

Abstract
Apolipoprotein B mRNA-editing enzyme catalytic subunit 3 (APOBEC-3) enzymes are cytidine deaminases that are broadly and constitutively expressed. They are often up-regulated during carcinogenesis and candidate genes for causing the major single-base substitution in cancer-associated DNA mutations. Moreover, APOBEC-3s are involved in host innate immunity against many viruses. However, how APOBEC-3 mutational activity is regulated in normal and pathological conditions remains largely unknown. Heat shock protein levels are often elevated in both carcinogenesis and viral infection and are associated with DNA mutations. Here, using mutational analyses of hepatitis B virus (HBV), we found that Hsp90 stimulates deamination activity of APOBEC-3G (A3G), A3B, and A3C during co-expression in human liver HepG2 cells. Hsp90 directly stimulated A3G deamination activity when the purified proteins were used in in vitro reactions. Hsp40, -60, and -70 also had variable stimulatory effects in the cellular assay, but not in vitro. Sequencing analyses further demonstrated that Hsp90 increased both A3G cytosine mutation efficiency on HBV DNA and total HBV mutation frequency. In addition, Hsp90 shifted A3G's cytosine region selection in HBV DNA and increased A3G's 5′ nucleoside preference for deoxycytidine (5′-CC). Furthermore, the Hsp90 inhibitor 17-N-allylamino-17-demethoxygeldanamycin dose dependently inhibited A3G and A3B mutational activity on HBV viral DNA. Hsp90 knockdown by siRNA or by Hsp90 active-site mutation also decreased A3G activity. These results indicate that heat shock proteins, in particular Hsp90, stimulate APOBEC-3–mediated DNA deamination activity, suggesting a potential physiological role in carcinogenesis and viral innate immunity.
Keywords: cancer; cytidine deaminase; heat shock protein 90 (Hsp90); hepatitis B virus (HBV, Hep B); mutagenesis; APOBEC-3G; cofactor
Introduction
Heat shock proteins (Hsps)2 are constitutively expressed molecular chaperones that guide the normal folding, intracellular disposition, and proteolytic turnover of many of the key regulators of cell growth and survival (1, 2). Hsps are expressed at high levels in a wide range of tumors and are closely associated with resistance to therapy (3–5). The chaperone functions of Hsps, especially Hsp90, are often subverted during oncogenesis, allowing mutant proteins to retain or even gain function (1, 2, 6). Many mutations associated with human disease, in particular cancer, are predicted to affect protein stability (7). Hsp stabilization of these mutant proteins is the major mechanism by which cells counteract the potentially deleterious effects of misfolded proteins. The Hsp family members play overlapping, essential roles in tumor growth both by promoting autonomous cell proliferation and by inhibiting apoptotic pathways (8). The ATP-driven heat shock protein 90 (Hsp90) is especially important in this process.
Hsp90 can constitute as much as 5% of total protein in transformed cells, and the increased heat shock levels correlate with poor prognosis in many cancer types (9). Together with other Hsp co-chaperones, Hsp90 catalyzes folding of many proteins that regulate cell growth and development (10). These Hsp90-mediating kinases, transcription factors, and ubiquitin ligases often play critical roles in cancer. Hsp90 allows cancer cells to harbor potentially lethal mutations and continue to propagate (11, 12). Much evidence using Hsp90 inhibitors suggests that inhibiting Hsp90 causes oncogene depletion and inhibition of tumor growth (13, 14).
Recent advances in next-generation sequencing technologies have revealed the existence of hundreds to thousands of mutations in most cancers (15). The predominant type of genetic alteration found in cancer is a somatic single-base substitution (15). Aberrant APOBEC-3B (A3B) deaminase activity has been suggested as a likely cause of single-base substitution in cancer because A3B is often up-regulated in cancer and its preferred target sequence (TC) is frequently mutated and clustered in multiple human cancers (16–20). Both A3A and A3B can actually induce base substitutions in the DNA genome when transfected into cells (21–23). APOBEC-3s (A, B, C, D, F, G, and H) are zinc-dependent cytidine deaminases that catalyze the conversion of cytidine to uracil in single-stranded DNA (24, 25). These enzymes are capable of introducing C-to-U mutations in viral cDNA intermediates that manifest as G-to-A mutations in proviral genomes (26–28). They have also been implicated in inhibiting the replication of many other DNA-based “parasites,” including transposable elements (28). The mutational activities of APOBEC-3s play a critical role in minimizing viral infectivity as part of the host innate viral defense (24).
It is important to note that most APOBEC-3s are expressed broadly and constitutively (29). Some APOBEC-3s also can be further up-regulated, such as A3A by interferon-α (up to 100-fold) (30); A3B by non-canonical NF-κB activation and HPV infection (31, 32); and A3C, A3D, A3F, A3G, and A3H by the combination of T cell activation and HIV-1 infection (33). Aberrant regulation of APOBEC-3s may result in cancer formation (16, 18, 19). These results indicate that APOBEC-3 proteins need to be stringently controlled under physiological conditions to prevent them from harming the host while still remaining ready to respond when needed as part of the innate immune system. However, how APOBEC-3s are physiologically regulated under normal or pathologic states remains largely unknown, especially regarding the role of host protein factors. Hsps are a family of factors known to be elevated in cancer and associated with DNA mutation. However, whether APOBEC-3–induced mutation activity is related to Hsps in normal and diseased states is not known.
In mutation analyses of HBV viruses from chronically infected cirrhotic liver, it has been shown that APOBEC-3 mutated up to 86% of cytosines in the minus-strand of HBV DNA, whereas the total mutated HBV population was only 2–35% (34). This result demonstrates that APOBEC-3s selectively mutated up to 35% of HBV viral DNA, but up to 86% of cytosines within the selectively mutated HBV DNAs were deaminated, indicating APOBEC-3's high-efficiency mutational activity in selected molecules in vivo. However, this efficacy has not been observed in vitro (25). The contrast between in vivo and in vitro mutational activity also suggests the presence of host factor(s) in vivo that enable APOBEC-3 proteins to be highly efficient mutational agents. Because increased Hsp expression under conditions such as viral infection and carcinogenesis is associated with increased APOBEC-3–induced DNA mutational activity, Hsps may potentially be involved in regulating APOBEC-3's deamination activity. In this paper, we investigated the potential role of Hsps on APOBEC-3s mutation activity, utilizing a HBV cellular replication system in HepG2 cells. We found that Hsp90 stimulates APOBEC-3G mutation activity, suggesting a potential modulatory role in vivo.
Results
APOBEC-3 DNA mutational activity in HBV can be quantitatively determined by a combination of differential DNA denaturation-PCR (3D-PCR) and primer extension analyses
HBV replication in the human liver cell line HepG2 cells induced by transfecting a HBV genome encoding plasmid has been commonly used to mimic the HBV viral process in vivo (35, 36) and has been used to investigate APOBEC-3 mutational activity on HBV DNA (34, 37). HBV viruses replicate through a capsid intermediate that contains pregenomic RNA, core protein, HBV polymerase, and various host proteins (35). HBV DNA mutations induced by APOBEC-3s occur within the capsid when pregenomic HBV RNA is converted by HBV polymerase into partial double-stranded DNA through cDNA. We utilized the same cellular system to investigate APOBEC-3 mutational activities on HBV viral genomic DNA. Briefly, a HBV genomic DNA-encoding plasmid was co-transfected with plasmids encoding APOBEC-3s and Hsps into HepG2 cells in a quantitative ratio of 4:1.5:0.5 (Hsp/APOBEC-3/HBV). The resultant HBV DNAs were extracted from the viral capsid, and the frequency of DNA mutations was analyzed. The plasmid ratio was empirically determined to ensure that each HepG2 cell transfected with HBV had co-expression of APOBEC-3 and Hsp. Although HepG2 cells have relatively low transfection efficiency, HBV capsids generated from 6-well plates were enough for HBV DNA mutation analyses. The HBV-induced DNA mutation results described below demonstrate that APOBEC-3s and Hsps were present in the viral capsids where the DNA mutation occurred.
The C-to-T mutations induced by APOBEC-3s result in AT-rich DNAs that have a lower denaturing temperature for PCR and can be selectively amplified by 3D-PCR (38). Sequencing analyses of cloned DNA after the enrichment by 3D-PCR have been generally used to determine APOBEC-3 mutational activity (38). However, the clonal sequencing method is labor-intensive and is not sensitive when HBV mutation levels are low. Consequently, it is difficult to accurately compare different protein effects on HBV-induced DNA mutation by this method. Another approach is the primer extension method that uses a specific primer to anneal to the PCR gene amplicons, followed by an extension reaction in the presence of ddGTP, resulting in variable length oligonucleotides due to extension stoppage at cytosines. This method has been used as a very sensitive method for quantitating apoB mRNA C-to-T mutation frequency at cytosine site 6666 (39). To quantitatively determine APOBEC-3's mutational activity, we first enriched the HBV-mutated DNA by 3D-PCR and then determined the mutation rates in the 3D-PCR mixture by primer extension analyses at selected HBV DNA cytosine sites. After a series of trials, we found that APOBEC-3's mutational activity on HBV DNA can be sensitively quantitated by this combination of 3D-PCR and primer extension at three cytosine sites, 1674, 1664, and 1453 (named pe1674, pe1664, and pe1453, respectively).
Utilizing this quantitation method, we first investigated the mutational activities of APOBEC-3A, -3B, -3C, -3F, -3G, and -3H on HBV DNA by their co-transfection with an HBV plasmid in HepG2 cells to select suitable APOBEC-3s for further evaluating the effect of Hsps. After a 2-day transfection, HBV DNA generated from the intracellular HBV capsids was extracted and amplified by PCR with a standard denaturing temperature of 94 °C first. HBV DNA mutations were then further amplified by 3D-PCR at various denaturing temperatures, followed by primer extension at cytosine sites 1674, 1664, and 1453. As shown in Fig. 1 (A and B), APOBEC-3s were expressed at variable levels, but HBV DNA was uniformly amplified in the 94 °C PCR and 90 °C 3D-PCR for all APOBEC-3 treatments. However, different amplification levels were observed in the 88 or 86 °C 3D-PCRs, reflecting potential mutational differences after treatment.
Figure 1.

APOBEC-3 mutational activity and quantitation using HBV. An APOBEC-3 plasmid was co-transfected with HBV into HepG2 cells. Cells were lysed after 2 days of transfection, and HBV DNA was extracted from HBV capsids in the cell lysate. HBV DNA was amplified by PCR, followed by DNA mutant enrichment using 3D-PCR. APOBEC-3s' mutational activities on HBV were quantitatively determined by primer extension. A, APOBEC-3s' protein expression analyses by Western blotting used an antibody against the FLAG tag in A3A and A3B or the HA tag in A3C, A3F, and A3G. B, PCR and 3D-PCR detection of HBV at different denaturing temperatures after treatment of APOBEC-3s. C–E, HBV mutational levels in the PCR or 3D-PCR were determined by primer extension reactions with primers specific for cytosine at sites 1674, 1664, and 1453, named pe1674, pe1664, and pe1453, respectively. The products of primer extension were separated by a 8% sequencing gel and quantitated with a PhosphorImager. The cytosine positions on the right of gels are numbered according to their sites in the HBV genome reference V01460.1 and are in reverse order relative to the oligonucleotide length. The mutation frequency was the percentage of these oligonucleotides run through the targeted cytosine due to C-to-T mutation divided by the total product counts of the lane. The data are also presented graphically on the right. Blank, background control without APOBEC-3 co-transfection. Vector, another background control with mock vector.
After removal of dNTP through PCR purification, the HBV DNA mutation frequencies in the PCR or 3D-PCR amplicons were analyzed by primer extension that actually was a PCR with a single sequence-specific primer labeled with 32P in the presence of ddGTP (39). The primer extension stopped when ddGTP was incorporated at the target site cytosine. When the cytosine at the target site was mutated, the primer extension continued to the next cytosine site downstream. As a result, the primer extension reaction generated a group of 32P-labeled oligonucleotides with lengths that varied based on C-to-T mutations starting from the target site. The 32P-labeled oligonucleotides were then separated on a 8% denaturing sequencing gel and quantitated by a PhosphorImager to determine the mutation frequencies at the site using the percentage of these run through the target site (representing these mutated at the target site) over the total extension products.
As shown in Fig. 1C, A3B induced a detectable mutation level (6.4%) in the 94 °C PCR by pe1674. The cytosine at this site has a sequence of TCG, the A3B-preferred sequence (24). The A3B mutation level increased from 6.4% in the 94 °C PCR to 65.8% in the 88 °C 3D-PCR, whereas the mock vector background control increased from 1.2 to 18.4%. These data demonstrate that mutated HBV DNA are enriched by the 88 °C 3D-PCR, and a combination of 88 °C 3D-PCR and primer extension significantly increases the detection sensitivity. In addition to A3B, A3C and A3H also had detectable mutation levels of 27.0 and 22.4% in the 88 °C 3D-PCR (background 18.4%), whereas others were lower. A similar pattern was also observed with the 86 °C 3D-PCR, but the effect for lower mutation levels as with A3H became obscure due to the low amount of amplification seen with the 86 °C 3D-PCR. The mock vector background control of HBV DNA mutation reflected the mutational effect of endogenous APOBEC-3s expressed in HepG2 cells. The endogenous expression levels of APOBEC-3s in HepG2 cells were too low to be detected by antibodies in Western blotting, but their mRNAs were detectable by RT-PCR (data not shown).
As shown in Fig. 1D, A3G-induced mutations were detectable in the 94 °C PCR (4.5%; vector background 1.1%) by the pe1664 assay with a sequence of CCG, the A3G-preferred sequence (24). The A3G mutation rate increased to 53.1% in the 88 °C 3D-PCR, whereas A3B increased to 40.6% and A3C increased to 17.6% (vector background 14.1%). The cytosine mutation rate at site 1453 by pe1453 is shown in Fig. 1E. A3C and A3G had 15.9 and 15.6% mutation levels, respectively, in the 94 °C PCR (background 7.8%). The A3G mutation rate increased to 36.4% in the 88 °C 3D-PCR, whereas the vector background increased from 7.8 to 9.7%. The A3C mutation level also increased from 15.9 to 19.5%. In contrast, A3B-induced mutations were not detectable at the 1453 site. This is because the primer-specific binding region was mutated extensively by A3B (data not shown).
These data demonstrate that HBV-induced DNA mutations can be quantitatively analyzed by a combination of 3D-PCR and primer extension at cytosine site 1674, 1664, or 1453. A3B- and A3G-induced HBV mutations were readily detectable by pe1674 and pe1664 or pe1453, respectively. A3C also had sufficiently high mutation levels for detection. Because all three APOBEC-3s' mutational activity was readily detectable, they were used to investigate the effect of Hsps on APOBEC-3 mutation activity.
To further verify the reliability of the quantitation method, we analyzed the mutational activities of A3B mutants with point mutations against A3B's conserved catalytic sites, including H66R, E68Q, and C97S for domain 1 and H253R, E255Q, and C284S for domain 2. As shown in Fig. 2, the catalytic mutation in domain 2 eliminated A3B activity, whereas the activity with mutants in domain 1 was reduced significantly but not eliminated. The protein expression levels of these mutants were comparable, as shown by the Western blotting, except for H66R, which mainly had a degraded form. These data are consistent with those previously reported (40) and verified that pe1674 can be reliably used for the analyses of A3B mutational activity on HBV.
Figure 2.

Verification of the quantitative method by A3B mutant activity. Plasmids encoding A3B wild type or point mutation were co-transfected with HBV into HepG2 cells. HBV DNA was extracted after 2 days of transfection and was amplified by 94 °C PCR. A, A3B mutational activity on the HBV DNAs was determined by pe1674 with the 94 °C PCR. The products of primer extension were separated by an 8% sequencing gel and quantitated with a PhosphorImager. B, A3B mutational activities reflected by the mutation frequencies are presented graphically. Each bar represents the average of triplicates for each treatment. C, A3B mutant protein expression analyses. A3B mutants were transfected into HepG2 cells, and the whole cell proteins were extracted after 2 days of transfection. A3B in the cell lysates were analyzed by Western blotting with an antibody against native A3B.
Hsps increase A3G mutational activity on HBV in HepG2 cells
Hsp40, Hsp70, Hsp90α, Hsp90β, Hsp104, Hsp60, and Hsp70-Hsp90 organizing protein (HOP) (co-chaperone) were selected to investigate how Hsps affected APOBEC-3 mutational activity in HepG2 cells. Hsps and A3G were first co-transfected with an HBV plasmid in a plasmid ratio of 4:1.5:0.5 (Hsp/A3G/HBV) to ensure co-expression of Hsp and A3G in cells that generated HBV capsids. After 2 days of transient co-transfection, the HepG2 cell lysates were analyzed for protein expression of A3G and Hsps. As shown in Fig. 3A, both A3G and Hsps were readily detected by Western blotting with antibodies against A3G or the FLAG tag in Hsps. The endogenous A3G in HepG2 cells reflected by the blank control was not detectable. The protein expression levels of the transfected A3G and Hsps were comparable among treatments with vector control, Hsp40, Hsp90α, or Hsp90β. However, the co-expression of A3G and Hsp70 resulted in lower A3G and higher Hsp70 expression compared with others. This may be due to incorporation differences of the plasmid amount during co-transfection. It is important to note that the detected A3G or Hsp came from limited transiently transfected cells in the whole plate lysate. The transient cellular transfection efficiency was about 5% when estimated by GFP plasmid under the same conditions (data not shown). So the actual expression levels of A3G and Hsps in these <5% co-transfected cells should be much higher than their endogenous expression in HepG2 cells. This was confirmed by the significant functional change in HBV DNA mutations from the co-expression of A3G with different Hsps as described below.
Figure 3.

Hsp effects on A3G mutational activity using HBV. Hsp-encoding plasmids were co-transfected with A3G and HBV into HepG2 cells. After 2 days of transfection, the cells were lysed, and HBV DNA in the HBV capsids was extracted and analyzed as in Fig. 1. A, the A3G and Hsps protein expression in the cell lysates was analyzed by Western blotting with antibodies against A3G or the FLAG tag in Hsps. B, the HBV DNA amplification by PCR and 3D-PCR at different denaturing temperatures was analyzed by 1% agarose gel. C–E, HBV mutational levels in the PCR or 3D-PCR were determined by primer extension analyses at cytosine site 1664 (pe1664), followed by gel separation and quantitation with a PhosphorImager. The data are presented graphically on the right. Each bar represents the average of triplicates for each treatment. Blank, background control without A3G co-transfection. Vector, A3G + mock vector as the A3G alone control to compare with A3G + Hsps. Asterisks, statistically significant differences comparing treatments with mock vector control: **, 0.01 < p < 0.001; *, 0.05 < p < 0.01. The one-factor ANOVA statistical test was used.
To determine the Hsp effect on A3G activity, HBV DNAs were isolated from intracellular HBV capsids after treatments. HBV DNA was amplified by 94 °C PCR, and the mutated DNA was further enriched by 88 or 87 °C 3D-PCR. HBV DNA mutation levels were then analyzed by primer extension at cytosine site 1664 (pe1664). As shown in Fig. 3C, A3G alone (labeled as Vector to represent A3G + mock vector) had a barely detectable 1.4% mutation frequency in the 94 °C PCR, whereas the blank (no A3G or Hsps) control was 0.7%. A small but detectable increase from 1.4 to 2.6% was observed when Hsp90β was co-expressed with A3G, although overall mutation rates were still relatively low. After enrichment by 3D-PCR, Hsp90β increased A3G mutational activity from 12.6 to 34.2% in 88 °C 3D-PCR or from 11.1 to 35.3% in 87 °C 3D-PCR as shown in Fig. 3, D and E, respectively. An up to 3-fold increase was observed by the co-expression of Hsp90β. The DNA mutation analyses of the serial PCR and 3D-PCR demonstrated that the small Hsp90β stimulatory effect on A3G mutation observed in the initial 94 °C PCR (Fig. 3C) was amplified and confirmed by an up to 3-fold increase after the mutated DNA enrichment process using the 88 °C 3D-PCR or 87 °C 3D-PCR.
Among the tested Hsps, Hsp90β was the most potent at stimulating A3G mutation activity. In addition to Hsp90β, Hsp90α, -104, and -60 also had stimulatory effects. Hsp40 was less effective but still had an up to 2-fold increase in the 88 °C 3D-PCR. A slight increase was also observed for HOP, whereas no effect was observed for Hsp70 with the 88 °C 3D-PCR. Importantly, the relative potencies of the various Hsps were similar using regular PCR or the 3D-PCR at either 88 or 87 °C, confirming the Hsp effect on A3G. In addition, no significant additive effect was observed when other Hsps were added to Hsp90β (data not shown), indicating the unique role of Hsp90 among Hsps. Among 3D-PCRs at different denaturing temperatures, the 88 °C 3D-PCR was the most effective condition for mutated DNA amplification and detection. There was no further improvement by 3D-PCR at either a higher or lower denaturing temperature compared with the preset conditions (Fig. 3E) (data not shown).
Hsp90 affects the A3G mutational efficiency and changes the HBV DNA cytosine mutation selection pattern
Sequencing analysis is a conventional method to verify mutation changes, although it is less sensitive when the mutation frequency is low. The effect of Hsp90 on A3G was further investigated by HBV DNA clonal sequencing analyses. HBV DNA amplification samples from the 88 °C 3D-PCR as in Fig. 3 were cloned into a TA-cloning vector and sequenced using random clones. Sixty clones were selected from A3G alone (A3G + mock vector), and 10 of 60 clones had C-to-T mutations. On the other hand, 13 or 10 of a total of 40 clones had C-to-T mutations for A3G + Hsp90α or A3G + Hsp90β, respectively. The C-to-T mutation frequency at individual cytosine sites was calculated using the collective C-to-T mutation clonal data. The distribution of C-to-T mutations in each clone is graphically presented in Fig. 4 (A–C), and the C-to-T mutation frequency at individual sites is presented in Fig. 4 (D–F). As shown in Fig. 4A, only one clone in the A3G alone group had extensive C-to-T mutations, whereas the rest of the clones only had a few. The Hsp90α treatment not only increased the positive clone number, reflecting the increased mutation frequency, but also the C-to-T mutation number in individual clones, reflecting single mutation event efficiency. The average number of C-to-T mutations per clone increased from 5 to 9.3 by the co-expression of Hsp90α, indicating that Hsp90α affected A3G mutational efficiency (see Table 1). Compared with the A3G alone, A3G + Hsp90α significantly increased the cytosine mutation frequency in the region between 1515 and 1815 nt, as shown in Fig. 4 (E versus D), indicating that Hsp90α altered the A3G target selection in the HBV region. Like Hsp90α, Hsp90β also increased both the number of the positive clones and the C-to-T mutational efficiency in individual clones, with the average C-to-T per clone increasing from 5 to 7.4 (Table 1). However, the overall C-to-T distribution induced by Hsp90β was similar to the A3G alone, as shown in Fig. 4 (F versus D). These data demonstrate that Hsp90 increases A3G mutational activity on HBV DNA and modulates the A3G selection of cytosines in the region, suggesting a stimulatory effect of Hsp90 on A3G mutational activity.
Figure 4.

Sequencing evaluation of the Hsp90α and Hsp90β stimulation on A3G mutational activity. A3G was co-transfected with mock vector, Hsp90α, or Hsp90β into HepG2 cells as in Fig. 3. HBV DNA in the HBV capsids was extracted and amplified by regular PCR followed by 88 °C 3D-PCR. The amplicons of 88 °C 3D-PCR were cloned into the TA cloning vector, and randomly selected clones of each sample were sequenced. A–C, distribution of C-to-T mutations in each positive clone after treatment with A3G, A3G + Hsp90α, or A3G + Hsp90β, respectively. Mutation-positive clone data from a total of 60 sequenced clones for A3G and 40 for both A3G + Hsp90α and Hsp90β are presented. C-to-T mutations in each clone are represented by markers (+) on each line against their cytosine sites in the HBV PCR region. D–F, C-to-T mutation frequencies at individual cytosine sites in the HBV region were calculated based on their sequence data in A–C, respectively, and presented as bars against their sites in the HBV region. The cytosine sites are numbered according to their positions in HBV by reference to V01460.1.
Table 1.
Hsp90 effect on APOBEC-3 mutational efficiency by sequencing analyses
| Treatment | C-to-T mutation numbers in each positive clone under treatmenta | Average C-to-T mutations per clone | Average mutated cytosine percentage in the evaluated regionb | Increase compared with control |
|---|---|---|---|---|
| % | -fold | |||
| A3G | 27, 7, 5, 3, 2, 2, 1, 1, 1, 1 | 5 | 4.3 | 1 |
| A3G + Hsp90α | 20, 20, 18, 11, 10, 9, 9, 9, 5, 5, 3, 1, 1 | 9.3 | 8.0 | 1.9 |
| A3G + Hsp90β | 30, 11, 10, 9, 5, 4, 2, 1, 1, 1 | 7.4 | 6.3 | 1.5 |
| A3B | 34, 12, 1, 1, 1 | 9.8 | 8.4 | 1 |
| A3B + Hsp90α | 87, 81, 70, 67, 66, 18, 6 | 56.4 | 48.2 | 5.7 |
| 3G60B | 54, 26, 25, 22, 10, 9, 8, 5, 4, 3, 2, 2, 1, 1, 1 | 11.5 | 9.9 | 1 |
| 3G60B + Hsp90β | 52, 51, 50, 46, 42, 41, 37, 37, 35, 26, 24, 24, 15, 12, 11, 11, 10, 8, 4, 4, 2, 2 | 24.7 | 21.1 | 2.1 |
| In vitro 0.5-μl A3G | 8, 4, 4, 4, 4, 3 | 4.5 | 3.8 | 1 |
| In vitro 0.5-μl A3G + Hsp90β | 11, 11, 10, 10, 10, 10, 10, 10, 10, 8, 7, 7, 7, 7, 2, 1 | 8.2 | 7.0 | 1.8 |
| In vitro 4-μl A3G | 7, 7, 7, 6, 5, 5, 5, 5, 5, 5, 5, 4, 3, 3 | 5.1 | 4.4 | 1 |
| In vitro 4-μl A3G + Hsp90β | 15, 15, 15, 15, 15, 14, 13, 10, 10, 10, 10, 9, 6, 3, 2 | 10.8 | 9.2 | 2.1 |
a The C-to-T mutation numbers for each mutation positive clone were input from the sequencing data that are graphically presented in Fig. 4 for A3G, A3G + Hsp90α, and A3G + Hsp90β; Fig. 6 for A3B, A3B + Hsp90α, 3G60B, and 3G60B + Hsp90β; and Fig. 9 for 0.5-μl A3G, 0.5-μl A3G + Hsp90β, 4-μl A3G, and 4-μl A3G + Hsp90β.
b There are a total of 117 cytosines in the HBV evaluated region.
A3B potently mutates and Hsp further increases A3B mutational potency
In addition to A3G, A3B and A3C also displayed readily detectable HBV DNA mutational activity (Fig. 1). Thus, the effect of Hsps was further evaluated by co-transfection with A3B and A3C in HepG2 cells. A3C is structurally different from A3G. A3C has only one zinc-dependent cytidine deaminase domain, whereas A3G has two. Nevertheless, a similar stimulatory effect of Hsps on A3C was also observed for Hsp90β, -90α, -104, and -60 despite its structural difference from A3G. Up to a 2-fold increase by pe1453 in the 88 °C 3D-PCR was observed with Hsp90 treatment (see supplemental Fig. 1).
A3B was the most potent APOBEC-3 protein for HBV DNA mutagenesis when tested by co-transfection in HepG2 cells (Fig. 1). We found that A3B mutational activity varied significantly, depending upon its structural form. There are two native mRNA variants of A3B in human liver, the long and short forms, resulting from alternative RNA splicing at the C terminus. The short form A3B, named A3B-WT here, is the predominant one in human liver (data not shown). We tried initially to clone A3B from the human macrophage but found that this A3B (named A3B-mutant here) had multiple point mutations. A3B-WT was not commercially available at the time, but its long form was available from Origen Co. We then tried to generate A3B wild type from its long form by internal deletion. We found that A3B wild type was very toxic to bacteria. No correct clone was obtained, and only a few recombinant clones with unexpected sequences survived during the cloning process. Bacterial clones with the A3B wild type could not be amplified due to its protein expression via a leaky T7 promoter in Escherichia coli, as observed by other laboratories (23). Only one correct clone survived when the internal deletion was performed using the bacterial expression vector p-DEST15 (Thermo Fisher Scientific) that has a tightly controlled T7 promoter. We finally obtained a mammalian expression clonal form very close to the A3B wild type, where the last 4 amino acids at the C terminus were replaced with different tails of 40 or 16 amino acids (named A3B-WT here; no obvious functional differences were found with either the 40- or 16-aa tail; data not shown). To compare the activities of these A3B variants, we also generated A3B-d7, a truncated A3B with a deletion of the last 7 amino acids at the C terminus, and 3G60B, a recombinant A3B formed by replacing the first 60 amino acids of A3B N terminus with the corresponding part of A3G by reference to its improved mutational activity reported on HIV-1 (41). We evaluated the effect of Hsp90β on A3B using variant forms, including A3B-long form, A3B-WT, A3B-d7, A3B-mutant, and A3G60B, by co-transfection in HepG2 cells. These A3B variant forms had comparable protein expression levels in HepG2 cells by Western blotting with an antibody against endogenous A3B despite having different protein sizes (data not shown).
As shown in Fig. 5A, A3B-WT had the highest HBV mutation frequency, 37.7%, among the tested A3B variants when using the 88 °C 3D-PCR followed by pe1674. Hsp90β further increased the A3B-WT mutational activity from 37.7 to 49.2%. The A3B-long form also induced readily detectable mutations (27.2%), but the addition of Hsp90β had no significant stimulatory effect. A3B-d7 had a 33.4% mutation frequency, but Hsp90β had a slightly inhibitory effect, opposite to what was observed with A3B-WT. The A3B-mutant had the weakest HBV mutational activity among the A3Bs tested, yet a 3.7-fold increase from 6.2 to 22.9% was observed in the presence of Hsp90β. A3G60B also responded to Hsp90β and had a 2.2-fold increase from 20.4 to 45.3%. These data demonstrate that A3B has potent mutagenesis activity, and A3B C-terminal variability results in different sensitivities to Hsp90β.
Figure 5.

Hsp effects on A3B mutational activity in HBV. Hsps and A3B variants were co-transfected into HepG2 cells followed by HBV DNA mutational analyses as in Fig. 3. A, the mutational activities of A3B variants with or without Hsp90β co-expression were determined by primer extension analyses at cytosine site 1674 (pe1674) with the 88 °C 3D-PCR. B and C, the effect of Hsps on A3B-WT or A3G60B mutation activity was analyzed by primer extension using pe1674 with the 88 °C 3D-PCR. The data are presented graphically on the right. Each bar represents the average of triplicates for each treatment. Blank, background control without APOBEC-3 co-transfection. Vector, A3 + mock vector as the A3 alone control to compare with A3 + Hsps. Asterisks, statistically significant differences comparing an individual treatment with a vector or corresponding non-Hsp treatment control: **, 0.01 < p < 0.001; *, 0.05 < p < 0.01. The one-factor ANOVA statistical test was used.
The effect of various Hsps on A3B-WT and A3G60B activity was further investigated. As shown in Fig. 5B, the blank control representing native mutational activity without A3B-WT transfection was 2.6%. The A3B-WT alone (labeled as Vector representing co-transfection of A3B-WT + mock vector) had a 24.8% mutation frequency by pe1674 analyses in a 88 °C 3D-PCR. Hsps increased A3B-WT mutational activity up to >2-fold compared with the vector control. Like A3G, A3B-WT had a similar stimulatory response to Hsp40, -90, -104, and -60. However, A3B-WT's response to Hsp70 and HOP was significantly different from that of A3G. Hsp70 stimulated A3B-WT activity almost 2-fold from 24.8 to 47.4% while having no effect on A3G. HOP, a co-chaperone connecting Hsp90 and Hsp70, more significantly increased A3B-WT than A3G. These data suggest that A3B is sensitive to both Hsp90 and Hsp70.
As shown in Fig. 5C, A3G60B alone represented by Vector (representing A3G60B + mock vector) had a 23.1% mutation frequency, whereas the blank background was 3.0%. Hsps' co-expression also stimulated A3G60B mutation activity up to 2-fold. As a recombinant A3B that has A3G N-terminal first 60-aa sequence, A3G60B's overall response was similar to that of A3G, with Hsp90β being the most potent. Interestingly, neither Hsp70 nor HOP had significant effects on A3G60B, indicating that the N-terminal first 60 aa altered the A3B and A3G responses to Hsp70. Taken together, these data demonstrate that Hsps stimulated APOBEC-3 mutation activity despite the significant structural difference among A3G, A3B, and A3C.
A3B had readily detectable HBV mutational activity even with regular PCR at 94 °C (Fig. 1) (data not shown). To confirm the effect of Hsps on A3B-WT, the A3B-WT 94 °C PCR as in Fig. 5B was cloned and evaluated by direct sequencing. As shown in Fig. 6 (A and B), 5 of 60 clones had C-to-T mutations for A3B alone, but 7 of 30 clones were positive for A3B + Hsp90α, indicating that A3B mutation frequency was increased by the co-expression of Hsp90. As shown by the graphic distribution of mutated cytosines in Fig. 6 (A and B), A3B-WT had extensive C-to-T mutations in each clone, especially in the presence of Hsp90α. A3B alone averaged 9.8 C-to-T mutations per clone (Fig. 6A and Table 1). Hsp90α further increased the A3B average C-to-T mutations per clone significantly from 9.8 for A3B alone to 56.4 for A3B + Hsp90α (Fig. 6B and Table 1). There were 117 cytosines in the HBV PCR-evaluated region. A3B and A3B + Hsp90α mutated on average 8.4 and 48.2% of the total of 117 cytosines in the HBV region (Table 1). One clone from A3B + Hsp90α in Fig. 6B had 87 of 117 cytosines converted to uridines, with a percentage of 74.4%. In contrast, A3G had an average of 5 C-to-U mutations per clone, and the average number was increased to 9.3 and 7.4 in the presence of Hsp90α and Hsp90β, respectively (see Table 1). These data demonstrate that A3B very potently mutated DNA, especially in the presence of Hsp90. This is consistent with the observation that A3B-WT was very toxic to host bacteria due to protein expression from a leaky T7 promoter. Based on the collective sequencing data, the mutation frequency at individual cytosine sites also increased from 2–3% up to 16–20% in the presence of Hsp90 as shown in Fig. 6 (E and F), indicating generally high mutation levels across the HBV region.
Figure 6.

Sequencing evaluation of the Hsp effects on A3B or A3G60B mutational activity. A3B or A3G60B was co-transfected with mock vector, Hsp90α, or Hsp90β into HepG2 cells as in Fig. 5. HBV DNA in the HBV capsids was extracted and amplified by 94 °C PCR and 88 °C 3D-PCR. A and B, distribution of C-to-T mutations in each mutation-positive clone after treatment with A3B or A3B + Hsp90α. 94 °C PCR amplicons were cloned into the TA cloning vector. Sixty clones for A3B and 30 for A3B + Hsp90α were randomly selected and sequenced. C-to-T mutations in each clone are represented by markers (+) on each line against their cytosine sites in the HBV PCR region. C and D, distribution of C-to-T mutations in each mutation-positive clone after treatment with A3G60B or A3G60B + Hsp90β. 88 °C 3D-PCR amplicons were cloned into the TA cloning vector, and 32 clones were randomly selected and sequenced. E–H, C-to-T mutation frequencies at individual cytosine sites in the HBV region were calculated based on their sequence data in A–D, respectively, and are presented as bars against their sites in the HBV region. The cytosine sites were numbered according to their positions in HBV by reference to V01460.1.
The Hsp90 effect on A3G60B revealed by primer extension analyses in Fig. 5C was also further verified by direct TA-cloning sequencing. Thirty-two clones were randomly selected and sequenced for A3G60B control and A3G60B + Hsp90β with the 88 °C 3D-PCR. The sequencing data are graphically presented in Fig. 6 (panels C and D and panels G and H). As shown in Fig. 6 (C and D), A3G60B alone had C-to-T mutations in 16 of 32 clones, and the average C-to-T mutations per clone were 11.5 (see Table 1). In contrast, A3G60B + Hsp90β had C-to-T mutations in 22 of 32 clones, and the average C-to-T mutations per clone were 24.7. As shown in Fig. 6 (G and H), the collective mutation frequency at individual cytosine sites in the HBV region also increased from 3–18% for A3G60B to 15–60% for A3G60B + Hsp90β across the region. These data further demonstrated that Hsp90 had a significant stimulating effect on both APOBEC-3's mutation frequency and efficiency.
Hsp90 directly interacts with A3G in vitro
HBV DNA mutation analyses demonstrated that Hsps stimulated A3G and A3B mutational activity by co-transfection in HepG2 cells. However, the mechanism remains to be clarified because mutating HBV is complex, occurring after multiple steps with many potential variations. These include plasmid co-transfection, transcription, and translation of the genes; encapsulation of the proteins together with viral RNA into capsids; and HBV viral RNA conversion to DNA. To confirm the Hsp90 stimulatory effect on A3G and to further investigate the interaction between them, we performed in vitro HBV mutation reactions using A3G.
We first investigated whether there was a direct protein interaction between Hsps and A3G. According to a previous report (42), co-immunoprecipitation (co-IP) of Myc-Hsp90 with A3G was not detected when Myc-Hsp90 and A3G were co-expressed in HEK293T cells, whereas it was readily detected for AID. However, the analyses did not consider that high levels of endogenous Hsp90 may compete with Myc-Hsp90 for binding to A3G and interfere with A3G detection when using anti-Myc antibody for co-IP. Hsp90 might have a direct protein interaction with A3G, although the interaction could be much weaker than that of AID. Therefore, we evaluated the co-IP of Hsp90 with A3G in HepG2 cells using an antibody against endogenous Hsp90β. As shown in Fig. 7A, Hsp90β was detected in the complex immunoprecipitated by an antibody against the HA tag at the C terminus of A3G. Because A3G tended to form RNP complexes once translated (43), that could make A3G less available for Hsp90 interaction due to the cellular sorting process. Therefore, we further tested the co-IP of endogenous Hsp90 with A3G using an in vitro translation system. As shown in Fig. 7B, Hsp90 was clearly detected in the A3G co-IP, indicating that there was a direct protein interaction between Hsp90 and A3G.
Figure 7.
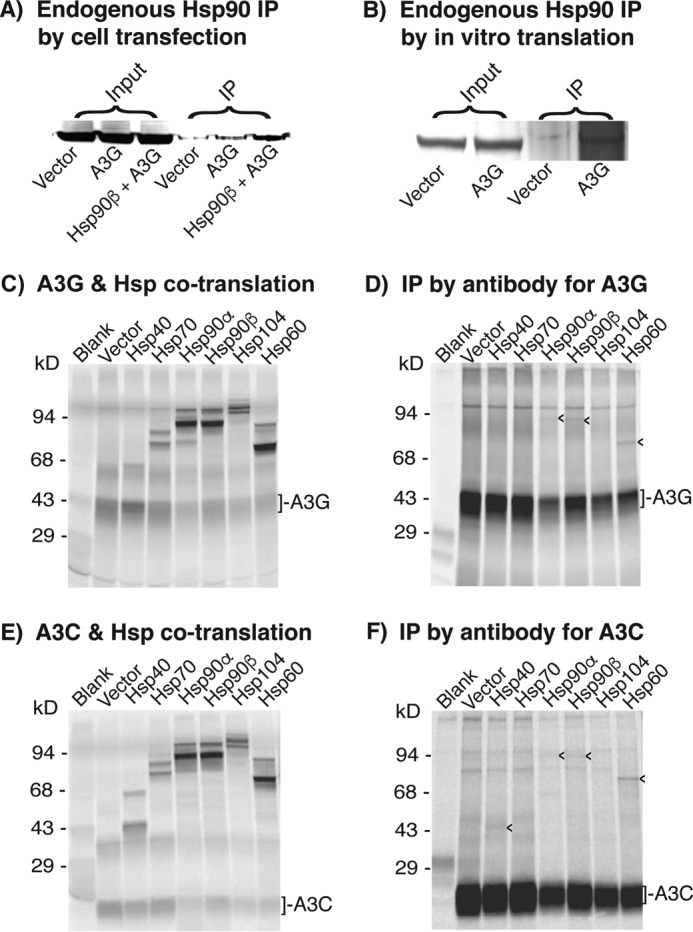
Protein interaction between Hsps and A3G or A3C. A, co-IP analyses of Hsp90β with A3G from cells. Mock vector, A3G, or A3G + Hsp90β was transfected into HepG2 cells. The whole-cell proteins were extracted after 2 days of transfection. A3G complexes in the cell lysates were immunoprecipitated by an antibody against the HA tag in A3G. The resultant A3G co-IP complexes were analyzed by Western blotting using an antibody against endogenous Hsp90β. B, co-IP analyses of Hsp90β with A3G from in vitro translation. Mock vector or A3G was translated by an in vitro coupling transcription and translation system for 1.5 h at 30 °C. A3G in the in vitro translations was immunoprecipitated by an antibody against the HA tag in A3G. The resultant A3G co-IP complexes were analyzed by Western blotting with an antibody against endogenous Hsp90β. C and E, Hsps and A3G or A3C were co-translated by an in vitro coupling transcription and translation system in the presence of l-[35S]methionine for 2 h at 30 °C. The protein expression of Hsps with A3G or A3C in the translation reactions was analyzed by 12% SDS-PAGE. D and F, the co-translated protein mixtures as in C and E were immunoprecipitated by an antibody against HA in A3G or A3C, followed by 12% polyacrylamide gel separation and detection with a PhosphorImager. The arrowheads (<) indicate Hsps that were immunoprecipitated with A3G or A3C. Blank, background control without APOBEC-3 plasmid. Vector, A3 + mock vector as the A3 alone control to compare with A3 + Hsps.
To further investigate the potential protein interaction of A3G with other Hsps, we utilized the in vitro TNT quick coupled transcription and translation system to label the newly translated proteins by [35S]methionine to increase the detection sensitivity of labeled Hsps relative to endogenous Hsps. The protein-to-protein direct interactions between Hsps and A3G or A3C were then analyzed by IP with an antibody against the HA tag of A3G or A3C. As shown in Fig. 7 (D and F), Hsp90α, -90β, and -60 consistently co-precipitated with both A3G and A3C. Hsp40 co-precipitation with A3C was also detected, although it could not be distinguished from A3G due to their similar molecular weights. Although the immunoprecipitated bands were weak due to the dilution effect of endogenous Hsps as expected, the consistent results suggest that there were direct protein interactions between Hsps and A3G or A3C.
Because the purified proteins of A3G and Hsps were available from Abnova Co., we used the purified proteins to perform in vitro mutation reactions by mixing them in a buffer containing HBV cDNA substrate and 100 μg/ml BSA. After incubating at 30 °C for 2 h, the HBV DNAs was extracted, and mutation levels were determined by a combination of 3D-PCR and primer extension at cytosine site 1453. As shown in Fig. 8A, HBV DNAs were uniformly amplified in both 94 °C PCR and 88 °C 3D-PCR. However, the amplification became barely detectable in 87 °C 3D-PCR (data not shown). Therefore, 88 °C 3D-PCR was chosen for further mutation analyses. As shown in Fig. 8B, A3G had a 5.3% mutation frequency in the 88 °C 3D-PCR, whereas the background water control was 2.2%. The A3G mutation activity was increased 2-fold from 5.3% to 10.7 and 10.9% in the presence of Hsp90α and Hsp104, respectively. However, there was no significant change observed in the presence of Hsp40, -70, and -60. These data demonstrate that Hsp90 and -104 directly stimulate A3G mutation activity in vitro. The absence of an Hsp40 effect on A3G in vitro contrasts to its results in vivo, suggesting that Hsp40 may work through Hsp90 as co-chaperone in vivo.
Figure 8.
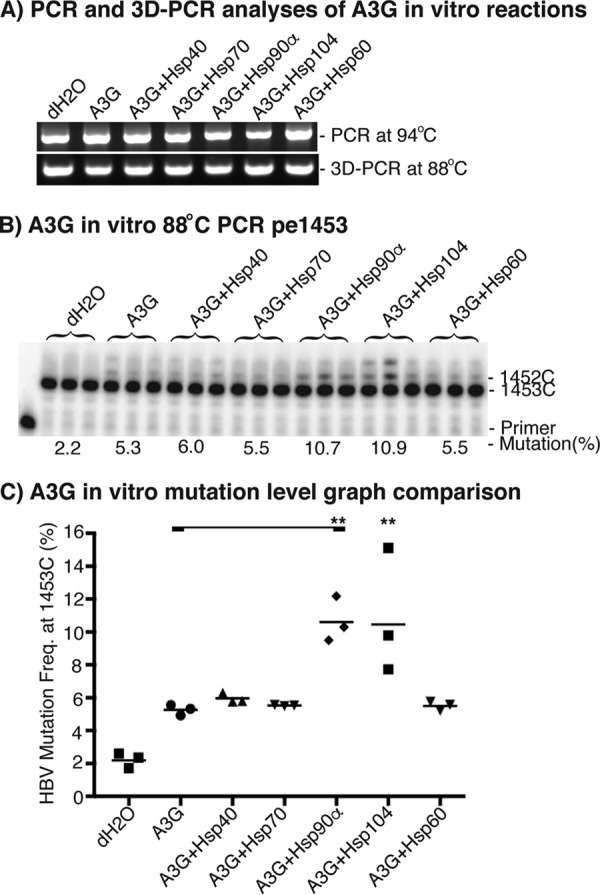
In vitro Hsp effects on A3G mutational activity. Purified A3G (0.07 μg) and Hsps (0.7–1.0 μg) were incubated with HBV cDNA substrate (20 ng) in a buffer containing 100 μg/ml BSA for 2 h at 30 °C. After the in vitro mutation reaction, the HBV cDNA was extracted and amplified by regular PCR followed by 88 °C 3D-PCR. A, PCR and 3D-PCR amplifications of HBV DNA were analyzed by 1% agarose gel. B, A3G-induced mutational activities in 88 °C 3D-PCR were determined by pe1453; C, the data as in B are presented graphically. Each bar represents the average of triplicates for each treatment. Asterisks, statistically significant differences comparing treatments with A3G control: **, 0.01 < p < 0.001. The one-factor ANOVA statistical test was used.
In vitro HBV DNA mutations by 0.5 and 4 μl of A3G with or without Hsp90β were further investigated by TA clonal sequencing. The distribution of C-to-T mutations in individual clones and the collective mutation frequency at individual cytosine sites were presented graphically in Fig. 9. As shown in Fig. 9A, there were only 5 C-to-T mutation clones in 20 randomly selected clones, and the average number of C-to-T mutations per clone was 4.5 in the 0.5-μl A3G reaction. Hsp90β increased the mutation-positive clones from 5 to 16, and the average number of C-to-T mutation per clone increased from 4.5 to 8.2 (Fig. 9B and Table 1). In addition, Hsp90β significantly increased the cytosine mutation frequency in the nt 1425–1530 region and 1664 site in the 0.5-μl A3G reaction as shown in Fig. 9 (E and F).
Figure 9.

Sequencing analyses of in vitro Hsp90β effects on A3G. The in vitro reactions of A3G-induced mutation on HBV cDNA with or without Hsp90β were performed as in Fig. 8. The mutated HBV DNA was enriched by 88 °C 3D-PCR and cloned into a TA cloning vector. Twenty clones were randomly selected and sequenced for each sample. A–D, distribution of C-to-T mutations in each clone after treatment with 0.5 μl of A3G, 0.5 μl of A3G + Hsp90β, 4 μl of A3G, and 4 μl of A3G + Hsp90β, respectively. C-to-T mutations in each clone are presented by markers (+) on each line against their cytosine sites in the HBV PCR region. E–H, C-to-T mutation frequencies at individual cytosine sites in the HBV region were calculated from their sequence data in A–D and are presented as bars against their sites. The cytosine sites are numbered according to their positions in HBV by reference to V01460.1.
There was no difference on the frequency of mutated clones in a total of 20 randomly selected clones in the 4-μl A3G reactions with or without Hsp90β treatment. There were 14 C-to-T mutated clones of 20 randomly selected clones for 4 μl of A3G alone, whereas 15 were positive for 4 μl of A3G + Hsp90β, as shown in Fig. 9 (C and D). However, a significant difference was observed in the distribution of C-to-T mutation sites. As shown in Fig. 9 (C, D, G, and H), 4 μl of A3G alone heavily mutated the cytosines in the nt 1710–1815 region, whereas 4 μl of A3G + Hsp90β caused a shift to the nt 1415–1710 region. In addition, Hsp90β increased the average number of C-to-T mutations per clone from 5.1 to 10.8 in the 4-μl A3G reaction (see Table 1), indicating an increased mutation efficiency in the presence of Hsp90β. These data are consistent with the in vivo HepG2 results (Fig. 6) and demonstrate that Hsp90β stimulates A3G with both the frequency and efficiency of cytosine mutation.
Hsp90 modulates A3G cytosine mutation preference
Hsp90 stimulated A3G mutation activity and modulated A3G's selection of HBV DNA cytosines for mutation both in vivo and in vitro (Figs. 4 and 9). We further investigated the effect of Hsp90 on A3G cytosine mutation preference for adjacent 5′ and 3′ nucleosides using both in vitro and in vivo data as in Figs. 9 and 4, respectively. There were 117 cytosines in the HBV PCR-tested region. If cytosine mutation is random, the frequency for 5′-GC, 5′-AC, 5′-TC, and 5′-CC (where C represents mutated cytosine) should be 24.8, 21.4, 23.1, and 30.8%, respectively, whereas the frequency for CG-3′, CA-3′, CT-3′, and CC-3′ should be 26.5, 27.4, 15.4, and 30.8%, respectively. Mutated cytosines from all positive clones under different treatments were collected and sorted into four groups according to their 5′ or 3′ adjacent nucleoside of G, A, T, or C. The frequency for each adjacent nucleoside relative to the total number of edited cytosines was calculated as a percentage and is graphically presented in Fig. 10.
Figure 10.
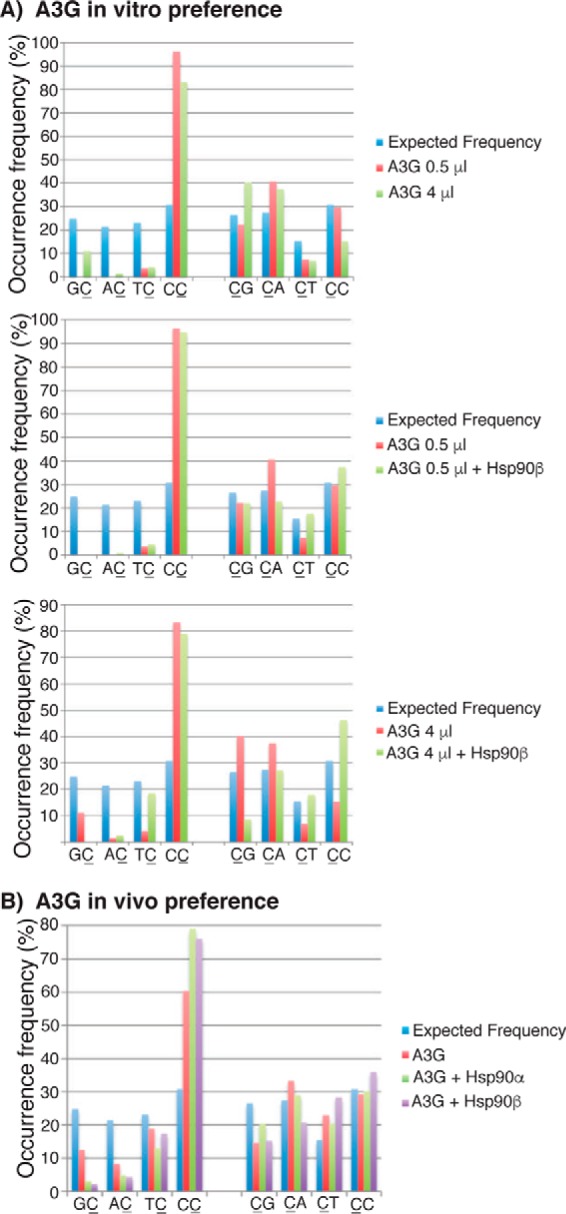
Hsp90 alters the A3G preference for 5′ and 3′ nucleosides. C-to-T mutations from all positive clones in Figs. 4 and 9 were collected and analyzed for frequency (%) relative to 5′ nucleosides (GC, AC, TC, and CC) or 3′ nucleosides (CG, CT, CA, and CC)(where C represents mutated cytosine) in the HBV sequencing region. The expected frequencies were calculated assuming an equal chance of mutation among a total of 117 cytosines in the HBV region. The data are presented as graphs for comparison. A, A3G in vitro preference data obtained from Fig. 9; B, A3G in vivo preference data obtained from Fig. 4.
As shown in Fig. 10A (top left), 0.5-μl A3G in vitro HBV DNA mutation results showed a very strong 5′-C preference (96.3% 5′-CC), consistent with previous preference reports (24). When the amount of A3G increased from 0.5 to 4 μl, the A3G preference for 5′-C decreased from 96.3 to 83.3%, whereas the preference for 5′-G increased from 0 to 11.1%. This indicates that the A3G-specific preference for 5′-C decreases toward the expected random distribution as the amount of A3G is increased in the in vitro reaction. Hsp90β had no significant effect on the 0.5-μl A3G 5′ nucleoside preference but had a significant effect on the 4-μl A3G. As shown in the bottom left of Fig. 11A, 11.1% 5′-G in the 4-μl A3G reaction was decreased to 0% by Hsp90β, whereas the major preference for 5′-C was unchanged. A3G had no strong 3′ nucleoside preference. An increase of A3G from 0.5 to 4 μl caused an increase of 3′-G from 22.2 to 40.3% and a decrease of 3′-C from 29.6 to 15.3%, as shown in Fig. 10A (top right). However, these changes were reversed by Hsp90β, with 3′-G decreasing from 40.3 to 8.6% and 3′-C increasing from 15.3 to 46.3%, as shown in the bottom right of Fig. 10A. These data indicate that Hsp90 partially reverses the A3G 5′ nucleoside preference induced by the increased amount of A3G in vitro.
Figure 11.
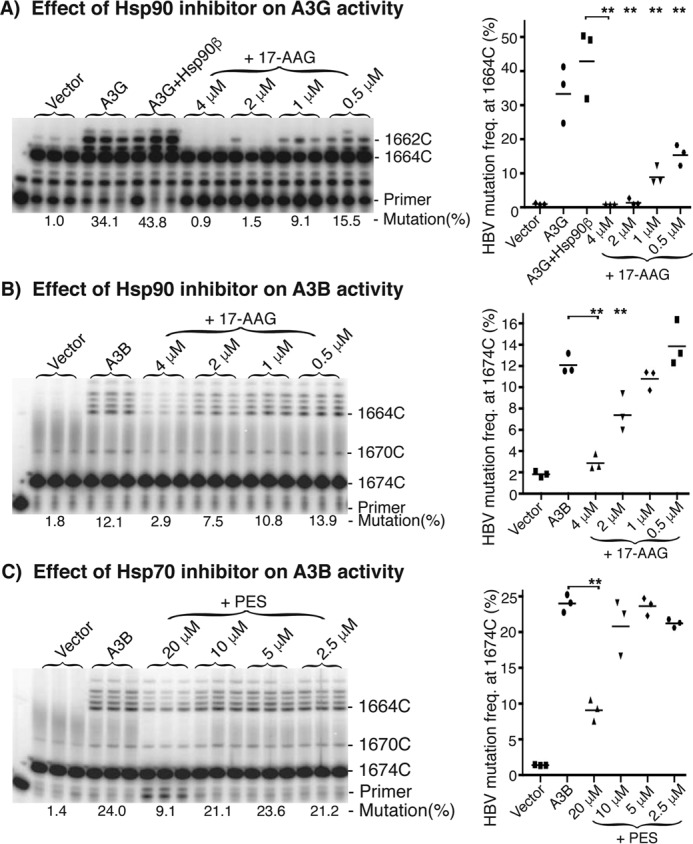
Effect of Hsp90 or Hsp70 inhibitor on A3G and A3B activity. Hsp inhibitors were used to evaluate the Hsp90 or Hsp70 effect on A3G and A3B. APOBEC-3s with or without Hsp90β were co-transfected with HBV into HepG2 cells in the presence or absence of Hsp90 inhibitor 17-AAG or Hsp70 inhibitor PES. After 2 days of treatment, HBV DNA mutation rates were determined by primer extension with the 88 °C 3D-PCR for A3G or 94 °C PCR for A3B. A, effect of Hsp90 inhibitor 17-AAG on A3G + Hsp90β mutation activity by pe1664; B, effect of Hsp90 inhibitor 17-AAG on A3B mutation activity by pe1674; C, effect of Hsp70 inhibitor PES on A3B mutational activity by pe1674. The data are presented graphically on the right. Each bar represents the average of triplicates for each treatment. Vector, background control with mock vector co-transfection; asterisks, statistically significant differences comparing treatments with controls: **, 0.01 < p < 0.001; *, 0.05 < p < 0.01. The one-factor ANOVA statistical test was used.
As shown in Fig. 10B, A3G alone expression in HepG2 cells had predominantly a 5′-C preference (60.4% 5′-CC). However, when Hsp90α was co-transfected, the 5′-C preference increased from 60.4 to 79%, whereas the 5′-G, 5′-A, and 5′-T preferences decreased variably. These findings suggest that Hsp90α significantly increases the specificity of A3G-induced mutation for 5′-CC. Hsp90β had an effect similar to Hsp90α on A3G 5′ nucleoside preference. Neither Hsp90α nor Hsp90β significantly affected the 3′ nucleoside preference. Taken together, these data demonstrate that Hsp90 significantly increases the specificity of A3G preference for 5′-C in vivo.
Hsp90 may have a modulatory effect on A3G and A3B in vivo
Hsp90 and Hsp104 stimulated A3G mutational activity in both cellular assays and in vitro reactions, whereas Hsp40 and 60 stimulated A3G in vivo but not in vitro. These data suggest that Hsp90 may have a modulatory effect on A3G, whereas Hsp40 may work through Hsp90 as a co-chaperone. In addition, Hsp70 uniquely affected A3B, but not A3G.
The potential role of Hsp90 and Hsp70 on A3G or A3B induced HBV mutation in HepG2 cells was further investigated using specific inhibitors. As shown in Fig. 11A, A3G and A3G + Hsp90 resulted in 34.1 and 43.8% mutation rates in the 88 °C 3D-PCR by pe1664, whereas the background control by vector alone was 1.0%. When A3G + Hsp90 was co-expressed in the presence of the Hsp90-specific inhibitor 17-AAG, A3G + Hsp90β mutational activity was inhibited in a dose-dependent manner. The A3G-induced HBV DNA mutation frequency was very sensitive to 17-AAG and was essentially totally inhibited at concentrations >2 μm (Fig. 11A). These data suggest that Hsp90 may be essential for A3G mutational activity on HBV DNA and that the inhibition of Hsp90 resulted in dose-dependent decreases of A3G mutational activity. On the other hand, why >2 μm 17-AAG totally inhibits A3G-induced HBV mutational activity is unclear because A3G alone had mutational activity in vitro. This may be due to the complex HBV viral replication process that requires the presence of Hsp90 from encapsulation of viral or host factors to P protein function for ssDNA generation (35, 36). To clarify the specificity of the Hsp90 effect, A3B-induced HBV mutational activity was evaluated using Hsp90 inhibitor 17-AAG. As shown in Fig. 11B, the background control with mock vector was 1.8%, and A3B co-expression resulted in 12.1% mutational activity in the 94 °C PCR followed by pe1674 analyses. When A3B was co-expressed in the presence of the Hsp90 inhibitor 17-AAG at the indicated concentrations, a dose-dependent inhibition of A3B HBV mutational activity was also observed. A3B-induced mutational activity using 0.5 μm 17-AAG was comparable with the A3B control without inhibitor. 17-AAG inhibited A3B activity by 11% at 1 μm 17-AAG and further up to 38 and 76% at 2 and 4 μm 17-AAG, respectively. Unlike A3G, the A3B-induced mutational activity was less sensitive to the Hsp90 inhibitor and was incompletely inhibited even when using 17-AAG as high as 4 μm, a concentration toxic to HepG2 cells. Together with the A3G results, these data suggest that existing endogenous Hsp90 in HepG2 cells supports the mutational function of A3G and A3B on HBV, and Hsp90 may have a modulatory effect on A3G- and A3B-induced mutational activity in vivo. In addition, the Hsp70 inhibitor PES also inhibited A3B-induced mutational activity, as shown in Fig. 11D, consistent with the increased mutation rate observed when Hsp70 was co-transfected with A3B (Fig. 5B). These data suggest that Hsp70 may also be involved in the A3B-induced mutational activity in vivo.
Specificity investigation for the interaction between Hsp90 and A3G
ATPase activity is required for the Hsp90 stimulatory effect on A3G and A3B, as described above, with the Hsp90 inhibitor 17-AAG (Fig. 11). To further investigate the nature of the interaction between Hsp90 and A3G or A3B, we evaluated whether the Hsp90 protective effect on A3G or A3B that Hsp90 typically has for its client proteins (42) was lost or not when Hsp90 function was inhibited by 17-AAG. As shown in Fig. 12A, up to a 60% decrease was observed for both A3G and A3B protein levels after a 24-h treatment, indicating that the presence of Hsp90 provided functional protection for A3G and A3B.
Figure 12.

Specificity of the interaction between Hsp90 and A3G. A, A3G and A3B protein stability analyses by Western blotting. A3G or A3B were co-transfected with Hsp90 into HepG2 cells, and the Hsp90 inhibitor 17-AAG was added to the indicated concentrations on the day after the transfection. The whole-cell proteins were extracted after an 8- or 24-h inhibitor treatment. A3G and A3B protein levels were determined by Western blotting using an antibody against the HA in A3G or FLAG tag in A3B, respectively. GAPDH was analyzed for the protein loading reference. B, A3G or Hsp mutant protein expression level analyses. Plasmids encoding A3G or Hsp mutants were co-transfected into HepG2 cells, and whole-cell proteins were extracted for Western blotting analyses with an antibody against the HA tag in A3G or the FLAG tag in Hsps. C, A3G specificity analyses by HBV DNA mutation. A3G mutants were co-transfected into HepG2 cells in the presence or absence of Hsp90. HBV DNAs were extracted from the cell lysates after 2 days of transfection, and viral DNA mutations were determined by pe1453 using an 88 °C 3D-PCR. D, Hsp specificity analyses by HBV DNA mutation. The Hsp mutants were transfected into HepG2 cells in the presence of A3G. The resultant HBV DNA mutations were determined by pe1453 using an 88 °C 3D-PCR. The data are presented graphically on the right. Each bar represents the average of triplicates for each treatment. Blank, background control without A3G co-transfection; asterisks, statistically significant differences comparing treatments with their corresponding wild-type control: **, 0.01 < p < 0.001; *, 0.05 < p < 0.01. The one-factor ANOVA statistical test was used.
Hsp90 often functions in a complex in vivo by cooperating with other co-chaperones (44). For example, in the Hsp90-dependent steroid hormone receptor activation, Hsp40 binds client protein first and then forms a complex with Hsp70. The client protein in the Hsp40 and Hsp70 complex is then delivered to Hsp90 through a HOP-mediated interaction between Hsp70 and Hsp90 (1, 2). However, Hsp90 action on A3G could be even more complicated because A3G-induced HBV DNA mutation occurs in the viral capsids. Hsp90 and A3G have to be taken into the capsid during capsid formation for the ssDNA to undergo A3G-induced mutation. To further explore the specificity for the interaction between Hsp90 and A3G, we evaluated the effect of both A3G and Hsp mutants with known functional alterations.
It is known that A3G is inactivated by a catalytic site Glu-259 mutation and that the phosphorylation of A3G Thr-218 decreases its mutational activity (45). As shown in Fig. 12 (B and C), A3G and its mutants had comparable protein expression in HepG2 cells. Nonspecific protein expression by EGFP had no stimulatory effect, unlike what was observed with Hsp90. The A3G E259Q mutation reduced A3G mutational activity to background levels, indicating dependence upon A3G for the observed HBV DNA mutational activity. On the other hand, mutant A3G T218A or T218E that mimicked site dephosphorylation or phosphorylation, respectively, resulted in an increased (5.9%) or decreased (4.2%) HBV mutation frequency compared with A3G wild type (4.5%), as expected. When these mutants were analyzed in the presence of Hsp90β co-expression, there were increases of 2.5-fold for A3G-WT (from 4.5 to 11.3%), 2.8-fold for T218A (from 5.9 to 16.8%), and 1.9-fold for T218E (from 4.2 to 7.8%), respectively. The relative activities of mutants compared with wild type remained the same in the presence or absence of Hsp90β, indicating that the increased A3G mutation activity was a specific response to Hsp90β stimulation.
It is known that the ATPase activity of Hsp90 is achieved through the conserved arginine and glutamine residues in the middle domain catalytic loop (Arg-380 and Gln-384 in yeast) that interact with the ATP bound in the N-terminal domain and promote ATP hydrolysis (44, 46). ATPase activity plays an essential role in Hsp90 functions. R380A, Q384A, and F349A mutations in yeast Hsp90 had substantially lower ATPase activity than wild-type protein, whereas F332A displayed nearly wild-type ATPase activity (46). These mutations in yeast Hsp90 correspond to R400A, Q404A, F369A, and F352A in human Hsp90α. In addition, it is known that the Hsp40 H32Q mutation disrupts the cooperation between Hsp40 and Hsp70 (47) and that the Hsp70 E175S mutation abolishes both Hsp70 ATPase and refolding activities, working as a dominant negative Hsp70 mutant (48). All of these Hsp mutants were evaluated for their effect on A3G mutation.
As shown in Fig. 12 (B and D), Hsp mutants had expression levels comparable with the wild-type proteins. Hsp90 R400A, Q404A, and F369A mutations resulted in decreased A3G stimulation compared with wild-type Hsp90, consistent with their decreased ATPase activity, whereas the Phe-352 mutation with comparable ATPase activity only had a slight decrease. These data indicate that A3G mutational activity is modulated according to Hsp90 ATPase activity. Similarly, the Hsp40 H32Q mutation also resulted in a statistically significant decrease, consistent with the observed increased effect by Hsp40. On the other hand, the Hsp70 E175S mutation resulted in an increased effect, in contrast to wild-type Hsp70, for which no obvious effect was observed (Fig. 3). These changes resulting from functional mutations suggest that both Hsp40 and Hsp70 were actually involved in A3G mutational activity. On the other hand, these mutants still had a stimulatory effect on A3G compared with the nonspecific protein co-expression control by EGFP, indicating that there could be a potential compensatory effect among Hsp proteins, including these endogenous Hsps.
The functional role of Hsps on A3G or A3B described above was mainly investigated by overexpressing Hsps. Another way of addressing this question is to evaluate the effect of endogenous Hsps on A3G mutational activity that has been suggested by the effect of Hsp90 inhibitor (Fig. 11). To further verify the Hsp90 potential role in physiological conditions, we investigated the effect of endogenous Hsp expression knockdown by siRNAs on HBV mutation frequency.
As shown in Fig. 13A, Hsp90β or Hsp90α gene expression could be knocked down to 25 or 35% of blank control by their siRNAs, respectively. However, the 25% Hsp90β knockdown resulted in a Hsp90α expression increase up to 33% of the control, and the 35% Hsp90α knockdown led to an up to 35% Hsp90β increase. In addition, either Hsp70 or Hsp40 knockdown also led to up to 50% increases for both Hsp90β and Hsp90α expression. These data demonstrate that there is a dynamic compensatory regulation among Hsps. Nevertheless, the endogenous Hsp90β and Hsp90α expression can be knocked down simultaneously to 29 and 43% of the control, respectively, by the siRNAs against both Hsp90β and Hsp90α at the same time. The Hsp mRNA expression knockdowns paralleled their protein levels, as verified by Western blotting in Fig. 13B.
Figure 13.

Effect of Hsp knockdown through siRNA on HBV mutation frequency. Each Hsp siRNA was transfected into HepG2 cells by a reverse transfection method with Lipofectamine RNAiMax. After a 24-h siRNA treatment, the cells were transfected with an HBV viral genome-encoding plasmid with or without A3G. The HepG2 cells were harvested for RNA or HBV extraction 24 h after HBV transfection. A, Hsp mRNA levels after siRNA treatment by quantitative RT-PCR determination. The relative mRNA levels for each siRNA treatment were determined by quantitative RT-PCR, using GAPDH as an internal reference. The mRNA levels relative to control were calculated and are represented graphically as percentages with the control as 100%. Each bar represents the average of triplicates for each treatment. Blank, background control by a scrambled negative siRNA without A3G co-transfection. Control, another control by the scrambled negative siRNA with A3G co-transfection. B, Hsp protein expression level analyses after siRNA treatment. Total cellular proteins were extracted from HepG2 cells after siRNA treatment, and Hsp protein levels in the cell lysates were analyzed by Western blotting with antibodies against endogenous Hsp90β, Hsp90α, Hsp70, and Hsp40. GAPDH was analyzed as the protein loading reference. C, HBV DNA mutation analyses. HBV DNAs were extracted from the cell lysates after a 24-h HBV transfection, and the resultant HBV DNA mutations were determined by pe1453 using an 88 °C 3D-PCR. The data are presented graphically on the right. Each bar represents the average of triplicates for each treatment. Asterisks, statistically significant differences comparing treatments with their corresponding control: **, 0.01 < p < 0.001; *, 0.05 < p < 0.01. The one-factor ANOVA statistical test was used.
HBV viral production was performed under the siRNA treatments, and the resultant HBV mutation frequencies were analyzed. Due to the need to remove siRNA after 24 h of treatment for initiation of HBV plasmid transfection, HBV viral production in HepG2 cells had to be shortened from the usual 2 days to 1 day to observe the effect of dynamically knocked down Hsps on HBV mutation. This change led to lower but still detectable HBV mutations. As shown in Fig. 13C, the blank control by a negative scrambled siRNA had a 0.9% HBV mutation frequency, reflecting the activity of the endogenously expressed APOBEC-3s in HepG2 cells. A small but repeatedly detectable decrease from the control 0.9% to 0.6 or 0.7% was observed upon siRNA treatment against Hsp90β, Hsp90α, or Hsp90α+β that did not reach statistical significance. The HBV mutation activity was increased from 0.9 to 13.0% by A3G co-expression. Statistically significant decreases from the control 13.0% to 5.5 or 7.6% were observed for siHsp90β or siHsp90α treatment, respectively, in the presence of elevated A3G expression. The siHsp90α+β treatment also resulted in a decrease from 13.0 to 10.9%, although there was no statistically significant difference due to sample variation. In addition, siHsp40 knockdown also resulted in A3G activity decrease from 13.0 to 7.2%, whereas siHsp70 had no apparent effect. These data demonstrate that endogenous Hsp90 expression knockdown leads to a decrease of HBV mutation frequency in the absence and presence of A3G co-expression, indicating that Hsp90 may have a modulatory effect on A3G activity under physiological conditions.
Discussion
APOBEC-3 proteins play an important role in single-base substitution mutations targeting viruses and producing cancers (15–17, 24). APOBEC-3 mutational activity on various viral genomes has been extensively investigated due to their important contribution to innate immunity against viruses. APOBEC-3 proteins are highly efficient in mutating their viral DNA cytosine targets (34, 38, 49). As a result, the mutated DNAs contain many C-to-T transitions that can be selectively amplified by 3D-PCR due to the lower denaturing temperature associated with the C-to-T mutation (34, 38, 49). The 3D-PCR amplification itself demonstrates the presence of highly mutated viral DNAs in the total population. APOBEC-3's highly efficient mutational activity is important for host defense against viruses because sublethal mutagenesis may not disable viruses but rather contribute to viral variation, leading to viral immune escape or drug resistance (50). Similarly, C-to-T mutations are also frequently observed as clusters within small regions of genomes in cancer named kataegis (“thunderstorm”) (51–53). Kataegis probably reflects the mutational characteristics of APOBEC-3 proteins in cancers (51–55). APOBEC-3 proteins alone have deaminase activity in in vitro reactions, but they do not achieve the highly efficient mutational activity that is often observed in vivo. We postulate that there are host protein factors that help APOBEC-3s to achieve the highly efficient mutational activity in vivo. It appears that Hsps could be such a candidate because Hsps are often elevated along with somatic mutations occurring in cancer. Therefore, we investigated how Hsps affected the mutational activity of APOBEC-3 proteins utilizing HBV viral DNA mutation in HepG2 cells.
We utilized the HBV viral system for the investigation because of its relative simplicity. APOBEC-3s and Hsps can be encapsulated into HBV viral capsids by co-transfection with the HBV genome in HepG2 cells. HBV DNA mutation occurring in capsids is dependent on the activity of APOBEC-3 proteins (24, 35). The effect of Hsps on APOBEC-3 mutation activity can be determined by a side-by-side comparison of the HBV DNA mutation analyses. However, the HBV mutation frequency resulting from APOBEC-3 deamination is generally low. We found that a combination of 3D-PCR and primer extension allowed a quantitative comparative determination of the HBV mutation frequency even at low levels. The mutation levels are significantly increased and can be easily detected by primer extension analyses after 3D-PCR enrichment. The primer extension uses a single specific primer to anneal randomly to a template in the PCR or 3D-PCR pool, and then a population ratio is determined through the analyses of primer-extended products. Although 3D-PCR is not a linear amplification reflecting the actual DNA mutational levels like regular PCR, the 3D-PCR followed by primer extension analyses provides a sensitive measurement of Hsps' effects on APOBEC-3s through side-by-side comparisons. On the other hand, conventional clonal sequencing analyses are also important, although they are labor-intensive and less sensitive. Sequencing analyses provide a landscape view of multiple simultaneous C-to-T mutations over a region, whereas primer extension analyses provide a mutation frequency at a single point. Together, these two approaches provide comprehensive and confirmative mutation data. Because multiple cytosine mutations often occur simultaneously in a single molecule, especially with A3B, primer selection for both PCR amplification and primer extension becomes critical for successful analyses. The primers need to bind in regions without mutations or with infrequent mutations so that this detection approach can accurately determine APOBEC-3 mutations.
With a combination of 3D-PCR amplification and primer extension analyses, we found that multiple Hsps, including Hsp40, -60, -90, and -104, stimulated the mutation activity of A3G, A3C, and A3B on HBV DNA in HepG2 cells, whereas Hsp70 had a unique effect on A3B (Figs. 3 and 5). This stimulation consistently occurs despite the structural variation among A3G, A3C, and A3B, indicating a common mechanism for the interaction between Hsps and APOBEC-3s. Hsps are normally expressed in the human liver cell line HepG2. Although the actual levels of different kinds of Hsps in HepG2 cells are unknown, the observed Hsps' stimulatory APOBEC-3 activity demonstrates that these Hsps are highly overexpressed above endogenous levels to result in the difference. On the other hand, we found that Hsp40, -60, or -70 had no significant effect on A3G mutation activity when tested by in vitro mutation reactions. Only Hsp90 and Hsp104 had a consistent stimulating effect both in vivo and in vitro (Fig. 8). The contrast between in vivo and in vitro results suggests that Hsp90 may be a common factor that other Hsps work through as co-chaperones. Hsp104 has an effect comparable with Hsp90 in the HBV mutation analyses both in vitro and in vivo. However, Hsp104 expression and availability in vivo are not comparable with Hsp90. Therefore, Hsp90 is likely to be more important in stimulating APOBEC-3 mutational activity. In fact, the Hsp90-specific inhibitor 17-AAG inhibited the mutational activity of both A3G and A3B in a dose-dependent manner (Fig. 11), and Hsp90 had a protective effect on A3G and A3B protein stability (Fig. 12). Hsp90 mutations with reduced ATPase activity decreased the Hsp90 stimulatory effect on A3G (Fig. 12). Hsp90 expression knockdown through siRNA led to a decreased HBV mutation frequency with or without A3G co-expression. These data suggest that Hsp90 may have a modulatory effect on APOBEC-3 mutation activity in vivo.
In addition to the quantitative HBV mutation analyses by primer extension mentioned above, conventional clonal sequencing provides further insight on C-to-T mutation distribution and frequency in the HBV region evaluated. We found that Hsp90 increased the A3G and A3B mutational efficiency in individually targeted HBV DNAs as well as in the overall total population mutational frequency. In vitro HBV mutation reactions with purified A3G and Hsp90β showed that A3G alone had an average of 4.5 C-to-T mutations per individual clone. The average number of C-to-T mutations per clone remained about the same, 4.5 versus 5.1 when A3G was increased from 0.5 to 4 μl (Fig. 9 and Table 1). However, the average number increased about 2-fold when Hsp90β was added to the in vitro A3G reactions, indicating that Hsp90β increased the A3G mutation efficiency in individually targeted HBV DNAs (Fig. 9 and Table 1). A similar Hsp90 effect was also observed by the cellular co-expression of Hsp90 with A3G or A3B in HepG2 cells. A3G alone had an average of 5 C-to-T mutations in individual HBV DNA clones with mutations (Fig. 4 and Table 1). However, the average number of C-to-T mutations per clone was increased from 5 to 9.3 and 7.4 by the co-expression of Hsp90α and Hsp90β, respectively. Hsp90α and Hsp90β also significantly increased the A3G mutation preference for 5′-C in vivo (Fig. 10) and shifted the A3G regional location of edited cytosines in HBV both in vivo and in vitro (Figs. 4 and 9). On the other hand, the A3G mutational frequency in the total population by primer extension analyses at cytosine site 1664 was increased from 12.6% by A3G alone to 32.7 or 34.2% by the co-expression of A3G with Hsp90α or Hsp90β, respectively (Fig. 3). Together, these data demonstrate that Hsp90 increased the A3G mutational efficiency in individual targeted HBV DNAs, mutational frequency in the total population, 5′-C cytosine preference, and reginal selection of cytosine, suggesting an important modulatory effect of Hsp90 on APOBEC-3 protein mutational activity.
APOBEC-3B is considered as the most likely candidate to play an important role in cancer DNA mutation because of its nuclear location and the frequent presence of its 5′ nucleotide mutation preference, 5′-TC in cancer DNA mutations (18–20, 56). We found that A3B alone had an average of 9.8 C-to-T mutations per HBV DNA clone (see Table 1). In contrast, A3G only had an average 5 C-to-T mutations per clone, indicating that A3B has much higher mutational efficiency than A3G. The A3B average mutated cytosine per clone can be further increased from 9.8 to 56.4 by the co-expression of Hsp90α (Fig. 6 and Table 1). A3B can mutate up to 74% of the cytosines in the HBV region containing a total of 117 cytosines. When the A3B N-terminal first 60 amino acids were replaced with the corresponding part of A3G (i.e. A3G60B), the average C-to-T mutations per clone decreased from 56.4 to 24.7 (Fig. 6 and Table 1). These data further demonstrate that A3B is a very potent mutational agent in the presence of elevated Hsp90. The potent A3B mutational activity may be the reason why A3B is especially associated with mutagenesis in cancer.
It has been reported that a highly prevalent chimeric A3A-A3B deletion allele exists in up to 30–40% of people in Southeast Asia and China and >90% in Oceania and links A3B expression absence to a greater risk of developing breast, ovarian, and liver cancer (23). Although the chimeric A3A-A3B deletion allele should lead to a higher A3A expression by in vitro investigation, cellular genomic DNA is not affected even when A3A expression is increased up to 100-fold by interferon (30), suggesting that the absence of A3B expression instead of A3A increased expression might play a role in the clinical outcome of the chimeric A3A-A3B deletion allele. A3B is up-regulated in many kinds of cancer (16–20). During ontogenesis, Hsp expression levels increase in response to various protein-denaturing stresses (1, 2). Taken together, both A3B and Hsp expression are increased during cancer development. As a result, the A3B mutational activity in the cancer cells could be enhanced by increased Hsp expression, as we found in this report. These data suggest that A3B could have a defensive role against cancer cells by inducing mutations through the interaction with other host factors, including Hsp90.
In summary, we found that multiple Hsps stimulate the HBV mutational activity of APOBEC-3s, including A3G, A3C, and A3B. Among them, Hsp90 stimulates both in vivo and in vitro. Hsp90 not only increases the overall mutational frequency of APOBEC-3s, but also their cytosine-mutating efficiency in affected individual DNA molecules. Inhibition of Hsp90 diminishes A3G and A3B mutational activity dose-dependently, suggesting that Hsp90 may have an important modulatory effect on APOBEC-3 protein deamination activity in vivo.
Experimental procedures
Plasmid DNA constructs
The plasmid encoding the HBV viral genome was a gift from Dr. Josef Kock (37). APOBEC-3A, A3B-mutant, A3C, A3F, A3G, and A3H were PCR-amplified from human cells or tissue and cloned into a pcDNA3.2 vector. Briefly, the RT-PCR amplicons of full-length open reading frame cDNA from the human acute monocytic leukemia cell line THP-1 were used for A3G and A3C; human liver for A3F; and human macrophage for A3A, A3B-mutant, and A3H. APOBEC-3s were constructed with a 9-amino acid HA tag preceded by a 3-alanine spacer at the C terminus of each gene and cloned into a pENTR1A vector (Thermo Fisher Scientific) using appropriate restriction enzyme sites. After the inserted genes were confirmed by plasmid DNA sequencing, the gene inserts were transferred into the expression vector pcDNA3.2-DEST (Thermo Fisher Scientific) by a DNA recombination reaction.
The A3B long form variant was purchased from Origene. The A3B wild type form was generated by internal deletion of the A3B long form using the QuikChange II XL kit (Stratagene) according to the manufacturer's instructions. The modified A3B containing the N-terminal 60 aa from A3G, named A3G60B, was generated by a two-step PCR of the fragments and cloned into the pcDNA3.2 vector (57). In addition, A3A and A3B wild type in the pCMV6-Entry vector from Origene and EGFP from Clontech were also purchased for comparison and control studies.
Hsp40 (DNAJB1), Hsp70 (HSPA1A), Hsp90α, Hsp90β, Hsp104, and Hsp60 genes were purchased from Origene or Addgene. All Hsps were constructed with a FLAG tag (DYKDDDDK) immediately after the N-terminal methionine and cloned into pcDNA3.2 expression vectors through an intermediate vector pENTR1A, as described above. All of the plasmid constructs were verified by DNA sequencing, in vitro protein translation, Western blotting, and functional analyses in HepG2 cells. The purified proteins, including Hsp40, Hsp70, Hsp90, Hsp104, Hsp60, and A3G, were purchased from Abnova. The siRNAs used in this investigation, including Hsp90β (ID no. 289652), Hsp90α (ID no. 122317), Hsp70 (ID no. 145248), and Hsp40 (ID no. 145306), were purchased from Thermo Fisher Scientific.
Cell culture and transfection
The human liver cell line HepG2 was purchased from ATCC and was maintained in EMEM containing 10% fetal bovine serum. For the investigation of APOBEC-3 mutational activity on HBV viral DNA, HepG2 cells were plated on 35-mm 6-well collagen-coated plates the day before transfection. Plasmids of Hsps, APOBEC-3s, and HBV in a ratio of 4:1.5:0.5 were mixed and co-transfected into HepG2 cells by X-tremeGENE HP (Roche Applied Science) according to the manufacturer's instructions. A total of 2 μg of plasmid DNA and 6 μl of transfection reagents were added to HepG2 cells in each well. Three separate samples were prepared for each evaluation. An empty pcDNA3.1 vector was used as a control. After 48 h post-transfection, the intracellular HBV viral capsids were harvested by cell lysis in a buffer containing 50 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, and 0.5% Nonidet P-40. After pretreatment with 10 units of micrococcal nuclease (New England Biolabs) in the presence of 2 mm CaCl2 at 37 °C for 1 h, the cell lysate was digested with 20 μl of 20 mg/ml proteinase K for 15 min at 55 °C, and the HBV DNA was extracted by a Pure Link genomic DNA purification kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
For the protein expression analyses, APOBEC-3s and Hsps were co-transfected into HepG2 cells under the same conditions as described above. The cells were lysed in the presence of proteinase inhibitor mixtures (Roche Applied Science) or directly by SDS sample buffer. The expressed protein levels were determined by electrophoresis in a 10% NuPAGE BisTris gel followed by Western blotting analyses using antibodies against native A3G, native A3B, native Hsp90β, HA tag in APOBEC-3s, or FLAG tag in Hsps (purchased from Sigma or Santa Cruz Biotechnology).
For siRNA-treated HBV mutational analyses, siRNAs (20 nm) were transfected into HepG2 cells by Lipofectamine RNAiMax (Thermo Fisher Scientific), using a reverse transfection method according to the manufacturer's instruction. After 24 h of siRNA transfection, the siRNA-containing medium was replaced by fresh medium, and the HepG2 cells were transfected with HBV viral genome-encoding plasmid with or without A3G as described above, and the resultant HBV viral capsids were harvested 24 h after HBV transfection.
HBV DNA mutation analyses
HBV mutational levels were quantitatively determined by the primer extension method as described previously (39, 49) after PCR or 3D-PCR amplifications. Briefly, HBV DNA was first amplified by PCR from the HBV DNA extracts after treatments and denatured for 2 min at 94 °C with primers X-F (ccggaattccgcaaatatacatcgtatccat) and X-R (aagaatgcggccgctaaaaagttgcatggtgctggtg), followed by 38 cycles of 30 s at 94 °C, 1 min at 58 °C, 2 min at 72 °C, and a final 7 min at 72 °C. The 94 °C PCRs were diluted 1:10, and a 2-μl aliquot was used for further mutated HBV DNA enrichment by 3D-PCR on a RoboCycler (Stratagene) using X-F and X-R primers. The 3D-PCR was performed by 5 min at denaturing temperatures (88 °C for example), followed by 35 cycles of 1 min at the denaturing temperature, 1 min at 58 °C, 2 min at 72 °C, and a final 7-min extension at 72 °C. The PCR amplicons were detected by 1% agarose gel electrophoresis.
For HBV mutational analyses by primer extension, PCR or 3D-PCR amplifications were purified by a QIAEX II gel extraction kit (Qiagen). An aliquot of the purified DNA amplicons was subjected to a thermal cycle primer extension reaction in the presence of ddGTP with a 5′-32P-end-labeled primer: tccccttctccgtctgccgtt for the cytosine at site 1674 (pe1674), caaagactgtttgtttaaaga for site 1453 (pe1453), or tccgtctgccgttccgaccga for site 1664 (pe1664). The cytosine sites were numbered according to their positions in HBV by reference to V01460.1. The primer extension reactions were performed by 1 min at 95 °C followed by 50 cycles of 15 s at 95 °C, 30 s at 56 °C, 45 s at 72 °C, and a final 4 min at 72 °C. The primer extension products were separated by electrophoresis in an 8% polyacrylamide-urea sequencing gel and quantitated by a PhosphorImager. Primer extension stopped at the target cytosine site due to ddGTP incorporation. When the target cytosine was mutated, the primer extension continued through the site until the next cytosine downstream and resulted in longer oligonucleotide products on the sequencing gel. The HBV mutational frequency at the target cytosine site was determined by the percentage of these bands above the target cytosine oligonucleotide divided by the total product counts in the lane.
A3G in vitro mutation reactions
An HBV fragment (nt 1175–2089 by reference to V01460.1) was PCR-amplified from the parent HBV plasmid and cloned into a pcDNA3.2 vector using X-FII (ccggaattcttctcgccaacttacaaggcctttc) and X-RII (aagaatgcggccgctagagctgaggcggtatctagaag). After being linearized by NotI, the HBV DNA fragment was transcribed into RNA by T7 RNA polymerase using an in vitro RNA transcription kit (Promega) according to the manufacturer's instructions. Upon removal of the DNA by treatment with DNase, the HBV RNA was then reverse-transcribed into HBV cDNA by RTase with the reverse primer. After treatment with RNase H and purification by ethanol precipitation, the HBV cDNA preparation was used as substrate for in vitro APOBEC-3 mutation tests.
For the in vitro A3G mutation analyses, purified A3G (0.07 μg) and Hsps (0.7–1.0 μg) (purchased from Abnova) were incubated with the HBV cDNA substrate (20 ng) for 2 h at 30 °C in SmartCut buffer (New England Biolabs) containing 20 mm Tris acetate, pH 7.9, 50 mm potassium acetate, 10 mm magnesium acetate, and 100 μg/ml BSA. After the in vitro mutational reactions, the HBV cDNA was then recovered by extraction with phenol/chloroform (1:1) twice, followed by ethanol precipitation. The HBV DNA was amplified by PCR at 94 °C using X-FII and X-RII primers, and the HBV-mutated DNA was further enriched by 88 °C 3D-PCR using X-F and X-R primers. The effect of Hsps on A3G mutational activity was determined by pe1453 primer extension analyses with 88 °C 3D-PCR.
For A3G dose-dependent mutational analyses in vitro, purified proteins of Hsp90β (0.17 μg/μl × 5 μl) and A3G (0.07 μg/μl × 0.5 or 4 μl) were mixed with HBV cDNA substrate (20 ng) and incubated for 3 h at 30 °C. The effect of Hsp90β on A3G mutational activity was determined by TA cloning of the 88 °C 3D-PCR amplicons. Twenty clones were randomly selected for each treatment and evaluated by sequencing analyses.
Protein interaction analyses
The protein interaction between Hsp90 and A3G was analyzed by immunoprecipitation from co-transfection in HepG2 cells or co-translation in vitro using the TNT quick coupled transcription and translation systems (Promega). Briefly, the cell lysates or in vitro translation products were immunoprecipitated using an antibody against the HA tag in A3G (Sigma) with a 60-min incubation at room temperature, followed by precipitation with Protein G-Dynabeads (Thermo Fisher Scientific). The immunoprecipitated protein complexes were analyzed by electrophoresis in a 10% NuPAGE BisTris gel followed by Western blotting analyses using antibodies against native Hsp90β.
For additional protein interaction analyses between Hsps and A3G or A3C, Hsps were co-translated with A3G or A3C in a plasmid ratio (1:1) by TNT quick coupled transcription and translation systems at 30 °C for 2 h in the presence of l-[35S]methionine. The protein complexes formed during the in vitro co-translation were immunoprecipitated using an antibody against the HA tag in A3G or A3C with incubation for 60 min at room temperature followed by precipitation with Protein G-Dynabeads. The immunoprecipitated protein complexes were analyzed directly by 12% SDS-polyacrylamide gel separation followed by detection with a PhosphorImager. The empty vector, pcDNA3.1, was used as a blank control. As a verification of protein translation, an aliquot before immunoprecipitation was denatured directly in SDS-sample buffer and analyzed by a 12% SDS-polyacrylamide gel for a parallel reference of each translated protein.
Quantitative RT-PCR for mRNA expression analyses
The total cellular RNA in HepG2 cells was extracted using TRIzol reagent according to the manufacturer's instructions (Thermo Fisher Scientific). Fifteen-microgram total RNA aliquots were taken and denatured briefly at 90 °C for 2 min. The RNAs were reverse-transcribed into cDNA at 37 °C for 1 h using random primer and Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific). The cDNA preparation was diluted to 80 μl, and 5-μl aliquots were used for quantitative PCR analyses using the SYBR Select master mix kit (Thermo Fisher Scientific) on a StepOne Plus machine. The PCR was performed for 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 45 s at 58 °C, and 1 min at 72 °C. GAPDH was used as an internal reference for the relative gene expression level quantitation. The primers used in this investigation were as follows: Hsp90β-F, gaaccattgccaagtctggt; Hsp90β-R, tttgacccgcctctcttcta; Hsp90α-F, aaacacctggagataaaccc; Hsp90α-R, gtatcatcagcagtagggtca; Hsp70-F, cgacctgaacaagagcatca; Hsp70-R, ctcgtacacctggatcagca; Hsp40-F, aagaagcaagatcccccagt; Hsp40-R, gctggaatgttgttggaggt; GAPDH-F, cgaccactttgtcaagctca; and GAPDH-R, aggggtctacatggcaactg.
The relative Hsp mRNA levels were calculated by a ΔΔCt method according to their Ct values determined by quantitative RT-PCR as described by the equipment manufacturer (Thermo Fisher Scientific). The Ct values for each sample were subtracted from their corresponding GAPDH Ct first. The resultant ΔCt for each sample was then subtracted from the ΔCt mean value of blank control to yield ΔΔCt. The gene expression level in each sample was finally presented as a percentage relative to the blank control by the power of 2 of its negative ΔΔCt multiplied by 100 for a graphic side-by-side comparison.
Author contributions
Z. C., T. L. E., and A. P. P. designed the study and wrote the paper. A. V. B., I. N. B., T. G. V., and R. K. provided technical assistance, data analyses, and consulting. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. 1.
- Hsp
- heat shock protein
- APOBEC-3
- apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3
- A3A
- -B, -C, -F, -G, and -H, APOBEC-3A, -3B, -3C, -3F, -3G, and -3H
- HBV
- hepatitis B virus
- 3D-PCR
- differential DNA denaturation-PCR
- pe
- primer extension
- siHsp90
- siRNA against Hsp90
- AID
- activation-induced cytidine deaminase
- EGFP
- enhanced green fluorescent protein
- PES
- 2-phenylethynesulfonamide
- aa
- amino acid(s)
- IP
- immunoprecipitation
- 17-AAG
- 17-N-allylamino-17-demethoxygeldanamycin
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- HOP
- Hsp70-Hsp90 organizing protein.
References
- 1. Lianos G. D., Alexiou G. A., Mangano A., Mangano A., Rausei S., Boni L., Dionigi G., and Roukos D. H. (2015) The role of heat shock proteins in cancer. Cancer Lett. 360, 114–118 [DOI] [PubMed] [Google Scholar]
- 2. Whitesell L., and Lindquist S. L. (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 3. Calderwood S. K., Khaleque M. A., Sawyer D. B., and Ciocca D. R. (2006) Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem. Sci. 31, 164–172 [DOI] [PubMed] [Google Scholar]
- 4. Ciocca D. R., and Calderwood S. K. (2005) Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10, 86–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ciocca D. R., Arrigo A. P., and Calderwood S. K. (2013) Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch. Toxicol. 87, 19–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khurana N., and Bhattacharyya S. (2015) Hsp90, the concertmaster: tuning transcription. Front. Oncol. 5, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sahni N., Yi S., Taipale M., Fuxman Bass J. I., Coulombe-Huntington J., Yang F., Peng J., Weile J., Karras G. I., Wang Y., Kovács I. A., Kamburov A., Krykbaeva I., Lam M. H., Tucker G., et al. (2015) Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jarosz D. (2016) Hsp90: a global regulator of the genotype-to-phenotype map in cancers. Adv. Cancer Res. 129, 225–247 [DOI] [PubMed] [Google Scholar]
- 9. Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., and Lindquist S. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pearl L. H. (2016) Review: the HSP90 molecular chaperone: an enigmatic ATPase. Biopolymers 105, 594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maisnier-Patin S., Roth J. R., Fredriksson A., Nyström T., Berg O. G., and Andersson D. I. (2005) Genomic buffering mitigates the effects of deleterious mutations in bacteria. Nat. Genet. 37, 1376–1379 [DOI] [PubMed] [Google Scholar]
- 12. Masel J., and Trotter M. V. (2010) Robustness and evolvability. Trends Genet. 26, 406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trepel J., Mollapour M., Giaccone G., and Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Workman P., Burrows F., Neckers L., and Rosen N. (2007) Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N.Y. Acad. Sci. 1113, 202–216 [DOI] [PubMed] [Google Scholar]
- 15. Roberts S. A., and Gordenin D. A. (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 14, 786–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swanton C., McGranahan N., Starrett G. J., and Harris R. S. (2015) APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 5, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henderson S., and Fenton T. (2015) APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol. Med. 21, 274–284 [DOI] [PubMed] [Google Scholar]
- 18. Burns M. B., Lackey L., Carpenter M. A., Rathore A., Land A. M., Leonard B., Refsland E. W., Kotandeniya D., Tretyakova N., Nikas J. B., Yee D., Temiz N. A., Donohue D. E., McDougle R. M., Brown W. L., et al. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burns M. B., Temiz N. A., and Harris R. S. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cescon D. W., Haibe-Kains B., and Mak T. W. (2015) APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc. Natl. Acad. Sci. U.S.A. 112, 2841–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shinohara M., Io K., Shindo K., Matsui M., Sakamoto T., Tada K., Kobayashi M., Kadowaki N., and Takaori-Kondo A. (2012) APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caval V., Bouzidi M. S., Suspène R., Laude H., Dumargne M. C., Bashamboo A., Krey T., Vartanian J. P., and Wain-Hobson S. (2015) Molecular basis of the attenuated phenotype of human APOBEC3B DNA mutator enzyme. Nucleic Acids Res. 43, 9340–9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caval V., Suspène R., Shapira M., Vartanian J. P., and Wain-Hobson S. (2014) A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat. Commun. 5, 5129. [DOI] [PubMed] [Google Scholar]
- 24. Harris R. S., and Dudley J. P. (2015) APOBECs and virus restriction. Virology 479, 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suspène R., Sommer P., Henry M., Ferris S., Guétard D., Pochet S., Chester A., Navaratnam N., Wain-Hobson S., and Vartanian J. P. (2004) APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32, 2421–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris R. S., and Liddament M. T. (2004) Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4, 868–877 [DOI] [PubMed] [Google Scholar]
- 27. Harris R. S., Bishop K. N., Sheehy A. M., Craig H. M., Petersen-Mahrt S. K., Watt I. N., Neuberger M. S., and Malim M. H. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809 [DOI] [PubMed] [Google Scholar]
- 28. Koito A., and Ikeda T. (2013) Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Refsland E. W., Stenglein M. D., Shindo K., Albin J. S., Brown W. L., and Harris R. S. (2010) Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stenglein M. D., Burns M. B., Li M., Lengyel J., and Harris R. S. (2010) APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17, 222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard B., McCann J. L., Starrett G. J., Kosyakovsky L., Luengas E. M., Molan A. M., Burns M. B., McDougle R. M., Parker P. J., Brown W. L., and Harris R. S. (2015) The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 75, 4538–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vieira V. C., Leonard B., White E. A., Starrett G. J., Temiz N. A., Lorenz L. D., Lee D., Soares M. A., Lambert P. F., Howley P. M., and Harris R. S. (2014) Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 10.1128/mBio.02234-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hultquist J. F., Lengyel J. A., Refsland E. W., LaRue R. S., Lackey L., Brown W. L., and Harris R. S. (2011) Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vartanian J. P., Henry M., Marchio A., Suspène R., Aynaud M. M., Guétard D., Cervantes-Gonzalez M., Battiston C., Mazzaferro V., Pineau P., Dejean A., and Wain-Hobson S. (2010) Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 6, e1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beck J., and Nassal M. (2007) Hepatitis B virus replication. World J. Gastroenterol. 13, 48–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Glebe D., and Bremer C. M. (2013) The molecular virology of hepatitis B virus. Semin. Liver Dis. 33, 103–112 [DOI] [PubMed] [Google Scholar]
- 37. Köck J., and Blum H. E. (2008) Hypermutation of hepatitis B virus genomes by APOBEC3G, APOBEC3C and APOBEC3H. J. Gen. Virol. 89, 1184–1191 [DOI] [PubMed] [Google Scholar]
- 38. Suspène R., Henry M., Guillot S., Wain-Hobson S., and Vartanian J. P. (2005) Recovery of APOBEC3-edited human immunodeficiency virus G → A hypermutants by differential DNA denaturation PCR. J. Gen. Virol. 86, 125–129 [DOI] [PubMed] [Google Scholar]
- 39. Patterson A. P., Chen Z., Rubin D. C., Moucadel V., Iovanna J. L., Brewer H. B. Jr., and Eggerman T. L. (2003) Developmental regulation of apolipoprotein B mRNA editing is an autonomous function of small intestine involving homeobox gene Cdx1. J. Biol. Chem. 278, 7600–7606 [DOI] [PubMed] [Google Scholar]
- 40. Bonvin M., and Greeve J. (2007) Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J. Gen. Virol. 88, 3270–3274 [DOI] [PubMed] [Google Scholar]
- 41. Pak V., Heidecker G., Pathak V. K., and Derse D. (2011) The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J. Virol. 85, 8538–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orthwein A., Patenaude A. M., Affar el B., Lamarre A., Young J. C., and Di Noia J. M. (2010) Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J. Exp. Med. 207, 2751–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soros V. B., Yonemoto W., and Greene W. C. (2007) Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prodromou C. (2016) Mechanisms of Hsp90 regulation. Biochem. J. 473, 2439–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Demorest Z. L., Li M., and Harris R. S. (2011) Phosphorylation directly regulates the intrinsic DNA cytidine deaminase activity of activation-induced deaminase and APOBEC3G protein. J. Biol. Chem. 286, 26568–26575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meyer P., Prodromou C., Hu B., Vaughan C., Roe S. M., Panaretou B., Piper P. W., and Pearl L. H. (2003) Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658 [DOI] [PubMed] [Google Scholar]
- 47. Michels A. A., Kanon B., Bensaude O., and Kampinga H. H. (1999) Heat shock protein (Hsp) 40 mutants inhibit Hsp70 in mammalian cells. J. Biol. Chem. 274, 36757–36763 [DOI] [PubMed] [Google Scholar]
- 48. Fontaine S. N., Rauch J. N., Nordhues B. A., Assimon V. A., Stothert A. R., Jinwal U. K., Sabbagh J. J., Chang L., Stevens S. M. Jr., Zuiderweg E. R., Gestwicki J. E., and Dickey C. A. (2015) Isoform-selective genetic inhibition of constitutive cytosolic Hsp70 activity promotes client Tau degradation using an altered co-chaperone complement. J. Biol. Chem. 290, 13115–13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen Z., Eggerman T. L., Bocharov A. V., Baranova I. N., Vishnyakova T. G., Kurlander R. J., Csako G., and Patterson A. P. (2012) Hypermutation of ApoB mRNA by rat APOBEC-1 overexpression mimics APOBEC-3 hypermutation. J. Mol. Biol. 418, 65–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sadler H. A., Stenglein M. D., Harris R. S., and Mansky L. M. (2010) APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 84, 7396–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lada A. G., Dhar A., Boissy R. J., Hirano M., Rubel A. A., Rogozin I. B., and Pavlov Y. I. (2012) AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol. Direct 7, 47; discussion 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sakofsky C. J., Roberts S. A., Malc E., Mieczkowski P. A., Resnick M. A., Gordenin D. A., and Malkova A. (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 7, 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taylor B. J., Nik-Zainal S., Wu Y. L., Stebbings L. A., Raine K., Campbell P. J., Rada C., Stratton M. R., and Neuberger M. S. (2013) DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2, e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lada A. G., Kliver S. F., Dhar A., Polev D. E., Masharsky A. E., Rogozin I. B., and Pavlov Y. I. (2015) Disruption of transcriptional coactivator Sub1 leads to genome-wide re-distribution of clustered mutations induced by APOBEC in active yeast genes. PLoS Genet. 11, e1005217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lada A. G., Stepchenkova E. I., Waisertreiger I. S., Noskov V. N., Dhar A., Eudy J. D., Boissy R. J., Hirano M., Rogozin I. B., and Pavlov Y. I. (2013) Genome-wide mutation avalanches induced in diploid yeast cells by a base analog or an APOBEC deaminase. PLoS Genet. 9, e1003736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leonard B., Hart S. N., Burns M. B., Carpenter M. A., Temiz N. A., Rathore A., Vogel R. I., Nikas J. B., Law E. K., Brown W. L., Li Y., Zhang Y., Maurer M. J., Oberg A. L., Cunningham J. M., et al. (2013) APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Z., Eggerman T. L., Bocharov A. V., Baranova I. N., Vishnyakova T. G., Csako G., and Patterson A. P. (2010) Hypermutation induced by APOBEC-1 overexpression can be eliminated. RNA 16, 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.