

Abstract
The nickel–iron hydrogenase enzymes efficiently and reversibly interconvert protons, electrons, and dihydrogen. These redox proteins feature iron–sulfur clusters that relay electrons to and from their active sites. Reported here are synthetic models for nickel–iron hydrogenase featuring redox-active auxiliaries that mimic the iron–sulfur cofactors. The complexes prepared are NiII(μ-H)FeIIFeII species of formula [(diphosphine)Ni(dithiolate)(μ-H)Fe(CO)2(ferrocenylphosphine)]+ or NiIIFeIFeII complexes [(diphosphine)Ni(dithiolate)Fe(CO)2(ferrocenylphosphine)]+ (diphosphine = Ph2P(CH2)2PPh2 or Cy2P(CH2)2PCy2; dithiolate = −S(CH2)3S−; ferrocenylphosphine = diphenylphosphinoferrocene, diphenylphosphinomethyl(nonamethylferrocene) or 1,1′-bis(diphenylphosphino)ferrocene). The hydride species is a catalyst for hydrogen evolution, while the latter hydride-free complexes can exist in four redox states – a feature made possible by the incorporation of the ferrocenyl groups. Mixed-valent complexes of 1,1′-bis(diphenylphosphino)ferrocene have one of the phosphine groups unbound, with these species representing advanced structural models with both a redox-active moiety (the ferrocene group) and a potential proton relay (the free phosphine) proximal to a nickel–iron dithiolate.
Introduction
Dihydrogen (H2) is present in our atmosphere at only half a part per million. Knowing this, one might be surprised that H2 is important to many prokaryotes, which metabolize H2 by way of the hydrogenases (H2ases), metalloenzymes expressed to either consume or generate reducing equivalents according to the surrounding chemical activities.[1] Two major enzyme classes, the nickel–iron and diiron hydrogenases, are responsible for the reversible generation or oxidation of H2, that is 2H+ + 2e− ⇌ H2. Of these redox enzymes, nickel–iron hydrogenases ([NiFe]-H2ases) are the more common, typically existing as heterodimers (63 + 29 kDa)[2–4] with bimetallic cores of the type (Cys-S)2Ni(μ-S-Cys)2Fe(CN)2CO. The active site can exist in several states, all of which feature low-spin metal centres on account of the strong field ligands present. Some forms feature substrate bound to the metal sites, as in the Ni-R state of [NiFe]-H2ase (Fig. 1).[3,5,6]
Fig. 1.
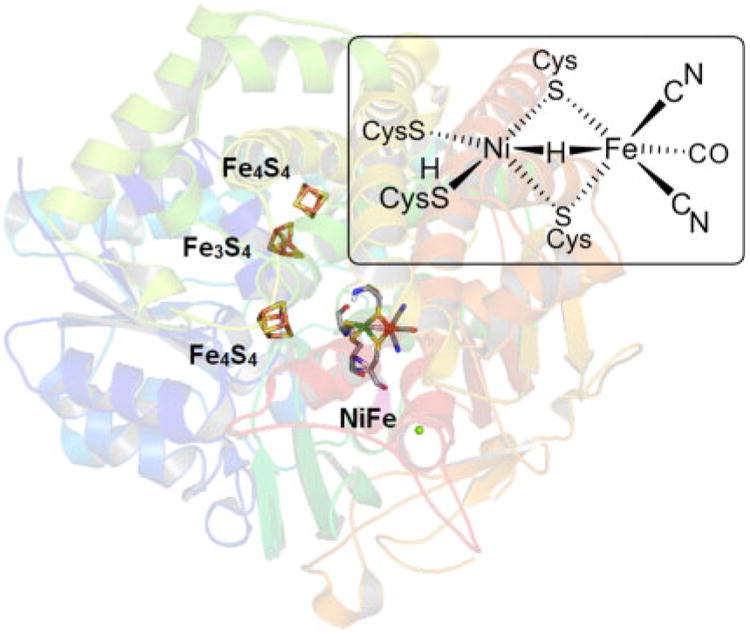
X-Ray structure of the Ni-R state of [NiFe]-H2ase from Desulfovibrio vulgaris Miyazaki F (PBD ID: 4U9H). Electrons are relayed to/from the NiFe active site by three iron–sulfur clusters.
Notably, the active site of at least one isoelectronic Ni-R substate features both an ionizable proton (bound to a terminal cysteinato ligand) and a hydride (bridging Ni and Fe), which together constitute H2. The substrates H2,H+, and e− are transported to and from the active site by very finely tuned processes. A series of hydrophobic channels conveys H2 between the NiFe site and the protein surface,[7] while a hydrogen-bonded H2O network[8] and several acidic/basic residues (among them a terminal Cys at Ni and a proximal Arg)[9] facilitate H+ transfer. The smallest substrate, e−, requires the largest machinery, namely a series of three iron– sulfur clusters, the closest of which is poised 12 Å from the NiFe centre. The trio of clusters forms a collinear chain of electron relays, each separated by ∼13 Å.[10] Returning to the active site, it was noted above that substates of Ni-R (NiII(μ-H)FeII, S = 0) can contain H+ and H−; the union of these liberates H2 and brings the protein into an electrophilic, coordinatively unsaturated state known as Ni-SIa (NiIIFeII, S = 0, Scheme 1).
Scheme 1.
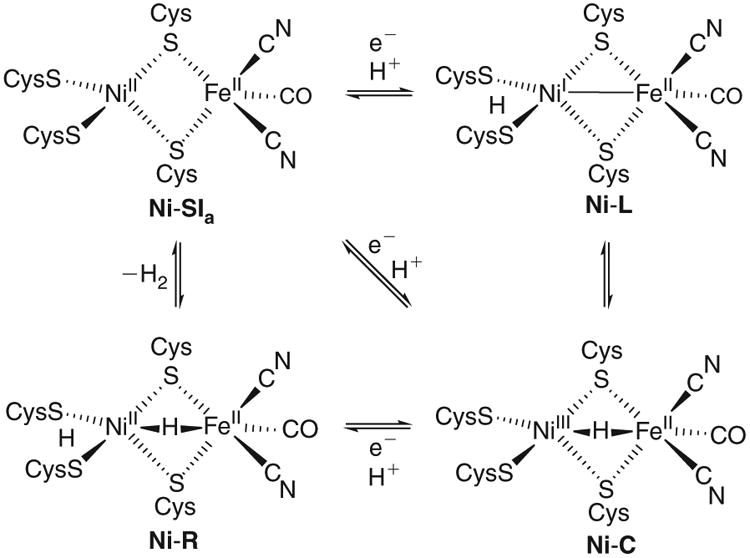
Catalytic cycle proposed for [NiFe]-H2ase-mediated H2 evolution (clockwise) and oxidation (anticlockwise).
The 1e− reduction and protonation of Ni-SIa affords hydride Ni-C (NiIII(μ-H)FeII, S = 1/2),[11,12] whose formation may proceed via an intermediate thiol tautomer Ni-L (NiIFeII, S = 1/2).[13–16] Turnover occurs upon 1e− reduction and protonation of Ni-C to give Ni-R.[11,17] Overall, H2ases mediate H2 evolution or oxidation extremely rapidly and at low over-potentials,[18–20] with the efficiency of these electrocatalysts often being ascribed to fast transfer of H+ and e− to or from the active site.
The remarkable activity of the [NiFe]-H2ases has inspired the synthesis of active site analogues, such pursuits often being motivated by the quest to obtain a more accurate picture of the native mechanism, as well as the promise of applications in renewable energy and antibiotics.[21] There are now many examples of small molecules that reproduce the topical Ni(μ-SR)2Fe core, although −CN ligands and terminal RS− groups are difficult to install owing to their propensity to bridge metals. This problem has led chemists to employ synthetic ‘surrogates’ for the native donors. Common surrogates for terminal RS− groups are tertiary amines and phosphines, while the strongly donating CN− ligands are approximated (with varying success) by using CNR, −CNBAr3, CO, and tertiary phosphines. Cyclopentadienide (Cp−) has also found its way into models, including the isomeric NiIIFeII species [(dppe)Ni(pdt)Fe(Cp)(CO)]+ and [(Cp)Ni(pdt)Fe(dppe)(CO)]+ (dppe = Ph2P(CH2)2PPh2; pdt2− = −S(CH2)3S−).[22,23] The latter reversibly accepts 1e− to give [(Cp)Ni(pdt)Fe(dppe)(CO)], a complex whose NiIFeII description matches that of the 33e− Ni-L state. While the neutral bimetallic, as well as the recently reported [(N,N′−diethyl-3,7-diazanonane-1,9-dithiolato)NiFe(CNtBu)4]+ are both NiIFeII species,[24] they contrast Ni-L in that they are 35e− complexes. This discrepancy prompted us to revisit the ‘triphosphine’ platform, with the archetypal mixed-valent example [(dppe)Ni(pdt)Fe(CO)2(PPh3)]+ ([a]+, Fig. 2)[25] featuring an unusual NiIIFeI core yet having the correct 33e− count. The oxidation states in such complexes are not obvious a priori, but become evident upon consideration of electron paramagnetic resonance (EPR) and IR spectroscopic data and density functional theory (DFT) calculations. Synthetic models for Ni-L are now in the spotlight, in no small part due to the recent observation of Ni-L during catalysis.[14] The mechanistic importance of Ni-L had previously been in question, and these new findings bring heightened relevance to the study of paramagnetic (S = 1/2) NiFe species.
Fig. 2.
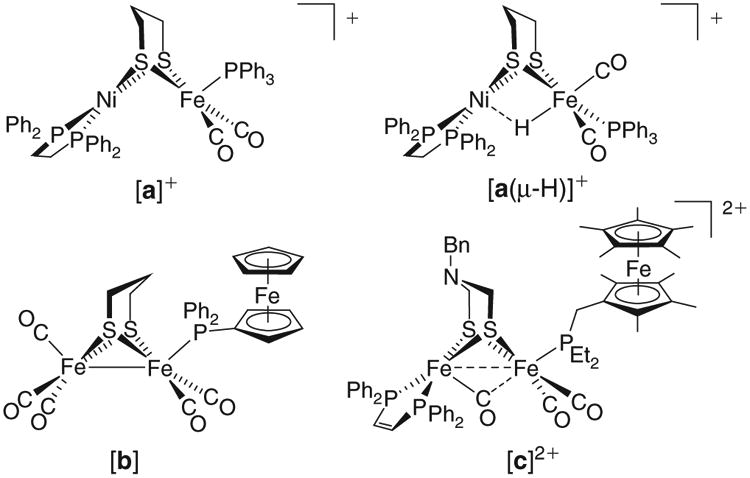
Structures of NiFe ‘triphosphine’ complexes (top) and [FeFe]-H2ase models bearing ferrocene-based redox-active ligands (bottom).
Aside from Ni-L modelling, the present study also describes new synthetic analogues for the hydride-bearing Ni-R state. In two independent breakthroughs, it was found that hydrides of formula {(N,N′-diethyl-3,7-diazanonane-1,9-dithiolato) Ni(μ-H)Fe[P(OEt)3]3}+ and [(diphosphine)Ni(pdt)(μ-H)Fe (CNBAr3)2(CO)]− could be prepared from H2.[26,27] In terms of structure, members of the latter class reproduce the native inner coordination sphere and are the highest fidelity Ni-R models to date. Anions of the type [(diphosphine)Ni(pdt)(μ-H) Fe(CNBAr3)2(CO)]− are rare examples of synthetic NiFe species that catalyse H2 oxidation, yet despite the structural similarities they share with the Ni-R active site, they are very poor H2 evolution catalysts. In this regard, the less sophisticated model [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]+ ([a(μ-H)]+) and its analogues are superior.[28,29]
The importance of Fe–S electron transport cofactors in the hydrogenases has inspired the synthesis of model complexes bearing redox-active ligands. Popular ligands include 1,2-dithiolenes, 2,3-bis(diphenylphosphino)maleic anhydride,[30] and 1,2-diimines, with a notable application of the latter described in a recent report of a NiFe complex bearing a Ni-bound 2,2′-bipyridine ligand. This complex catalyses proton reduction via an intermediate bearing an anionic bipyridine ligand; in this regard bipyridine can be considered an electron relay.[31] However, it must be noted that the basicities of the ligands described above may vary substantially from one redox state to another. The redox state of the proximal Fe–S clusters in the native [NiFe]-H2ases affect chemistry at the active sites to a lesser degree, and many chemists seek to assemble models featuring ligands whose donicities are largely independent of redox. Such ligands include ferrocenylphosphines, with diphenylphosphinoferrocene (mppf) and 1,1′-bis(diphenylphosphino)ferrocene (dppf) having been incorporated into the [FeFe]-H2ase models [(OC)3Fe(pdt)Fe(CO)2(mppf)] ([b]),[32] [(OC)2Fe(pdt)(μ-dppf)Fe(CO)2],[33] and {[(OC)3Fe(pdt)Fe (CO)2]2(μ-dppf)}.[34] Trimetallic [b] has three reversible oxidations (E1/2 = 0.77, 0.47, 0.16 V versus ferrocenium/ferrocene, Fc+/0), the mildest wave being assigned to the mppf metalloligand. The complex [(OC)3Fe(pdt)Fe(CO)2(PPh3)], in contrast, does not bear a ferrocene bound to the phosphine and exhibits only a single oxidation.[32] A later application of a ferrocenylphosphine as an electron source/sink allowed for the first example of H2 oxidation catalyzed by a [FeFe]-H2ase model.[35,36] The conversion was mediated by a triiron complex [c]2+ (Fig. 2) featuring both a H+ relay (the amino group poised near the pyramidal Fe centre for substrate transfer) as well as a diethylphosphinomethyl(nonamethylferrocene) redox-active ligand. Notably, the interaction of the FeIFeIIFeIII species [c]2+ with H2 affords a hydride complex described as FeII(μ-H)FeIIFeII, demonstrating the utility of the ferrocenium fragment (whose metal centre we will denote in italics) to mildly oxidize the diiron core and induce H2 heterolysis.
The present study elaborates on the ‘triphosphine’ motif in [NiFe]-H2ase models [(dppe)Ni(pdt)Fe(CO)2(PPh3)]+ ([a]+) and [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]+ ([a(μ-H)]+) by introducing ferrocene-containing phosphines, a theme that has found much success in the [FeFe]-H2ase modelling arena. The redox-active ligands employed are thus designed to mimic the proximal [Fe4S4]2+/+ cluster in the [NiFe]-H2ase protein; accordingly, we report here the synthesis, characterization and reactivity of trimetallic models for this most ancient, yet active, class of catalysts.
Results and Discussion
A Trimetallic Hydride Model for Ni-R
The low-valent NiIFeI tricarbonyl [(dppe)Ni(pdt)Fe(CO)3]([1], Scheme 2), while more reduced than any [NiFe]-H2ase state, represents a useful precursor to many NiFe species of relevance to the biological system. The electron-rich complex [1] is both basic (pKa(MeCN) 10.7) and reducing (E1/2 −0.520 V).[28,37] With respect to the former, the hydride [(dppe)Ni(pdt)(μ-H)Fe (CO)3]+ ([1(μ-H)]+) is readily accessible from acids of high (e.g. HBF4·Et2O, HCl·Et2O) and intermediate strength (e.g. HCO2H).
Scheme 2.
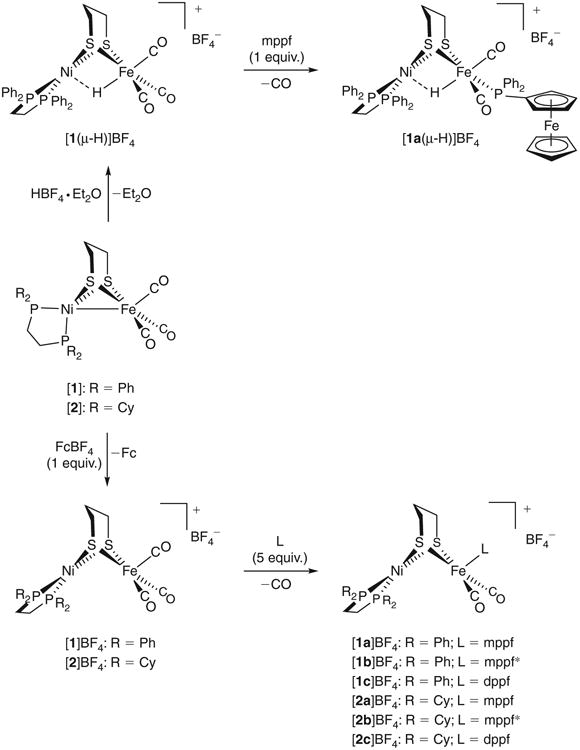
Synthesis of the hydride-containing NiII(μ-H)FeIIFeII Ni-R model (top) and the mixed-valent NiIIFeIFeII Ni-L models (bottom).
The substitution of one CO ligand with a tertiary phosphine PR3 (a much better surrogate for the native CN− ligands) is typically a facile thermal reaction, yet the use of redox-active phosphines introduces complications. Following a protocol analogous to that for [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]BF4, heating [1(μ-H)]BF4 with mppf (5 equiv.) in THF afforded not only the targeted [(dppe)Ni(pdt)(μ-H)Fe(CO)2(mppf)]BF4 ([1a (μ-H)]BF4), but also [1] and unreacted [1(μ-H)]BF4. Given that neither PPh3 nor mppf are sufficiently basic to deprotonate [1(μ-H)]+, it is instead likely that these forcing conditions induce e− transfer from mppf to [1(μ-H)]+ such that [1], H2, and [mppf]+ form. Although mppf is only a weak reductant, it is important to note that the present mixture is not at equilibrium because: (i) the H2 evolved leaves the headspace of the mixture and (ii) [1]+ does not activate H2 to give [1(μ-H)]+. When the reaction was instead performed at 40°C using a single equivalent of mppf, [1a(μ-H)]BF4 could be isolated as a brown solid whose sole positive ion in the electrospray ionization (ESI) mass spectrum (m/z 1045.1) is consistent with ligand substitution. The νCO bands at 2014 and 1962 cm−1 (versus 2016, 1965 cm−1 for [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]BF4,[a(μ-H)]BF4) are assigned to A and B symmetry species, respectively, the symmetric and anti-symmetric stretches of a cis dicarbonyl.[28] Characteristic of the hydrido ligand, 1H NMR spectra of [1a(μ-H)]BF4 feature a high-field resonance at −3.11 ppm, whose multiplicity indicates coupling to the 31P nucleus in mppf ([2]JHP 36.0), with the weaker interaction to the nuclei in dppe ([2]JHP 4.9) indicating that the μ-H− ligand resides primarily at Fe. Overall, the well resolved signals are consistent with a diamagnetic, low-spin NiII(μ-H)FeIIFeII description for [1a(μ-H)]+.
As with [a(μ-H)]+,the 31P resonance for the dppe group in [1a (μ-H)]BF4 is not evident at room temperature, although three signals (67.3 and 64.8 (dppe); 63.6 ppm (mppf)) are clear at −20°C, when rotation of dppe – through a transition state in which the NiS2P2 moiety is tetrahedral[38] – is slowed. The signals indicate that mppf occupies a ‘basal’ location trans to S, and the rapid rotation of(dppe)Ni is consistent with the μ-H− ligand interacting only weakly with Ni. A preliminary X-ray structure of [1a(μ-H)]BF4 suggests that it is isostructural to [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]BF4,[28] with mppf indeed in a basal site at an octahedral Fe centre, which is linked to square-pyramidal Ni through the dithiolate and (weakly) through the hydrido. Owing to the poor data quality, details beyond the connectivity of heavy atoms are unclear, although the proposed structure is consistent with the donor–acceptor properties of the ligands at Fe (see Supplementary Material).
Although mppf can apparently reduce [1(μ-H)]BF4 to give H2 under forcing conditions, mppf is unable to induce reduction at ambient temperature (E1/2[1(μ-H)]+/0 = 1.34 V). When its phosphine group binds a metal, mppf would be an even weaker reductant, and indeed trimetallic triphosphine [1a(μ-H)]BF4 does not spontaneously evolve H2 through an intramolecular electron transfer from the FeII ligand to the NiII(μ-H)FeII core. It appears that the present system is almost too robust, such that it is unreactive towards excess strong acid (e.g. no H2 is evolved upon treatment with [H(OEt2)2][B(3,5-(CF3)2C6H3)4]). Nevertheless, [1a(μ-H)]+ exhibits three redox waves, these appearing at Epa +0.71 ([1a(μ-H)]3+/2+, NiII(μ-H)FeIII/IIFeIII), E1/2 +0.22 (ipa/ipc 0.82, [1a(μ-H)]2+/+, NiII(μ-H)FeIIFeIII/II) and −1.63 V versus Fc+/0 (ipa/ipc 0.38, [1a(μ-H)]+/0,NiII/I(μ-H)FeIIFeII, Fig. 3). While the middle wave is reversible, the anodic and cathodic waves are less so, and one can thus write the following reactions: NiII(μ-H) FeIIIFeIII ← NiII(μ-H)FeIIFeIII ⇌ NiII(μ-H)FeIIFeII → NiI (μ-H)FeIIFeII. The poor reversibility of the cathodic wave for [1a(μ-H)]+ (as well as that for the prototypical triphosphine [a(μ-H)]+) is associated with a chemical step following reduction, of pertinence to the hydrogen evolution reaction. The reductive current for the NiII/I(μ-H)FeIIFeII couple increases linearly with the concentration of CF3CO2H added, consistent with a catalytic hydrogen evolution process second order in [CF3CO2H] (see Fig. S7, Supplementary Material). Under the conditions employed, the ratio of the reductive current in the presence and absence of acid (ic/ip 42.6) for the peak at Epc −1.37 V (potential at ic/2 = Ecat/2 = −1.33 V), corresponds to a turnover frequency of 350 s−1. The voltage required, beyond that required thermodynamically, to drive the reaction at this rate is 0.63 V. To put this in context, [a(μ-H)]+ can evolve H2 at only 50 s−1,[28] and it does so at a similar overpotential of 0.60 V, as one might expect given both complexes are triphosphines with similar electronics at their NiII(μ-H)FeII cores. In terms of turnover frequency, catalyst [1a(μ-H)]+ is even superior to [(dppe)Ni(1,2-ethanedithiolate)(μ-H)Fe(CO)3]+ (240– 310 s−1), although the latter electron-deficient complex performs at a lower overpotential (0.49 V).[29]
Fig. 3.
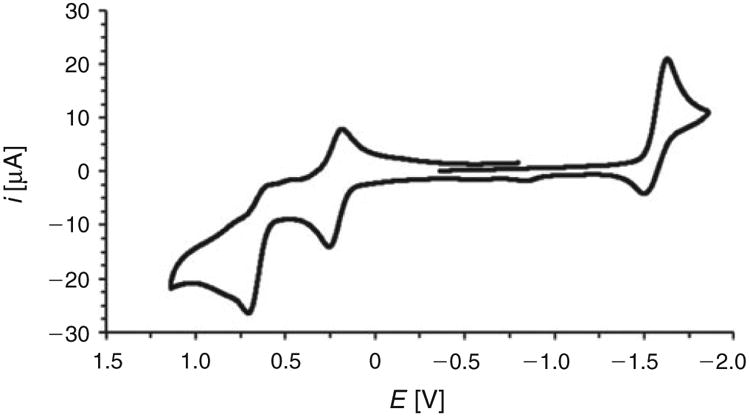
Cyclic voltammogram of [1a(μ-H)]BF4. The potential (shown here relative to Fc+/0) was swept at 50 mV s−1.
It is conceivable that the next step in improving the catalytic rates of [1a(μ-H)]+ might call for a more reducing redox agent to be attached to the NiII(μ-H)FeII core. However, attempts at installing diphenylphosphinomethyl(nonamethylferrocene) (mppf*) resulted in electron transfer to afford intractable mixtures of [1], [mppf*]+, and some of the targeted dicarbonyl. The incorporation of mppf* into trimetallic complexes can, however, be achieved when substitution is performed on mixed-valent NiIIFeI cores instead of the present NiII(μ-H)FeII systems. The remainder of this report details this chemistry.
Trimetallic Mixed-Valent Models for Ni-L
In parallel with work on ferrocene-containing triphosphine hydrides, syntheses of analogous mixed-valent triphosphines were also performed. While the CO ligands in [1] are not readily substituted, the NiIIFeI cation [1]+ is a strong electrophile that reacts instantly with most tertiary phosphines in CH2Cl2 to liberate CO and give substituted complexes such as [(dppe)Ni (pdt)Fe(CO)2(PPh3)]+ ([a]+).[25] The reaction scope is shown here to also include ferrocenylphosphines, with the crystalline salts [(dppe)Ni(pdt)Fe(CO)2(mppf)]BF4 ([1a]BF4), [(dppe) Ni(pdt)Fe(CO)2(mppf*)]BF4 ([1b]BF4), and [(dppe)Ni(pdt) Fe(CO)2(κ1-dppf)]BF4 ([1c]BF4) each being isolable in good yield. Employing the electron-rich dcpe (1,2-bis(dicyclohexylphosphino)ethane) derivative [(dcpe)Ni(pdt)Fe(CO)3]BF4 ([2] BF4) afforded the analogues [(dcpe)Ni(pdt)Fe(CO)2(mppf)]BF4 ([2a]BF4), [(dcpe)Ni(pdt)Fe(CO)2(mppf*)]BF4 ([2b]BF4), and [(dcpe)Ni(pdt)Fe(CO)2(κ1-dppf)]BF4 ([2c]BF4) in a similar fashion. The compounds were isolated as yellow-brown solids that are highly sensitive to H2O and O2, particularly when in solution.
When subjected to ESI-MS, the compounds each ionize to give intact dicarbonyl cations [(dppe)Ni(pdt)Fe(CO)2L]+ as well as some decarbonylated species, with the observed isotopic distributions matching those predicted. The IR spectra of the mixed-valent salts [1a–c]BF4 and [2a–c]BF4 are similar to those of the prototypical [(dppe)Ni(pdt)Fe(CO)2(PPh3)]BF4 and [(dcpe)Ni(pdt)Fe(CO)2(PPh3)]BF4, respectively. The νCO energies among individual members of the dppe- and dcpe-containing series are similar, although a trend in the donicities of the monophosphines could be identified (mppf* > mppf ≈ dppf > PPh3), with the alkyldiarylphosphine mppf* being the strongest σ-donor (Table 1). While band energies report on the electron density at the Fe centre, the relative band intensities are related to the C–Fe–C angle.[39] In what is a subtle effect, it is noted that the symmetric/anti-symmetric stretching intensity ratio follows the above trend, being greatest for the mppf* complexes. Thus, strong donors in such systems destabilize square-pyramidal geometries such that a distorted stereochemistry results. In the limit, the use of the highly basic ligand PCy3 gives complexes whose ‘rotated’ structures feature a semi-bridging CO, with Fe adopting square-pyramidal/trigonalbipyramidal coordination.[40]
Table 1. IR data for salts of type [(dxpe)Ni(pdt)Fe(CO)2L]BF4 in CH2Cl2 solution.
| Species | Formula | νCO [cm−1] | E1/2NiII/I [V]C |
|---|---|---|---|
| [1a]+ | [(dppe)Ni(pdt)Fe(CO)2(mppf)]+ | 1984, 1925 | −0.84 |
| [1b]+ | [(dppe)Ni(pdt)Fe(CO)2(mppf*)]+ | 1980, 1920 | −0.82 |
| [1c]+ | [(dppe)Ni(pdt)Fe(CO)2(κ1-dppf)]+ | 1984, 1925 | −0.67 |
| [a]+ | [(dppe)Ni(pdt)Fe(CO)2(PPh3)]+ | 1988, 1929A | −0.80 |
| [2a]+ | [(dcpe)Ni(pdt)Fe(CO)2(mppf)]+ | 1980, 1919 | −1.18 |
| [2b]+ | [(dcpe)Ni(pdt)Fe(CO)2(mppf*)]+ | 1976, 1914 | −1.14 |
| [2c]+ | [(dcpe)Ni(pdt)Fe(CO)2(κ1-dppf)]+ | 1980, 1919 | −1.31 |
| [(dcpe)Ni(pdt)Fe(CO)2(PPh3)]+ | 1984, 1925B | −1.11 |
Selected mixed-valent derivatives were characterized by X-band EPR spectroscopy; representative data for [1a]BF4, collected at 110 K and room temperature, are presented in Fig. 4. The spectrum of [1a]BF4 in frozen CH2Cl2/PhMe comprises two similar overlapping signals, the rhombicity of these indicating orbital asymmetry owing to distortion of [1a]+ away from an idealized axial (Cs-symmetric) structure. Hyperfine interaction of the singly occupied molecular orbital (SOMO) with a single 31P nucleus is evident for both frozen and liquid solutions, with g values in both cases being in the vicinity of ge 2.0023, the value for the free electron. Collectively, these data confirm the presence of an FeI centre in a complex described as NiIIFeIFeII, with the low g-values suggesting minimal spin-orbital coupling, consistent with a d(z[2])-like SOMO at the low-spin 3d[7] site. The observation of two almost identical signals stems from the presence of two [1a]+ conformations differing in the location of the central CH2 group in the pdt2− ligand. This ring-flipping occurs in each of the complexes studied as well as the archetypal [(dppe)Ni(pdt)Fe (CO)2(PPh3)]+,[25] whose spectra are similar to those reported here (see Supplementary Material for more examples).
Fig. 4.
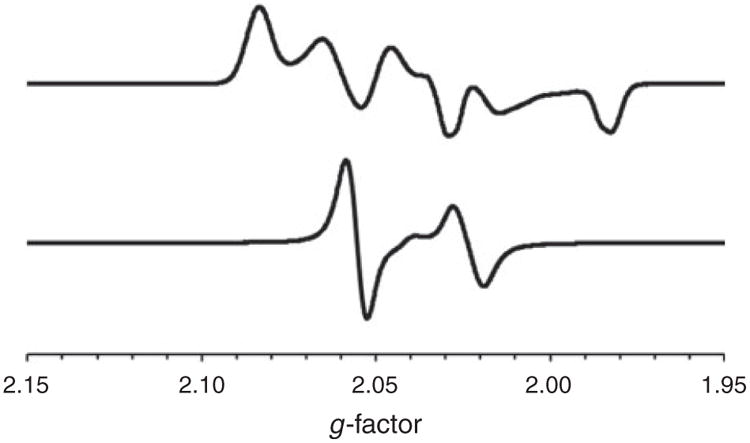
X-Band EPR spectra of [1a]BF4 in CH2Cl2/PhMe recorded at 110 K (top) and room temperature (bottom).
The X-ray structure of mixed-valent salt [2b]BF4 features a Ni–Fe distance (2.787 Å, Fig. 5) comparable to that in the tricarbonyl precursor [2]BF4 (2.818 Å), but significantly shorter than that in the PCy3 analogue [(dcpe)Ni(pdt)Fe(CO)2(PCy3)] BF4 (2.990 Å). The Ni centre in [2b]BF4 exists in a distorted square planar coordination environment, the twist angle between the NiP2 and NiS2 planes being 6.88. The metal ions are bridged by the pdt2− ligand, whose S atoms, along with the C atoms of the carbonyls, define the basal plane of the distorted square-pyramidal Fe site (Addison τ parameter = 0.28).[41] The apical position is occupied by the mppf* metalloligand, the decamethylferrocenyl fragment of which adopts a staggered conformation, with the mean Fe2-centroid distance being 1.648 Å. Overall, the Fe1 coordination environment is very similar to that for the FeI centre in [2]BF4, with slight contractions (∼0.04 Å) in the Fe1–C30 and Fe1–C31 distances due to increased π-backbonding in [2b]+. The X-ray structure of [2b]+ contrasts that of [(dcpe)Ni(pdt)Fe(CO)2(PCy3)]+, which features the strongly σ-donating PCy3 ligand.[40] While the latter is also a FeI complex, its Fe centre is ‘rotated’ such that PCy3 moves away from the apical position (see above). Significantly, [2b]+ is the first ‘unrotated’ [(dxpe)Ni(pdt)Fe(CO)2L]+ complex to be crytallographically characterized, giving credence to the apical nature of the monophosphines in previous complexes, which was instead inferred from EPR data (including isotopic labelling) and DFT calculations.
Fig. 5.
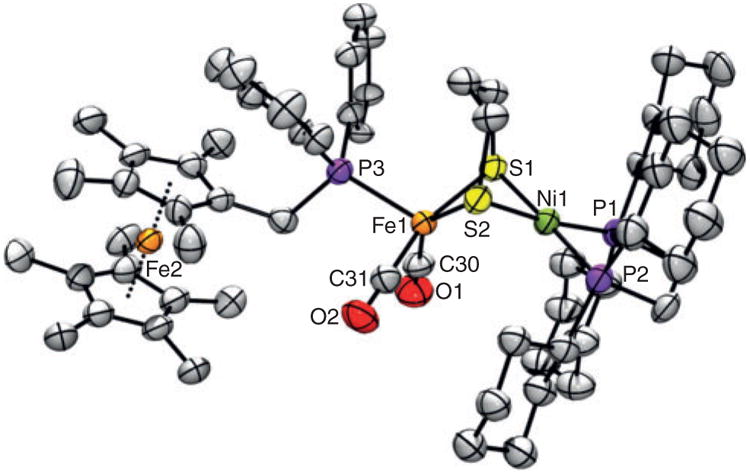
ORTEP of [2b]BF4 3CH2Cl2 with ellipsoids drawn at the 50 % probability level. The H atoms, disordered CH2Cl2 solvate molecules, and BF4− anion are omitted for clarity. Selected distances (Å): Ni1–Fe1 2.787, Ni1–P1 2.188, Ni1–P2 2.200, Ni1–S1 2.239, Ni1–S2 2.233, Fe1–S1 2.286, Fe1–S2 2.307, Fe1–C30 1.758, Fe1–C31 1.761, Fe1–P3 2.292.
The present NiIIFeIFeII complexes have rich electrochemistry, and in principle four redox states are accessible: NiIIFeIIFeIII ⇌ NiIIFeIFeIII ⇌ NiIIFeIFeII ⇌ NiIFeIFeII. Accordingly, cyclic voltammograms of [1a]+,[1b]+,[2a]+, and [2b]+ feature three prominent waves, and in each case the most positive of these is of limited reversibility. For example, the electron-rich species [(dcpe)Ni(pdt)Fe(CO)2(mppf*)]+ ([2b]+) exhibits redox peaks at +0.03 ([2b]3+/2+, NiIIFeII/IFeIII), 0.41 ([2b]2+/+, NiIIFeIFeIII/II), and −1.14 V versus Fc+/0([2b]+/0, NiII/IFeIFeII, Fig. 6, solid trace). The assignments are made by analogy with [(dcpe)Ni(pdt)Fe(CO)2(PPh3)]+, a ferrocene-free triphosphine complex whose oxidation at −0.01 (NiIIFeII/I) and reduction at −1.11 V (NiII/IFeI) are well studied. The comparable potentials and reversibilities of the more cathodic waves suggest that a NiII/IFeIFeII assignment is appropriate for [2b]+/0, whose slightly more donating ligand (mppf* versus PPh3) may account for the 30 mV cathodic shift (Table 1). Taking the similarities of the anodic waves into account, it appears that NiIIFeII/IFeIII redox is at play in [2b]3+/2+, with the 40 mV anodic shift probably arising from the greater charges in [2b]3+/2+ compared with [(dcpe)Ni(pdt)Fe(CO)2(PPh3)]2+/+ (i.e. oxidizing a dication is more difficult than a monocation). Lastly, the intermediate wave at −0.41 V for the trimetallic must correspond to the metalloligand couple NiIIFeIFeIII/II. This feature lies more positive than that for the free ligand couple [mppf*]+/0 (−0.50 V), consistent with coordination of the diphenylphosphino group inducing only subtle changes in the 3d orbital energies of the sandwiched Fe centre.
Fig. 6.
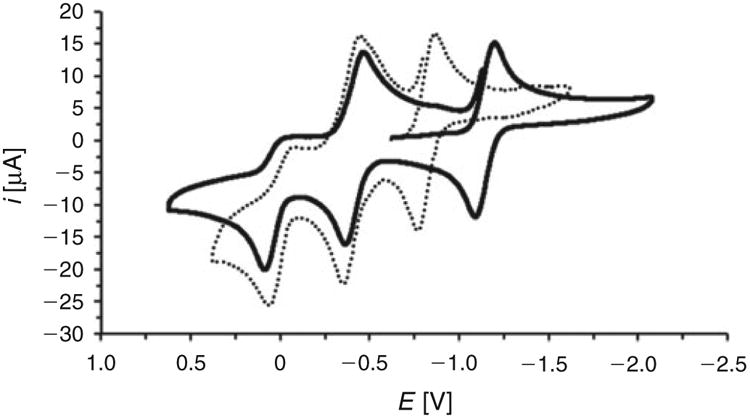
Cyclic voltammogram of [1b]BF4 (dotted trace) and [2b]BF4 (solid trace). The potentials (shown here relative to Fc+/0) were swept at 100 mV s−1.
The analysis of [1b]+ (Fig. 6, dotted trace), a complex identical to [2b]+ but with the less basic diphosphine dppe in place of dcpe, is in line with the assignments above. The potentials of the two waves assigned to redox at the Fe centres in [1b]+ (Epa = 0.06 V, E1/2 = 0.40 V) are virtually identical to those for [2b]+, but the cathodic wave for [1b]+ occurs at a less negative potential (E1/2 −0.82 V), consistent with the Ni centre in [1b]+ bearing a weaker donor. The electrochemistry is more complicated for the mppf complexes [1a]+ and [2a]+, for which even the NiIIFeIFeIII/II couple, which involves redox at the ligated mppf ligand, is not fully reversible. Indeed, it appears that the lack of an ‘insulating’ methylene separating the diphenylphosphino and ferrocene groups results in significant communication between the metal centres. Regardless of the ligand, the NiIIFeII/IFeIII oxidation is irreversible in each case, in agreement with studies indicating that pentacoordinate, 16e− FeII centres in NiIIFeII dithiolates can be highly reactive.
It is conceivable that a NiIIFeIFeIII species could split H2 to afford NiII(μ-H)FeIIFeII and H+. However, [2b]2+ (generated from [2b]+ and Fc+ or acetylferrocenium) was inert towards H2 even in the presence of 2,6-di(tert-butyl)pyridine as a proton sink. Similarly, the voltammograms of [2b]+ are unperturbed when the proton sink and H2 are present. These results indicate at least one of the following: (i) the NiIIFeI core is insufficiently electrophilic to bind H2, and/or (ii) a pendant base is necessary to deprotonate the transient NiII(Z[2]-H2)FeI complex. With respect to electrophilicity, the problem with present systems is that NiIIFeIFeIII oxidizes only irreversibly to the ostensibly more electrophilic NiIIFeIIFeIII form. This latter species represents a model for the H2-activating Ni-SIa enzyme state (NiIIFeII), although the instability of the synthetic species precluded further H2 activation experiments.
The reluctance of [2b]2+ to bind H2 prompted the use of better 2e− donor ligands to bind the electrophilic FeI centre. Consider that the FeIFeIIFeIII complex in Fig. 2 binds exogenous CO to form the adduct (OC)FeIIFeIIFeII in a process involving electron transfer to the decamethylferrocenium group. Cationic NiFe triphosphines do undergo reaction with CO (1 atm), although this results in displacement of the monosphosphine to give the corresponding diphosphine tricarbonyl. When PMe2Ph is used instead of CO, ligand displacement also occurs, with [2b]2+ losing mppf* to give [(dcpe)Ni(pdt)Fe (CO)2(PMe2Ph)2]2+, a complex that forms even when strictly one equivalent of PMe2Ph is used. While [(dcpe)Ni(pdt)Fe (CO)2(PMe2Ph)2]2+ is indeed a NiIIFeII species, this synthetic 34e− complex in which Fe is octahedrally coordinated represents a model for the CO-inhibited state Ni-SCO (the CO adduct of Ni-SIa)[1] rather than the unsaturated 32e− state Ni-SIa.
Dppf-Containing Models for Ni-L and Ni-SCO
Of the mixed-valent complexes [1a–c]+ and [2a–c]+, undoubtedly the most curious are [1c]+ and [2c]+, which each feature a dppf ligand bound through a single phosphine group with the second phosphine unbound. Complexes of κ1-dppf are relatively rare,[42] and [1c]+ and [2c]+ are to date arguably the most advanced structural models for [NiFe]-H2ase. They bear not only a redox-active ligand (modelling the proximal Fe4S4 cluster in the enzyme) but also a free phosphine, a weak Brønsted base that, at least in principle, could serve as a potential proton relay (a role played in the enzyme by terminal Cys residues or proximal Arg residues). When dppe is used in place of dppf one cannot observe the analogue [(dppe)Ni(pdt)Fe (CO)2(κ1-dppe)]+.[25] This species forms as a putative intermediate upon treatment of dppe with the NiIIFeI tricarbonyl [1]+, which, upon ‘dispropotionation’, gives 0.5 equivalents each of the NiIFeI species 1 and NiIIFeII dication [(dppe)Ni(pdt) Fe(CO)(κ2-dppe)]2+ (even in the presence of excess dppe).[25] This can be rationalized by considering the reactions:
| (1) |
| (2) |
The oxidation coupled with chelation of dppe (Eqn 1) is instantaneous, perhaps due to the stability of the five-membered Fe(κ2-dppe) ring formed. In contrast, the Fe(κ1-dppf) species depicted in Eqn 2 is not readily oxidized by [1]+, perhaps on account of the entropic penalty on forming a large chelate ring from a flexible ligand (see below). Presented below is the diverse chemistry associated with Fe(κ1-dppf) derivatives, in particular [1c]+ (Scheme 3).
Scheme 3.
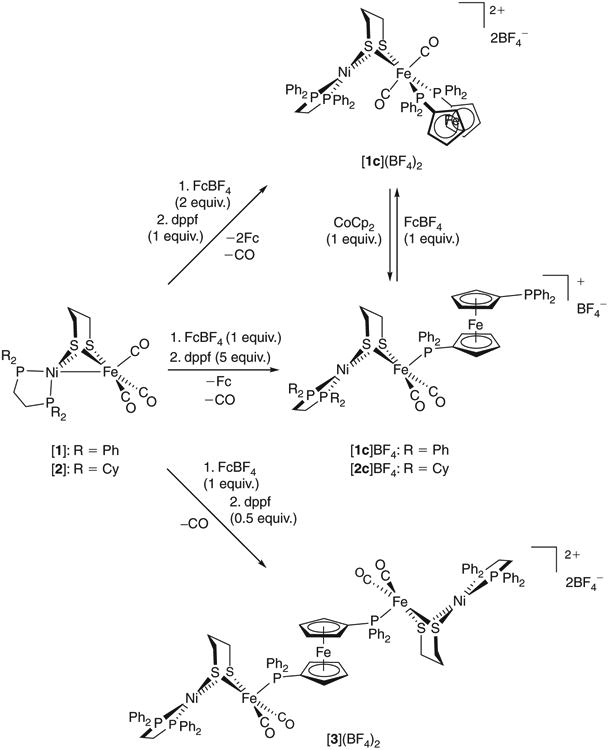
Reactions affording multimetallic complexes with dppf bound in κ1, κ2 and μ2 modes.
As with the NiIIFeIFeII triphosphines discussed thus far, [1c]+ is prepared by addition of in situ generated [1]+ to an excess of dppf. The use of excess dppf is necessary as it curbs a side reaction in which the free phosphine in [1c]+ bridges to unreacted [1]+ such that a pentanuclear complex formulated as {[(dppe)Ni(pdt)Fe(CO)2]2(μ-dppf)}2+ ([3]2+) is observed. This dication is best prepared by slow addition of [1]+ to dppf (0.5 equiv.), a process that first affords [1c]+ en route to [3]2+. The νCO bands of [3]2+ are similar in shape and energy (1984, 1925 cm−1) to those of trimetallic mixed-valent species such as [1c]BF4, suggesting that the donicity of the free phosphine in [1c]+ is comparable to that of free dppf. Further evidence supporting the formula of [3]2+ came from ESI-MS, which allowed for the detection of intact [3]2+ (m/z 952.5). Even under low cone voltage conditions, the decarbonylated ions [3 – CO]2+ and [3 – 2CO]2+ (m/z 938.5 and 924.6, respectively) were also observed. In addition to Fe–C bond scission, it appears that Fe–P scission also occurs, such that [1c]+ and [1c – CO]+ are also detected. Given the lability of mixed-valent triphosphine complexes under these conditions,[25,40] the fragility of [3]2+ is unsurprising given its large and dicationic nature. Overall, the similarity of the IR data for [3]2+ to NiIIFeIFeII triphosphines indicates a NiIIFeIFeIIFeINiII description for the former complex. Its two FeI centres give rise to an EPR spectrum similar to those of NiIIFeIFeII triphosphines, although with some broadening consistent with weak communication between the FeI radicals and/or the presence of several conformers. In terms of structure, [3]2+ bears some resemblance to the pentairon species [(OC)3Fe(pdt)Fe(CO)2]2(μ-dppf),[34] although the latter species is diamagnetic (FeIFeIFeIIFeIFeI) and more robust.
Like the NiIIFeIFeII complexes discussed so far, [1c]+ undergoes reversible 1e− reduction (E1/2− 0.67 V) to the charge-neutral NiIFeIFeII species. The 1e− oxidation of [1c]+ is more complicated: an irreversible wave (Epa 0.07 V) involves generation of a NiIFeIIFeII species whose five-coordinate FeII centre readily binds the free phosphine to complete its coordination sphere. In principle, the oxidation could initially occur at the ferrocenyl FeII, but then one might expect a similar potential for this wave in the electron-rich dcpe analogue [2c]+. Yet, the latter oxidizes at −0.11 V, consistent with the nature of the diphosphine having a small, albeit noticeable influence on redox. The dppf metalloligand certainly has diverse chemistry.[42]
Having considered the κ1 and μ[2] binding of dppf, we now turn to the more typical κ2 chelating mode in NiIIFeIIFeII complex [1c]2+, whose diamagnetic 34e− NiIIFeII core resembles that in the CO-inhibited Ni-SCO enzyme state. Addition of [1]+ to excess dppf affords [(dppe)Ni(pdt)Fe(CO)2(κ1-dppf)] ([1c]+), which does not spontaneously convert into the chelate [1c]2+. Indeed, while [1]+ is too weak an oxidant to induce ring closure, Fc+ does allow for the clean generation of [(dppe) Ni(pdt)Fe(CO)2(κ2-dppf)]2+ ([1c]2+). Similar chemistry has been observed for the isolable species [(dxpe)Ni(pdt)Fe (CO)2(κ1-(2-pyridyl)PPh2)]+, whose (2-pyridyl)PPh2P ligand is bound through P in this NiIIFeI state, but is thought to chelate through P and N atoms when in the NiIIFeII complex.[40]
Accordingly, a cleaner and more rapid (one-pot) synthesis of [1c](BF4)2 involves partial dissolution of 1 and FcBF4 (2 equiv.) in CH2Cl2, followed by treatment with dppf (1 equiv.) in CH2Cl2. This method is analogous to the preparation of [(dppe)Ni(pdt)Fe(CO)2(κ2-dppe)](BF4)2,[25] with the solid product being robust and air-stable. When in solution, [1c] (BF4)2 gives rise to a strong νCO band, whose energy (1975 cm −1) is consistent with a trans-FeII(CO)2 moiety.[43] Merged with this band, and also at 2040 cm−1, are two much weaker bands that are assigned to a cis isomer. 31P NMR resonances for this minor isomer could not be resolved, with the only two signals observed being those for the Cs-symmetric trans species. Although broad, four 1H signals are resolved for the C5H4 groups, indicating that, on the NMR timescale, the dppf ligand does not rapidly rotate 180° along the Fe2-Fe1 vector. Thus, the atoms P3 and P4 will not exchange places rapidly. Attenuated total reflectance IR data point to solid [1c] (BF4)2 existing as a single trans isomer, whose structure was also confirmed by X-ray diffraction (Fig. 7).
Fig. 7.
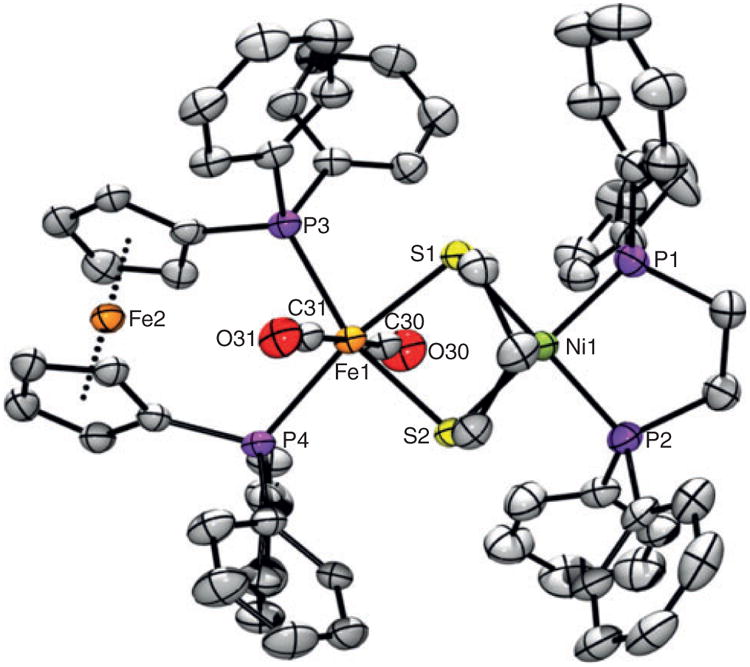
ORTEP of [1c](BF4)2·6CH2Cl2 with the H atoms, BF4− anions, and CH2Cl2 solvate molecules omitted for clarity. Selected distances (Å): Ni1– Fe1 3.205, Ni1–P1 2.178, Ni1–P2 2.183, Ni1–S1 2.219, Ni–S2 2.214, Fe1–S1 2.342, Fe1–S2 2.322, Fe1–C30 1.810, Fe1–C31 1.811, Fe1–P3 2.303, Fe1–P4 2.351.
As was found spectroscopically, the structure of [1c]2+ indeed features trans CO ligands at an octahedral Fe site, which is far enough from Ni (3.205 Å) such that no bonding between the metal ions is possible. The Fe–C distances (1.811, 1.810 Å) are almost identical, as are the two Ni–P (2.183, 2.178 Å), Ni–S (2.214, 2.219 Å), and Fe–S bond lengths (2.322, 2.342 Å). Very slight asymmetry arises from the Fe–P distances (2.303, 2.351 Å) being somewhat different and the cyclopentadienyl rings adopting a staggered conformation. These metrics are similar to those reported for [(dppe)Ni(pdt)Fe(CO)2(κ2−dppe)]2+,[25] although a notable difference is the P–Fe–P bite angle, which is much larger in the dppf complex [1c]2+ (106.6°) than in its dppe congener (87.9°). Overall, the observation of a κ1 complex for dppf but not for dppe may be rationalized in terms of this disparity: the dppf complex is finely poised, whereas chelate ring closure in the dppe case is overwhelmingly favourable.
With the oxidation of [1c]+ triggering chelation of dppf in the dication [1c]2+, it is of interest to consider what might occur upon reduction of [1c]2+. One motivation for this is that addition of 1e− to an 18e− FeII centre will likely induce cleavage of one Fe–ligand bond, given the propensity for FeI to adopt pentacoordinate structures. Liberation of a CO ligand would afford [(dppe)Ni(pdt)Fe(CO)(κ2-dppf)]+, a complex which, given the Fe(CO)(CN)2 core present in the enzyme, represents a good structural model for the NiIFeII Ni-L state (taking κ2-dppf as a surrogate for the two CN− ligands). Indeed, the related complex [(dppe)Ni(pdt)Fe(CO)(κ2-dppe)]+ is thought to exist as a mixtures of two conformers, one described as NiIFeII, the other as NiIIFeI.[44] However, treatment of the NiIIFeIIFeII species [1c]2+ with cobaltocene (1 equiv.) does not induce decarbonylation but rather leads to cleavage of a Fe–P bond to regenerate, according to FT-IR analysis, the NiIIFeIFeII species [1c]+.
It is clear that NiIIFeI triphosphines, including NiIIFeIFeII species discussed here, feature the same spin (S = 1/2) and 33e− count as the Ni-L form of [NiFe]-H2ase. Yet, aside from the latter's description as NiIFeII, it also differs from the present complexes in that it binds CO to give a 35e− adduct Ni-CO, an enzyme state in which CO is thought to bind NiI.[45] This reactivity is not reproduced in models, as reflected in the failure of the metal centres in [1c]+ to bind the other arm of pendant κ1-dppf.
Lastly, given that [1c]+ features both a proton relay (the free phosphine) and an electron relay (the ferrocene), it appears to be an advanced structural model of Ni-L. Yet, its susceptibility to ring-closure precludes H2 oxidation catalysis, as removal of 1e− from [1c]+ results in the unsaturated Fe site readily binding the free arm of κ1-dppf in preference to H2. Furthermore, [1c]+ was found not to be an electrocatalyst for H2 evolution from the strong acid [H(OEt2)2][B(3,5-(CF3)2C6H3)4], perhaps due to the acid inducing outer-sphere oxidation to [1c]2+, in which FeII is coordinatively saturated. In these systems, we note in general that FeI shows a preference for five donors, and is insufficiently electrophilic to bind further ligands, as required for H2 heterolysis. Among such compounds, no system yet exists that presents an unsaturated FeII centre for H2 binding in tandem with a proximal proton acceptor.
Conclusion
The remarkable activity of the [NiFe]-H2ases owes a lot to the exquisitely designed machinery by which protons and electrons are relayed to their active sites. We have paid particular attention to electron transport here, and a series of trimetallic complexes, each featuring a topical NiFe dithiolate core and a redox-active ferrocenyl group, has been prepared. The trimetallic [1a(μ-H)]+ evolves H2 at a higher rate than does the related triphosphine [(dppe)Ni(pdt)(μ-H)Fe(CO)2(PPh3)]+, the reason for this being unclear although likely related to redox at the ferrocene in [1a(μ-H)]+. Aside from this hydride, redox-active ligands were also incorporated into the new families of mixed-valent complexes [1a–c]+ and [2a–c]+. When a ferrocene-containing monophosphine is employed, the resulting trimetallics exhibit rich electrochemistry such that up to four redox states can be observed. With a ferrocene-based diphosphine, one can build advanced models with putative proton and electron relays. The chemistry of such systems is complicated by the free phosphine also being able to coordinate Fe such that the diamagnetic trimetallic [1c]2+ and the pentanuclear species [3]2+ can form. Overall, our investigations into NiFe complexes featuring redox-active ligands break new ground towards preparing high-fidelity models better able to mimic both the structure and function of natural systems to play a role in a renewable future.
Experimental
Unless otherwise stated, all chemicals were purchased from commercial sources and used as received. CD2Cl2 was distilled from CaH2 under N2. The complexes 1,[28] 2,[29] and mppf*[35] were prepared according to the literature methods. All reactions were conducted in an MBraun glovebox equipped with a solvent purification system; the concentrations of O2 and H2O in the N2 atmosphere were less than 1 ppm. The mixed-valent salts were stored at −28°C. IR spectra of complexes (in CH2Cl2) were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer. EPR spectra of complexes (∼1 mM in 1: 1 CH2Cl2/PhMe) were recorded on a Varian E-line 12″ Century Series X-band CW spectrometer. A Waters Micromass Quattro II spectrometer was used to acquire ESI-MS data for analytes in dilute CH2Cl2 solution. Analytical data were acquired using an Exeter Analytical CE-440 elemental analyzer. Unless otherwise stated, NMR data were acquired at room temperature, with samples under an atmosphere of N2. 1H and 31P{1H} NMR spectra were recorded on a Varian Unity 500 spectrometer at 500 and 202 MHz, respectively. Chemical shifts (δ/ppm) are referenced to CHDCl2 (5.32 ppm for 1H) and external 85 % H3PO4 (0 ppm for 31P). Cyclic voltammetry was carried out in a single compartment glass cell using a CH Instruments CHI600D electrochemical analyzer. The working, counter, and pseudo-reference electrodes were glassy C, Pt, and Ag, respectively. The analyte (1 mM) and NBu4PF6 (100 mM) were dissolved in CH2Cl2, and potentials (reported here relative to internal Fc/Fc+) were typically swept at 0.1 V s−1. Crystallographic data for [1a(μ-H)] BF4 and [2b]BF4 were collected using a Bruker X8 (λ(Cu Kα) 1.54178 Å) diffractometer, while data for [3](BF4)2 were collected on a Siemens SMART (δ(Mo Kα) 0.71073 Å). Each instrument was equipped with an Apex II detector.
[(dppe)Ni(pdt)(μ-H)Fe(CO)2(mppf)]BF4 ([1a(μ-H)]BF4)
[1H]BF4 (118.6 mg, 150 μmol) and mppf (55.5 mg, 150 μmol) were stirred in THF (5 mL) at 40°C for 2 h, after which the brown solution was evaporated to dryness. The residue was taken up in CH2Cl2 (5 mL), and pentane (50 mL) was added to induce precipitation of a solid, which was isolated by filtration, washed with pentane (2 × 2 mL), and dried to afford the product as a brown powder (131.4 mg, 116 μmol, 77 %).
νCO/cm−1 2014, 1962. δH (CD2Cl2, 500 MHz) 8.00–6.75 (m, 30H, Ph), 4.52 (m, 1H, PC5H4), 4.48 (m, 1H, PC5H4), 4.25 (m, 1H, PC5H4), 4.14 (m, 1H, PC5H4), 3.7 (s, 5H, C5H5), 2.63 (m, 4H, PCH2CH2P), 2.50–1.34 (m, 6H, SCH2CH2CH2S), −3.11 (dt, [2]JHP 36.0, 4.9). dP (CD2Cl2, 202 MHz, −20°C) 67.3 (dppe), 64.8 (dppe), 63.6 (mppf). m/z (ESI) 1045.1 [M – BF4−]+. Anal. Calc. for C53H50BF4Fe2NiO2P3S2: C 54.66, H 4.37, N 0.00. Found: C 54.69, H 4.34, N 0.00 %.
Brown single crystals of [1a(μ-H)]BF4 CH2Cl2 formed upon slow diffusion of pentane vapour into a concentrated CH2Cl2 solution of the title compound. Results of the crystallographic analysis are presented in the Supplementary Material.
[1a-c]BF4 and [2a-c]BF4
[(dppe)Ni(pdt)Fe(CO)3] or [(dcpe)Ni(pdt)Fe(CO)3] (20 μmol) and FcBF4 (5.5 mg, 20 μmol) were dissolved in CH2Cl2 (1 mL) with rapid stirring. After 1 min the solution was added dropwise to a stirred solution of mppf, mppf*, or dppf (100 μmol) in CH2Cl2 (0.5 mL). The solution was stirred for a further 0.5 min and pentane (−28°C, 15 mL) was added and the mixture allowed to stand at −28°C for 10 min. The solids were isolated by filtration, washed with pentane (−28°C, 2 × 2 mL), and dried briefly to afford the phosphine-substituted derivatives as brown or yellow-brown solids.
[(dppe)Ni(pdt)Fe(CO)2(mppf)]BF4 ([1a]BF4)
Yield: 90 %, yellow-brown powder. νCO/cm−1 1984, 1925. m/z (ESI) 1044.2 [M – BF4−]+,1016.3 [M – CO – BF4−]+. Anal. Calc. for C53H49BF4Fe2NiO2P3S2: C 56.22, H 4.36, N 0.00. Found: C 55.81, H 4.27, N 0.00 %.
[(dppe)Ni(pdt)Fe(CO)2(mppf*)]BF4 ([1b]BF4)
Yield: 97 %, yellow-brown powder. νCO/cm−1 1980, 1920. m/z (ESI) 1184.5 [M – BF4−]+. Anal. Calc. for C63H69BF4 Fe2NiO2P3S2·0.5CH2Cl2: C 58.00, H 5.37, N 0.00. Found: C 58.10, H 5.28, N 0.02 %.
[(dppe)Ni(pdt)Fe(CO)2(κ1-dppf)]BF4 ([1c]BF4)
Yield: 78 %, yellow-brown powder. νCO/cm−1 1984, 1925. m/z (ESI) 1229.3 [M – BF4−]+, 1201.3 [M – CO – BF4−]+, 953.5 {[(dppe)Ni(pdt)Fe(CO)2]2(dppf)}2+, 938.9 {[(dppe) Ni(pdt)Fe(CO)2]2(dppf) – CO}2+, 924.8 {[(dppe)Ni(pdt) Fe(CO)]2(dppf) – 2CO}2+. Anal. Calc. for C65H58BF4Fe2-NiO2P4S2·0.33CH2Cl2: C 58.37, H 4.40, N 0.00. Found: C 58.17, H 4.27, N 0.00 %.
[(dcpe)Ni(pdt)Fe(CO)2(mppf)]BF4 ([2a]BF4)
Yield: 80 %, yellow powder. νCO/cm−1 1980, 1919. m/z (ESI) 1068.4 [M – BF4−]+, 1040.4 [M – CO – BF4−]+. Anal. Calc. for C53H73BF4Fe2NiO2P3S2·2CH2Cl2: C 49.81, H 5.85, N 0.00. Found: C 49.52, H 5.81, N 0.02 %.
[(dcpe)Ni(pdt)Fe(CO)2(mppf*)]BF4 ([2b]BF4)
Yield: 70 %, yellow powder. νCO/cm−1 1976, 1914. m/z (ESI) 1209.1 [M – BF4−]+, 1183.0 [M – CO – BF4−]+. Anal. Calc. for C63H93BF4Fe2NiO2P3S2·1.5CH2Cl2: C 54.40, H 6.79, N 0.00. Found: C 54.49, H 6.90, N 0.00 %.
Brown single crystals of [2b]BF4 3CH2Cl2 were grown by layering a concentrated CH2Cl2 solution with pentane and allowing the mixture to stand at −28°C. One plate-like crystal (0.607 × 0.229 × 0.070 mm3) was subjected to X-ray diffraction at 193 K over the range 2.17°≤ θ ≤ 68.15°. Its space group was determined to be monoclinic P21/n (Z 4) with cell parameters: a 12.8326(4), b 40.6746(11), c 14.7590(4) Å, α 90.008, β 105.021(2)8, γ 908. Integration of 4182 reflections and solution by direct methods using SHELXTL V6.12[46,47] afforded a model with goodness-of-fit 1.039, R1 0.0584, and wR2 0.1462.
[(dcpe)Ni(pdt)Fe(CO)2(κ1-dppf)]BF4 ([2c]BF4)
Yield: 86 %, yellow-brown powder. νCO/cm−1 1980, 1919. m/z (ESI) 1268.6 [M + O–BF4−]+, 1252.6 [M – BF4−]+, 1240.6 [M + O – CO – BF4−]+, 1224.6 [M – CO – BF4−]+, 976.5 [{(dppe)Ni(pdt)Fe(CO)2}2dppf]2+, 962.6 [{(dppe)Ni(pdt)Fe (CO)2}2dppf – CO]2+, 726.4 [(dcpe)Ni(pdt)Fe(CO)3]+. Anal. Calc. for C65H82BF4Fe2NiO2P4S2·3.5CH2Cl2: C 50.23, H 5.48, N 0.00. Found: C 50.22, H 5.72, N 0.34 %.
{[(dppe)Ni(pdt)Fe(CO)2]2(μ-dppf)}(BF4)2 ([3](BF4)2)
This compound was prepared analogously to [1c]BF4, instead using 10 μmol of dppf.
Yield: 92 %, yellow-brown powder. νCO/cm−1 1984, 1925. m/z (ESI) 1964.2 [M – CO – BF4−]+, 1229.7 [M – (dppe) Ni(pdt)Fe(CO)2+ – 2BF4−]+, 1200.7 [M – (dppe)Ni(pdt) Fe(CO)2+ – CO – 2BF4−]+, 952.5 [M – 2BF4−]2+, 938.5 [M – CO – 2BF4−]2+, 924.6 [M – 2CO – 2BF4−]2+. Anal. Calc. for C96H88B2F8Fe3Ni2O4P6S4·0.5CH2Cl2: C 54.65, H 4.23, N 0.00. Found: C 54.45, H 4.41, N 0.28 %.
[(dppe)Ni(pdt)Fe(CO)2(κ2-dppf)](BF4)2 ([1c](BF4)2)
[(dppe)Ni(pdt)Fe(CO)3] (14.1 mg, 20 μmol) and FcBF4 (10.9 mg, 40 μmol) were partially dissolved in CH2Cl2 (2 mL) with rapid stirring. After 1 min the solution was treated dropwise with dppf (11.1 mg, 20 μmol) in CH2Cl2 (0.5 mL). The solution was stirred for a further 0.5 min and pentane (−28°C, 15 mL) was added and the mixture allowed to stand at −28°C for 10 min. The solids were isolated by filtration, washed with pentane (−28°C, 2 × 2 mL), and dried briefly to afford the title compound as an orange powder (24.8 mg, 17.6 μmol, 88 %).
νCO/cm−1 1975. δH (CD2Cl2, 500 MHz) 8.00–7.10 (m, 40H, Ph), 4.93 (m, 2H, C5H4), 4.77 (m, 2H, C5H4), 4.60 (m, 2H, C5H4), 4.27 (m, 2H, C5H4), 2.70–2.00 (m, 4H, PCH2CH2P), 1.75–1.37 (m, 6H, SCH2CH2CH2S). δP (CD2Cl2, 202 MHz) 58.6 (dppe), 42.7 (dppf). m/z (ESI) 1315.1 [M – BF4−]+, 614.2 [M – 2BF4−]2+. Anal. Calc. for C65H58B2F8 Fe2NiO2P4S2: C 55.64, H 4.17, N 0.00. Found: C 55.56, H 4.15, N 0.00 %. Orange prismatic single crystals of [1c] (BF4)2·6CH2Cl2 were grown by layering a concentrated CH2Cl2 solution with pentane and allowing the mixture to stand at −28°C. One crystal (0.304 × 0.151 × 0.142 mm3) was subjected to X-ray diffraction at 193 K over the range 1.34° ≤ θ ≤ 26.49°. Its space group was determined to be triclinic P-1 (Z 2) with cell parameters: a 13.8769(4), b 15.8982(4), c 20.2444(7) Å, α 104.920(2)8, β 106.982(2)8, γ 92.545(2)8. Integration of 7861 reflections and solution by direct methods using SHELXTL V6.12[46,47] afforded a model with goodness-of-fit 0.874, R1 0.0447, and wR2 0.1061.
Crystallographic data
Crystallographic data for [2b]BF4·3CH2Cl2 (CCDC 1511540) and [1c](BF4)2 (CCDC 1511513) are available online.
Supplementary Material
Acknowledgments
The authors thank Dr James Camara for the preparation of mppf* and Dr Mark Nilges for assistance with EPR spectroscopy. This work was supported by the National Institutes of Health (GM46441).
Footnotes
This paper is dedicated to Emeritus Professor Len Lindoy on the occasion of his 80th birthday.
Supplementary Material: ESI-MS and electrochemical data, as well as IR and NMR spectra, are available on the Journal's website.
References
- 1.Lubitz W, Ogata H, Rüdiger O, Reijerse E. Chem Rev. 2014;114:4081. doi: 10.1021/CR4005814. [DOI] [PubMed] [Google Scholar]
- 2.Higuchi Y, Yagi T, Yasuoka N. Structure. 1997;5:1671. doi: 10.1016/S0969-2126(97)00313-4. [DOI] [PubMed] [Google Scholar]
- 3.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580. doi: 10.1038/373580A0. [DOI] [PubMed] [Google Scholar]
- 4.Volbeda A, Darnault C, Parkin A, Sargent F, Armstrong FA, Fontecilla-Camps JC. Structure. 2013;21:184. doi: 10.1016/J.STR.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 5.Volbeda A, Martin L, Cavazza C, Matho M, Faber BW, Roseboom W, Albracht SPJ, Garcin E, Rousset M, Fontecilla-Camps JC. J Biol Inorg Chem. 2005;10:239. doi: 10.1007/S00775-005-0632-X. [DOI] [PubMed] [Google Scholar]
- 6.Schilter D. ChemBioChem. 2015;16:1712. doi: 10.1002/CBIC.201500270. [DOI] [PubMed] [Google Scholar]
- 7.Montet Y, Amara P, Volbeda A, Vernede X, Hatchikian EC, Field MJ, Frey M, Fontecilla-Camps JC. Nat Struct Biol. 1997;4:523. doi: 10.1038/NSB0797-523. [DOI] [PubMed] [Google Scholar]
- 8.Ogata H, Nishikawa K, Lubitz W. Nature. 2015;520:571. doi: 10.1038/NATURE14110. [DOI] [PubMed] [Google Scholar]
- 9.Evans RM, Brooke EJ, Wehlin SAM, Nomerotskaia E, Sargent F, Carr SB, Phillips SEV, Armstrong FA. Nat Chem Biol. 2015;12:46. doi: 10.1038/NCHEMBIO.1976. [DOI] [PubMed] [Google Scholar]
- 10.Ogata H, Lübitz W, Higuchi Y. Dalton Trans. 2009:7577. doi: 10.1039/B903840J. [DOI] [PubMed] [Google Scholar]
- 11.Fichtner C, Laurich C, Bothe E, Lubitz W. Biochemistry. 2006;45:9706. doi: 10.1021/BI0602462. [DOI] [PubMed] [Google Scholar]
- 12.Greene BL, Wu CH, Vansuch GE, Adams MWW, Dyer RB. Biochemistry. 2016;55:1813. doi: 10.1021/ACS.BIOCHEM.5B01348. [DOI] [PubMed] [Google Scholar]
- 13.Kampa M, Pandelia ME, Lubitz W, van Gastel M, Neese F. J Am Chem Soc. 2013;135:3915. doi: 10.1021/JA3115899. [DOI] [PubMed] [Google Scholar]
- 14.Hidalgo R, Ash PA, Healy AJ, Vincent KA. Angew Chem Int Ed. 2015;54:7110. doi: 10.1002/ANIE.201502338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy BJ, Hidalgo R, Roessler MM, Evans RM, Ash PA, Myers WK, Vincent KA, Armstrong FA. J Am Chem Soc. 2015;137:8484. doi: 10.1021/JACS.5B03182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai H, Nishikawa K, Inoue S, Higuchi Y, Hirota S. J Phys Chem B. 2015;119:13668. doi: 10.1021/ACS.JPCB.5B03075. [DOI] [PubMed] [Google Scholar]
- 17.Greene BL, Wu CH, McTernan PM, Adams MWW, Dyer RB. J Am Chem Soc. 2015;137:4558. doi: 10.1021/JACS.5B01791. [DOI] [PubMed] [Google Scholar]
- 18.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273. doi: 10.1021/CR050195Z. [DOI] [PubMed] [Google Scholar]
- 19.Jones AK, Sillery E, Albracht SPJ, Armstrong FA. Chem Commun. 2002:866. doi: 10.1039/B201337A. [DOI] [PubMed] [Google Scholar]
- 20.Pershad HR, Duff JLC, Heering HA, Duin EC, Albracht SPJ, Armstrong FA. Biochemistry. 1999;38:8992. doi: 10.1021/BI990108V. [DOI] [PubMed] [Google Scholar]
- 21.Schilter D, Camara JM, Huynh MT, Hammes-Schiffer S, Rauchfuss TB. Chem Rev. 2016;116:8693. doi: 10.1021/acs.chemrev.6b00180. doi:10.1021/ACS.CHEM REV.6B00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu W, Marr AC, Wang Q, Neese F, Spencer DJE, Blake AJ, Cooke PA, Wilson C, Schröder M. Proc Natl Acad Sci USA. 2005;102:18280. doi: 10.1073/PNAS.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chambers GM, Huynh MT, Li Y, Hammes-Schiffer S, Rauchfuss TB, Reijerse E, Lubitz W. Inorg Chem. 2016;55:419. doi: 10.1021/ACS.INORGCHEM.5B01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perotto CU, Marshall G, Jones GJ, Davies ES, Lewis W, McMaster J, Schröder M. Chem Commun. 2015;51:16988. doi: 10.1039/C5CC05881C. [DOI] [PubMed] [Google Scholar]
- 25.Schilter D, Nilges MJ, Chakrabarti M, Lindahl PA, Rauchfuss TB, Stein M. Inorg Chem. 2012;51:2338. doi: 10.1021/IC202329Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogo S, Ichikawa K, Kishima T, Matsumoto T, Nakai H, Kusaka K, Ohhara T. Science. 2013;339:682. doi: 10.1126/SCIENCE.1231345. [DOI] [PubMed] [Google Scholar]
- 27.Manor BC, Rauchfuss TB. J Am Chem Soc. 2013;135:11895. doi: 10.1021/JA404580R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barton BE, Rauchfuss TB. J Am Chem Soc. 2010;132:14877. doi: 10.1021/JA105312P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. Inorg Chem. 2011;50:9554. doi: 10.1021/IC2012759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Si Y, Charreteur K, Capon JF, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J. J Inorg Biochem. 2010;104:1038. doi: 10.1016/J.JINORGBIO.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Brazzolotto D, Gennari M, Queyriaux N, Simmons TR, Pécaut J, Demeshko S, Meyer F, Orio M, Artero V, Duboc C. Nat Chem. 2016;8:1054. doi: 10.1038/NCHEM.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu YC, Lee CH, Lee GH, Chiang MH. Eur J Inorg Chem. 2011:1155. doi: 10.1002/EJIC.201000972. [DOI] [Google Scholar]
- 33.Ghosh S, Hogarth G, Hollingsworth N, Holt KB, Kabir SE, Sanchez BE. Chem Commun. 2014;50:945. doi: 10.1039/C3CC46456C. [DOI] [PubMed] [Google Scholar]
- 34.Song LC, Li QS, Yang ZY, Hua YJ, Bian HZ, Hu QM. Eur J Inorg Chem. 2010:1119. doi: 10.1002/EJIC.200901023. [DOI] [Google Scholar]
- 35.Camara JM, Rauchfuss TB. Nat Chem. 2011;4:26. doi: 10.1038/NCHEM.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lansing JC, Camara JM, Gray DL, Rauchfuss TB. Organometallics. 2014;33:5897. doi: 10.1021/OM5004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton BE, Whaley CM, Rauchfuss TB, Gray DL. J Am Chem Soc. 2009;131:6942. doi: 10.1021/JA902570U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huynh MT, Schilter D, Hammes-Schiffer S, Rauchfuss TB. J Am Chem Soc. 2014;136:12385. doi: 10.1021/JA505783Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braterman PS. Metal Carbonyl Spectra. Academic Press; New York, NY: 1975. [Google Scholar]
- 40.Schilter D, Rauchfuss TB, Stein M. Inorg Chem. 2012;51:8931. doi: 10.1021/IC300910R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349. doi: 10.1039/DT9840001349. [DOI] [Google Scholar]
- 42.Onaka S, Moriya T, Takagi S, Mizuno A, Foruta H. Bull Chem Soc Jpn. 1992;65:1415. doi: 10.1246/BCSJ.65.1415. [DOI] [Google Scholar]
- 43.Carroll ME, Chen J, Gray DE, Lansing JC, Rauchfuss TB, Schilter D, Volkers PI, Wilson SR. Organometallics. 2014;33:858. doi: 10.1021/OM400752A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ulloa OA, Huynh MT, Richers CP, Bertke JA, Nilges MJ, Hammes-Schiffer S, Rauchfuss TB. J Am Chem Soc. 2016;138:9234. doi: 10.1021/JACS.6B04579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandelia ME, Ogata H, Currell JJ, Flores M, Lubitz W. Biochim Biophys Acta, Bioenerg. 2010;1797:304. doi: 10.1016/J.BBABIO.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 46.Sheldrick GM. Acta Crystallogr Sect A: Found Adv. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 47.Bruker. SHELXTL v6. Vol. 12. Bruker AXS, Inc.; Madison, WI: 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.