal similar trends when bound to (S)-6. The largest and most correlated of these regions occurs in the center of the DCCMs. In the orientation of the protein kinase domains in Fig. 7d, this region occurs near the ATP binding pocket.
DISCUSSION
Here, in an effort to identify small-molecules inhibitors of members of the CaMK family, we resorted to virtual screening using a target-specific re-scoring approach that we had previously developed. The method, known as SVMSP, uses Support Vector Machine (SVM) to train an algorithm using statistical pair potentials as features obtained from three-dimensional protein-compound structures. The positive set consists of protein-compound co-crystal structures obtained from the PDB, while the negative set consists of a randomly selected set of decoy molecules docked to the target of interest. The SVMSP scoring approach will favor docked compounds that adopt a binding mode that is similar to those found in the positive set while rejecting compounds that are similar to those found in the decoy set. The scoring approach does not take binding affinity in consideration, so its ability to identify active compounds is based solely on binding mode. The use of a target of interest (in this case CaMKIIδ) to create the negative set trains the algorithm to reject compounds that are decoy-like. We believe that the use of CaMKIIδ enables the algorithm to learn more about the features of the CaMKIIδ binding site, making it possible for the scoring approach to favor compounds that are more specific to CaMKIIδ. The performance of SVMSP was previously evaluated using the Directory of Useful Decoys (DUD),51 which provides a list of targets along with a library containing a set of known ligands and decoy molecules for validation of scoring methods. The performance of SVMSP was compared to several other scoring methods, which includes Glide,23, 24 GoldScore,25 ChemScore,25 X-Score,26, 52 and PMF.53 We found that overall, SVMSP performed better than these scoring functions. More importantly, SVMSP showed consistently good performance across several protein families that included kinases, proteases, metalloenzymes and nuclear receptors.
We applied SVMSP re-scoring towards the search for small molecules that inhibit members of the CaMK family. Our long-term objective is to identify selective and potent inhibitors of individual members of the CaMK family that play an important part in cancer initiation, progression and metastasis. Here, a member of the CaMKII sub-group of the large CaMK family (CaMKIIδ) was used. First, 149,979 compounds were docked to the ATP-binding pocket of CaMKIIδ followed by re-scoring of the resulting protein-compound structures with SVMSP. We identified the top 500 candidates and further clustered them into a set of 50 compounds. We tested 35 of these compounds that were commercially-available. Three active compounds were found, 1, 2, and 3. Compound 3’s activity is unlikely to be due to specific inhibition of CaMKIIδ since it is considered a pan-assay interference compound (PAINS).44 The fact that this compound emerged among the top candidates confirms that virtual screening is not immune to these compounds. Compound 2, while not considered a PAINS compound, contains an acylhydrazone moiety. This moiety, while robust at higher pH, is considered unstable at lower pH. A recent study revealed that these compounds can be successfully modified to obtain more stable derivatives.54
A question of interest is whether the binding mode of 1 that emerged from virtual screening is close to the real binding mode of the compound. Re-docking of the compounds using a different and robust docking program (Glide) led to a similar binding mode. The position of the quinazoline ring on this Glide-predicted binding pose was nearly exactly the position of quinazoline moieties found in the crystal structure of other small molecules bound to kinases. This suggests that the Glide binding mode is likely closer to the real binding mode of the compound. The fact that SVMSP identified 1 as an active compound despite the fact that it adopts the correct arrangement but does not likely adopt the binding mode of the compound within 1–2 Å may actually be a positive aspect of these scoring methods. Virtual screening does not always lead to binding poses that are in perfect agreement with what is found in the co-crystallized complex.22, 55 Since SVMSP is not as sensitive to the binding mode of a compound, this suggests that the re-scoring method can identify active compounds that would otherwise be ignored by other scoring functions.
To explore the inhibition profile of 1 across the human kinome, we pursued 1 and profiled an analog 6 for activity against 337 kinases initially at a single concentration of 20 μM. Remarkably, CaMKIIδ, the kinase that was used in the virtual screening, was among the top 10 most potently inhibited by 6 among 337 kinases. In addition, the kinase that was most potently inhibited by 6 was CaMKIIγ, a close homolog of CaMKIIδ. In fact, all four of the serine/threonine kinases among the top 25 kinases were members of the CaMK family. One belongs to the CaMK1 sub-group (CaMK1α), and another belongs to the CaMKL sub-group (maternal leucine embryonic zipper kinase or MELK). The remaining targets among the top 25 were all tyrosine kinases. Analysis of the ATP binding site among the top targets of 6 revealed that the majority of residues were conserved across all the kinases (e.g. Lys-43 and Asn-141) or the alternate residue at the position did not result in a large difference in the hydrophobicity (e.g. Leu-20 to Ile and Val-74 to Ile). In some cases, hydrophobic residues in the CaMKIIs are replaced by less hydrophobic residues in the tyrosine kinases (e.g. Leu-92 and Val-93). The fact that 6 exhibited some specificity towards CaMKIIδ may be attributed to the target-specific nature of the SVMSP scoring method. The use of CaMKIIδ in the negative training set may have led to an algorithm that enriches for compounds that bind to CaMKIIδ.
Comparatively, two CaMKII inhibitors found in the literature were among 178 kinase inhibitors recently profiled at Reaction Biology Corporation.56 Both KN-6257 (Ki = 0.9 μM) and KN-9358–60 (Ki = 0.37 μM, IC50 ~ 1–4 μM) were systematically tested against 300 kinases at lower concentrations for the compounds (20 μM versus 0.5 μM) than our kinome-wide screening of 6 and 14. Both CaMKII inhibitors showed 90% kinase activity to the α, β, γ, and δ subtypes of CaMKII. A more recent screening effort from EMD Millipore profiled these two compounds at both 1 and 10 μM against 234 kinases, including the β, γ, and δ subtypes of CaMKII.61 Neither compound showed activity at 1 μM against the 3 tested subtypes. At 10 μM, KN-62 inhibits the β, γ, and δ subtypes by 22, 13, and 9 percent, respectively, while KN-93 inhibits the same subtypes by 80, 33, and 53 percent, respectively. Both of these inhibitors are non-competitive ATP inhibitors and bind allosterically to CaMKII.62 Two additional inhibitors have been co-crystallized with CaMKIIδ at the ATP binding site: SU665663 and K00606.64 SU6656 (PDB ID: 2WEL, 1.90 Å) inhibits kinase activity to the α, β, and δ subtypes of CaMKII by ~30% but not CaMKIIγ at 0.5 μM in the RBC study. In addition, SU6656 also shows more than 60 and 80 percent inhibition to 38 and 19 different kinases at 0.5 μM, respectively. The other co-crystallized inhibitor K00606 (PDB ID: 2VN9, 2.30 Å) was not profiled in either of the aforementioned studies.
In addition to 1, we also tested a derivative of compound 2, another hit that emerged from the virtual screen. Although 2 may not be suitable for studies in cell culture considering the presence of an acylhydrazone moiety in its structure, the compound is suitable for binding and inhibition studies at the near neutral pH values used to conduct the enzyme activity studies for this work. We selected an analog of 2, namely 14, and tested it for activity against 337 kinases. The compound inhibited 46 kinases by 50% or more at 20 μM. Concentration-dependent studies of the top 25 kinases most potently inhibited by 14 show that all 25 kinases were inhibited with IC50s of 10 μM of less. Interestingly, when the 337 kinases are ranked by percent inhibition at 20 μM, CaMKIIδ is rank 46 on the list. It is interesting to note that 14 inhibited two kinases with sub-micromolar IC50s, BLK (IC50 = 0.51 μM) and BMX/ETK (IC50 = 0.98 μM).
To further gain insight into the basis for the preference of 6 towards CaMKIIs, we resorted to explicit-solvent molecular dynamics simulations followed by end-point free energy calculations of 1 bound to 6 kinases selected from among the top 25 targets of its close analog 6. We find that among the most highly favorable interactions of the compound are Val-93 and Glu-97. A valine residue at position 93 is only present in the three CaMKs. In the other tyrosine kinases, this position is replaced with less hydrophobic residues and the interaction energy is less favorable. Position 97 also appears to be unique among CaMKs. A glutamic acid occupies this position in contrast to the overwhelming majority of the other kinases that possess other neutral residues at that position (IR is the exception). Glu-97 interacts with the flexible N-alkylamine moiety of 1.
To explore the moieties on the compound that may be responsible for activity, we investigated the activity of 1 and its derivatives against 11 of the top kinases that were inhibited by 6. The data overall showed striking specificity of compounds towards members of the CaMK family, consistent with the kinome profiling above. Compounds lacking the alkylamine moiety of 6 and its parent structure 1 had significantly less activity against members of the CaMK family. This suggests that this moiety, which forms a salt-bridge with the conserved DFG motif in our modeled binding pose, is essential for activity. Molecular dynamics simulations followed by end-point free energy calculations confirmed these results. The excellent correlation coefficient between the experimental IC50 and predicted binding affinities provides additional support for the accuracy of the predicted binding mode. The calculations show that the electrostatic component of the free energy of binding becomes dramatically more favorable when the salt-bridge of the alkylamine moiety is present. Generally, there is a large penalty associated with desolvation of charged groups from solution. However, the fact that the salt bridge is nearly completely solvent exposed means that the binding of 1 will result in minimal loss of solvation energy and a significant gain in free energy as a result of the favorable intermolecular interaction of the salt bridge.
Decomposition energies, which measure the internal and solvation components between the compound and individual residues, reveal the importance of three hydrophobic residues: Leu-20, Val-28, and Leu-143. Leu-20 interacts with the benzene ring substituent that is attached to the core quinazoline, while Val-28 and Leu-143 form hydrophobic interactions with the quinazoline core. Examination of the dynamics of the compounds on CaMKIIδ reveal relatively similar dynamics across 1 and its derivatives. Positive correlations between the motion of the protein and the compound are observed at residues surrounding the binding pocket. At the DFG motif of the activation loop where the salt-bridge is formed, a region of positive correlation becomes smaller when the N-alkylamine moiety is removed.
In addition to free energy calculations, we also explored the effect of 6 on the dynamics of the 6 kinases. This was done by determining the correlation of the motion of 6 with each residue of the target. Correlation coefficients provide a measure of global dynamical changes. Every protein exhibits unique collection of global dynamics that are driven by the cross-correlation of all residues within the protein. Even proteins that possess different sequences but adopt the same fold are expected to exhibit different global dynamics. Interestingly, it appeared that CaMKIIγ showed the most distinct pattern of correlation. Hence, it is likely that the specificity observed for 6 towards CaMKII across the kinases tested may be partly due to the differences in the global dynamical changes of the target proteins.
MATERIALS AND METHODS
Virtual Screening
The virtual screening workflow is described in Fig. 1a. The Protein Preparation Wizard workflow from Schrödinger (Schrödinger LLC, New York, NY) was employed to prepare protein structures. The crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) was imported into the workspace from the Protein Data Bank (PDB).65 The auto-inhibitory and calmodulin-binding domains of CaMKIIδ (residues 276–334), calmodulin (chain D), solvent molecules, and inhibitors were removed. The structure was protonated, neutralized, and minimized using Impref. Both the protein and ligand structures were loaded into AutoDockTools,21 where the structures were converted to united-atom models and saved in PDBQT format. The docking box was centered on the co-crystallized ligand in the ATP binding site with dimensions of 21 Å × 17.25 Å × 18.75 Å. All other parameters of the docking program were set to default values. The docked conformations from AutoDock Vina (version 1.1.2)22 were converted with in-house Python scripts into mol2 format for additional analysis. Approximately 150,000 compounds from the ChemDiv commercial library42 were docked to the ATP-binding site of CaMKIIδ using Vina and the above parameters. This library was generated by Chemdiv by clustering the entire ChemDiv library using the SUBSET66 algorithm at 90% Tanimoto similarity. The library can be accessed by clicking on the “Distribution & Clustering” tab at the following URL: http://zinc.docking.org/catalogs/cdiv.
Docked receptor-ligand complexes were rescored using the previously-described 76-feature SVMSP scoring approach.33, 34, 38 Briefly, this machine learning approach uses predictive models to rank-order compounds by using knowledge-based pairwise potentials from various atom types.33, 39 Using these pairwise potentials as descriptors, SVMSP classification model was trained to discriminate active compounds from inactive compounds based on its 3D complex structure. The outcome of the SVMSP scoring function is the distance from the hyperplane in the SVM model, which is used to rank-order compounds. This model has demonstrated promising performance on the DUD validation set34 and was validated by in vitro assays.33 389 co-crystallized kinase complexes from the PDB (Table S1) and 4,992 randomly selected ligands docked to the CaMKIIδ binding pocket (Table S2) were used for the positive and negative training sets of the SVM model, respectively. The atom triplet fingerprints of the top 500 compounds from the SVM predictions were hierarchically clustered in Canvas67 to 50 clusters. The compound corresponding to each cluster center was selected as the representative compound for that cluster.
Validation of Scoring Functions Against CaMKIIδ
A set of approximately 2300 compounds were identified to contain biological data against CaMKIIδ in ChEMBL.43 Compounds were filtered by the type of activity data and limited to those with direct binding studies (Assay Type = ‘B’) that reported results as IC50 or Kd values. Compounds with activities ≤ 1 μM were classified as active and compounds with activities ≥ 10 μM were classified as inactive. Compounds containing pan-assay interference (PAINS)44 moieties were discarded. In total, we identified a set of 33 active and 58 inactive compounds against CaMKIIδ as SMILES strings (Table S3). Compounds were prepared using LigPrep in Schrödinger at a pH of 7.0. All stereoisomers were generated for each SMILES string, regardless of existing stereochemistry already present in the string. Compounds were docked to the prepared CaMKIIδ structure using AutoDock Vina22 and Glide in Standard Precision (SP) mode.68 Docked poses were rescored using Glide and the previously generated SVMSP model. Performance was assessed using receiver operator characteristic (ROC) curves and the area under the curve (ROC-AUC) as implemented in the Python scikit-learn package.69 Performance was assessed for docked poses with and without energy minimization. Energy minimization of docked poses was carried out using the “Refine Protein-Ligand Complex” workflow in the Prime module in Schrödinger.70 The VSGB solvation model71 and OPLS3 force field72 were used for energy minimization. Only residues within 5 Å of the docked compound were included. The local optimization algorithm was used to sample the docked structure.73, 74
Screening Top Candidates for Activity
Initial high-throughput screens for inhibitors against CaMKIIδ was carried out using the LANCE Ultra ULight® kinase assays from Perkin Elmer (Perkin Elmer Inc., Waltham, MA) according to the manufacturer’s recommendation. This is a time-resolved fluorescence energy transfer (TR-FRET) assay with a Europium labeled antibody that recognizes a phosphorylated Ulight-labeled peptide (ULight is a red-shifted dye); TR-FRET is measured by excitation at 320 nm of Europium and emission at 665 nm from the ULight acceptor dye using an Envision plate reader. Purified kinases were obtained from Carna Biosciences (CaMKIIδ) and were used at a final concentration of 20 nM. Testing for compound activity against all other kinases was done at Reaction Biology Corp (http://www.reactionbiology.com, Malvern, PA). In order to determine which peptide to use for the different kinases, the LANCE discovery kit (Perkin Elmer) was employed, which includes five different peptide substrates and covers about 80% of all Ser/Thr kinases. Based on the initial screen using the LANCE discovery kit of all five substrates against all four kinases, we settled with the ULight-CREBtide peptide for all kinases. Kinase assay buffer consisted of 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM DTT and 0.01% Tween-20. The screen with inhibitors was done using 384-well plates for all kinases against compounds at a final concentration of 20 μM, using 400 μM ATP and 100 nM peptide concentration. Kinase reactions were incubated for 60 min at 23 °C and stopped by the addition of 10 mM EDTA. The Eu-anti-phospho-MBP antibody diluted in detection buffer was then added to a final concentration of 2 nM. The addition of all components for the high-throughput screen was done using a Freedom Evo robot from Tecan. All curves were fitted with the program SigmaPlot 11 (Systat Software, Inc, San Jose, CA, 2008), which provides an estimate of the IC50 values.
Identification of Compound Analogs
The ZINC42 (http://zinc.docking.org) website was used to identify analogs of active compounds. The ZINC website uses Extended Connectivity Fingerprints (ECFP4) and Tanimoto coefficient to identify analogs. A similarity search was conducted using compound SMILES strings and an 80% similarity threshold. Several analogs were selected and purchased.
Binding Mode Prediction
The program AutoDock Vina was used as the docking program for all the virtual screening carried out in this work. To further validate the binding modes that emerged from AutoDock Vina during the virtual screening, we used the Glide docking program. Glide is a robust docking program that has been extensively validated in previous studies. The structures of 1 (Scheme 1) and its 8 derivatives (Scheme 3) were prepared using LigPrep in Schrödinger. Compounds were protonated at pH 7. Additionally, the structures of CaMKIIγ (PDB ID: 2V7O, 2.25 Å), and MELK (PDB ID: 4CQG, 2.57 Å) were prepared following the protocol used to prepare CaMKIIδ for virtual screening. Grid boxes were generated for the three proteins for docking in Glide,68 with a box size of 21 Å and an inner box of 14 Å. All other parameters were set to default values. A series of binding poses were generated by docking 1 and its derivatives against the ATP binding site of each of the three proteins using Glide in standard precision (SP) mode. All other parameters were left to default values. A consensus binding mode was identified by visual inspection. 43 co-crystallized quinazoline-containing compounds that bound to the ATP site of kinases were identified using a substructure search against the PDB. Structures were retrieved and aligned against the CaMKIIδ structure using the built-in align function in PyMOL (The PyMOL Molecular Graphics System, Version 1.5 Schrödinger, LLC.). The orientation of the quinazoline ring of each co-crystallized complex was visually inspected and compared against the Glide-predicted binding mode of 1.
Analysis of Binding Mode Across Multiple Kinases
Structural representatives of the protein kinase domain of each kinase was identified from the PDB. Each structure was aligned to a common protein kinase domain structure (PDB ID: 1ATP, 2.20 Å) and prepared using the Protein Preparation Wizard workflow in Schrödinger. Any additional chains in the PDB entry were removed and missing sidechains and loops were added using Prime.70 Solvent molecules and existing ligands were removed. Structures were protonated at pH 7 using PROPKA.75 A grid box centered at the ATP binding site was generated using Glide. We generated 50 binding poses for 6 in its S configuration for each of the kinase structures by using Glide. For each kinase structure, we visually identified a binding pose that shared the orientation and position of the predicted binding pose in CaMKIIδ and used it as the initial model for subsequent molecular dynamics simulations.
Molecular Dynamics Simulations
Docked binding poses were used to run molecular dynamics simulations using the AMBER14 software package.76 Each compound was assigned AM1-BCC77 charges and gaff78 atom types using antechamber.79 Complexes were immersed in a box of TIP3P80 water molecules. No atom on the complex was within 14 Å from any side of the box. The solvated box was further neutralized with Na+ or Cl− counterions using the tleap program. Simulations were carried out using the GPU accelerated version of the pmemd program with ff14SB81 and gaff78 force fields in periodic boundary conditions. All bonds involving hydrogen atoms were constrained by using the SHAKE algorithm,82 and a 2 femtoseconds (fs) time step was used in the simulation. The particle mesh Ewald83 (PME) method was used to treat long-range electrostatics. Simulations were run at 298 K under 1 atm in NPT ensemble employing Langevin thermostat and Berendsen barostat. Water molecules were first energy-minimized and equilibrated by running a short simulation with the complex fixed using Cartesian restraints. This was followed by a series of energy minimizations in which the Cartesian restraints were gradually relaxed from 500 kcal·Å−2 to 0 kcal·Å−2, and the system was subsequently gradually heated to 298 K via a 48 ps molecular dynamics run. By assigning different initial velocities, 10 independent simulations that are 10 ns in length each were carried out for the protein-compound structures.
Free Energy Calculations
In each of the 10 trajectories (10 ns in length), the first 2 ns were discarded for equilibration. Molecular dynamics snapshots were saved every 1 ps yielding 8000 structures per trajectory. A total of 80000 snapshots were generated per 100 ns of simulation. 1000 snapshots were selected at regular intervals from the 80000 snapshots for free energy calculations using the cpptraj program.84 The Molecular Mechanics-Generalized Born Surface Area (MM-GBSA)85 method was used to calculate the free energy using the MMPBSA.py script.86 The calculation using the GB method was performed with the Onufriev’s GB model.87, 88 SASA calculation was switched to ICOSA method, surface area was computed by recursively approximating a sphere around an atom, starting from an icosahedra. The entropy was estimated by normal mode calculations89 with the nmode module from 100 of the 1000 snapshots used in the free energy calculations. The maximum number of cycles of minimization was set to 10000. The convergence criterion for the energy gradient to stop minimization was 0.5.
Dynamic Cross-Correlation Matrix
Matrices were calculated from the set of 1000 snapshots from the free energy calculations using the matrix correl function in cpptraj. Correlation matrices were calculated by grouping together atoms of the same residues with no averaging by atom mass. In this n×n matrix, each row and column corresponds to a single residue in the protein-compound complex. We retrieve the 1×n vector of the compound, where each item in the vector corresponds to the Pearson correlation between the dynamics of the compound with the dynamics of a residue on the protein receptor. A multiple sequence alignment of the protein kinase domains was generated using Clustal Omega48 to align individual residues across the kinase targets. We then map the vector to the multiple sequence alignment. Gaps introduced in the alignment were assigned a correlation of 0. This resulted in a vector of correlation coefficients of equal length corresponding to the dynamics of the ligand on the protein kinase. A distance matrix between the vectors was calculated using Euclidean distance and used to hierarchically cluster the kinases using average linkage in R.90
Per-Residue Flexibility
Per-residue flexibility was calculated from the set of 1000 snapshots for the apo CaMKIIδ protein and three docked poses of compound 1: the (R)-1 pose from the initial AutoDock Vina-docked virtual screen and the (S) and (R) configuration poses of 1 from the follow up binding mode analysis using Glide. Per-residue flexibility is adapted using the scheme from Fuchs et. al.91 Per-residue B-factor is calculated using the atomicfluc function in cpptraj. The per-residue B-factors of the apo structure are used to normalize the B-factors of the bound complexes. The quotient of the bound and apo structures is thus a ratio of the relative flexibility at a residue. A flexibility of 1 corresponds to a residue’s flexibility equal to that of the apo protein. Values greater than 1 corresponds to residues that show greater mobility/flexibility, while values less than 1 correspond to increased stability upon ligand binding.
Statistical Analysis
Values are expressed as mean ± standard error, unless otherwise specified. Correlation analysis was performed using the SciPy92 package in Python, respectively.
Kinome Profiling and Concentration-Dependent Studies
Kinome profiling of 6 was carried out at Reaction Biology Corp using their Kinase HotSpot assay platform. We selected 6 over 1 due to availability. Considering the high level of structural similarity between 1 and 6, as well as the nearly identical IC50s for inhibition of CaMKIIδ, we felt that 6 will likely share the same targets as 1. Follow-up concentration-dependent studies for 6 against the top 25 targets that emerged from the kinome profiling was also carried out at Reaction Biology Corp. Follow-up studies for analogs of 1 were done at Reaction Biology Corp. The assay is a radiometric assay based on conventional filter-binding, which directly measures kinase catalytic activity toward a specific substrate.93
General Methods for Synthesis
Compound 1 was synthesized (Scheme 2) and all other compounds were purchased from ChemDiv Inc. The compounds were re-ordered to confirm activity. All chemicals were purchased from either Sigma-Aldrich or Acros. Column chromatography was carried out with silica gel GF254 (25–63 μm). Mass Spectra were measured on an Agilent 6520 Mass Q-TOF instrument. 1H NMR and 13C NMR spectra were recorded on a BRUKER 500 MHz spectrometer, using TMS as an internal standard and CDCl3 or DMSO-d6 as solvents. Chemical shifts (δ values) and coupling constants (J values) are reported in ppm and hertz, respectively. Anhydrous solvent and reagents were all analytically pure and dried through routine protocols. All compounds that were evaluated in biological essays had greater than 95% purity using HPLC.
7-chloro-2-methylquinazolin-4(3H)-one (12)
2-Amino-4-chlorobenzoic acid (3.43 g, 20 mmol) was dissolved in acetic anhydride (30 mL) and the mixture was heated to 110 °C for 5 h. The reaction was cooled to 0 °C in an ice-bath to yield yellow precipitate. The resulting yellow solid was filtered, washed with saturated NaHCO3 (aq) and dried under high vacuum. Then the solid was dissolved in ammonium hydroxide solution (40 mL) and the mixture was stirred at 50–60 °C for 10 h. The reaction was cooled to ambient temperature and the precipitate was filtered and washed with water (3 × 15 mL). The solid was dried under high vacuum to afford 1 as an off-white solid (2.72g, 70%). 1H NMR (500 MHz, DMSO-d6) δ 12.29 (brs, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.57 (s, 1H), 7.44 (d, J = 8.0 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 161.11, 156.03, 150.04, 138.83, 127.74, 126.09, 125.67, 119.44, 21.48; HRMS calcd for C9H8ClN2O [M+H]+ 195.0320, found 195.0322.
7-chloro-2-styrylquinazolin-4(3H)-one (13)
Compound 12 (388 mg, 2 mmol) and sodium acetate (246.1 mg, 3 mmol) were dissolved in glacial acetic acid (4 mL). The mixture was stirred at ambient temperature and aldehyde (4 mmol) was added slowly. After stirring at 110 °C for 30h, the resulting mixture was cooled to 0 °C and yellow precipitate was filtered and washed with copious amounts of water. The solid was dried under high vacuum to give 13 as yellow solids. Yield 92%, pale yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.09 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 16.0 Hz, 1H), 7.70 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 7.0 Hz, 2H), 7.50 (d, J = 2.0 Hz, 1H), 7.48 (t, J = 7.5 Hz, 2H), 7.43 (d, J = 7.5 Hz, 1H), 7.00 (d, J = 16 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 161.22, 152.90, 150.12, 139.11, 139.06, 134.82, 129.96, 129.09, 127.93, 127.73, 126.35, 126.10, 120.77, 119.91; HRMS calcd for C16H12ClN2O [M+H]+ 283.0633, found 283.0624.
(E)-N4-(7-chloro-2-styrylquinazolin-4-yl)-N1,N1-diethylpentane-1,4-diamine (1)
Compound 13 (282.1 mg, 1 mmol) was dissolved in anhydrous toluene (5 mL) and the mixture was cooled to 0 °C. POCl3 (0.28 mL, 3 mmol) and N, N-dimethylaniline (0.38 mL, 3 mmol) was added slowly to the solution, respectively. The resulting mixture was stirred at 115–120 °C for 8 h until the solution became dark red-purple color. The reaction was cooled to room temperature and solvent was removed in vacuo. The crude mixture was purified by flash column chromatography gradient eluting with 5% to 20% Et2O/hexanes to give the chlorinated intermediate (Rf = 0.3, 10% Et2O/hexanes) as light yellow solid (138 mg, 46%). The resulting product was identified by LC/MS: ESI-MS (m/z): 301.0 [M+1]+. This intermediate was continuously dissolved in anhydrous toluene (5 mL) at ambient temperature. Triethylamine (0.20 mL, 1.38 mmol) and DMAP (5.6 mg, 10% eq) were added and the mixture was stirred at room temperature for 10 minutes. 2-Amino-5-diethylaminopentane (0.18 mL, 0.92 mmol) was then added and the resulting mixture was stirred at 110 °C for 15 h. Solvent was removed in vacuo and the crude residue was purified by flash column chromatography (30: 1, CHCl3/CH3OH) to afford the desired product 1 as light grey solid (117 mg, 60%). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 16.0 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 8 Hz, 2H), 7.36 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 2.0 Hz, 1H), 7.32 (s, 1H), 7.18 (d, J = 16.0 Hz, 1H), 4.66–4.67 (m, 1H), 2.68–2.76 (m, 6H), 2.02–2.06 (m, 1H), 1.78–1.79 (m, 4H), 1.41 (d, J = 6.5 Hz, 3H), 1.15 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 161.96, 158.93, 158.34, 151.62, 138.54, 137.62, 136.69, 129.29, 128.88, 127.81, 127.74, 127.38, 125.96, 112.66, 77.74, 57.13, 52.80, 47.24, 46.79, 34.13, 21.03; HRMS calcd for C25H32ClN4 [M+H]+ 423.2310, found 423.2337.
Supplementary Material
Figure S1. (a–b) Receiver operator characteristic (ROC) curves of four docking program and scoring function combinations on a set of 33 active and 58 inactive compounds against CaMKIIδ. In (a), docked poses were used to calculate the ROC curves and ROC-AUC. In (b), the docked pose of the compound and surrounding residues on CaMKIIδ were energy minimized. (c–e) uses the 33 actives and 58 inactive compounds from (a). (c) The absolute difference in GlideScore and SVMSP between Vina-docked and Glide-docked poses. (d) RMSD between the Vina-docked and Glide-docked poses versus the absolute difference in GlideScore between the two poses. (e) RMSD between the Vina-docked and Glide-docked poses versus the absolute difference in SVMSP between the two poses.
Figure S2. (a) Chemical structure of 14 (KIN-332), a derivative of the parent compound 2 found via virtual screening against CaMKIIδ. (b) Kinome-wide profiling of 14 against 337 kinases at 20 μM. 14 inhibited 46 kinases by 50% or more at 20 μM. (c) Concentration-dependent study of 14 against the top 25 targets.
Figure S3. Stereoview of different orientations of a quinazoline core in co-crystallized compounds at the ATP binding site of protein kinases. In (a–c), a crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) is shown as cartoon (gray) with the hinge loop (red), DFG-motif (green), and αC helix (orange) highlighted. The protein is shown with the glycine-rich loop removed (Residues Glu-19 to Arg-29) to expose the ATP binding pocket. ATP (PDB ID: 1ATP, 2.20 Å) is superimposed onto the binding pocket with carbons colored in pale green. (a) Example of a compound (PDB ID: 4OBQ, 2.19 Å, colored pink) with the quinazoline ring mimicking the quinazoline of ATP. (b) Example of a compound (PDB ID: 1M17, 2.60 Å, colored pink) with the quinazoline ring mirroring that of the predicted binding mode of 1. (c) Example of a compound (PDB ID: 4F7S, 2.20 Å, colored pink) with the quinazoline ring in the same orientation as that of the predicted binding mode of 1.
Figure S4. Per-residue relative flexibility from the MD simulations of compound 1 against CaMKIIδ. The normalized flexibility describes the relative flexibility of a set of atoms compared to the apo receptor. A flexibility of 1 corresponds to a residue’s flexibility equal to that of the apo protein. Values greater than 1 corresponds to residues that show greater mobility/flexibility, while values less than 1 correspond to increased stability upon ligand binding. Contact residues within 5 Å of the ligand binding site are shown at the bottom of each panel using green squares. Flexibility is calculated on (a) all atoms on the residue, (b) the backbone residues (N, Cα, and C), and (c) the atoms in the sidechain.
Table S1. Crystal Structures in the Positive Set of the SVMSP Model.
Table S2. Decoy Compounds in the Negative Set of the SVMSP Model.
Table S3. Active and Inactive Compounds with Bioactivities Against CaMKIIδ.
Table S4. Physiochemical Properties of Hit Compounds from SVMSP.
Table S5. QikProp ADMET Properties for Compounds 1 (KIN-1) and 6 (KIN-236).
Table S6. RMSD Statistics for MD Simulations.
Table S7. Calculated Free Energies (± Standard Error) of (S)-6 (KIN-236) Against Select Kinases (kcal mol−1).
Acknowledgments
The research was supported by the National Institutes of Health (CA135380) (SOM) and the American Cancer Society Research Scholar Grant RSG-12-092-01-CDD (SOM). Computer time on the Big Red, Big Red II, and Karst supercomputers at Indiana University is supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Fedorov O, Muller S, Knapp S. The (Un)Targeted Cancer Kinome. Nat Chem Biol. 2010;6:166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. Protein Kinases--The Major Drug Targets of the Twenty-First Century? Nat Rev Drug Discovery. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG. LKB1 is a Master Kinase That Activates 13 Kinases of the AMPK Subfamily, Including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyer BS, Warsowe J, Solter D, Knowles BB, Ackerman SL. New Member of the Snf1/AMPK Kinase Family, Melk, is Expressed in the Mouse Egg and Preimplantation Embryo. Mol Reprod Dev. 1997;47:148–156. doi: 10.1002/(SICI)1098-2795(199706)47:2<148::AID-MRD4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 6.Gil M, Yang Y, Lee Y, Choi I, Ha H. Cloning and Expression of a cDNA Encoding a Novel Protein Serine/threonine Kinase Predominantly Expressed in Hematopoietic Cells. Gene. 1997;195:295–301. doi: 10.1016/s0378-1119(97)00181-9. [DOI] [PubMed] [Google Scholar]
- 7.Paris J, Philippe M. Poly(A) Metabolism and Polysomal Recruitment of Maternal mRNAs During Early Xenopus Development. Dev Biol. 1990;140:221–224. doi: 10.1016/0012-1606(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 8.Bright N, Thornton C, Carling D. The Regulation and Function of Mammalian AMPK-Related Kinases. Acta Physiol. 2009;196:15–26. doi: 10.1111/j.1748-1716.2009.01971.x. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Lee Y-M, Baitsch L, Huang A, Xiang Y, Tong H, Lako A, Choi C, Lim E, Min J. MELK is an Oncogenic Kinase Essential for Mitotic Progression in Basal-like Breast Cancer Cells. eLife. 2014:e01763–e01763. doi: 10.7554/eLife.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kig C, Beullens M, Beke L, Van Eynde A, Linders JT, Brehmer D, Bollen M. Maternal Embryonic Leucine Zipper Kinase (MELK) Reduces Replication Stress in Glioblastoma Cells. J Biol Chem. 2013;288:24200–24212. doi: 10.1074/jbc.M113.471433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Shi L, Zhang B, Sun X, Lu S, Liu Z, Liu Y, Li H, Wang L, Wang X, Zhao C. MiR-204 Inhibits Human NSCLC Metastasis Through Suppression of NUAK1. Br J Cancer. 2014;111:2316–2327. doi: 10.1038/bjc.2014.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X, Lv W, Zhang JH, Lu DL. miR-96 Functions as a Tumor Suppressor Gene by Targeting NUAK1 in Pancreatic Cancer. Int J Mol Med. 2014;34:1599–1605. doi: 10.3892/ijmm.2014.1940. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee S, Buhrlage SJ, Huang HT, Deng X, Zhou W, Wang J, Traynor R, Prescott AR, Alessi DR, Gray NS. Characterization of WZ4003 and HTH-01-015 as Selective Inhibitors of the LKB1-Tumour-Suppressor-Activated NUAK Kinases. Biochem J. 2014;457:215–225. doi: 10.1042/BJ20131152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Si J, Mueller L, Collins SJ. CaMKII Regulates Retinoic Acid Receptor Transcriptional Activity and the Differentiation of Myeloid Leukemia Cells. J Clin Invest. 2007;117:1412–1421. doi: 10.1172/JCI30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ang ES, Zhang P, Steer JH, Tan JW, Yip K, Zheng MH, Joyce DA, Xu J. Calcium/calmodulin-dependent Kinase Activity is Required for Efficient Induction of Osteoclast Differentiation and Bone Resorption by Receptor Activator of Nuclear Factor Kappa B Ligand (RANKL) J Cell Physiol. 2007;212:787–795. doi: 10.1002/jcp.21076. [DOI] [PubMed] [Google Scholar]
- 16.Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Targeting of a Novel Ca+2/Calmodulin-Dependent Protein Kinase II Is Essential for Extracellular Signal-Regulated Kinase–Mediated Signaling in Differentiated Smooth Muscle Cells. Circ Res. 2005;97:541–549. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- 17.Bouallegue A, Pandey NR, Srivastava AK. CaMKII Knockdown Attenuates H2O2-induced Phosphorylation of ERK1/2, PKB/Akt, and IGF-1R in Vascular Smooth Muscle Cells. Free Radical Biol Med. 2009;47:858–866. doi: 10.1016/j.freeradbiomed.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 18.Gu Y, Chen T, Meng Z, Gan Y, Xu X, Lou G, Li H, Gan X, Zhou H, Tang J, Xu G, Huang L, Zhang X, Fang Y, Wang K, Zheng S, Huang W, Xu R. CaMKII gamma, a Critical Regulator of CML Stem/Progenitor Cells, is a Target of the Natural Product Berbamine. Blood. 2012;120:4829–4839. doi: 10.1182/blood-2012-06-434894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Liu Q, Zhang Y, Liu K, Yu P, Liu K, Luan J, Duan H, Lu Z, Wang F, Wu E, Yagasaki K, Zhang G. Suppression of Growth, Migration and Invasion of Highly-Metastatic Human Breast Cancer Cells by Berbamine and Its Molecular Mechanisms of Action. Mol Cancer. 2009;8:81. doi: 10.1186/1476-4598-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM Serine/Threonine Kinases in the Pathogenesis and Therapy of Hematologic Malignancies and Solid Cancers. Haematologica. 2010;95:1004–1015. doi: 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trott O, Olson AJ. Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halgren T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem Biol Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 24.Halgren TA. Identifying and Characterizing Binding Sites and Assessing Druggability. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 25.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved Protein-ligand Docking Using GOLD. Proteins: Struct, Funct Bioinf. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Lu Y, Wang S. Comparative Evaluation of 11 Scoring Functions for Molecular Docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 27.Korb O, Stutzle T, Exner TE. Empirical Scoring Functions for Advanced Protein–Ligand Docking with PLANTS. J Chem Inf Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 28.Aqvist J, Medina C, Samuelsson JE. A New Method for Predicting Binding Affinity in Computer-Aided Drug Design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 29.Mukherjee S, Balius TE, Rizzo RC. Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J Chem Inf Model. 2010;50:1986–2000. doi: 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballester PJ, Mitchell JB. A Machine Learning Approach to Predicting Protein-ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics. 2010;26:1169–1175. doi: 10.1093/bioinformatics/btq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-Knowledge-Based Scoring Function Derived from Small Molecule Crystal Data with Superior Recognition Rate of Near-Native Ligand Poses and Better Affinity Prediction. J Med Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 32.Gohlke H, Klebe G. DrugScore Meets CoMFA: Adaptation of Fields for Molecular Comparison (AFMoC) or How to Tailor Knowledge-Based Pair-Potentials to a Particular Protein. J Med Chem. 2002;45:4153–4170. doi: 10.1021/jm020808p. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Khanna M, Jo I, Wang F, Ashpole NM, Hudmon A, Meroueh SO. Target-Specific Support Vector Machine Scoring in Structure-Based Virtual Screening: Computational Validation, In Vitro Testing in Kinases, and Effects on Lung Cancer Cell Proliferation. J Chem Inf Model. 2011;51:755–759. doi: 10.1021/ci100490w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Wang B, Meroueh SO. Support Vector Regression Scoring of Receptor–Ligand Complexes for Rank-Ordering and Virtual Screening of Chemical Libraries. J Chem Inf Model. 2011;51:2132–2138. doi: 10.1021/ci200078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin EJ, Sullivan DC. AutoShim: Empirically Corrected Scoring Functions for Quantitative Docking with a Crystal Structure and IC50 Training Data. J Chem Inf Model. 2008;48:861–872. doi: 10.1021/ci7004548. [DOI] [PubMed] [Google Scholar]
- 36.Martin EJ, Sullivan DC. Surrogate AutoShim: Predocking into a Universal Ensemble Kinase Receptor for Three Dimensional Activity Prediction, Very Quickly, without a Crystal Structure. J Chem Inf Model. 2008;48:873–881. doi: 10.1021/ci700455u. [DOI] [PubMed] [Google Scholar]
- 37.Ortiz AR, Pisabarro MT, Gago F, Wade RC. Prediction of Drug Binding Affinities by Comparative Binding Energy Analysis. J Med Chem. 1995;38:2681–2691. doi: 10.1021/jm00014a020. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Li L, Hurley TD, Meroueh SO. Molecular Recognition in a Diverse Set of Protein–Ligand Interactions Studied with Molecular Dynamics Simulations and End-Point Free Energy Calculations. J Chem Inf Model. 2013;53:2659–2670. doi: 10.1021/ci400312v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Li J, Khanna M, Jo I, Baird JP, Meroueh SO. Docking Small Molecules to Predicted Off-Targets of the Cancer Drug Erlotinib Leads to Inhibitors of Lung Cancer Cell Proliferation with Suitable In vitro Pharmacokinetic Properties. ACS Med Chem Lett. 2010;1:229–233. doi: 10.1021/ml100031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YY, Zhao R, Zhe H. The Emerging Role of CaMKII in Cancer. Oncotarget. 2015;6:11725–11734. doi: 10.18632/oncotarget.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erickson JR, He BJ, Grumbach IM, Anderson ME. CaMKII in the Cardiovascular System: Sensing Redox States. Physiol Rev. 2011;91:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Kruger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014;42:D1083–1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 45.Shahin R, Taha MO. Elaborate Ligand-Based Modeling and Subsequent Synthetic Exploration Unveil New Nanomolar Ca2+/Calmodulin-Dependent Protein Kinase II Inhibitory Leads. Bioorg Med Chem. 2012;20:377–400. doi: 10.1016/j.bmc.2011.10.071. [DOI] [PubMed] [Google Scholar]
- 46.Jorgensen WL, Duffy EM. Prediction of Drug Solubility from Structure. Adv Drug Delivery Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Yang PL, Gray NS. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 48.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 50.Xing L, Rai B, Lunney EA. Scaffold Mining of Kinase Hinge Binders in Crystal Structure Database. J Comput-Aided Mol Des. 2014;28:13–23. doi: 10.1007/s10822-013-9700-4. [DOI] [PubMed] [Google Scholar]
- 51.Huang N, Shoichet BK, Irwin JJ. Benchmarking Sets for Molecular Docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang R, Lai L, Wang S. Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J Comput-Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 53.Muegge I, Martin YC. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 54.Catrow JL, Zhang Y, Zhang M, Ji H. Discovery of Selective Small-Molecule Inhibitors for the beta-Catenin/T-Cell Factor Protein-Protein Interaction through the Optimization of the Acyl Hydrazone Moiety. J Med Chem. 2015;58:4678–4692. doi: 10.1021/acs.jmedchem.5b00223. [DOI] [PubMed] [Google Scholar]
- 55.Xu D, Meroueh SO. Effect of Binding Pose and Modeled Structures on SVMGen and GlideScore Enrichment of Chemical Libraries. J Chem Inf Model. 2016;56:1139–1151. doi: 10.1021/acs.jcim.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hidaka H, Kobayashi R. Pharmacology of Protein Kinase Inhibitors. Annu Rev Pharmacol Toxicol. 1992;32:377–397. doi: 10.1146/annurev.pa.32.040192.002113. [DOI] [PubMed] [Google Scholar]
- 58.Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The Newly Synthesized Selective Ca2+Calmodulin Dependent Protein Kinase II Inhibitor KN-93 Reduces Dopamine Contents in PC12h Cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- 59.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an Inhibitor of Multifunctional Ca++/Calmodulin-Dependent Protein Kinase, Decreases Early Afterdepolarizations in Rabbit Heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 60.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N -methylbenzylamine), A Calcium/Calmodulin-Dependent Protein Kinase II Inhibitor, is a Direct Extracellular Blocker of Voltage-Gated Potassium Channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- 61.Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, Harvey KJ. A Broad Activity Screen in Support of a Chemogenomic Map for Kinase Signalling Research and Drug Discovery. Biochem J. 2013;451:313–328. doi: 10.1042/BJ20121418. [DOI] [PubMed] [Google Scholar]
- 62.Pellicena P, Schulman H. CaMKII Inhibitors: From Research Tools to Therapeutic Agents. Front Pharmacol. 2014;5:21. [Google Scholar]
- 63.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a Selective Src Family Kinase Inhibitor, Used to Probe Growth Factor Signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pike AC, Rellos P, Niesen FH, Turnbull A, Oliver AW, Parker SA, Turk BE, Pearl LH, Knapp S. Activation Segment Dimerization: A Mechanism for Kinase Autophosphorylation of Non-consensus Sites. EMBO J. 2008;27:704–714. doi: 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voigt JH, Bienfait B, Wang S, Nicklaus MC. Comparison of the NCI Open Database with Seven Large Chemical Structural Databases. J Chem Inf Model. 2001;41:702–712. doi: 10.1021/ci000150t. [DOI] [PubMed] [Google Scholar]
- 67.Duan JX, Dixon SL, Lowrie JF, Sherman W. Analysis and Comparison of 2D Fingerprints: Insights into Database Screening Performance Using Eight Fingerprint Methods. J Mol Graphics Modell. 2010;29:157–170. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 69.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. scikit-learn: Machine Learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 70.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct, Funct Bioinf. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins: Struct, Funct Bioinf. 2011;79:2794–2812. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 73.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 74.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force Field Validation Using Protein Side Chain Prediction. J Phys Chem. 2002;106:11673–11680. [Google Scholar]
- 75.Sondergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 76.Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham TEI, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, York DM, Kollman PA. AMBER 2015. University of California; San Francisco: 2015. [Google Scholar]
- 77.Jakalian A, Jack DB, Bayly CI. Fast, Efficient Generation of High-quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and Testing of a General Amber Force Field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graphics Modell. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 80.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 81.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ryckaert JP, Ciccotti G, Berendsen JJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 83.Darden T, York D, Pedersen L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 84.Roe DR, Cheatham TE., 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 85.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 86.Miller BR, 3rd, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 87.Onufriev A, Bashford D, Case DA. Exploring Protein Native States and Large-scale Conformational Changes with a Modified Generalized Born Model. Proteins: Struct, Funct Bioinf. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 88.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL., 3rd Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J Comput Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 89.Brooks B, Karplus M. Harmonic Dynamics of Proteins: Normal Modes and Fluctuations in Bovine Pancreatic Trypsin Inhibitor. Proc Natl Acad Sci U S A. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.R Core. Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2013. [Google Scholar]
- 91.Fuchs JE, Waldner BJ, Huber RG, von Grafenstein S, Kramer C, Liedl KR. Independent Metrics for Protein Backbone and Side-Chain Flexibility: Time Scales and Effects of Ligand Binding. J Chem Theory Comput. 2015;11:851–860. doi: 10.1021/ct500633u. [DOI] [PubMed] [Google Scholar]
- 92.van der Walt S, Colbert SC, Varoquaux G. The NumPy Array: A Structure for Efficient Numerical Computation. IEEE Comput Sci Eng. 2011;13:22–30. [Google Scholar]
- 93.Ma H, Deacon S, Horiuchi K. The Challenge of Selecting Protein Kinase Assays for Lead Discovery Optimization. Expert Opin Drug Discovery. 2008;3:607–621. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a–b) Receiver operator characteristic (ROC) curves of four docking program and scoring function combinations on a set of 33 active and 58 inactive compounds against CaMKIIδ. In (a), docked poses were used to calculate the ROC curves and ROC-AUC. In (b), the docked pose of the compound and surrounding residues on CaMKIIδ were energy minimized. (c–e) uses the 33 actives and 58 inactive compounds from (a). (c) The absolute difference in GlideScore and SVMSP between Vina-docked and Glide-docked poses. (d) RMSD between the Vina-docked and Glide-docked poses versus the absolute difference in GlideScore between the two poses. (e) RMSD between the Vina-docked and Glide-docked poses versus the absolute difference in SVMSP between the two poses.
Figure S2. (a) Chemical structure of 14 (KIN-332), a derivative of the parent compound 2 found via virtual screening against CaMKIIδ. (b) Kinome-wide profiling of 14 against 337 kinases at 20 μM. 14 inhibited 46 kinases by 50% or more at 20 μM. (c) Concentration-dependent study of 14 against the top 25 targets.
Figure S3. Stereoview of different orientations of a quinazoline core in co-crystallized compounds at the ATP binding site of protein kinases. In (a–c), a crystal structure of CaMKIIδ (PDB ID: 2WEL, 1.90 Å) is shown as cartoon (gray) with the hinge loop (red), DFG-motif (green), and αC helix (orange) highlighted. The protein is shown with the glycine-rich loop removed (Residues Glu-19 to Arg-29) to expose the ATP binding pocket. ATP (PDB ID: 1ATP, 2.20 Å) is superimposed onto the binding pocket with carbons colored in pale green. (a) Example of a compound (PDB ID: 4OBQ, 2.19 Å, colored pink) with the quinazoline ring mimicking the quinazoline of ATP. (b) Example of a compound (PDB ID: 1M17, 2.60 Å, colored pink) with the quinazoline ring mirroring that of the predicted binding mode of 1. (c) Example of a compound (PDB ID: 4F7S, 2.20 Å, colored pink) with the quinazoline ring in the same orientation as that of the predicted binding mode of 1.
Figure S4. Per-residue relative flexibility from the MD simulations of compound 1 against CaMKIIδ. The normalized flexibility describes the relative flexibility of a set of atoms compared to the apo receptor. A flexibility of 1 corresponds to a residue’s flexibility equal to that of the apo protein. Values greater than 1 corresponds to residues that show greater mobility/flexibility, while values less than 1 correspond to increased stability upon ligand binding. Contact residues within 5 Å of the ligand binding site are shown at the bottom of each panel using green squares. Flexibility is calculated on (a) all atoms on the residue, (b) the backbone residues (N, Cα, and C), and (c) the atoms in the sidechain.
Table S1. Crystal Structures in the Positive Set of the SVMSP Model.
Table S2. Decoy Compounds in the Negative Set of the SVMSP Model.
Table S3. Active and Inactive Compounds with Bioactivities Against CaMKIIδ.
Table S4. Physiochemical Properties of Hit Compounds from SVMSP.
Table S5. QikProp ADMET Properties for Compounds 1 (KIN-1) and 6 (KIN-236).
Table S6. RMSD Statistics for MD Simulations.
Table S7. Calculated Free Energies (± Standard Error) of (S)-6 (KIN-236) Against Select Kinases (kcal mol−1).