

Abstract
Disulfide bonds play a crucial role in proteins, modulating their stability and constraining their conformational dynamics. A particularly important case is that of proteins that need to withstand forces arising from their normal biological function and that are often disulfide bonded. However, the influence of disulfides on the overall mechanical stability of proteins is poorly understood. Here, we used single-molecule force spectroscopy (smFS) to study the role of disulfide bonds in different mechanical proteins in terms of their unfolding forces. For this purpose, we chose the pilus protein FimG from Gram-negative bacteria and a disulfide-bonded variant of the I91 human cardiac titin polyprotein. Our results show that disulfide bonds can alter the mechanical stability of proteins in different ways depending on the properties of the system. Specifically, disulfide-bonded FimG undergoes a 30% increase in its mechanical stability compared with its reduced counterpart, whereas the unfolding force of I91 domains experiences a decrease of 15% relative to the WT form. Using a coarse-grained simulation model, we rationalized that the increase in mechanical stability of FimG is due to a shift in the mechanical unfolding pathway. The simple topology-based explanation suggests a neutral effect in the case of titin. In summary, our results indicate that disulfide bonds in proteins act in a context-dependent manner rather than simply as mechanical lockers, underscoring the importance of considering disulfide bonds both computationally and experimentally when studying the mechanical properties of proteins.
Keywords: atomic force microscopy (AFM), disulfide, molecular dynamics, protein folding, single-molecule biophysics
Introduction
Disulfide bonds are present in ∼10% of all proteins and, together with peptide bonds, they virtually represent their only covalent intramolecular interaction (1). Disulfide bonds have long been known to influence the thermodynamic stability of proteins (2), on one hand reducing the entropy of the unfolded state (3), and on the other hand enthalpically stabilizing the folded form (4). Which of these effects dominates seems to vary from case to case (2, 5, 6). More recently, regulatory roles for protein disulfides have also been invoked (7). In the case of proteins that bear mechanical loads in physiological conditions, the influence of disulfide bonds in the modulation of the mechanical stability has not been addressed systematically.
Contrary to thermodynamic stability, defined as the free energy difference between the folded and unfolded states, ΔGNU, mechanical stability is kinetically controlled and hence related to the free energy barrier that must be overcome by a pulling force, ΔGN‡(F), and the distance to the transition state, ΔxN‡(F) (8). Overcoming this barrier in the presence of disulfide bonds that sequester amino acid residues requires higher forces, and for this reason disulfides are often seen as mechanical lockers (9). But additional factors may also contribute to the net effect in the mechanical stability. Computational studies have highlighted the importance of the disulfide bond geometry (10). Single-molecule experiments and computer simulations have demonstrated that a mechanical load may affect the redox state of disulfides (11–16). The applied forces can make disulfide bonds accessible for solvents (17) and oxidoreductase enzymes involved in the control of the redox state (18, 19). This mechanochemical regulation is biologically crucial for several cellular processes, some of them related with diseases such as heart aging or failing (20) or HIV viral attachment (21).
In this work we study the effects of disulfides in the overall mechanical stability of proteins, focusing on two immunoglobulin-like domains. Our first case study is a self-complemented form of the bacterial pilus FimG protein, with well conserved disulfide forming cysteines (22, 23). Our second example is a mutant of the I91 (formerly termed I27) human cardiac titin polyprotein (I91G32C/A75C), which has its engineered Cys residues far in sequence, but close enough in structure to spontaneously form disulfide bonds (9). We measure their mechanical properties using single-molecule force spectroscopy (smFS)6 in the constant speed mode (24–27) using reducing and oxidative conditions that enhance the presence of the corresponding forms of each protein. We interpret the experimental results using a well established coarse-grained simulation model (28–32). We find that disulfide bonds in proteins act in a context-dependent manner rather than just behaving as mechanical lockers. When the disulfide bond compromises the unfolding pathway, transitions will necessarily induce a change in forced unfolding pathways. However, in the absence of such hard constraint the mechanical stability of proteins may remain unchanged and even decrease.
Results
Constant speed pulling experiments
In the smFS experiments we report, a gold substrate with the polyprotein attached moves away from the cantilever and sensor induced by a piezo-electric actuator (see Fig. 1A and “Experimental procedures”). Protein unfolding results in a trace with a sawtooth pattern, where each of the peaks corresponds to the unfolding of a single domain (Fig. 1B). For FimG, the protein is flanked with four I91 domains that unfold first and are used as a molecular fingerprint. In the case of the I91G32C/A75C chimera, an ideal sawtooth pattern is composed of eight individual peaks. From fits to the worm-like chain model to the data we extract characteristic contour lengths, and from the maximum of the individual sawtooth peaks we obtain the unfolding forces for both FimG and the titin Ig domain. When these domains are disulfide bonded we measure a shorter contour length as corresponds to the sequestered region of the protein sequence (see Fig. 1B).
Figure 1.
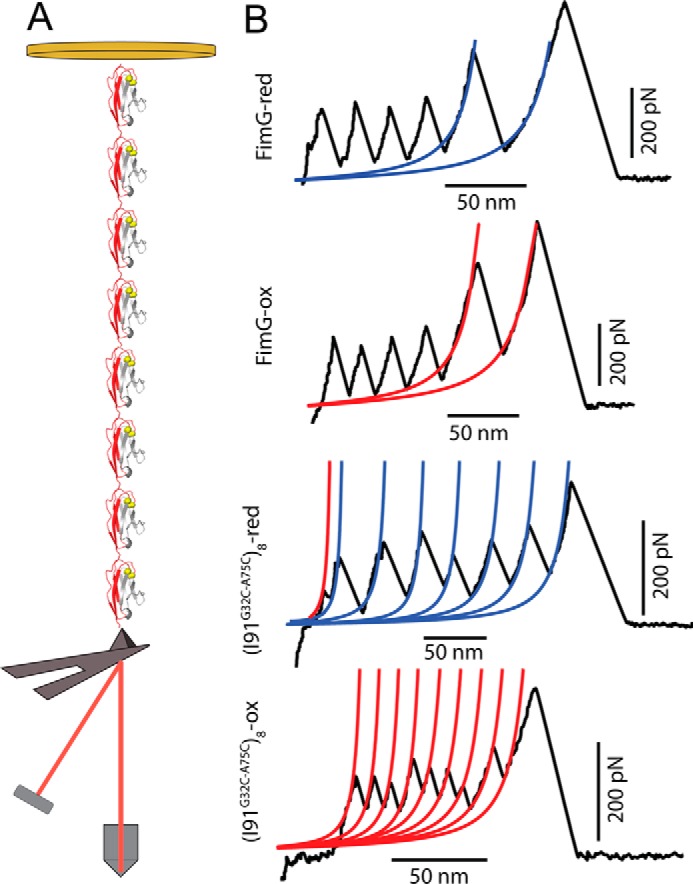
Single molecule pulling experiments on polyprotein constructs. A, schematic illustration of a smFS experiment with the(I91G32C/A75C)8 polyprotein. The Ig domains unfold due to the mechanical force applied between the gold substrate and the cantilever (not to scale). B, typical FimG and (I91G32C/A75C)8 construct experimental force-extension traces for their oxidized and reduced states. The contour lengths of oxidized and reduced domains are shown in red and blue, respectively. In all the traces the last peak corresponds to the detachment of the protein from the cantilever or surface.
Disulfide bonds increase the mechanical stability of FimG
In Fig. 2 we show scatter plots of the unfolding forces and contour lengths for FimG, and histograms of the forces and extensions to highlight observed differences between oxidized and reduced domains. The scatter plot of extension versus unfolding force shows two populations at high (52 ± 1 nm) and low (40 ± 2 nm) contour lengths, corresponding to the reduced and oxidized forms of the protein, respectively. These values compare well with the expectation of 54 nm for the reduced form of the protein and 39 nm for the oxidized domain (see supplemental Table S1 for details). When FimG is disulfide-bonded its mechanical stability raises drastically, up to 440 ± 36 pN, whereas the reduced state has an average unfolding force of 339 ± 27 pN. This represents an increment in the mechanical stability of 101 pN (∼30%) for the oxidized domains.
Figure 2.
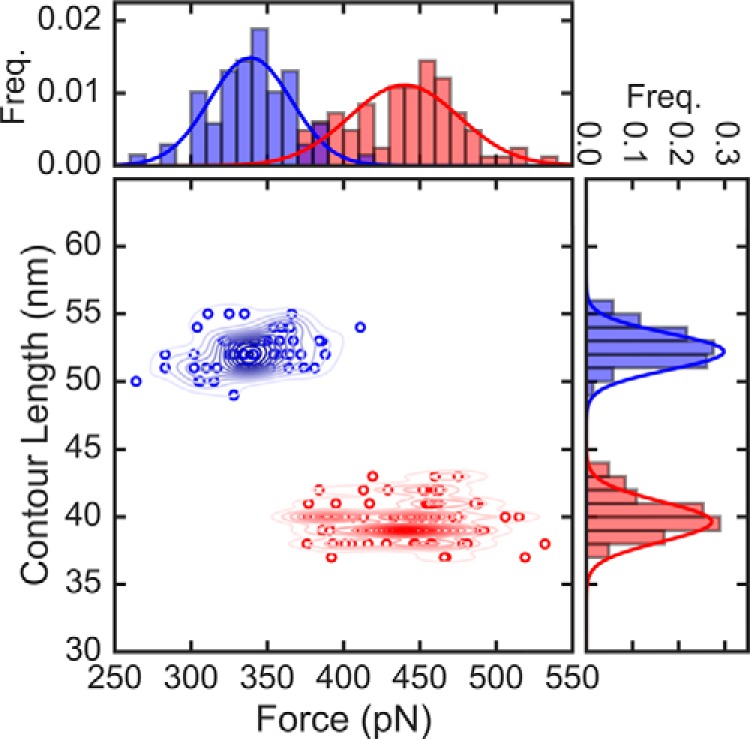
Unfolding forces and contour lengths of FimG from constant force pulling. Scatter plot of force versus contour length with corresponding histograms (top and right, respectively) of the self-complemented FimG. The total number of data points are n = 83 and 69 for oxidized domains and reduced domains, respectively. Lines in the histograms are Gaussian fits to the data. The red and blue colors denote the oxidized and reduced states, respectively. Contour lines in the scatter plot were generated using kernel density estimates.
Decrease in the mechanical stability of titin upon disulfide bond formation
In the case of the I91G32C/A75C titin domain (Fig. 3A), the oxidized form displays an average extension of 12 ± 1 nm (mean ± S.D.) and an unfolding force of 176 ± 32 pN (mean ± S.D.), whereas the reduced form of the protein has an average contour length of 29 ± 1 nm and a mechanical stability of 191 ± 31 pN. The contour lengths are in good agreement with previously reported values (see details in supplemental Table S1). With respect to the forces, on average, there is a subtle decrease (15 pN or 8%) in the mechanical stability of the protein upon disulfide bonding. A value of 15 pN difference between the oxidized and reduced forms can be considered within the experimental uncertainty of measured forces.
Figure 3.
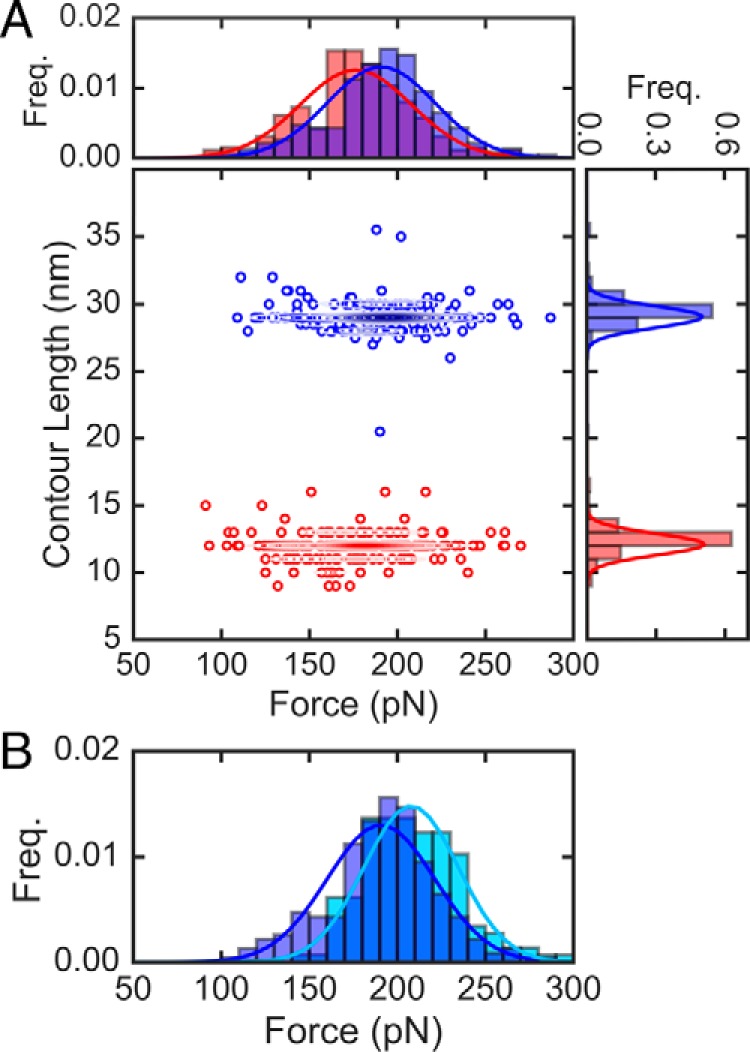
Unfolding forces and contour lengths of titin I91 from constant force pulling. A, scatter plot of force versus contour length with corresponding histograms (top and right, respectively) of the (I91G32C/A75C)8 polyprotein. Lines in the histograms are Gaussian fits to the data. The red and blue colors denote the oxidized and reduced states, respectively. The total number of data points are n = 262 and 232 for oxidized domains and reduced domains, respectively. Contour lines in the scatter plot were generated using kernel density estimates. B, comparison of unfolding forces for I91G32C/A75C (blue) and I91 WT (light blue), with n = 146.
We also compared the unfolding force between the reduced I91G32C/A75C form and the WT form. The value we have obtained for the unfolding force of WT 191 is 208 ± 27 pN, which is consistent with previous studies (9). In Fig. 3B we show the comparison of unfolding forces for I91 WT and the reduced form of the I91G32C/A75C double mutant, which are also within experimental uncertainty. This is indicative that these mutations have little effect on the overall mechanical stability of the protein, a non-trivial result considering that mutations are on average not neutral for the thermodynamic stability (33). Although the pairwise comparison of I91 mechanical stability between oxidized/reduced and reduced/WT forms yields a subtle effect on mechanical stability within the experimental uncertainty, the comparison between oxidized I91G32C/A75C and WT I91 forms yields a more significant decrease on stability of 15%, which is beyond the experimental uncertainty.
Hence, depending on the context in which they appear, disulfide bonds seem to act differently. Although the mechanical stability in I91 decreases when comparing oxidized I91G32C/A75C and WT form, for the FimG domain the unfolding force raises considerably for the oxidized state. Many different contributions to protein energetics may be required to explain the context dependence of the effects of disulfides on mechanical stability. In particular, the position where the disulfide is located within the protein topology seems to be of key importance. In the case of titin Ig domains, disulfides are cryptic and are located in the middle region of the domain. This is consistent with other titin domains for which the structure has been solved (34). On the other hand, for FimG the disulfide is found at the very start of the sequence. This observation is reminiscent of previous findings on the de novo designed protein Top7 (35, 36). For this synthetic protein the disulfide-bonded cysteines positioned early in the sequence result in a 23% increase in the mechanical stability for the oxidized state (172 pN) relative to the reduced one (140 pN). However, this is a novel observation in a natural protein.
Molecular simulations explain FimG results via a shift in unfolding mechanism
To gain more detailed insight on this result we have run molecular dynamics simulations of the oxidized and reduced FimG using a coarse-grained, structure-based (i.e. Gō) simulation model (29), used many times before in the context of atomic force microscopy pulling (28, 30–32) (see “Experimental procedures” for details). Although coarse-grained models sacrifice much of the structural detail from atomistic molecular dynamics, recent work has come to support the central assumption of Gō models. As it turns out, folding mechanisms in atomistic simulations are determined by native contacts, which are the only energetically favorable interactions in Gō models. Given the computational efficiency of coarse-grained models we have been able to run 300 simulation trajectories at constant pulling speed for FimG and the I91 mutant, allowing for good statistical estimates of average unfolding forces.
We show representative force-extension curves from the pulling simulations of FimG in Fig. 4A. Unfolding forces were estimated as the highest value of the force concomitant with the unfolding transition path, as identified by a decrease in the fraction of native contacts (Q). In line with the experiments, we find that in the case of the oxidized protein, the unfolding force increases by ∼30% (Fig. 4B). Examining the simulations we find that this effect is due to a change in the unfolding pathway that the protein follows during unfolding. In the case of the reduced state unfolding occurs via the rupture of hydrogen bonds between β-strands 1 and 2, whereas in the case of the oxidized state these are stapled together by a disulfide bond. Necessarily, unfolding occurs via a higher energy route (i.e. rupture from the C terminus as opposed to the N terminus, see Fig. 4C). It is known that altering the unfolding pathway changes the rupture sequence of hydrogen bonds thus changing the unfolding force (27).
Figure 4.
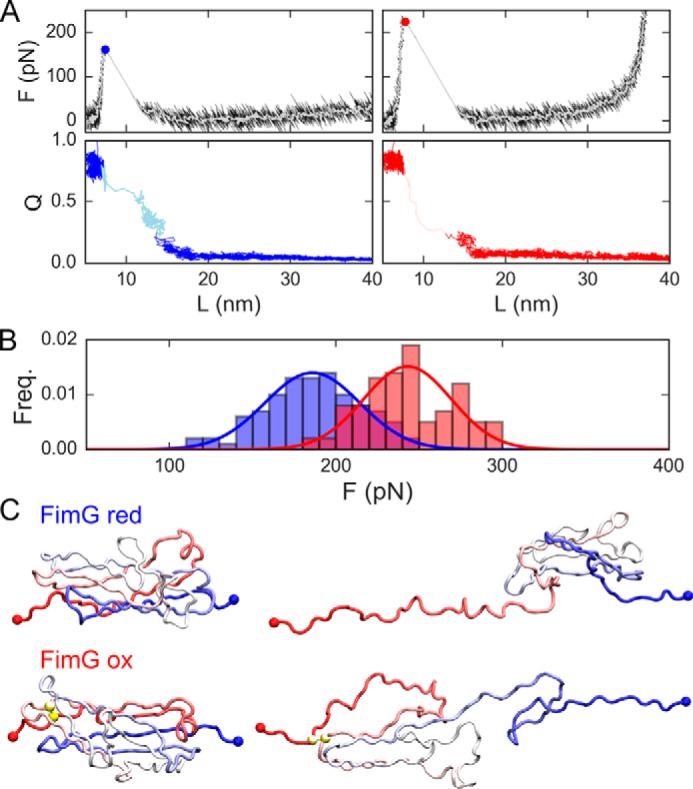
Coarse-grained simulation results for oxidized and reduced FimG. A, force (top) and fraction of native contacts (Q, bottom) versus the protein extension (L) for representative trajectories of the model for the oxidized and reduced protein (blue and red, respectively). White lines are running averages of the forces. Filled circles represent the estimated unfolding force. In the QvsL plots, the lighter shade of color indicates the unfolding transition path. B, cumulative frequencies of the unfolding forces. C, representative snapshots from the simulations for the folded state (left) and the unfolded state reached right after the unfolding force peak (right) for reduced (top) and oxidized (bottom) FimG models. In the latter, yellow spheres indicate the position of disulfide bonded cysteines.
The small decrease in unfolding force for titin is not captured by the simulation model
This simple explanation based on the protein topology and the restrictions imposed by the disulfide bonds does not explain the somewhat smaller (15 pN or 8%) decrease observed for unfolding forces of the titin Ig domain. Simulations using the same coarse-grained model for I91G22C/A75C suggest that unfolding forces remain largely unchanged upon disulfide bonding (see Fig. 5), as expected for a structure-based model where the force-bearing region of the protein (i.e. the “mechanical clamp”) remains uncompromised by the presence of the disulfide bond.
Figure 5.
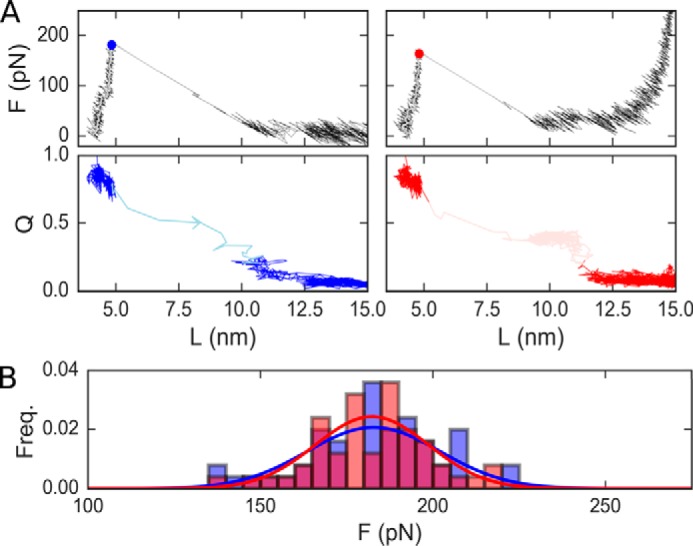
Coarse-grained simulation results for oxidized and reduced titin I91G32C/A75C. A, force/Q versus extension plots for the reduced (blue) and oxidized (red) forms of the protein. Circles mark the unfolding force. Transition paths are shown in a lighter shade of each color. B, cumulative histograms of the simulated unfolding forces for the oxidized (red) and reduced forms (blue).
Discussion
Overall, these results suggest that disulfide bonds in proteins act in a context-dependent manner rather than simply as mechanical lockers. When the disulfide bond compromises the unfolding pathway, transitions will necessarily occur via higher energy routes (35). This is akin to the effect of disulfide bonds connecting parts of the protein that unfold before the rate-limiting step, which were found to slow down the overall unfolding process (37). However, in the absence of such hard constraint, the effects in the energetics will be more subtle, depending on both enthalpic and entropic contributions, as happens in the case of thermodynamic stability (2, 5, 38, 39).
In the case of titin we find that there is a modest reduction of the mechanical stability of the protein, which may be too subtle for a simple structure-based simulation model to capture. We note that atomistic simulations of the I1 domain of titin showed an increase of the unfolding when a disulfide bond was present (40), a different trend to that observed here for the I91 domains of titin. Hence, higher order effects are required to explain the smaller changes identified for the titin Ig domain. One possibility is that side chain interactions in the core of I91, which have also been shown to have a role in the protein mechanical stability (41), are contributing in different ways in the oxidized and reduced forms, although the mutated residues are far from the A–B and E–F loops. Another contribution will come from the constrain introduced in the native state vibrational entropy by a disulfide bond (39), which has been found to affect the subpicosecond to 100-ps dynamics of proteins (38). Another possibility is the presence of mechanical prestress in the disulfide-bonded titin (10). Measuring the dihedral angles of the disulfide bonded cysteines in our model for I91G32C/A75C produced with the Modeler software, we find that the χ1 and χ′2 angles are statistically highly unlikely. It has been previously reported that the spatial configuration of the disulfide bonds of intrachain Ig domains could exert a tensile stress on the order of 100 pN (42). The type of disulfide bond could affect in a different manner the mechanical stability of the protein. The disulfide bond in FimG involves the A and B β-strands in a “staple” configuration, whereas disulfides in the titin I91G32C/A75C are cryptic and of the “hook” type. Indeed for all other titin Ig-like domains solved experimentally we also find that disulfide bonds are cryptic and “spiral.” There may exist a correlation between the disulfide bond configuration, its position in the sequence, and its importance remodeling the unfolding pathway.
Future work involving a systematic analysis of alternative locations of disulfide bonds across the β-strands in different proteins will enable to establish a consistent theory about the regulation of the mechanical stability induced by disulfide bonds. Finally, these experiments stress the importance of considering disulfide bonds both computationally and experimentally when studying the mechanical properties of proteins, as they regulate the mechanical stability beyond their nature as simple covalent bonds.
Experimental procedures
Protein expression and purification
Genes of the (I91G32C/A75C)8 titin segment and FimG protein were codon-optimized for expression in Escherichia coli cells (see sequences in supplemental information). Plasmid pQE80L-(I91G32C/A75C)8 and the I91 wild-type gene were a kind gift from Julio M. Fernández (Columbia University). FimG was bought from Life Technologies and designed as a self-complemented variant lacking its N-terminal donor-strand and carrying the sequence of the donor β-strand of the protein FimF connected through a 4-amino acid linker (DNKQ) in the C-terminal end. We have cloned FimG into Qiagen pQE80L vector and transformed both plasmids onto E. coli BL21 from Merck Millipore. Bacterial cultures were incubated overnight in LB medium at 37 °C. To induce protein expression 1 mm isopropyl 1-thio-β-d-galactopyranoside (IPTG) was added when the optical density of the culture was higher than 0.6. Bacterial culture was centrifuged at 4,000 × g during 20 min. After centrifugation, cell pellets were lysed by means of a French pressure cell press and the His6-tagged proteins were loaded onto His GraviTrap affinity column (GE Healthcare). We promoted the oxidation of proteins by adding 0.3–0.5% H2O2 overnight at room temperature. Reduction was triggered by adding 1 mm DTT overnight for FimG and 5 mm DTT for I91G32C/A75C for 1 h at 50 °C. After incubation, the proteins were purified by size exclusion chromatography using a Superdex 200HR column (GE Healthcare). The buffer used was 10 mm HEPES, pH 7.2, 150 mm NaCl, 1 mm EDTA at pH 7.0. The purified proteins were checked with SDS-PAGE.
Single-molecule force spectroscopy experiments
smFS experiments were carried out on a commercial Atomic Force Spectrometer (AFS-1) from Luigs and Neumann. We used cantilever models MLCT and OBL-10 of silicon nitride from Bruker. The cantilevers have been calibrated using the equipartition theorem with a typical spring constant of 0.02 N m−1 for MLCT and 0.006 N m−1 for OBL-10. The pulling speed and amplitude in force-extension mode were 400 nm/s and 400 nm, respectively. The buffer used in the experiment was the same as for the purification mentioned above. The experiments were performed by placing 10–20 μl of protein solution at ∼0.2 mg ml−1 onto a gold-covered slide. A custom-written software in Igor Pro 6.37 (Wavemetrics) was used to collect and analyze the traces.
Coarse grained, structure-based model
Simulations of the self-complemented FimG and the titin I91G32C/A75C domain using the Karanicolas and Brooks structure-based (i.e. Gō) model (29) were preformed. In the simulations, the protein geometry is reduced to the Cα trace and the solvent degrees of freedom are not considered, resulting in a great computational efficiency. The potential energy is defined as the following.
| (Eq. 1) |
In Equation 1, Vbond and Vangle are native-centric harmonic terms for bonds and angles, respectively, whereas Vdihedral is based on statistical preferences for torsion angles in the Protein Data Bank (29). The non-bonded contribution is favorable for pairs of residues that are in contact in the reference (i.e. experimental) structure. Two residues i and j are defined to be in contact when any pair of atoms closer than 5 Å, and their contribution to the energy is,
| (Eq. 2) |
where σij is the distance between the Cα of residues i and j in the reference structure, rij is the same distance but in the instantaneous configuration, and ϵij is the strength of the pairwise interaction (29).
Because experimental structures are not available for neither the FimG self-complemented variant nor the titin I91G32C/A75C we used molecular models produced with the Modeler (43) software in both cases. In the case of FimG we used as reference the usher plus pilus tip experimental structure (Protein Data Bank code 4J3O (44)), selecting the relevant atoms from FimG and those from FimF that complement FimG, and then adding the linker. For titin, the WT structure (Protein Data Bank code 1tit) was used as a template and Cys mutations were introduced.
Molecular simulations
Simulations were run using an modified version of Gromacs 4.0.5 (45). We propagated the dynamics at a constant temperature of 300 K using a Langevin integrator with a time-step of 10 fs, and a friction constant of 0.2 ps−1. Pulling experiments were mimicked using the pull-code from Gromacs, by defining the pulling groups in the protein ends and pulling in a single dimension at a constant speed of 0.1 nm/ps and with a harmonic force constant of 20 kJ/mol/nm2. The total number of simulation runs at constant pulling speed was 50 for the I91 domain and 100 for FimG, both for the reduced and the disulfide bonded variants. In the latter disulfide bonds were modeled as an additional bond with the same harmonic constant as Cα–Cα bonds within the model.
Analysis of the simulations
For each of the simulation trajectories we produced force-extension plots. Additionally, we calculated the fraction of native contacts Q (28). Force peaks were identified automatically using the find_peaks_cwt algorithm from the signal processing library from SciPy (https://www.scipy.org).7 The unfolding force was estimated as the maximum force peak identified in a bracket of the time starting 5 ps before the start of the transition path and finishing at the end of the transition path. We define transition paths as the parts of the trajectory that connect two dividing lines without recrossings (QF and QU). These were set at QF = 0.9 and QU = 0.5 for titin I91 and QF = 0.8 and QU = 0.2 for FimG.
Author contributions
R. P. -J. and D. D. conceived and supervised research. A. M., A. A.-C., and S. P. performed experiments. M. F. contributed to protein expression and purification. D. D. performed the simulations. A. M., A. A. C., and D. D. analyzed the experiments. All the authors contributed to writing the paper and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
Plasmid pQE80L-(I91–32/75)8 was a present from Julio M. Fernández (Columbia University). D. D. wishes to thank Robert B. Best for the use of computational tools.
This work was supported in part by Ministry of Economy and Competitiveness (MINECO) Grant BIO2016-77390-R, BFU2015-71964 (to R. P. -J.), and CTQ2015-65320-R (to D. D.), and European Commission grant CIG Marie Curie Reintegration program FP7-PEOPLE-2014 (to R. P. -J.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- smFS
- single-molecule force spectroscopy
- pN
- piconewton.
References
- 1. Fass D. (2012) Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 41, 63–79 [DOI] [PubMed] [Google Scholar]
- 2. Betz S. F. (1993) Disulfide bonds and the stability of globular proteins. Protein Sci. 2, 1551–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flory P. J. (1956) Theory of elastic mechanisms in fibrous proteins. J. Am. Chem. Soc. 78, 5222–5235 [Google Scholar]
- 4. Doig A. J., and Williams D. H. (1991) Is the hydrophobic effect stabilizing or destabilizing in proteins? J. Mol. Biol. 217, 389–398 [DOI] [PubMed] [Google Scholar]
- 5. Santiveri C. M., León E., Rico M., and Jiménez M. A. (2008) Context-dependence of the contribution of disulfide bonds to β-hairpin stability. Chemistry 14, 488–499 [DOI] [PubMed] [Google Scholar]
- 6. Beeser S. A., Oas T. G., and Goldenberg D. P. (1998) Determinants of backbone dynamics in native BPTI: cooperative influence of the 14–38 disulfide and the Tyr35 side-chain 1. J. Mol. Biol. 284, 1581–1596 [DOI] [PubMed] [Google Scholar]
- 7. Hogg P. J. (2003) Disulfide bonds as switches for protein function. Trends Biochem. Sci. 28, 210–214 [DOI] [PubMed] [Google Scholar]
- 8. Crampton N., and Brockwell D. J. (2010) Unravelling the design principles for single protein mechanical strength. Curr. Opin. Struct. Biol. 20, 508–517 [DOI] [PubMed] [Google Scholar]
- 9. Ainavarapu S. R., Brujic J., Huang H. H., Wiita A. P., Lu H., Li L., Walther K. A., Carrion-Vazquez M., Li H., and Fernandez J. M. (2007) Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 92, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou B., Baldus I. B., Li W., Edwards S. A., and Gräter F. (2014) Identification of allosteric disulfides from prestress analysis. Biophys. J. 107, 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wiita A. P., Ainavarapu S. R., Huang H. H., and Fernandez J. M. (2006) Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci. U.S.A. 103, 7222–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liang J., and Fernández J. M. (2011) Kinetic measurements on single-molecule disulfide bond cleavage. J. Am. Chem. Soc. 133, 3528–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia-Manyes S., Kuo T. L., and Fernández J. M. (2011) Contrasting the individual reactive pathways in protein unfolding and disulfide bond reduction observed within a single protein. J. Am. Chem. Soc. 133, 3104–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosuri P., Alegre-Cebollada J., Feng J., Kaplan A., Inglés-Prieto A., Badilla C. L., Stockwell B. R., Sanchez-Ruiz J. M., Holmgren A., and Fernández J. M. (2012) Protein folding drives disulfide formation. Cell 151, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin M., Wang W., and Thirumalai D. (2015) Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 112, 11241–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baldus I. B., and Gräter F. (2012) Mechanical force can fine-tune redox potentials of disulfide bonds. Biophys. J. 102, 622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ainavarapu S. R., Wiita A. P., Huang H. H., and Fernandez J. M. (2008) A single-molecule assay to directly identify solvent-accessible disulfide bonds and probe their effect on protein folding. J. Am. Chem. Soc. 130, 436–437 [DOI] [PubMed] [Google Scholar]
- 18. Kahn T. B., Fernández J. M., and Perez-Jimenez R. (2015) Monitoring oxidative folding of a single protein catalyzed by the disulfide oxidoreductase DsbA. J. Biol. Chem. 290, 14518–14527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perez-Jimenez R., Li J., Kosuri P., Sanchez-Romero I., Wiita A. P., Rodriguez-Larrea D., Chueca A., Holmgren A., Miranda-Vizuete A., Becker K., Cho S. H., Beckwith J., Gelhaye E., Jacquot J. P., et al. (2009) Diversity of chemical mechanisms in thioredoxin catalysis revealed by single-molecule force spectroscopy. Nat. Struct. Mol. Biol. 16, 890–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grützner A., Garcia-Manyes S., Kötter S., Badilla C. L., Fernandez J. M., and Linke W. A. (2009) Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys. J. 97, 825–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perez-Jimenez R., Alonso-Caballero A., Berkovich R., Franco D., Chen M. W., Richard P., Badilla C. L., and Fernandez J. M. (2014) Probing the effect of force on HIV-1 receptor CD4. ACS Nano 8, 10313–10320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen W. J., Phan G., Hultgren S. J., and Waksman G. (2013) Dissection of pilus tip assembly by the FimD usher monomer. J. Mol. Biol. 425, 958–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vetsch M., Puorger C., Spirig T., Grauschopf U., Weber-Ban E. U., and Glockshuber R. (2004) Pilus chaperones represent a new type of protein-folding catalyst. Nature 431, 329–333 [DOI] [PubMed] [Google Scholar]
- 24. Rief M., Oesterhelt F., Heymann B., and Gaub H. E. (1997) Single molecule force spectroscopy on polysaccharides by atomic force microscopy. Science 275, 1295–1297 [DOI] [PubMed] [Google Scholar]
- 25. Jobst M. A., Milles L. F., Schoeler C., Ott W., Fried D. B., Bayer E. A., Gaub H. E., and Nash M. A. (2015) Resolving dual binding conformations of cellulosome cohesin-dockerin complexes using single-molecule force spectroscopy. Elife 4, e10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y., Lang P., and Linke W. A. (2016) Titin stiffness modifies the force-generating region of muscle sarcomeres. Sci. Rep. 6, 24492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perez-Jimenez R., Garcia-Manyes S., Ainavarapu S. R., and Fernandez J. M. (2006) Mechanical unfolding pathways of the enhanced yellow fluorescent protein revealed by single molecule force spectroscopy. J. Biol. Chem. 281, 40010–40014 [DOI] [PubMed] [Google Scholar]
- 28. Graham T. G., and Best R. B. (2011) Force-induced change in protein unfolding mechanism: discrete or continuous switch? J. Phys. Chem. B 115, 1546–1561 [DOI] [PubMed] [Google Scholar]
- 29. Karanicolas J., and Brooks C. L. 3rd. (2002) The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci. 11, 2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Best R. B., and Hummer G. (2008) Protein folding kinetics under force from molecular simulation. J. Am. Chem. Soc. 130, 3706–3707 [DOI] [PubMed] [Google Scholar]
- 31. Best R. B., Paci E., Hummer G., and Dudko O. K. (2008) Pulling direction as a reaction coordinate for the mechanical unfolding of single molecules. J. Phys. Chem. B 112, 5968–5976 [DOI] [PubMed] [Google Scholar]
- 32. de Sancho D., and Best R. B. (2016) Reconciling intermediates in mechanical unfolding experiments with two-state protein folding in bulk. J. Phys. Chem. Lett. 7, 3798–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tokuriki N., and Tawfik D. S. (2009) Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604 [DOI] [PubMed] [Google Scholar]
- 34. von Castelmur E., Marino M., Svergun D. I., Kreplak L., Ucurum-Fotiadis Z., Konarev P. V., Urzhumtsev A., Labeit D., Labeit S., and Mayans O. (2008) A regular pattern of Ig super-motifs defines segmental flexibility as the elastic mechanism of the titin chain. Proc. Natl. Acad. Sci. U.S.A. 105, 1186–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sharma D., Perisic O., Peng Q., Cao Y., Lam C., Lu H., and Li H. (2007) Single-molecule force spectroscopy reveals a mechanically stable protein fold and the rational tuning of its mechanical stability. Proc. Natl. Acad. Sci. U.S.A. 104, 9278–9283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuhlman B., Dantas G., Ireton G. C., Varani G., Stoddard B. L., and Baker D. (2003) Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 [DOI] [PubMed] [Google Scholar]
- 37. Clarke J., and Fersht A. R. (1993) Engineered disulfide bonds as probes of the folding pathway of barnase: increasing the stability of proteins against the rate of denaturation. Biochemistry 32, 4322–4329 [DOI] [PubMed] [Google Scholar]
- 38. Ishikawa H., Kim S., Kwak K., Wakasugi K., and Fayer M. D. (2007) Disulfide bond influence on protein structural dynamics probed with 2D-IR vibrational echo spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 104, 19309–19314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tidor B., and Karplus M. (1993) The contribution of cross-links to protein stability: a normal mode analysis of the configurational entropy of the native state. Proteins 15, 71–79 [DOI] [PubMed] [Google Scholar]
- 40. Gao M., Wilmanns M., and Schulten K. (2002) Steered molecular dynamics studies of titin I1 domain unfolding. Biophys. J. 83, 3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Best R. B., Fowler S. B., Herrera J. L., Steward A., Paci E., and Clarke J. (2003) Mechanical unfolding of a titin Ig domain: structure of transition state revealed by combining atomic force microscopy, protein engineering and molecular dynamics simulations. J. Mol. Biol. 330, 867–877 [DOI] [PubMed] [Google Scholar]
- 42. Anjukandi P., Dopieralski P., Ribas-Arino J., and Marx D. (2014) The effect of tensile stress on the conformational free energy landscape of disulfide bonds. PLoS ONE 9, e108812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Webb B., and Sali A. (2016) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1.–5.6.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Phan G., Remaut H., Wang T., Allen W. J., Pirker K. F., Lebedev A., Henderson N. S., Geibel S., Volkan E., Yan J., Kunze M. B., Pinkner J. S., Ford B., Kay C. W., Li H., Hultgren S. J., Thanassi D. G., and Waksman G. (2011) Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 474, 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.