

Abstract
Inducible nitric oxide synthase (iNOS) plays an important role in host defense. Macrophages expressing iNOS release the reactive nitrogen intermediates (RNI) nitrite and S-nitrosoglutathione (GSNO), which are bactericidal in vitro at a pH characteristic of the phagosome of activated macrophages. We sought to characterize the active intrabacterial forms of these RNI and their molecular targets. Peptide methionine sulfoxide reductase (MsrA; EC 1.8.4.6) catalyzes the reduction of methionine sulfoxide (Met-O) in proteins to methionine (Met). E. coli lacking MsrA were hypersensitive to killing not only by hydrogen peroxide, but also by nitrite and GSNO. The wild-type phenotype was restored by transformation with plasmids encoding msrA from E. coli or M. tuberculosis, but not by an enzymatically inactive mutant msrA, indicating that Met oxidation was involved in the death of these cells. It seemed paradoxical that nitrite and GSNO kill bacteria by oxidizing Met residues when these RNI cannot themselves oxidize Met. However, under anaerobic conditions, neither nitrite nor GSNO was bactericidal. Nitrite and GSNO can both give rise to NO, which may react with superoxide produced by bacteria during aerobic metabolism, forming peroxynitrite, a known oxidant of Met to Met-O. Thus, the findings are consistent with the hypotheses that nitrite and GSNO kill E. coli by intracellular conversion to peroxynitrite, that intracellular Met residues in proteins constitute a critical target for peroxynitrite, and that MsrA can be essential for the repair of peroxynitrite-mediated intracellular damage.
The phenotype of mice deficient in the inducible Ca2+-independent isoform of nitric oxide synthase (iNOS, NOS2) demonstrates that host defenses require reactive nitrogen intermediates (RNI) for successful control of certain pathogens, such as Mycobacterium tuberculosis (1). Moreover, the phenotype of mice deficient in both NOS2 and phagocyte oxidase suggests that RNI play a redundant role along with reactive oxygen intermediates (ROI) in controlling spontaneous infection by commensal microorganisms, such as Escherichia coli (2). Despite the importance of the process, little is known at the molecular level about how RNI kill bacteria.
Which RNI contribute to control of microbial pathogens is unclear,
because NO, the primary product of NOS2, can be converted in biological
settings to other RNI such as NO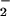 ,
⋅NO2,
N2O3,
NO−, and NO+. Such
conversions can take place before or after RNI enter the microorganism.
Equally unclear are the critical molecular lesions in bacteria, because
the potential targets of RNI are diverse. Considering only proteins,
RNI can nitrosylate cysteine sulfhydryls and heme prosthetic groups,
disrupt iron–sulfur clusters, and inactivate tyrosyl radicals (3). The
facile reaction of NO with superoxide (O
,
⋅NO2,
N2O3,
NO−, and NO+. Such
conversions can take place before or after RNI enter the microorganism.
Equally unclear are the critical molecular lesions in bacteria, because
the potential targets of RNI are diverse. Considering only proteins,
RNI can nitrosylate cysteine sulfhydryls and heme prosthetic groups,
disrupt iron–sulfur clusters, and inactivate tyrosyl radicals (3). The
facile reaction of NO with superoxide (O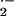 )
generates peroxynitrite (OONO−), whose reactive
derivatives nitrate tyrosine residues (4) and oxidize cysteines (5) and
methionines (Met) (6, 7).
)
generates peroxynitrite (OONO−), whose reactive
derivatives nitrate tyrosine residues (4) and oxidize cysteines (5) and
methionines (Met) (6, 7).
Study of the RNI sensitivity of mutants in specific repair pathways
offers a potentially powerful approach to help identify which forms of
RNI exert microbicidal actions within microbial pathogens and to
characterize their molecular targets (8). msrA encodes an
enzyme (EC 1.8.4.6) whose only known action is to reduce Met-O residues
back to Met (9). The reversible oxidation of key Met residues in
certain proteins can modulate the function of the protein (7, 10–12).
Mutant bacteria and yeast lacking msrA are more sensitive to
hydrogen peroxide (13, 14) than wild-type cells. The present work uses
an E. coli msrA-null mutant to evaluate whether
oxidation of Met plays a role in the killing of E. coli by
two forms of RNI secreted by iNOS-expressing macrophages,
NO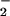 and S-nitrosoglutathione (GSNO) (1).
We began to explore the biology of msrA from M.
tuberculosis, a pathogen whose control by the murine host depends
on expression of iNOS.
and S-nitrosoglutathione (GSNO) (1).
We began to explore the biology of msrA from M.
tuberculosis, a pathogen whose control by the murine host depends
on expression of iNOS.
Materials and Methods
Reagents.
Reagents were as follows: GSNO was purchased from Alexis (San Diego, CA) or prepared as by Gibson et al. (15); isopropyl–d-thiogalactopyranoside (IPTG) and rapid ligation kit were purchased from Boehringer Mannheim; AlamarBlue was purchased from AccuMedInternational (West Lake, OH); restriction endonucleases, T4 DNA ligase, DNA polymerase I Klenow fragment, and calf intestinal alkaline phosphatase were all from New England Biolabs; Pfu DNA polymerase came from Stratagene; pT7-Blue blunt cloning kit was purchased from Novagene (Madison, WI); dNTPs came from Amersham Pharmacia; Ni2+-nitrilotriacetic acid (NTA) agarose beads, Miniprep, Maxiprep, and gel extraction kits all came from Qiagen (Chatsworth, CA); nitrocellulose came from Schleicher & Schuell; other chemicals were purchased from Sigma.
Cloning of MsrA from M. tuberculosis.
The M. tuberculosis genome contains a putative msrA (accession no. Rv0137c) (16). PCR primers were used to amplify a 0.8-kb DNA fragment containing the ORF (16) from a genomic library from M. tuberculosis strain CB3.3 (17). Forward primer (5′-GCGCGGGCCACGGTCTTCTCG) corresponded to base pairs 164579–164600, and reverse primer (5′-CCACTTCACCCTCGAGACCACCAC) corresponded to the complement of base pairs 165362–165386. The product was gel-purified and cloned into the SmaI site of the pT7-Blue vector to generate pMtb8.
Plasmids.
M. tuberculosis msrA was subcloned from pMtb8 by PCR with forward (5′-CATATGACGAGCAATCAGAAAGCG) and reverse (5′-GGCCCGCGTTCAGGTCCCGA) primers. The forward primer incorporated a NdeI site around the ATG start codon. The product was cloned into pT7-Blue such that the 5′-terminus of the insert was distal to the NdeI site within the vector, generating pT7MsrAMtb. A 240-bp region immediately upstream of E. coli msrA containing the msrA promoter was amplified by using primers (forward, 5′-AAGCTTACACAGCATAACTG; and reverse, 5′-CATATGGGTGTCGCTCTCC), creating an NdeI site encompassing the ATG start codon. The amplified promoter fragment was cloned into pT7-Blue, generating pEMP. M. tuberculosis msrA was subcloned from pT7MsrAMtb by means of an NdeI digestion and ligated into pEMP digested with the same enzyme, producing pEMPMsrAMtb. Medium-copy plasmid pRB3–273c (gift of Ferric Fang, Univ. of Colorado, Boulder) was used to introduce M. tuberculosis msrA and E. coli msrA into ΔmsrA E. coli. Thus pMsrAMtb was generated by subcloning a BamHI–SalI fragment of pEMPMsrAMtb into SmaI-digested pRB3–273c. Plasmid pAR100 (18) containing E. coli msrA was used to subclone this gene into pRB3–273c. To generate pMsrAEcoli a 1.3-kb fragment of pAR100 carrying the E. coli msrA gene and its promoter was subcloned by SacI–HindIII digestion and ligated into pRB3–273c digested with the same enzymes. The Cys → Ser (C52S) mutant of E. coli MsrA was created with the QuickChange Site-Directed Mutagenesis kit (Stratagene) using primer 5′-GCCATTTTTGCGATGGGTTCTTTCTGGGGTGTG and its complement.
Bacterial Strains.
Wild-type MC1061 E. coli and its congenic ΔmsrA E. coli (Tn903∷msrA) (13) were used for the functional characterization of msrA. XL-10 Gold (Stratagene) and HB101 (Life Technologies) E. coli strains were used for general genetic manipulation. Cultures were grown in LB broth or on LB agar with ampicillin (100 μg/ml) and/or kanamycin (50 μg/ml) as required. S. Ehrt (Weill Medical College of Cornell University) kindly provided lysates of M. tuberculosis strains CB3.3, H37Rv, and 1254 (ATCC 51910), Mycobacterium bovis bacillus Calmette–Guérin, Mycobacterium microti, and Mycobacterium chelonae.
Production of Recombinant M. tuberculosis MsrA and Antiserum.
An ApaI–BspEI fragment of pMtb8 carrying M. tuberculosis msrA was subcloned downstream of an IPTG-inducible promoter in pQE32 (Qiagen) and the resulting plasmid was used to transform E. coli M15 (pREP4) (Qiagen). The hexahistidine fusion protein was overexpressed after 3-h induction with 1 mM IPTG and purified on Ni-NTA agarose beads. The purified protein was subjected to SDS/PAGE. The Coomassie blue-stained band corresponding in mass to MsrA was homogenized in 25 mM Tris/75 mM glycine (pH 8.8/1% SDS/5 mM DTT/40% (vol/vol) glycerol, and the protein was electroeluted. Rabbits were injected each time with 50–75 μg of protein in Freund's adjuvant (complete, first injection; incomplete three times thereafter).
Immunoblot.
E. coli transformed with plasmid pMsrAMtb were sonicated on ice in 20 mM Tris (pH 7.4). Mycobacteria were lysed in a FastPrep beater. Protein concentrations were measured by the Bradford method (19). Proteins (100 μg) were separated on SDS/15% PAGE, transferred to nitrocellulose membranes, and reacted with antisera in (pH 7.5) Tris-buffered saline with 5% nonfat milk and 0.1% Tween-20 (TBS-T). Bound antibody was detected by enhanced chemiluminescence (NEN Life Science or Pierce). Blots to be reprobed were treated for 30 min at 55°C in 62.5 mM Tris, pH 6.8/100 mM 2-mercaptoethanol/2% SDS, then washed with several changes of TBS-T.
Enzymatic Activity.
MsrA activity was assayed as described (20) by using N-acetyl-[3H]Met-O as substrate and either DTT or thioredoxin plus thioredoxin reductase and NADPH as the reducing system.
Bacterial Survival.
Overnight cultures of transformed E. coli were diluted 1:200 or 1:500. Survival assays were carried out in LB (for assays with H2O2, ethanol, or urea), LB with 100 mM Mes, pH 5 (for assays with GSNO or sodium nitrite), or LB, pH 4. Bacteria were cultured at 37°C in 96-well microtiter plates with agitation. At the indicated times, aliquots were assayed for surviving bacteria by a fluorescence-based microplate assay (21). For anaerobic cultures, custom-made glass tubes with a side-arm on one wall and a stopcock on the opposite wall were fitted with an airtight stopper. After overnight aerobic growth, bacteria were added to the side-arm, while LB medium supplemented with 0.5% glucose and RNI was placed in the main compartment. The tubes were sparged with N2 through a submerged catheter for 30 min followed by 10 min of evacuation for three cycles. The sealed tubes were then tipped to inoculate the medium with bacteria. In controls, the stopcock was opened to readmit air before the tubes were tipped. Tubes were incubated at 37°C for the indicated times and assayed for surviving bacteria. Colony-forming units (CFU) are reported on a log10 scale.
Results
Cloning and Characterization of M. tuberculosis msrA.
The DNA sequence of the msrA gene cloned from M. tuberculosis strain CB3.3 was identical to the putative msrA sequence of strain H37Rv (16). M. tuberculosis MsrA (MsrAMtb) is 40% identical at the amino acid level to its counterpart in E. coli but shorter by 30 aa. The M. tuberculosis protein contains the active site consensus sequence, GCFWG (22–24), but preserves only 2 of the 4 cysteine residues found in the E. coli protein. Recombinant MsrAMtb was overexpressed in E. coli and purified to homogeneity. Although the apparent molecular mass of 27 kDa on SDS/PAGE (Fig. 1A) exceeded the expected mass for the recombinant protein including the histidine tag and linker (23,069 Da), the predicted mass was confirmed by mass spectrometry. The specific activity of MsrAMtb was comparable to that of E. coli MsrA (MsrAEcoli) when both were measured by using E. coli thioredoxin and thioredoxin reductase (Fig. 1B). Activity was not detectable when the following were individually omitted from the reaction mixture: thioredoxin (Fig. 1B), thioredoxin reductase, MsrA, or NADPH, or when BSA was substituted for MsrA (data not shown).
Figure 1.

Purification and characterization of MsrAMtb. (A) Coomassie blue-stained SDS/15% PAGE of MsrAMtb fusion protein after elution from Ni2+-NTA. Molecular masses (kDa) of markers are shown. Lane 1, 100 μg of protein from lysate of uninduced M15 E. coli transformed with pQE32 harboring M. tuberculosis msrA; lane 2, as in 1, but from bacteria grown in the presence of 1 mM IPTG for 3 h; lane 3, molecular weight markers; lane 4, 1.4 μg of MsrAMtb fusion protein after purification. (B) Recombinant MsrAMtb (▵) or MsrAEcoli (□) (14) was incubated with Tris⋅HCl (25 mM, pH 7.4), thioredoxin (10 μM), thioredoxin reductase (0.5 unit), NADPH (300 μM), and N-Ac[3H]Met-O (5.4 nmol, 132 cpm/pmol) in a final volume of 30 μl at 37°C. Controls are as follows: thioredoxin omitted (●), thioredoxin reductase omitted, NADPH omitted, MsrA omitted, and BSA substituted for MsrA. Values for controls were similar to value for thioredoxin omitted. Means are shown of two independent experiments, each performed in triplicate. SDs fall within the symbols.
Expression and Activity of MsrA in Wild-Type, Complemented, and ΔmsrA E. coli Strains.
Pure MsrAMtb was used to raise a specific antiserum, which detected a protein migrating at ≈27 kDa in E. coli transformed with M. tuberculosis msrA in the presence (Fig. 2A, lane 1) but not the absence of IPTG (Fig. 2A, lane 2).
Figure 2.

Expression of MsrA in E. coli and mycobacteria. (A Left) Immunoblot using MsrAMtb antiserum. Lane 1, 100 μg of protein from lysate of M15 E. coli transformed with pQE32 harboring M. tuberculosis msrA grown with IPTG for 3 h; lane 2, as in 1, but from bacteria grown without IPTG; lane 3, 1.4 μg of purified MsrAMtb. (A Right) Immunoblot as in Left, but probed with preimmune serum. (B) Detection of MsrA in E. coli. Wild-type E. coli was transformed with pRB3–273c containing no insert (lane 1). ΔmsrA E. coli was transformed with pRB3–273c containing no insert (lane 2), pMsrAEcoli (lane 3), or pMsrAMtb (lane 4). Lysates (100 μg of protein) from each strain were separated on SDS/15% PAGE, transferred to nitrocellulose, and immunoblotted with MsrAMtb antiserum (Upper) or MsrAEcoli antiserum (Lower). (C) Detection of MsrA in mycobacterial species. Lysates (100 μg of protein) were separated and transferred as in B, and immunoblotted with MsrAMtb antiserum (Upper) or preimmune serum (Lower).
In previous studies, an E. coli null mutant of msrA (ΔmsrA) was created through the insertion of a kanamycin-resistance cassette (Tn903) into the codon for amino acid 30 (13). The ΔmsrA strain was devoid of MsrA protein (Fig. 2B, and ref. 13) and enzyme activity (Table 1 and ref. 13). The msrA-deficient strain was complemented with msrA from either E. coli or M. tuberculosis by means of the low-copy plasmid pBR3–273c under the control of the E. coli msrA promoter. In addition, ΔmsrA E. coli was transformed with a C52S substitution mutant of E. coli msrA. As positive and negative controls, wild-type and msrA E. coli were transformed with empty vector.
Table 1.
Methionine sulfoxide reductase activity is restored in ΔmsrA E. coli by msrA from M. tuberculosis or E. coli
| E. coli strain | N-Ac-[3H]Met, pmol/μg/hr |
|---|---|
| Wild type | 1.0 ± 0.2 |
| ΔmsrA | 0.0 ± 0.1 |
| ΔmsrA(pMsrAEcoli) | 1.1 ± 0.2 |
| ΔmsrA(pMsrAMtb) | 0.9 ± 0.2 |
| ΔmsrA(pMsrAEcoliC52S) | 0.0 ± 0.0 |
Results are means ± SD from one experiment representative of three performed in triplicate.
MsrAMtb antiserum immunoblotted a single polypeptide (21 kDa) only in E. coli lysates prepared from msrA-pMsrAMtb (Fig. 2B). MsrAEcoli antiserum immunoblotted a single 22-kDa polypeptide in lysates from msrA-pMsrAEcoli, wild-type E. coli (Fig. 2B), and msrA-pMsrAEcoliC52S (data not shown).
Enzymatic specific activities for the complemented strains (ΔmsrA-pMsrAMtb and ΔmsrA-pMsrAEcoli) were the same as for wild-type E. coli. In contrast, no MsrA activity was detectable in lysates from ΔmsrA E. coli or ΔmsrA-pMsrAEcoliC52S (Table 1).
Detection of MsrA in Mycobacteria.
MsrAMtb antiserum, but not preimmune serum, immunoblotted a single polypeptide of 21 kDa in M. tuberculosis strains CB3.3 (Fig. 2C), 1254, and H37Rv (data not shown), and M. bovis bacillus Calmette–Guérin and M. microti (Fig. 2C). Lack of reactivity with M. chelonae may reflect limited crossreactivity of the MsrAMtb antiserum.
The Hypersensitive Phenotype of ΔmsrA E. coli to H2O2 Is Reversed by msrA from E. coli or M. tuberculosis, but Not by the E. coli C52S Mutant.
By using a qualitative assay (disk diffusion), ΔmsrA E. coli was found to be hypersensitive to H2O2 (13). We confirmed the hypersensitive phenotype of ΔmsrA E. coli to H2O2 by quantitating the number of viable bacteria over several hours in liquid culture (Fig. 3). The ΔmsrA mutant lost about 50-fold more CFU than wild type when both were challenged with H2O2. Hypersensitivity to H2O2 was reversed by complementation with msrA from M. tuberculosis as well as that from E. coli (Fig. 3B). Previous studies have shown that enzyme activity depends on a cysteine residue located in a conserved sequence, GCFWG, in the N-terminal domain in yeast, bovine, and E. coli homologs of MsrA (22–24), although the mutation of this residue to serine (C52S) has no detectable impact on tertiary structure (24). The C52S mutant failed to protect ΔmsrA E. coli against oxidative stress (Fig. 3C). Thus, the protection against H2O2 afforded by MsrA requires enzymatically active protein.
Figure 3.

Hypersensitive phenotype of ΔmsrA E. coli to H2O2 is reversed by msrA from M. tuberculosis or E. coli, but not by the E. coli C52S mutant. Survival of E. coli in LB with 0 mM (A), or 2 mM H2O2 (B and C). (A and B) Wild-type E. coli transformed with pRB3–273c containing no insert (□), or ΔmsrA E. coli transformed with pRB3–273c containing no insert (●), or containing pMsrAEcoli (▵) or pMsrAMtb (♦). (C) Survival of the E. coli C52S mutant after 12 h in the presence of 0 or 2 mM H2O2. Black bars, wild-type E. coli transformed with pRB3–273c containing no insert; white bars, ΔmsrA E. coli transformed with pRB3–273c containing no insert; checkered bars, pMsrAEcoli C52S; cross-hatched bars, pMsrAEcoli. Results are means ± SD from one experiment representative of three performed in triplicate.
ΔmsrA E. coli Is Highly Sensitive to Killing by RNI.
We next investigated the viability of ΔmsrA and wild-type E. coli exposed to two physiological forms of RNI secreted by activated macrophages, GSNO and nitrite. The medium was buffered at pH 5 because this approximates the hydrogen ion concentration in the phagosome of activated macrophages (25). The acidity of the phagosome is likely to be critical to the antibacterial action of RNI. Mild acidity stabilizes GSNO (26), and GSNO must be taken up to inhibit Salmonella typhimurium (8). Nitrite lacks antibacterial activity at neutrality, but is bactericidal at pH 5 (27) because the protonated form dismutates to NO and higher oxides of nitrogen (28).
The survival of all strains in the absence of RNI was similar (Fig. 4A). However, the survival of ΔmsrA in the presence of GSNO or NaNO2 was severely decreased (103- to 104-fold fewer surviving bacteria) compared with wild-type E. coli or ΔmsrA transformed with a plasmid expressing enzymatically active MsrA from either E. coli or M. tuberculosis (Fig. 4 B and C). Expression of msrA C52S failed to restore resistance to RNI (Fig. 4D).
Figure 4.

ΔmsrA E. coli is highly sensitive to killing by RNI. Survival of E. coli in LB (A), 4 mM GSNO (B), and 1 mM NaNO2 (C and D). In A–C, strains are as described for Fig. 3A. (D) Survival of the E. coli C52S substitution mutant after 12 h in the presence of NaNO2. Strains are as described for Fig. 3C. Results are means ± SD from one experiment representative of three performed in triplicate.
RNI Lack Bactericidal Activity Under Anaerobic Conditions.
The foregoing results suggested that addition of GSNO or
NO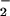 to the growth medium resulted in the oxidation of
critical Met residues within E. coli, even though GSNO and
NO
to the growth medium resulted in the oxidation of
critical Met residues within E. coli, even though GSNO and
NO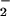 themselves do not oxidize Met. This paradox could
be explained if GSNO and NO
themselves do not oxidize Met. This paradox could
be explained if GSNO and NO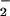 gave rise to more potent
oxidants on reacting with products of aerobic bacterial metabolism. To
test this hypothesis, we repeated the RNI challenge experiments under
anaerobic conditions. In the absence of molecular oxygen, neither
nitrite nor GSNO was bactericidal for E. coli, either wild
type or ΔmsrA (Fig. 5).
gave rise to more potent
oxidants on reacting with products of aerobic bacterial metabolism. To
test this hypothesis, we repeated the RNI challenge experiments under
anaerobic conditions. In the absence of molecular oxygen, neither
nitrite nor GSNO was bactericidal for E. coli, either wild
type or ΔmsrA (Fig. 5).
Figure 5.

RNI-induced toxicity to ΔmsrA E. coli requires aerobic conditions. Survival of E. coli grown in the presence or absence of 1 mM NaNO2 (A) or 4 mM GSNO (B) under anaerobic conditions for 24 h. Black bars, wild-type E. coli transformed with pRB3–273c containing no insert; white bars, ΔmsrA E. coli transformed with pRB3–273c containing no insert. Some cultures were prepared for anaerobic growth, but oxygen was readmitted before the bacteria were mixed with RNI. Results are means ± SD from 2 experiments in duplicate (A) or from a representative experiment in triplicate (B).
Survival of E. coli During Exposure to Stresses Other Than ROI or RNI.
To test whether MsrA specifically protects against Met-O accumulation induced by RNI and ROI, we tested the survival of ΔmsrA E. coli under other stressful conditions not thought to yield Met-O (Fig. 6). Viability was reduced after 3 h at pH 7 in ethanol or urea or at pH 4, but the presence of MsrA had no effect (Fig. 6).
Figure 6.

Survival of E. coli exposed to other stresses. E. coli was grown at pH 7, at pH 4, in 10% ethanol (EtOH), or in 250 mM urea. Shown are wild-type E. coli transformed with pRB3–273c containing no insert (black bars) and ΔmsrA E. coli transformed with pRB3–273c containing no insert (white bars), pMsrAEcoli (checkered bars), or pMsrAMtb (cross-hatched bars). Conditions are as in Fig. 3 except surviving bacteria were scored after 3 h. Results are means ± SD from one experiment representative of three in triplicate.
Discussion
It is remarkable that the fate of a bacterium confronted with hydrogen peroxide or RNI is dependent on the ability of the organism to reduce oxidized Met, given the multiplicity of other potential injuries and other pathways of protection, such as catalase, glutathione, glutaredoxins, thioredoxins, and peroxiredoxins. This is particularly striking in that there is no known reactivity of the added RNI with Met.
What explains this paradox? The demonstration that killing of E.
coli by NO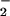 and GSNO requires molecular oxygen
appears to be of fundamental importance. Oxygen and the RNI tested were
both required for the bactericidal action, because bacteria were not
killed by RNI without oxygen or by oxygen without RNI. A likely
explanation is that nitrite and GSNO gave rise to NO, whereas
O2 was converted to superoxide through bacterial
metabolism, and together these products formed
OONO−, which can oxidize Met residues (29).
Because MsrA and its physiologic reductants (thioredoxin, thioredoxin
reductase, and NADPH) are all cytoplasmic, the critical Met residues,
the oxidation of which was reversed by MsrA, must likewise have been
intracellular. Because OONO− decomposes rapidly
and has a short diffusion distance, the intracellular localization of
target Met residues is most consistent with an intracellular site of
formation of OONO−.
and GSNO requires molecular oxygen
appears to be of fundamental importance. Oxygen and the RNI tested were
both required for the bactericidal action, because bacteria were not
killed by RNI without oxygen or by oxygen without RNI. A likely
explanation is that nitrite and GSNO gave rise to NO, whereas
O2 was converted to superoxide through bacterial
metabolism, and together these products formed
OONO−, which can oxidize Met residues (29).
Because MsrA and its physiologic reductants (thioredoxin, thioredoxin
reductase, and NADPH) are all cytoplasmic, the critical Met residues,
the oxidation of which was reversed by MsrA, must likewise have been
intracellular. Because OONO− decomposes rapidly
and has a short diffusion distance, the intracellular localization of
target Met residues is most consistent with an intracellular site of
formation of OONO−.
The role posited above for intracellular OONO−
is inferential, and other explanations must be considered. NO may
displace bound iron or copper ions such that they can interact with
H2O2 that arises from
aerobic metabolism, thereby generating OH⋅, which can oxidize
Met. NO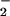 , when protonated to
HNO2, can give rise not only to NO but also to
higher oxides of nitrogen that might possibly oxidize Met. However,
these rearrangements are not dependent on molecular oxygen (26).
Nitrite-derived NO or NO+ can nitrosate
glutathione (GSH). This might deplete reserves of GSH that would
otherwise be available to react with
H2O2 generated during
aerobic metabolism. This could leave more
H2O2 available to oxidize
Met. However, the latter scenario would not explain the oxygen
dependence of the bactericidal action of GSNO itself, because
transnitrosation of endogenous GSH by exogenous GSNO would
generate an equimolar amount of free sulfhydryl. Finally, RNI and
O2 could each cause unrelated effects that were
individually innocuous but lethal in combination. However, this
explanation would not be consistent with the evidence that the
oxygen-dependent lethality of RNI arose not from two distinct forms of
killing, but from one, namely, the oxidation of Met residues. If the
RNI- and oxygen-dependent bacterial killing observed here is indeed
because of peroxynitrite, then the protection afforded by MsrA may be
the first example of the repair of peroxynitrite-mediated injury by a
defined enzymatic pathway.
, when protonated to
HNO2, can give rise not only to NO but also to
higher oxides of nitrogen that might possibly oxidize Met. However,
these rearrangements are not dependent on molecular oxygen (26).
Nitrite-derived NO or NO+ can nitrosate
glutathione (GSH). This might deplete reserves of GSH that would
otherwise be available to react with
H2O2 generated during
aerobic metabolism. This could leave more
H2O2 available to oxidize
Met. However, the latter scenario would not explain the oxygen
dependence of the bactericidal action of GSNO itself, because
transnitrosation of endogenous GSH by exogenous GSNO would
generate an equimolar amount of free sulfhydryl. Finally, RNI and
O2 could each cause unrelated effects that were
individually innocuous but lethal in combination. However, this
explanation would not be consistent with the evidence that the
oxygen-dependent lethality of RNI arose not from two distinct forms of
killing, but from one, namely, the oxidation of Met residues. If the
RNI- and oxygen-dependent bacterial killing observed here is indeed
because of peroxynitrite, then the protection afforded by MsrA may be
the first example of the repair of peroxynitrite-mediated injury by a
defined enzymatic pathway.
Peroxynitrite is emerging as a major toxin among RNI and ROI, not only
toward microbes but also toward mammalian cells (4, 30–32). Studies of
the oxidative damage of proteins by peroxynitrite have focused on
nitration of tyrosine residues and, to a lesser extent, oxidation of
cysteines. Our results suggest that protein Met residues may be another
important biological target of peroxynitrite, even if they are not a
quantitatively major route to its catabolism within the cell. Until
recently, the only way known for cells to protect themselves from
peroxynitrite was to reduce the concentration of one precursor,
superoxide, through the action of superoxide dismutase, thereby
preventing peroxynitrite formation. Subsequently, it was suggested that
some bacteria could also deplete the other precursor, NO, through NO
dioxygenase-catalyzed oxidation to NO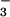 (33).
Recently, it was demonstrated that bacterial peroxiredoxins can
catabolize peroxynitrite fast enough to protect other molecular targets
(5). Here we suggest that a third way for cells to protect themselves
from the effects of peroxynitrite is to repair key lesions after they
are inflicted.
(33).
Recently, it was demonstrated that bacterial peroxiredoxins can
catabolize peroxynitrite fast enough to protect other molecular targets
(5). Here we suggest that a third way for cells to protect themselves
from the effects of peroxynitrite is to repair key lesions after they
are inflicted.
Why is the ability to reduce Met-O residues in proteins so important for the survival of E. coli (see also ref. 13) and yeast (14)? Three possible explanations are proposed. First, oxidation of Met residues alters the biological activity of specific proteins, such as ribosomal protein L12, α-1-proteinase inhibitor, calmodulin, and a voltage-gated K+ channel (34). In cells challenged with ROI or RNI, there may be specific proteins required for viability that contain a Met residue, whose oxidation by peroxynitrite inactivates the proteins. Inability of the ΔmsrA mutant to reduce Met-O in such proteins could explain the sensitivity of the bacterium to RNI and ROI.
Second, studies with methionine aminopeptidase (MAP) mutants have shown that the removal of the N-terminal Met from many newly synthesized proteins is essential for cell viability (35, 36). Synthetic peptides with Met-O at the N-terminal position either are not substrates for MAP (37) or are much poorer substrates than the corresponding Met peptide (38). Studies on the effect of H2O2 on protein oxidation in neutrophils showed that Met residues in nascent protein chains were more sensitive to oxidation than were Met residues in a mature protein (39). Thus, the oxidation of N-terminal Met residues in nascent chains could be a lethal event if there were no mechanism to reduce the N-terminal Met-O to Met. To test this hypothesis, we carried out Edman degradation to quantitate the content of N-terminal Met-O residues in lysates from wild-type and ΔmsrA E. coli before and after exposure to ROI and RNI. However, the high background contributed by preformed proteins precluded detecting small differences (G.S.J., H.E.-B., P.T., D. Wellner, C.N., and N.B., unpublished observations).
A third possibility comes from studies on the sensitivity of Met residues in glutamine synthetase to oxidation (40). Highly exposed Met residues were the most readily oxidized. It was suggested that these Met residues were part of a reversible oxidation/reduction mechanism that helped to protect the protein against oxidative damage. In this scheme some Met residues in proteins act as antioxidants by their oxidation to Met-O. Subsequent reduction by MsrA regenerates Met. The net result is that, in the presence of MsrA, the exposed Met residues act catalytically to remove ROI and RNI at the expense of NADPH. The concept that some proteins, in addition to their other roles, may also function catalytically to destroy ROI and RNI through Met oxidation followed by MsrA-catalyzed reduction raises the possibility that MsrA deficiency could sensitize cells to oxidant injury by permitting the accumulation of higher levels of intracellular ROI and RNI than in cells that express MsrA. However, the postulate that Met residues in proteins can act as antioxidants requires invoking an additional step, epimerization. Chemical oxidation of Met results in the formation of both Met-R-O and Met-S-O epimers. MsrA is specific for Met-S-O (24, 41). Therefore, after repeated exposure to oxidizing agents, proteins would accumulate Met-R-O. This would be deleterious to the cell unless there were a way to reduce Met-R-O directly to Met or an epimerase to convert Met-R-O to Met-S-O. Preliminary evidence suggests that E. coli extracts contain an epimerase that can catalyze this conversion (H.W., F. Etienne, and N.B., unpublished data). Such an enzyme would have to be closely coupled to MsrA for proteins to be fully reactivated after oxidation of Met residues.
Because RNI play a prominent role in the control of experimental tuberculosis, it is of interest that msrA from M. tuberculosis protected heterologous bacteria from RNI.
Acknowledgments
We thank R. Bryk and S. Ehrt for insightful discussions, Daniel Wellner for N-terminal sequencing, M. Fuortes for software, and Jo Ann Henry (Research Animal Resources Center) and Albert Marcus Morrishow (Mass Spectrometry Core Facility). This work was funded by National Institutes of Health Grant HL61241 (to C.N.) and by a fellowship (to G.S.J.) through National Institutes of Health Immunology Research Training Grant T32 A107621. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
Abbreviations
- CFU
colony forming units
- GSNO
S-nitrosoglutathione
- IPTG
isopropyl β-d-thiogalactopyranoside
- Met-O
methionine sulfoxide
- MsrA
peptide methionine sulfoxide reductase
- MsrAEcoli
E. coli MsrA
- MsrAMtb
M. tuberculosis MsrA
- NTA
Ni2+-nitrilotriacetic acid
- iNOS or NOS2
inducible Ca2+-independent nitric oxide synthase
- RNI
reactive nitrogen intermediates
- ROI
reactive oxygen intermediates
References
- 1.Nathan C, Shiloh M U. Proc Natl Acad Sci USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiloh M U, MacMicking J D, Nicholson S, Brause J E, Potter S, Marino M, Fang F, Dinauer M, Nathan C. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 3.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 4.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 5.Bryk R, Griffin P, Nathan C. Nature (London) 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 6.Pryor W A, Squadrito G L. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 7.Vogt W. Free Radical Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 8.De Groote M A, Testerman T, Xu Y, Stauffer G, Fang F C. Science. 1996;272:414–417. doi: 10.1126/science.272.5260.414. [DOI] [PubMed] [Google Scholar]
- 9.Brot N, Weissbach L, Werth J, Weissbach H. Proc Natl Acad Sci USA. 1981;78:2155–2158. doi: 10.1073/pnas.78.4.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciorba M A, Heinemann S H, Weissbach H, Brot N, Hoshi T. Proc Natl Acad Sci USA. 1997;94:9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abrams W R, Weinbaum G, Weissbach L, Weissbach H, Brot N. Proc Natl Acad Sci USA. 1981;78:7483–7486. doi: 10.1073/pnas.78.12.7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochrane C G, Spragg R, Revak S D. J Clin Invest. 1983;71:754–761. doi: 10.1172/JCI110823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moskovitz J, Rahman M A, Strassman J, Yancey S O, Kushner S R, Brot N, Weissbach H. J Bacteriol. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moskovitz J, Berlett B S, Poston J M, Stadtman E R. Proc Natl Acad Sci USA. 1997;94:9585–9589. doi: 10.1073/pnas.94.18.9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson A, Babbedge R, Brave S R, Hart S L, Hobbs A J, Tucker J F, Wallace P, Moore P K. Br J Pharmacol. 1992;107:715–721. doi: 10.1111/j.1476-5381.1992.tb14512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry C E, III, et al. Nature (London) 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 17.Friedman C R, Stoeckle M Y, Kreiswirth B N, Johnson W D, Jr, Manoach S M, Berger J, Sathianathan K, Hafner A, Riley L W. Am J Respir Crit Care Med. 1995;152:355–359. doi: 10.1164/ajrccm.152.1.7599845. [DOI] [PubMed] [Google Scholar]
- 18.Rahman M A, Brot N, Weissbach H. Cell Mol Biol. 1992;38:529–542. [PubMed] [Google Scholar]
- 19.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Brot N, Werth J, Koster D, Weissbach H. Anal Biochem. 1982;122:291–294. doi: 10.1016/0003-2697(82)90283-4. [DOI] [PubMed] [Google Scholar]
- 21.Shiloh M U, Ruan J, Nathan C. Infect Immun. 1997;65:3193–3198. doi: 10.1128/iai.65.8.3193-3198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 23.Lowther W T, Brot N, Weissbach H, Matthews B W. Biochemistry. 2000;39:13307–13312. doi: 10.1021/bi0020269. [DOI] [PubMed] [Google Scholar]
- 24.Moskovitz J, Poston J M, Berlett B S, Nosworthy N J, Szczepanowski R, Stadtman E R. J Biol Chem. 2000;275:14167–14172. doi: 10.1074/jbc.275.19.14167. [DOI] [PubMed] [Google Scholar]
- 25.Schaible U E, Sturgill-Koszycki S, Schlesinger P H, Russell D G. J Immunol. 1998;160:1290–1296. [PubMed] [Google Scholar]
- 26.Freelisch M, Stamler J S, editors. Methods in Nitric Oxide Research. West Sussex, England: Wiley; 1996. pp. 71–115. [Google Scholar]
- 27.Ehrt S, Shiloh M U, Ruan J, Choi M, Gunzburg S, Nathan C, Xie Q, Riley L W. J Exp Med. 1997;186:1885–1896. doi: 10.1084/jem.186.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuehr D J, Nathan C F. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pryor W A, Jin X, Squadrito G L. Proc Natl Acad Sci USA. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darrah P A, Hondalus M K, Chen Q, Ischiropoulos H, Mosser D M. Infect Immun. 2000;68:3587–3593. doi: 10.1128/iai.68.6.3587-3593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman-Davis J, Gibbs-Erwin J, Lindsey J R, Matalon S. Proc Natl Acad Sci USA. 1999;96:4953–4958. doi: 10.1073/pnas.96.9.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu K, Mitchell C, Xing Y, Magliozzo R S, Bloom B R, Chan J. Tuber Lung Dis. 1999;79:191–198. doi: 10.1054/tuld.1998.0203. [DOI] [PubMed] [Google Scholar]
- 33.Poole R K, Hughes M N. Mol Microbiol. 2000;36:775–783. doi: 10.1046/j.1365-2958.2000.01889.x. [DOI] [PubMed] [Google Scholar]
- 34.Brot N, Weissbach H. Biopolymers. 2000;55:288–296. doi: 10.1002/1097-0282(2000)55:4<288::AID-BIP1002>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 35.Chang S Y, McGary E C, Chang S. J Bacteriol. 1989;171:4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller C G, Strauch K L, Kukral A M, Miller J L, Wingfield P T, Mazzei G J, Werlen R C, Graber P, Movva N R. Proc Natl Acad Sci USA. 1987;84:2718–2722. doi: 10.1073/pnas.84.9.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Bassat A, Bauer K, Chang S Y, Myambo K, Boosman A, Chang S. J Bacteriol. 1987;169:751–757. doi: 10.1128/jb.169.2.751-757.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solbiati J, Chapman-Smith A, Miller J L, Miller C G, Cronan J E., Jr J Mol Biol. 1999;290:607–614. doi: 10.1006/jmbi.1999.2913. [DOI] [PubMed] [Google Scholar]
- 39.Fliss H, Weissbach H, Brot N. Proc Natl Acad Sci USA. 1983;80:7160–7164. doi: 10.1073/pnas.80.23.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine R L, Mosoni L, Berlett B S, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharov V S, Ferrington D A, Squier T C, Schoneich C. FEBS Lett. 1999;455:247–250. doi: 10.1016/s0014-5793(99)00888-1. [DOI] [PubMed] [Google Scholar]